

Abstract
Diabetes mellitus and the metabolic syndrome are becoming leading causes of death in the world. Identifying the etiology of diabetes is key to prevention. Despite the similarity in their structures, fructose and glucose are metabolized in different ways. Uric acid, a byproduct of uncontrolled fructose metabolism is known risk factor for hypertension. In the liver, fructose bypasses the two highly regulated steps in glycolysis, glucokinase and phosphofructokinase, both of which are inhibited by increasing concentrations of their byproducts. Fructose is metabolized by fructokinase (KHK). KHK has no negative feedback system, and ATP is used for phosphorylation. This results in intracellular phosphate depletion and the rapid generation of uric acid due to activation of AMP deaminase. Uric acid, a byproduct of this reaction, has been linked to endothelial dysfunction, insulin resistance, and hypertension. We present possible mechanisms by which fructose causes insulin resistance and suggest actions based on this association that have therapeutic implications.
1. Background
Type 2 diabetes mellitus is characterized by hyperglycemia, insulin resistance, and an impairment in insulin secretion. In the late nineteenth century, William Osler described diabetes as a rare disorder more likely to develop in obese people and patients with gout. He estimated its prevalence as approximately two to nine cases per 100,000 population in the USA and Europe being more common in the latter [1]. Diabetes, one of the leading causes of death in the United States, affects over 200 million people worldwide. The estimated prevalence of diabetes among adults in the United States ranges from 4.4 to 17.9 percent [2]. The community-based Framingham Heart Study, in a predominantly non-Hispanic white population, found a doubling in the incidence of type 2 diabetes over the last 30 years [3]. Identifying the etiology of type 2 diabetes is a key to its prevention. Obesity and intra-abdominal fat accumulation induce insulin resistance [4]. Studies have documented high rates of type 2 diabetes in the absence of classic obesity [5]. This suggests that other risk factors besides obesity might play a role in the epidemic of type 2 diabetes.
2. Fructose: Sources and Metabolism
Fructose is a simple sugar present in fruits and honey and is responsible for their sweet taste. However, the major source of fructose worldwide is sucrose or table sugar, which is derived from sugar cane and sugar beets. It is man-made, first developed in New Guinea and in the Indian subcontinent and was a rare and expensive commodity that was introduced into Europe via Venice, Italy, and other trading ports during the Middle Ages. Sucrose is a disaccharide that is comprised of fructose and glucose. After ingestion, sucrose is degraded in the gut by sucrase, releasing fructose and glucose that are then absorbed. In addition to sucrose, the other major source of fructose is high fructose corn syrup (HFCS), which was introduced in the early 1970s as an additional sweetener. HFCS consists of fructose and glucose mixed in a variety of concentrations, but most commonly as 55% fructose and 45% glucose. In the United States, HFCS and sucrose are the major sources of fructose in the diet, and HFCS is a major ingredient in soft drinks, pastries, desserts, and various processed foods [6, 7]. Despite the similarity in their chemical structures, fructose and glucose are metabolized in completely different ways and utilize different GLUT transporters [8]. In the liver, fructose bypasses the two highly regulated steps of glycolysis, catalyzed by glucokinase/hexokinase and phosphofructokinase both of which are inhibited by increasing concentrations of their byproducts. Instead, fructose enters the pathway at a level that is not regulated and is metabolized to fructose-1-phosphate primarily by fructokinase or ketohexokinase (KHK) (Figures 1 and 2). Fructose may also be metabolized by hexokinase; however, the Km for fructose is much higher than glucose, and hence minimal amounts of fructose are metabolized via this pathway [6]. Fructokinase has no negative feedback system, and ATP is used for the phosphorylation process. As a result, continued fructose metabolism results in intracellular phosphate depletion, activation of AMP deaminase, and uric acid generation which is harmful at the cellular level [6, 9, 10].
Figure 1.
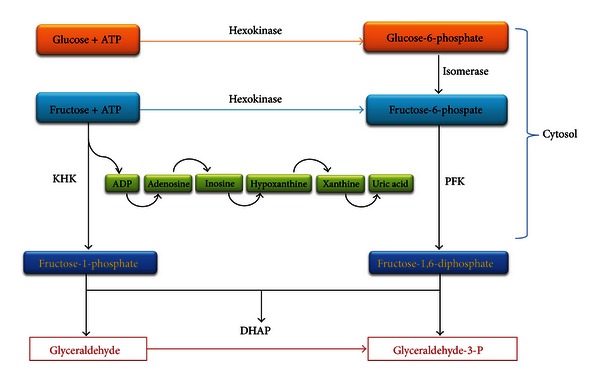
Fructose metabolism. Fructose is primarily metabolized to fructose-1-phosphate by KHK due to its lower Km for fructose compared with hexokinase. Uncontrolled consumption of ATP leads to intracellular phosphate depletion and activation of AMP deaminase leading to the increased production of uric acid. Fructose-1-phosphate is further metabolized by aldolase B and triokinase to glyceraldehyde-3-phosphate.
Figure 2.
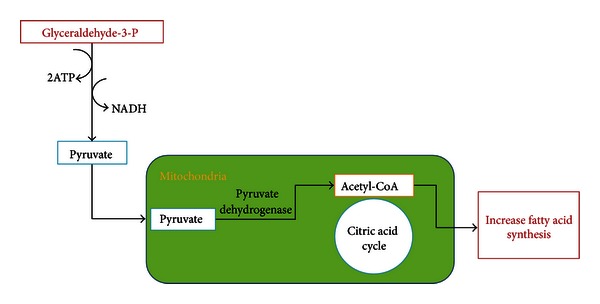
Role of fructose in lipogenesis. Glyceraldehyde-3-P continues downstream in the glycolysis pathway forming pyruvate which enters the mitochondria and is further metabolized to acetyl-CoA by pyruvate dehydrogenase. Acetyl-CoA enters the citric acid cycle by combining with oxaloacetate to form citrate. In the well fed state, citrate can be transported to the cytosol, providing CoA necessary for lipogenesis.
Fructose-1-phosphate is subsequently converted to dihydroxyacetone-phosphate and D-glyceraldehyde by the action of the aldolase B. D-glyceraldehyde is phosphorylated and continued downstream in the glycolysis pathway to form pyruvate. Two of the most energetic reactions of all organophosphates are in the pathway of fructose metabolism, catalyzed by phosphoglycerate and pyruvate kinases. Two ATP molecules as well as free energy, approximately 12 kcal/mole, are released [12]. Fructose controls the activity of glucokinase, the principle enzyme of glucose metabolism in the liver. Fructose is a potent and acute regulator of liver glucose uptake and glycogen synthesis. Inclusion of catalytic quantities of fructose in a carbohydrate meal improves glucose tolerance. This improvement is primarily mediated by the activation of hepatic glucokinase resulting in improved liver glucose uptake [13].
Uric acid, a byproduct of fructose degradation, stimulates KHK expression through the activation of the transcription factor ChREBP, which in turn results in the transcriptional activation of KHK by the binding to a specific sequence within its promoter [14]. Uric acid inhibits endothelial NO both in vivo and in vitro, [15] and directly induces adipocyte dysfunction [16]. Serum uric acid increases rapidly after ingestion of fructose, resulting in increases as high as 2 mg/dL within 1 hour [17–19]. Uncontrolled fructose metabolism leads to postprandial hypertriglyceridemia, which increases visceral adipose deposition. Visceral adiposity contributes to hepatic triglyceride accumulation, protein kinase C activation, and hepatic insulin resistance by increasing the portal delivery of free fatty acids to the liver [20].
A number of other furanose sugars can also act as KHK substrates [21]. KHK is expressed as two isoforms, KHK-C and KHK-A. KHK-C is primarily expressed in the liver, kidney, pancreas, and duodenum, while KHK-A is expressed more widely including adipose tissue, heart, and the adrenal gland [22]. The exact biologic function of KHK-A is unknown. KHK-A has a higher Km for fructose (7 mmol/L) than does KHK-C (0.8 mmol/L) suggesting that it phosphorylates fructose poorly at physiological concentrations [23]. Recently, it was found that adiposity and metabolic syndrome were prevented in mice lacking both KHK isoforms but exacerbated in mice lacking KHK-A [24]. It was also demonstrated that neither KHK isoform is required for normal growth and development in rats [25]. Serum leptin, triglycerides, and fasting blood glucose levels are higher in humans placed on a high fructose diet for four weeks compared with those on a starch-based diet [26].
