

Abstract
Autoimmune disease results from a loss of tolerance to self-antigens in genetically susceptible individuals. Completely understanding this process requires that targeted antigens be identified, and so a number of techniques have been developed to determine immune receptor specificities. We previously reported the construction of a phage-displayed synthetic human peptidome and a proof-of-principle analysis of antibodies from three patients with neurological autoimmunity. Here we present data from a large-scale screen of 298 independent antibody repertoires, including those from 73 healthy sera, using phage immunoprecipitation sequencing. The resulting database of peptide-antibody interactions characterizes each individual’s unique autoantibody fingerprint, and includes specificities found to occur frequently in the general population as well as those associated with disease. Screening type 1 diabetes (T1D) patients revealed a prematurely polyautoreactive phenotype compared with their matched controls. A collection of cerebrospinal fluids and sera from 63 multiple sclerosis patients uncovered novel, as well as previously reported antibody-peptide interactions. Finally, a screen of synovial fluids and sera from 64 rheumatoid arthritis patients revealed novel disease-associated antibody specificities that were independent of seropositivity status. This work demonstrates the utility of performing PhIP-Seq screens on large numbers of individuals and is another step toward defining the full complement of autoimmunoreactivities in health and disease.
Keywords: autoantigen discovery, high throughput screening, PhIP-Seq, proteomics
1. Introduction
Our understanding of autoimmunity is constrained by our inability to completely characterize the molecular targets of an adaptive immune system. To begin to address this limitation, we have developed an unbiased proteomic technology, phage immunoprecipitation sequencing (PhIP-Seq), which employs a synthetic version of the complete human peptidome (T7-Pep).[1] This technology can be used to define interactions between an individual’s antibody repertoire and each of over 400,000 overlapping 36 mer peptides. In the present work, we have improved upon the previously reported PhIP-Seq methodology in two ways. First, sample processing was made compatible with a 96-well plate format and automated on a liquid handling robot. Second, we developed a method to perform 96-plex analysis of individual PhIP-Seq experiments using just 2–3 lanes of an Illumina HiSeq, thus reducing the cost of each screen to about $25 per sample. This method was recently employed to unambiguously identify the target of autoantibodies associated with inclusion body myositis (IBM).[2] Furthermore, PhIP-Seq was used to localize the antigenic epitopes and to provide the first definitive evidence of antigen-driven autoimmunity in IBM.
There are several autoimmune diseases of relatively