Deficiency of KHK-C, an autosomal recessive inborn error of metabolism, results in essential fructosuria with an estimated incidence of 1 : 130,000 [27]. This condition was first recognized in 1876 [23]. It is an anomaly rather than a disease, since it does not lead to any outward signs or symptoms. Most cases of fructosuria have been described in Jewish families [28]. It has no metabolic or morbid manifestations other than having transient fructosuria after meals containing either sucrose or fructose. This condition used to be detected during routine medical examination when tests based on reducing properties of glucose like Benedict's solution and Clini test were used to diagnose diabetes. These tests have since been replaced by the more specific glucose oxidase method which does not react with fructose; therefore, patients with essential fructosuria are no longer being identified [28]. Moreover, the lack of treatment consideration and counseling for affected individuals and their family members, the absence of screening recommendations, and the lack of serum KHK assay makes it difficult to identify subjects with this anomaly. This is in contrast to hereditary fructose intolerance, a disease characterized by the deficiency of fructose-1-phosphate aldolase which has significant metabolic and developmental complications that manifest themselves as early as in the neonatal period. In a well-characterized family, in which three of eight siblings have fructosuria, all affected individuals are compound heterozygotes for the mutations Gly40Arg and Ala43Thr [29].
3. Epidemiological Evidence
Since 1970, the total availability of sugars has dramatically increased. Comparison of the 1977–1978 NFCS analysis with the analyses of NHANES for the period 1999–2004 indicated that, over the intervening period, mean individual intake of total fructose increased by ∼32% [30]. HFCS now represents nearly 50% of caloric sweeteners use in the United States [30, 31]. Increased total fructose consumption has been implicated in the development of the obesity epidemic in the United States with the consumption of HFCS increased >1000% between 1970 and 1990, far exceeding the changes in intake of any other food or food group [32].
Several reviews have concluded that intake of both fructose and HFCS by children and adults was associated with an increased risk of obesity and metabolic syndrome [33–37]. However, not all published meta-analyses have reported a statistically significant link [38–41]. Recently, Sievenpiper and colleagues concluded in a meta-analysis of controlled feeding trials that fructose does not cause weight gain when substituted for other carbohydrates in isocaloric trials [42]. This was criticized on the basis of fructose causing weight gain by altering appetite resulting in increased food intake, by inducing leptin resistance and by a direct effect on the brain indicating that isocaloric trials do not show a difference in weight gain between groups. Moreover, the use of weight gain as a marker is subject to debate, since other fructose effects, beyond body weight as central fat accumulation and insulin resistance, can be more important [43].
In children, intake of artificially sweetened beverages was found to be positively associated with adiposity [44]. A prospective cohort analyses conducted from 1991 to 1999 among nondiabetic women in the Nurses' Health Study II concluded that higher consumption of sugar-sweetened beverages was associated with greater magnitude of weight gain and an increased risk for the development of type 2 diabetes [45]. On the other hand, a recent analysis of NHANES 1999–2006 databases comparing data from >25,000 subjects showed no relation between daily fructose intake and the indicators of metabolic syndrome, uric acid, and BMI [46]. This study was based on a snapshot of one- or two-days recalls. Thus, the intake data may not truly represent the long term, consistent consumption of food. Moreover, due to no fructose intake data being available in NHANES databases and a lack of fructose content data for many food items in the USDA National Nutrient Database for Standard Reference, the fructose intake of individuals was indirectly estimated using several databases.
Interpretation of cross-sectional studies examining the relationship of sugar intake to obesity can be misleading due to the fact that subjects who become obese may well reduce their sugar intake, since sugar is widely recognized to cause weight gain. Therefore, examining the relationship of sugar to obesity is best performed with well controlled, long-term longitudinal studies. It is also important to recall that leptin resistance leading to liporegulatory failure, and, subsequently, insulin resistance can be perpetuated once obesity and intracellular lipid accumulation are manifest, especially in sites other than adipose tissue including pancreatic B cells and cardiomyocytes. Thus, reducing fructose intake may not fully reverse insulin resistance and diabetes. This further adds to the complexity and clouds the interpretation of these epidemiological data [6, 47, 48].
The relationship between fructose intake and hypertension was also examined in several clinical studies. In a randomized controlled trial, high dose fructose (200 gm/24 h) increased ambulatory blood pressure and elevated fasting insulin levels. In this study, allopurinol prevented the increase in mean arterial blood pressure [49]. In an analysis of the NHANES 2003–2006 data, fructose intake, in the form of added sugar, was independently associated with higher blood pressure levels [50]. Fructose consumption in the form of sugar-sweetened beverages was associated with hypertension and elevated uric acid level in USA adolescents [51] but not in adults [52]. Uric acid may raise systemic blood pressure by increasing inflammation, activating the renin-angiotensin system, and decreasing nitric oxide production contributing to renal vasoconstriction that results in salt insensitive hypertension [53]. Persistent vasoconstriction may contribute to arteriosclerosis and the subsequent development of salt-sensitive hypertension, even if the hyperuricemia is corrected [54]. This may explain the different results in the preceding two studies that looked at two different age groups. A recent meta-analysis of controlled feeding trials found that isocaloric substitution of fructose with other carbohydrates did not adversely affect blood pressure in humans suggesting that there is a need for long-term and large trials to clarify these findings [40].
Few studies have examined the role of naturally occurring sugars, for example, 100% fruit juice, in the origin of obesity and related end points. It is believed that fructose from natural sources can be less harmful because the presence of additional nutrients and antioxidants. On the other hand, crystalline fructose, as in table sugar, and HFCS are regarded as less safe, since glucose present in these sugars can accelerate fructose absorption. 100% fruit juice consumption among USA adults is associated with lower insulin resistance [55] and lower odds of obesity and metabolic syndrome. Obesity remained an independent factor after adjusting to other lifestyle factors. What is interesting in this study is that 100% juice consumers had significantly higher white milk intake which, on its own and among other dairy products, has been shown to enhance weight loss [43, 56, 57].
There is mounting evidence from studies looking at the association between fructose and obesity and metabolic syndrome that primary fructose malabsorption in children was negatively associated with obesity [58]. In obese African-Americans, high rates of fructose malabsorption were associated with reduced liver fat thought to be protective against fatty liver disease [59].
In conclusion, it is evident that there is a need for clinical trials with variable amounts of fructose intake to determine effects on metabolic outcomes rather than depending on meta-analyses of existing studies of mixed design and duration.
4. Effect of Fructose on Adipocyte Differentiation
Adipocyte development in mice and humans follows a well-defined pathway that begins with a common stem cell mediated adipocyte regeneration and is referred to as adipogenesis [60]. The first step of adipogenesis is the generation and commitment of mesenchymal stem cells (MSCs) to adipocyte lineage. The effect of fructose-mediated renin-Ang II activation is on the stages of cell differentiation (“commitment”) and involves local and systemic effects.
Figure 3 shows the mechanism of Ang II inhibition during adipogenesis. MSCs or preadipocytes differentiate into lipid-laden and insulin-sensitive adipocytes [61].
Figure 3.
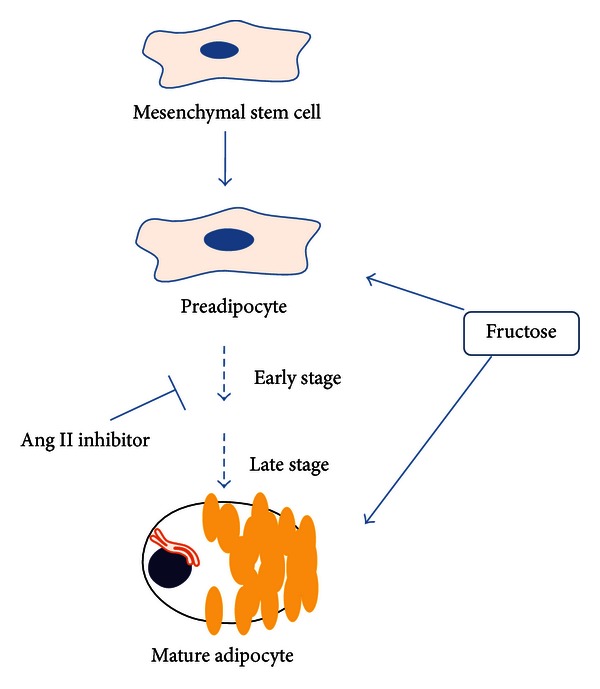
Overview of stages of adipocyte differentiation and the impact of angiotensin II inhibitors and elevated fructose levels.
Briefly, the stages of adipocyte differentiation are affected by increases in the levels of fructose and Ang II which cause MSC-derived adipocyte growth arrest, clonal expansion, and early differentiation. Ang II blockade can prevent terminal differentiation leading to the development of the mature adipocyte phenotype [62, 63].
Normal diets result in the activation of PPARγ followed by adipose expansion through adipocyte hyperplasia, resulting in an increased number of new preadipocytes. The resulting adipocytes are small in size and effectively store lipids, thereby reducing lipotoxicity in the liver and adipose tissue and release adiponectin [64, 65]. Activation of these genes leads to repartitioning of lipids resulting in an increased triglyceride content of adipose tissue, a lowered free fatty acid content in circulation and availability for liver and muscle use, thereby improving insulin sensitivity. Methylisobutylxanthine (MIX), an phosphodiesterases inhibitor, increases intracellular cAMP, activating adipocyte differentiation in a PKA-independent manner [66]. MIX also increased the expression of C/EBP-β, required for the subsequent expression of PPAR-γ [62].
Although, the mechanisms by which fructose controls adipogenesis in vivo are largely unknown, there are a number of candidates that mediate adipocyte differentiation in culture and are thought to control adipocyte accumulation and function in vivo. Two main factors fit this criterion: (1) high fructose diets, (2) increases in ROS. They have been implicated as the link between adipogenesis and metabolic diseases including T2DM.
Recent studies demonstrated that the induction of oxidative stress by high fructose or glucose increased NAD(P)H oxidase and the mitochondrial respiratory chain which is associated with diabetic complications [67]. Therefore, fructose diets may lead to adipocyte differentiation associated with adipocyte dysfunction and formation of adipocytes external to normal adipocyte depots, that is, muscle, liver, and pancreas leading to advanced diabetic complications.
The fructose-mediated increase in ROS via activation of the adipocyte renin-Ang II system may lead to adipocyte dysfunction and insulin resistance. Adipose tissue is a key endocrine organ, the function of which, via interaction with the vascular endothelium system, regulates lipid uptake, storage, synthesis, and secretion of paracrine and autocrine factors that regulate insulin sensitivity. However, fructose-mediated vascular dysfunction may have a negative effect on adipocyte function and the secretion of anti-inflammatory molecules such as adiponectin, IL-1, and IL-10. The glucose or fructose-mediated decrease in vascular function increases adipocyte size resulting in decreased levels of adiponectin, but increased levels of MCP-1, IL-6, and TNF-α that have systemic effects on β cells (Figure 4).
Figure 4.
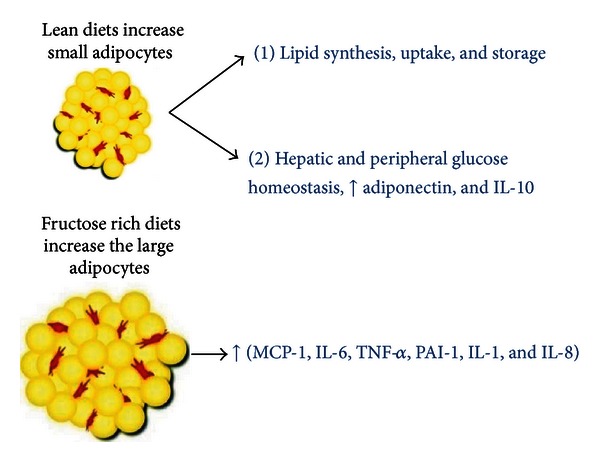
Enlargement of adipocytes causes alterations in secretion of adipokines. Under normal conditions, adipocyte is a site of lipid synthesis, uptake, and storage. Secreted adipokines function as endocrine, paracrine, or autocrine mediators. Increased adipocyte size can lead to deleterious alterations in insulin sensitivity caused by a decrease in adiponectin secretion and the induction of inflammatory mediators. Modified from [11].
The adipocyte-mediated increase in adipokine release plays a critical role in the regulation of blood pressure (angiotensinogen), vascular haemostasis, and angiogenesis. The release of these cytokines by adipocytes suggest that fructose-mediated diabetes may be related to systemic effects which include altered adiposity and insulin resistance. Adipocyte dysfunction occurs as a consequence of chronic overfeeding of fructose leading to adipocyte enlargement and inflammation and mitochondrial dysfunction (Figure 5).
Figure 5.
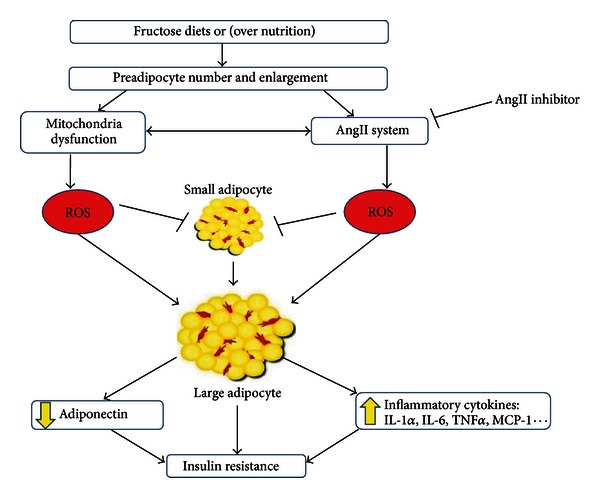
Molecular mechanisms by which fructose diets, inactivity, and gluttony increase preadipocyte number and enlargement via increases in ROS generated by the renin-AngII system and mitochondrial dysfunction leading to obesity, insulin resistance, and diabetes. Hyperglycemia results in increased ROS production within the mitochondria via a number of mechanisms including a reduction in the glutathione/glutathione disulfide ratio. ROS generation mediates a proinflammatory cascade resulting in increase of adipogenesis, release of inflammatory cytokines, and decrease in adiponectin leading to insulin resistance.
Mitochondria play an important role in adipocyte differentiation and function [68]. During the early stages of preadipocyte development, an increased number of mitochondria are required, resulting in small mature adipocytes, highly sensitive to insulin, and secreting high levels of adiponectin [69]. By contrast, mitochondrial dysfunction has also been linked to T2DM complications in fructose diets. The results of impaired mitochondrial function include increased FFA levels resulting in the accumulation of mitochondrial products including fatty acyl coenzyme A (CoA) and reduced insulin sensitivity [69].
In summary, fructose diets, inactivity, and gluttony results in adipocyte expansion with the resultant detrimental perturbations in the renin-Ang II system and in mitochondria, both of which undergo cellular changes that result in the increased generation of ROS and TNF-α, IL-1, and IL-6 and a decrease in adiponectin levels. Adiponectin is synthesized and released only by the adipocyte and has an essential role in vascular and renal function.
Increases in adipocyte release of adiponectin inhibits both the expression of hepatic gluconeogenic enzymes and the rate of endogenous glucose production in diabetic mice [70]. In adiponectin transgenic mice, adiponectin reduced the expression of phosphoenolpyruvate carboxylase and glucose-6-phosphatase, which are associated with elevated phosphorylation of hepatic AMPK and decreased glucose production [70, 71].
5. Prohypertensive Effects of Fructose and Putative Mechanisms
Hypertension, diabetes, and obesity were originally documented in England and France where sugar first became available to the public. The rise in sugar intake in the United Kingdom and the United States also correlated with the rise in obesity rates observed in these countries [7]. In the early 1900s, blood pressure in over 140,000 healthy adults who applied for life insurance in the New York region suggested that a blood pressure of 140 (systolic)/90 (diastolic) mm Hg was abnormal because it reflected only 5-6% of the population in the United States [7]. Subsequent studies over the past century showed a significant and dramatic rise in the prevalence of hypertension in the United States [72–74] (Figure 6(a)). This was paralleled by an increase in the rates of obesity and diabetes. Body mass index (BMI; in kg/m2) >30 was observed in only 3.4% of 50- to 59-year-old male veterans in 1890, compared with 30.4% in 1999–2002 [7] (Figure 6(b)). This is paralleled by the epidemic of sugar consumption that has worsened over the past 300 years.
Figure 6.
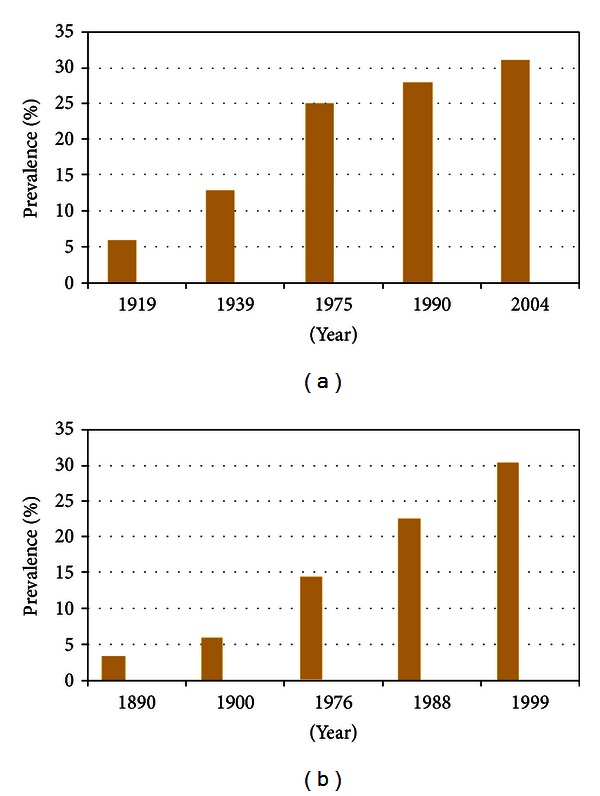
Changing prevalence of hypertension over 100 years in the USA (a). Increasing prevalence of obesity in the USA over 100 years. Obesity defined as body mass index (BMI Kg/m2) >30 (b).
During the last decade, emerging data have altered our perspective on the link between fructose, uric acid, and hypertension. Epidemiologic data, in the form of large, longitudinal studies strengthened the link between elevated uric acid and hypertension [50, 75–79]. In animals, mild hyperuricemia induced by the uricase inhibitor oxonic acid, mimicking levels in humans, increased blood pressure by crystal-independent mechanism resulting in stimulation of the renin-angiotensin system and inhibition of nitric oxide synthase [80]. Two-thirds of adolescents with newly diagnosed essential hypertension and elevated uric acid normalized their blood pressure when treated with the xanthine oxidase inhibitor allopurinol [81]. This study could not exclude the possibility that some or all of the observed effect could have been mediated by a reduction in superoxide production, a byproduct of xanthine oxidase function. A more recent study done by the same group confirmed these results by using two differently acting urate lowering drugs, allopurinol, and probenecid [82]. This study clearly implicated uric acid as the biochemical mediator of increased blood pressure. Animal data suggested that uric acid induced hypertension has two phases. The first is salt insensitive which is likely to be managed by urate lowering drugs, while the second phase is salt sensitive. Due to a paucity of outcome data, recommendations on how to treat uric acid associate hypertension cannot be made at this time although the mechanism appears clear, especially in the early stages before the development of salt sensitivity. Future clinical trials are required to include different levels of hypertension and different age groups before recommending urate lowering agents especially as they have an inferior efficacy profile when compared with antihypertensive medications presently in clinical use.
Consumption of high-fructose chow by mice produced nocturnal hypertension and autonomic imbalance which may be related to activation of the sympathetic and RAS systems [83]. Subsequent data suggested that changes in autonomic modulation may be an initiating mechanism underlying the cluster of symptoms associated with cardiometabolic disease [84]. The addition of clonidine to drinking water inhibited fructose-induced hypertension in rats [85].
6. Effect of Fructose on Dyslipidemia and Insulin Resistance
The earliest recorded metabolic perturbation resulting from fructose consumption is postprandial hypertriglyceridemia, which increased visceral adipose deposition. Visceral adiposity contributes to hepatic triglyceride accumulation, protein kinase C activation, and hepatic insulin resistance by increasing the portal delivery of free fatty acids to the liver. With insulin resistance, VLDL production is upregulated and this, along with systemic free fatty acids, increase lipid delivery to muscle. It is also possible that fructose initiates hepatic insulin resistance independently of visceral adiposity and free fatty acid delivery [20]. Splanchnic perfusion studies have shown that hepatic production of triglycerides is much greater with fructose compared with equimolar concentrations of glucose [86]. Unlike glucose, fructose does not stimulate insulin secretion, due to its hepatic metabolism and the low level of expression of the fructose transporter GLUT5 in pancreatic β-cells [87]. Consumption of fructose-sweetened beverages with meals produced a rapid and prolonged elevation of plasma triglycerides compared with glucose-sweetened beverages. Because insulin, leptin, and possibly ghrelin function as key signals to the central nervous system in the long-term regulation of energy balance, decreases of circulating insulin and leptin and increased ghrelin concentrations could lead to increased caloric intake and ultimately contribute to weight gain and obesity during chronic consumption of diets high in fructose [88]. Apolipoprotein B levels were found to be higher following fructose consumption compared with isocaloric amount of glucose [89].
Fructose increases the incidence of hypertension, NAFL, and diabetes [90]. In fact, countries electing to use HFCS in their food supply have a 20% higher prevalence of diabetes compared to countries that did not use HFCS independent of obesity [91]. Uric acid stimulates fructokinase and the development of NAFL [14] via an increase in fructose metabolism thereby increasing the development of type 2 diabetes in children. This may be related to an increase in SREBP-1c and reduced acyl-CoA oxidase during pregnancy [92]. Body size at birth is related to food intake and the content of fructose [93] or an elevation of estrogen during pregnancy in women with a family history of type 2 diabetes [94, 95]. Genetic morphism in the glucokinase regulatory protein, which binds to glucokinase and inhibits its activity in the presence of fructose-6-phosphate (F6P) is associated with ethnicity and may be responsible for different response rates to obesity and diabetes in different populations [96]. An increase of fructose-1,6-biophosphatase [97], or in angiotensin 1–7 [98] decreases fructose-mediated diabetes by an increase in pancreatic islet metabolism [99].
7. Prospective and Therapeutic Implications
Based on the present knowledge of fructose and its detrimental metabolic effects when in excess and the unique nature of KHK, it is clear that fructose is, at least, partially responsible for the pandemic of diabetes and the metabolic syndrome that is presently occurring. A number of therapeutic approaches appear viable as a result of the data outlined previously.
(1) Assessing KHK activity in human blood samples opens a new approach to the diagnosis as well as the treatment of type 2 diabetes. We hypothesize that individuals either completely or partially deficient in KHK activity are immune at variable levels from developing type 2 diabetes mellitus. In order to test this hypothesis, the following require clarification: (a) KHK-C is primarily expressed in the liver and kidney. Thus, the in vivo handling of fructose to assess enzyme expression and efficiency would be difficult to monitor. We propose that PMN expression will parallel hepatic expression, a series of pilot clinical studies in individuals selected across populations at varying risk for type 2 diabetes mellitus is required. (b) Once this association has been demonstrated, measurement of KHK-C activity in PMN's in young adults with type 2 diabetes and age/sex matched controls is required. If results support the hypothesis, new approach to the prediction as well as a therapeutic approach to treat type 2 diabetes mellitus is at hand.
(2) Interference with fructose transport at the GLUT transporter level diminishes the availability of fructose. Due to their hydrophilic nature, sugars must first traverse lipid bilayer membranes via carrier-mediated transport mechanisms. Several transporters have been identified, and these include the facilitative glucose transporter (GLUT) which primarily regulates the clearance of blood glucose along a concentration gradient and the sodium-coupled glucose cotransporter (SGLT) family members that are distinct at both the primary and secondary structural levels from the GLUT proteins. Expression of SGLT proteins is restricted to the gut and kidney, where their role is energy-dependent reabsorption of glucose from lumen. The GLUT transporter family comprises 13 members that exhibit tissue distribution, substrate specificity, and transport kinetics that reflect their physiologic role. These have not been fully defined for all 13 isoforms [100]. Fructose gains access to the circulation and hepatocytes via GLUT 5, GLUT 7, and GLUT 11 [8]. Interference with fructose transport at the level of these transporters represents a therapeutic approach to prevent fructose-induced adiposity and insulin resistance.
(3) Interference at the level of KHK is an attractive approach to modify the metabolism of fructose and, possibly, alleviating adiposity and vascular dysfunction. This can be implemented in two ways. (a) An inhibitor is currently being developed (Richard Johnson, patent number WO/2012/019188, public knowledge). It remains unclear how effective this approach will be, and if this intervention will be dependent on an individual's KHK activity profile as suggested previously. (b) The use of certain furanose sugars, which can also be substrates for KHK and compete with fructose for its metabolism to hexose-1-phosphate [21]. D-Tagatose, a furanose sugar metabolized by KHK, when compared with fructose, caused markedly higher levels of serum uric acid and lower of Pi [101]. This highlights the importance of the poorly regulated KHK in this pathway and necessitates further study.
(4) Therapeutic agents that decrease the effect of high fructose diets on diabetes and insulin resistance include statins [102], metformin [103], and renin inhibitor attenuated diabetes and insulin resistance [104]. The latter is due to a decrease in lipid peroxidation. In support of these observations, candesartan cilexetil [105] and losartan improve renal hemodynamic and insulin resistance [106] and lower blood pressure in diabetic rodents fed a high fructose diet [107] presumably by the activation of AMP-activated protein kinase [108]. Infusion of Ang II decreased adiponectin and potentiated fructose-mediated insulin resistance in fructose-fed rats [109]. The presence of renin-angiotensin aldosterone in adipose tissue has been described [110–112] in which Ang II increased NADPH oxidase activity and WAT mediated inflammation and blocked RAAS thereby preventing the onset of diabetes. Blocking ROS via the inhibition of Ang II converting enzyme reduced obesity in rats [113] and dysregulation of inflammatory cytokines released by adipocytes [114, 115]. Plasma Ang II is associated with markers of insulin resistance and obesity [116]. Renin angiotensin expression regulates mouse and human adipocyte differentiation [117, 118].
(5) Uric acid can be a byproduct of uncontrolled fructose metabolism due to the rapid consumption of ATP as noted previously. Uric acid has been linked to endothelial and adipocytes dysfunction. In contrast, uric acid also functions as an antioxidant [119]. Treating hyperuricemia in the absence of gout is not recommended at this time due to the lack of sufficient data.
(6) The ideal and logical, but most difficult, approach would be to modify fructose content in food. The reason that KHK is a poorly regulated enzyme may be due to the high levels of physical activity and limited fructose intake that was prevalent in humans before the introduction of refined cane sugar and high fructose corn syrup (HFCS). Negative feedback was unnecessary due to the limited supply of fructose. This does not exist today where calorie sweeteners, including HFCS, are part of the normal diet. Moreover, and of grave concern, is the increased consumption of sugar-sweetened beverages, and fresh and processed juices which provide an easy vehicle for excessive sugar intake, over very a short period of time and have been directly linked to obesity [35, 120] and type 2 diabetes [45, 121]. At the end of the day, we might conclude that “only drinking “milk and water” can prevent diabetes.” The present choice is either to continue with the currently high rates of fructose consumption which lead to adipocyte expansion, obesity, and hypertension or to minimize fructose intake and alleviate health care cost and the cost of managing hypertension and its complications, thereby improving the lots of health care professional, patients, and their families and the well-being of millions of individuals.
8. Conclusion
Fructose metabolism is very unique in a sense that it is not regulated. The consequences of uncontrolled fructose metabolism can be harmful at the cellular level resulting in intracellular ATP depletion, increased uric acid production, endothelial dysfunction, oxidative stress, and increased lipogenesis. High fructose consumption induces insulin resistance and other manifestations of metabolic syndrome in a series of animal models. These effects are not seen in animals fed either glucose or starch. Human epidemiological data are generally of poor quality due to the lack of consistency in study design, methodology, and length. It remains unclear if targeting fructose by interfering with its transport or metabolism can be of any clinical benefit.
Acknowledgments
This work was supported by National Institutes of Health Grant HL-55601 (N.G.A.) and by the BrickStreet Foundation (J.S., N.G.A.). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
References
- 1.Osler W. The Principles and Practice of Medicine. 2nd edition 1895. [Google Scholar]
- 2.Cory S, Ussery-Hall A, Griffin-Blake S, et al. Prevalence of selected risk behaviors and chronic diseases and conditions-steps communities, United States, 2006-2007. Morbidity and Mortality Weekly Report. 2010;59(8):1–37. [PubMed] [Google Scholar]
- 3.Fox CS, Pencina MJ, Meigs JB, Vasan RS, Levitzky YS, D’Agostino RB. Trends in the incidence of type 2 diabetes mellitus from the 1970s to the 1990s: The Framingham Heart Study. Circulation. 2006;113(25):2914–2918. doi: 10.1161/CIRCULATIONAHA.106.613828. [DOI] [PubMed] [Google Scholar]
- 4.Kahn BB, Flier JS. Obesity and insulin resistance. The Journal of Clinical Investigation. 2000;106(4):473–481. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan WH, Flegal KM, Chang HY, Yeh WT, Yeh CJ, Lee WC. Body mass index and obesity-related metabolic disorders in Taiwanese and US whites and blacks: implications for definitions of overweight and obesity for Asians. The American Journal of Clinical Nutrition. 2004;79(1):31–39. doi: 10.1093/ajcn/79.1.31. [DOI] [PubMed] [Google Scholar]
- 6.Johnson RJ, Perez-Pozo SE, Sautin YY, et al. Hypothesis: could excessive fructose intake and uric acid cause type 2 diabetes? Endocrine Reviews. 2009;30(1):96–116. doi: 10.1210/er.2008-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson RJ, Segal MS, Sautin Y, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease1-3. The American Journal of Clinical Nutrition. 2007;86(4):899–906. doi: 10.1093/ajcn/86.4.899. [DOI] [PubMed] [Google Scholar]
- 8.Zhao FQ, Keating AF. Functional properties and genomics of glucose transporters. Current Genomics. 2007;8(2):113–128. doi: 10.2174/138920207780368187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fox IH, Kelley WN. Studies on the mechanism of fructose-induced hyperuricemia in man. Metabolism. 1972;21(8):713–721. doi: 10.1016/0026-0495(72)90120-5. [DOI] [PubMed] [Google Scholar]
- 10.Mäenpää PH, Raivio KO, Kekomäki MP. Liver adenine nucleotides: fructose-induced depletion and its effect on protein synthesis. Science. 1968;161(3847):1253–1254. doi: 10.1126/science.161.3847.1253. [DOI] [PubMed] [Google Scholar]
- 11.Gustafson B. Adipose tissue, inflammation and atherosclerosis. Journal of Atherosclerosis and Thrombosis. 2010;17(4):332–341. doi: 10.5551/jat.3939. [DOI] [PubMed] [Google Scholar]
- 12.Murray R, Bender D, Botham KM, Kennelly PJ, Rodwell V, Weil PA. Harper's Illustrated Biochemistry. New York, NY, USA: 2003. [Google Scholar]
- 13.McGuinness OP, Cherrington AD. Effects of fructose on hepatic glucose metabolism. Current Opinion in Clinical Nutrition and Metabolic Care. 2003;6(4):441–448. doi: 10.1097/01.mco.0000078990.96795.cd. [DOI] [PubMed] [Google Scholar]
- 14.Lanaspa MA, Sanchez-Lozada LG, Cicerchi C, et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One. 2012;7(10) doi: 10.1371/journal.pone.0047948.e47948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khosla UM, Zharikov S, Finch JL, et al. Hyperuricemia induces endothelial dysfunction. Kidney International. 2005;67(5):1739–1742. doi: 10.1111/j.1523-1755.2005.00273.x. [DOI] [PubMed] [Google Scholar]
- 16.Sautin YY, Nakagawa T, Zharikov S, Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. American Journal of Physiology. 2007;293(2):C584–C596. doi: 10.1152/ajpcell.00600.2006. [DOI] [PubMed] [Google Scholar]
- 17.Fiaschi E, Baggio B, Favaro S, et al. Fructose-induced hyperuricemia in essential hypertension. Metabolism. 1977;26(11):1219–1223. doi: 10.1016/0026-0495(77)90114-7. [DOI] [PubMed] [Google Scholar]
- 18.Perheentupa J, Raivio K. Fructose-induced hyperuricaemia. The Lancet. 1967;2(7515):528–531. doi: 10.1016/s0140-6736(67)90494-1. [DOI] [PubMed] [Google Scholar]
- 19.Stirpe F, Della Corte E, Bonetti E, Abbondanza A, Abbati A, De Stefano F. Fructose-induced hyperuricaemia. The Lancet. 1970;2(7686):1310–1311. doi: 10.1016/s0140-6736(70)92269-5. [DOI] [PubMed] [Google Scholar]
- 20.Stanhope KL, Havel PJ. Fructose consumption: potential mechanisms for its effects to increase visceral adiposity and induce dyslipidemia and insulin resistance. Current Opinion in Lipidology. 2008;19(1):16–24. doi: 10.1097/MOL.0b013e3282f2b24a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raushel FM, Cleland WW. The substrate and anomeric specificity of fructokinase. The Journal of Biological Chemistry. 1973;248(23):8174–8177. [PubMed] [Google Scholar]
- 22.Diggle CP, Shires M, Leitch D, et al. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. Journal of Histochemistry and Cytochemistry. 2009;57(8):763–774. doi: 10.1369/jhc.2009.953190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asipu A, Hayward BE, O’Reilly J, Bonthron DT. Properties of normal and mutant recombinant human ketohexokinases and implications for the pathogenesis of essential fructosuria. Diabetes. 2003;52(9):2426–2432. doi: 10.2337/diabetes.52.9.2426. [DOI] [PubMed] [Google Scholar]
- 24.Ishimoto T, Lanaspa MA, Le MT, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(11):4320–4325. doi: 10.1073/pnas.1119908109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diggle CP, Shires M, McRae C, et al. Both isoforms of ketohexokinase are dispensable for normal growth and development. Physiological Genomics. 2010;42(4):235–243. doi: 10.1152/physiolgenomics.00128.2010. [DOI] [PubMed] [Google Scholar]
- 26.Lê KA, Faeh D, Stettler R, et al. A 4-wk high-fructose diet alters lipid metabolism without affecting insulin sensitivity or ectopic lipids in healthy humans. The American Journal of Clinical Nutrition. 2006;84(6):1374–1379. doi: 10.1093/ajcn/84.6.1374. [DOI] [PubMed] [Google Scholar]
- 27.Mayatepek E, Hoffmann B, Meissner T. Inborn errors of carbohydrate metabolism. Best Practice and Research. 2010;24(5):607–618. doi: 10.1016/j.bpg.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Froesch ER. Disorders of fructose metabolism. Clinics in Endocrinology and Metabolism. 1976;5(3):599–611. doi: 10.1016/s0300-595x(76)80042-4. [DOI] [PubMed] [Google Scholar]
- 29.Bonthron DT, Brady N, Donaldson IA, Steinmann B. Molecular basis of essential fructosuria: molecular cloning and mutational analysis of human ketohexokinase (fructokinase) Human Molecular Genetics. 1994;3(9):1627–1631. doi: 10.1093/hmg/3.9.1627. [DOI] [PubMed] [Google Scholar]
- 30.Marriott BP, Cole N, Lee E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. Journal of Nutrition. 2009;139(6):1228S–1235S. doi: 10.3945/jn.108.098277. [DOI] [PubMed] [Google Scholar]
- 31.Park YK, Yetley EA. Intakes and food sources of fructose in the United States. The American Journal of Clinical Nutrition. 1993;58(5, supplement):737S–747S. doi: 10.1093/ajcn/58.5.737S. [DOI] [PubMed] [Google Scholar]
- 32.Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. The American Journal of Clinical Nutrition. 2004;79(4):537–543. doi: 10.1093/ajcn/79.4.537. [DOI] [PubMed] [Google Scholar]
- 33.Bray GA. The epidemic of obesity and changes in food intake: the fluoride hypothesis. Physiology and Behavior. 2004;82(1):115–121. doi: 10.1016/j.physbeh.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 34.Hu FB, Malik VS. Sugar-sweetened beverages and risk of obesity and type 2 diabetes: epidemiologic evidence. Physiology and Behavior. 2010;100(1):47–54. doi: 10.1016/j.physbeh.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malik VS, Schulze MB, Hu FB. Intake of sugar-sweetened beverages and weight gain: a systematic review. The American Journal of Clinical Nutrition. 2006;84(2):274–288. doi: 10.1093/ajcn/84.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgan RE. Does consumption of high-fructose corn syrup beverages cause obesity in children? Pediatric Obesity. 2013 doi: 10.1111/j.2047-6310.2013.00173.x. [DOI] [PubMed] [Google Scholar]
- 37.Stanhope KL, Havel PJ. Fructose consumption: recent results and their potential implications. Annals of the New York Academy of Sciences. 2010;1190:15–24. doi: 10.1111/j.1749-6632.2009.05266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolan LC, Potter SM, Burdock GA. Evidence-based review on the effect of normal dietary consumption of fructose on development of hyperlipidemia and obesity in healthy, normal weight individuals. Critical Reviews in Food Science and Nutrition. 2010;50(1):53–84. doi: 10.1080/10408390903461426. [DOI] [PubMed] [Google Scholar]
- 39.Forshee RA, Anderson PA, Storey ML. Sugar-sweetened beverages and body mass index in children and adolescents: a meta-analysis. The American Journal of Clinical Nutrition. 2008;87(6):1662–1671. doi: 10.1093/ajcn/87.6.1662. [DOI] [PubMed] [Google Scholar]
- 40.Ha V, Sievenpiper JL, de Souza RJ, et al. Effect of fructose on blood pressure: a systematic review and meta-analysis of controlled feeding trials. Hypertension. 2012;59(4):787–795. doi: 10.1161/HYPERTENSIONAHA.111.182311. [DOI] [PubMed] [Google Scholar]
- 41.Mattes RD, Shikany JM, Kaiser KA, Allison DB. Nutritively sweetened beverage consumption and body weight: a systematic review and meta-analysis of randomized experiments. Obesity Reviews. 2011;12(5):346–365. doi: 10.1111/j.1467-789X.2010.00755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sievenpiper JL, de Souza RJ, Mirrahimi A, et al. Effect of fructose on body weight in controlled feeding trials: a systematic review and meta-analysis. Annals of Internal Medicine. 2012;156(4):291–304. doi: 10.7326/0003-4819-156-4-201202210-00007. [DOI] [PubMed] [Google Scholar]
- 43.Chen M, Pan A, Malik VS, Hu FB. Effects of dairy intake on body weight and fat: a meta-analysis of randomized controlled trials. The American Journal of Clinical Nutrition. 2012;96(4):735–747. doi: 10.3945/ajcn.112.037119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ledoux TA, Watson K, Barnett A, Nguyen NT, Baranowski JC, Baranowski T. Components of the diet associated with child adiposity: a cross-sectional study. Journal of the American College of Nutrition. 2011;30(6):536–546. doi: 10.1080/07315724.2011.10720000. [DOI] [PubMed] [Google Scholar]
- 45.Schulze MB, Manson JE, Ludwig DS, et al. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. Journal of the American Medical Association. 2004;292(8):927–934. doi: 10.1001/jama.292.8.927. [DOI] [PubMed] [Google Scholar]
- 46.Sun SZ, Anderson GH, Flickinger BD, Williamson-Hughes PS, Empie MW. Fructose and non-fructose sugar intakes in the US population and their associations with indicators of metabolic syndrome. Food and Chemical Toxicology. 2011;49(11):2875–2882. doi: 10.1016/j.fct.2011.07.068. [DOI] [PubMed] [Google Scholar]
- 47.Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144(12):5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 48.Zammit VA, Waterman IJ, Topping D, McKay G. Insulin stimulation of hepatic triacylglycerol secretion and the etiology of insulin resistance. Journal of Nutrition. 2001;131(8):2074–2077. doi: 10.1093/jn/131.8.2074. [DOI] [PubMed] [Google Scholar]
- 49.Perez-Pozo SE, Schold J, Nakagawa T, Sánchez-Lozada LG, Johnson RJ, Lillo JL. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. International Journal of Obesity. 2010;34(3):454–461. doi: 10.1038/ijo.2009.259. [DOI] [PubMed] [Google Scholar]
- 50.Jalal DI, Smits G, Johnson RJ, Chonchol M. Increased fructose associates with elevated blood pressure. Journal of the American Society of Nephrology. 2010;21(9):1543–1549. doi: 10.1681/ASN.2009111111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen S, Choi HK, Lustig RH, Hsu CY. Sugar-sweetened beverages, serum uric acid, and blood pressure in adolescents. Journal of Pediatrics. 2009;154(6):807–813. doi: 10.1016/j.jpeds.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim YH, Abris GP, Sung MK, Lee JE. Consumption of sugar-sweetened beverages and blood pressure in the United States: the national health and nutrition examination survey 2003–2006. Clinical Nutrition Research. 2012;1(1):85–93. doi: 10.7762/cnr.2012.1.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feig DI, Mazzali M, Kang DH, et al. Serum uric acid: a risk factor and a target for treatment? Journal of the American Society of Nephrology. 2006;17(4, supplement 2):S69–S73. doi: 10.1681/ASN.2005121331. [DOI] [PubMed] [Google Scholar]
- 54.Feig DI, Kang DH, Johnson RJ. Medical progress: uric acid and cardiovascular risk. The New England Journal of Medicine. 2008;359(17):1811–1821. doi: 10.1056/NEJMra0800885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pereira MA, Fulgoni VL., III Consumption of 100% fruit juice and risk of obesity and metabolic syndrome: findings from the national health and nutrition examination survey 1999–2004. Journal of the American College of Nutrition. 2010;29(6):625–629. doi: 10.1080/07315724.2010.10719901. [DOI] [PubMed] [Google Scholar]
- 56.Champagne CM, Broyles ST, Moran LD, et al. Dietary intakes associated with successful weight loss and maintenance during the Weight Loss Maintenance trial. Journal of the American Dietetic Association. 2011;111(12):1826–1835. doi: 10.1016/j.jada.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosado JL, Garcia OP, Ronquillo D, et al. Intake of milk with added micronutrients increases the effectiveness of an energy-restricted diet to reduce body weight: a randomized controlled clinical trial in Mexican women. Journal of the American Dietetic Association. 2011;111(10):1507–1516. doi: 10.1016/j.jada.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 58.Disse SC, Buelow A, Boedeker RH, et al. Reduced prevalence of obesity in children with primary fructose malabsorption: a multicentre, retrospective cohort study. Pediatric Obesity. 2013 doi: 10.1111/j.2047-6310.2013.00163.x. [DOI] [PubMed] [Google Scholar]
- 59.Walker RW, Le KA, Davis J, et al. High rates of fructose malabsorption are associated with reduced liver fat in obese African Americans. Journal of the American College of Nutrition. 2012;31(5):369–374. doi: 10.1080/07315724.2012.10720445. [DOI] [PubMed] [Google Scholar]
- 60.Vázquez-Vela MEF, Torres N, Tovar AR. White adipose tissue as endocrine organ and its role in obesity. Archives of Medical Research. 2008;39(8):715–728. doi: 10.1016/j.arcmed.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 61.Lefterova MI, Lazar MA. New developments in adipogenesis. Trends in Endocrinology and Metabolism. 2009;20(3):107–114. doi: 10.1016/j.tem.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 62.Gregoire FM, Smas CM, Sul HS. Understanding adipocyte differentiation. Physiological Reviews. 1998;78(3):783–809. doi: 10.1152/physrev.1998.78.3.783. [DOI] [PubMed] [Google Scholar]
- 63.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes and Development. 2000;14(11):1293–1307. [PubMed] [Google Scholar]
- 64.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nature Medicine. 2004;10(4):355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 65.Kim JY, van de Wall E, Laplante M, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. The Journal of Clinical Investigation. 2007;117(9):2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martini CN, Plaza MV, Vila MDC. PKA-dependent and independent cAMP signaling in 3T3-L1 fibroblasts differentiation. Molecular and Cellular Endocrinology. 2009;298(1-2):42–47. doi: 10.1016/j.mce.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 67.Shen GX. Oxidative stress and diabetic cardiovascular disorders: roles of mitochondria and NADPH oxidase. Canadian Journal of Physiology and Pharmacology. 2010;88(3):241–248. doi: 10.1139/Y10-018. [DOI] [PubMed] [Google Scholar]
- 68.De Pauw A, Tejerina S, Raes M, Keijer J, Arnould T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. American Journal of Pathology. 2009;175(3):927–939. doi: 10.2353/ajpath.2009.081155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocrine Reviews. 2010;31(3):364–395. doi: 10.1210/er.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nature Medicine. 2001;7(8):947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 71.Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nature Medicine. 2002;8(11):1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 72.Burt VL, Whelton P, Roccella EJ, et al. Prevalence of hypertension in the US adult population: results from the third National Health and Nutrition Examination Survey, 1988-1991. Hypertension. 1995;25(3):305–313. doi: 10.1161/01.hyp.25.3.305. [DOI] [PubMed] [Google Scholar]
- 73.Fields LE, Burt VL, Cutler JA, Hughes J, Roccella EJ, Sorlie P. The burden of adult hypertension in the United States 1999 to 2000: a rising tide. Hypertension. 2004;44(4):398–404. doi: 10.1161/01.HYP.0000142248.54761.56. [DOI] [PubMed] [Google Scholar]
- 74.Taylor J. The hypertension detection and follow-up program: a progress report. Circulation Research. 1977;40(5, supplement 1):I106–I109. [PubMed] [Google Scholar]
- 75.Alper AB, Jr., Chen W, Yau L, Srinivasan SR, Berenson GS, Hamm LL. Childhood uric acid predicts adult blood pressure: The Bogalusa Heart Study. Hypertension. 2005;45(1):34–38. doi: 10.1161/01.HYP.0000150783.79172.bb. [DOI] [PubMed] [Google Scholar]
- 76.Masuo K, Kawaguchi H, Mikami H, Ogihara T, Tuck ML. Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension. 2003;42(4):474–480. doi: 10.1161/01.HYP.0000091371.53502.D3. [DOI] [PubMed] [Google Scholar]
- 77.Mellen PB, Bleyer AJ, Erlinger TP, et al. Serum uric acid predicts incident hypertension in a biethnic cohort: The Atherosclerosis Risk in Communities Study. Hypertension. 2006;48(6):1037–1042. doi: 10.1161/01.HYP.0000249768.26560.66. [DOI] [PubMed] [Google Scholar]
- 78.Nagahama K, Inoue T, Iseki K, et al. Hyperuricemia as a predictor of hypertension in a screened cohort in Okinawa, Japan. Hypertension Research. 2004;27(11):835–841. doi: 10.1291/hypres.27.835. [DOI] [PubMed] [Google Scholar]
- 79.Sundström J, Sullivan L, D’Agostino RB, Levy D, Kannel WB, Vasan RS. Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension. 2005;45(1):28–33. doi: 10.1161/01.HYP.0000150784.92944.9a. [DOI] [PubMed] [Google Scholar]
- 80.Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;38(5):1101–1106. doi: 10.1161/hy1101.092839. [DOI] [PubMed] [Google Scholar]
- 81.Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. Journal of the American Medical Association. 2008;300(8):924–932. doi: 10.1001/jama.300.8.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soletsky B, Feig DI. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension. 2012;60(5):1148–1156. doi: 10.1161/HYPERTENSIONAHA.112.196980. [DOI] [PubMed] [Google Scholar]
- 83.Farah V, Elased KM, Chen Y, et al. Nocturnal hypertension in mice consuming a high fructose diet. Autonomic Neuroscience. 2006;130(1-2):41–50. doi: 10.1016/j.autneu.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 84.De AK, Senador DD, Mostarda C, Irigoyen MC, Morris M. Sympathetic overactivity precedes metabolic dysfunction in a fructose model of glucose intolerance in mice. American Journal of Physiology. 2012;302(8):R950–R957. doi: 10.1152/ajpregu.00450.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hwang I, Ho H, Hoffman BB, Reaven GM. Fructose-induced insulin resistance and hypertension in rats. Hypertension. 1987;10(5):512–516. doi: 10.1161/01.hyp.10.5.512. [DOI] [PubMed] [Google Scholar]
- 86.Wolfe BM, Ahuja SP. Effects of intravenously administered fructose and glucose on splanchnic secretion of plasma triglycerides in hypertriglyceridemic men. Metabolism. 1977;26(9):963–978. doi: 10.1016/0026-0495(77)90014-2. [DOI] [PubMed] [Google Scholar]
- 87.Sato Y, Ito T, Udaka N, et al. Immunohistochemical localization of facilitated-diffusion glucose transporters in rat pancreatic islets. Tissue and Cell. 1996;28(6):637–643. doi: 10.1016/s0040-8166(96)80067-x. [DOI] [PubMed] [Google Scholar]
- 88.Teff KL, Elliott SS, Tschöp M, et al. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. Journal of Clinical Endocrinology and Metabolism. 2004;89(6):2963–2972. doi: 10.1210/jc.2003-031855. [DOI] [PubMed] [Google Scholar]
- 89.Swarbrick MM, Stanhope KL, Elliott SS, et al. Consumption of fructose-sweetened beverages for 10 weeks increases postprandial triacylglycerol and apolipoprotein-B concentrations in overweight and obese women. British Journal of Nutrition. 2008;100(5):947–952. doi: 10.1017/S0007114508968252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rebollo A, Roglans N, Alegret M, Laguna JC. Way back for fructose and liver metabolism: bench side to molecular insights. World Journal of Gastroenterology. 2012;18(45):6552–6559. doi: 10.3748/wjg.v18.i45.6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goran MI, Ulijaszek SJ, Ventura EE. High fructose corn syrup and diabetes prevalence: a global perspective. Global Public Health. 2013;8(1):55–64. doi: 10.1080/17441692.2012.736257. [DOI] [PubMed] [Google Scholar]
- 92.Mukai Y, Kumazawa M, Sato S. Fructose intake during pregnancy up-regulates the expression of maternal and fetal hepatic sterol regulatory element-binding protein-1c in rats. Endocrine. 2012 doi: 10.1007/s12020-012-9815-8. [DOI] [PubMed] [Google Scholar]
- 93.Perala MM, Mannisto S, Kaartinen NE, et al. Body size at birth is associated with food and nutrient intake in adulthood. PLoS One. 2012;7(9) doi: 10.1371/journal.pone.0046139.e46139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koricanac G, Tepavcevic S, Romic S, et al. Estradiol enhances effects of fructose rich diet on cardiac fatty acid transporter CD36 and triglycerides accumulation. European Journal of Pharmacology. 2012;694(1–3):127–134. doi: 10.1016/j.ejphar.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 95.Zou M, Arentson EJ, Teegarden D, Koser SL, Onyskow L, Donkin SS. Fructose consumption during pregnancy and lactation induces fatty liver and glucose intolerance in rats. Nutrition Research. 2012;32(8):588–598. doi: 10.1016/j.nutres.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li H, Xu R, Peng X, Wang Y, Wang T. Association of glucokinase regulatory protein polymorphism with type 2 diabetes and fasting plasma glucose: a meta-analysis. Molecular Biology Reports. 2013;40(6):3935–3942. doi: 10.1007/s11033-012-2470-6. [DOI] [PubMed] [Google Scholar]
- 97.Masotti A. Comment on: Visinoni et al. The role of liver fructose-1,6-bisphosphatase in regulating appetite and adiposity, Diabetes, vol. 61, pp. 1122–1132, 2012. Diabetes. 2012;61(12, article e20) doi: 10.2337/db12-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marcus Y, Shefer G, Sasson K. Angiotensin 1-7 as means to prevent the metabolic syndrome: lessons from the fructose-fed rat model. Diabetes. 2013;62(4):1121–1130. doi: 10.2337/db12-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Merrins MJ, Bertram R, Sherman A, Satin LS. Phosphofructo-2-kinase/fructose-2,6-bisphosphatase modulates oscillations of pancreatic islet metabolism. PLoS One. 2012;7(4) doi: 10.1371/journal.pone.0034036.e34036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wilson-O’Brien AL, Patron N, Rogers S. Evolutionary ancestry and novel functions of the mammalian glucose transporter (GLUT) family. BMC Evolutionary Biology. 2010;10(1, article 152) doi: 10.1186/1471-2148-10-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Buemann B, Toubro S, Holst JJ, Rehfeld JF, Bibby BM, Astrup A. D-Tagatose, a stereoisomer of D-fructose, increases blood uric acid concentration. Metabolism. 2000;49(8):969–976. doi: 10.1053/meta.2000.7724. [DOI] [PubMed] [Google Scholar]
- 102.Schaalan MF. Effects of pioglitazone and/or simvastatin on circulating TNFalpha and adiponectin levels in insulin resistance. Journal of Immunotoxicology. 2012;9(2):201–209. doi: 10.3109/1547691X.2012.660998. [DOI] [PubMed] [Google Scholar]
- 103.Puyo AM, Borroni JS, Boudou S, et al. Metformin reduces vascular production of vasoconstrictor prostanoids in fructose overloaded rats. Autonomic and Autacoid Pharmacology. 2012;32(1, part 2):9–14. doi: 10.1111/j.1474-8673.2012.00469.x. [DOI] [PubMed] [Google Scholar]
- 104.Chou CL, Pang CY, Lee TJ, Fang TC. Direct renin inhibitor prevents and ameliorates insulin resistance, aortic endothelial dysfunction and vascular remodeling in fructose-fed hypertensive rats. Hypertension Research. 2013;36(2):123–128. doi: 10.1038/hr.2012.124. [DOI] [PubMed] [Google Scholar]
- 105.Li P, Koike T, Jiang HY, Wang ZH, Kawata Y, Oshida Y. Acute treatment with candesartan cilexetil, an angiotensin II type 1 receptor blocker, improves insulin sensitivity in high-fructose-diet-fed rats. Hormone and Metabolic Research. 2012;44(4):286–290. doi: 10.1055/s-0032-1304321. [DOI] [PubMed] [Google Scholar]
- 106.Abdulla MH, Sattar MA, Abdullah NA, Johns EJ. The effect of losartan and carvedilol on renal haemodynamics and altered metabolism in fructose-fed Sprague-Dawley rats. Journal of Physiology and Biochemistry. 2012;68(3):353–363. doi: 10.1007/s13105-012-0147-1. [DOI] [PubMed] [Google Scholar]
- 107.Guo Q, Mori T, Jiang Y, et al. Losartan modulates muscular capillary density and reverses thiazide diuretic-exacerbated insulin resistance in fructose-fed rats. Hypertension Research. 2012;35(1):48–54. doi: 10.1038/hr.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yoshida D, Higashiura K, Shinshi Y, et al. Effects of angiotensin II receptor blockade on glucose metabolism via AMP-activated protein kinase in insulin-resistant hypertensive rats. Journal of the American Society of Hypertension. 2009;3(1):3–8. doi: 10.1016/j.jash.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 109.Ran J, Hirano T, Fukui T, et al. Angiotensin II infusion decreases plasma adiponectin level via its type 1 receptor in rats: an implication for hypertension-related insulin resistance. Metabolism. 2006;55(4):478–488. doi: 10.1016/j.metabol.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 110.Jing F, Mogi M, Horiuchi M. Role of renin-angiotensin-aldosterone system in adipose tissue dysfunction. Molecular and Cellular Endocrinology. 2012 doi: 10.1016/j.mce.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 111.Kalupahana NS, Massiera F, Quignard-Boulange A, et al. Overproduction of angiotensinogen from adipose tissue induces adipose inflammation, glucose intolerance, and insulin resistance. Obesity. 2012;20(1):48–56. doi: 10.1038/oby.2011.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yvan-Charvet L, Quignard-Boulangé A. Role of adipose tissue renin-angiotensin system in metabolic and inflammatory diseases associated with obesity. Kidney International. 2011;79(2):162–168. doi: 10.1038/ki.2010.391. [DOI] [PubMed] [Google Scholar]
- 113.Weisinger HS, Begg DP, Egan GF, et al. Angiotensin converting enzyme inhibition from birth reduces body weight and body fat in Sprague-Dawley rats. Physiology and Behavior. 2008;93(4-5):820–825. doi: 10.1016/j.physbeh.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 114.Kurata A, Nishizawa H, Kihara S, et al. Blockade of angiotensin II type-1 receptor reduces oxidative stress in adipose tissue and ameliorates adipocytokine dysregulation. Kidney International. 2006;70(10):1717–1724. doi: 10.1038/sj.ki.5001810. [DOI] [PubMed] [Google Scholar]
- 115.Yvan-Charvet L, Even P, Bloch-Faure M, et al. Deletion of the angiotensin type 2 receptor (AT2R) reduces adipose cell size and protects from diet-induced obesity and insulin resistance. Diabetes. 2005;54(4):991–999. doi: 10.2337/diabetes.54.4.991. [DOI] [PubMed] [Google Scholar]
- 116.Saiki A, Ohira M, Endo K, et al. Circulating angiotensin II is associated with body fat accumulation and insulin resistance in obese subjects with type 2 diabetes mellitus. Metabolism. 2009;58(5):708–713. doi: 10.1016/j.metabol.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 117.Matsushita K, Wu Y, Okamoto Y, Pratt RE, Dzau VJ. Local renin angiotensin expression regulates human mesenchymal stem cell differentiation to adipocytes. Hypertension. 2006;48(6):1095–1102. doi: 10.1161/01.HYP.0000248211.82232.a7. [DOI] [PubMed] [Google Scholar]
- 118.Sydow M, Burchardi H, Zinserling J, Ische H, Crozier TA, Weyland W. Improved determination of static compliance by automated single volume steps in ventilated patients. Intensive Care Medicine. 1991;17(2):108–114. doi: 10.1007/BF01691433. [DOI] [PubMed] [Google Scholar]
- 119.Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(11):6858–6862. doi: 10.1073/pnas.78.11.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brown CM, Dulloo AG, Montani JP. Sugary drinks in the pathogenesis of obesity and cardiovascular diseases. International Journal of Obesity. 2008;32(supplement 6):S28–S34. doi: 10.1038/ijo.2008.204. [DOI] [PubMed] [Google Scholar]
- 121.Palmer JR, Boggs DA, Krishnan S, Hu FB, Singer M, Rosenberg L. Sugar-sweetened beverages and incidence of type 2 diabetes mellitus in African American women. Archives of Internal Medicine. 2008;168(14):1487–1492. doi: 10.1001/archinte.168.14.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]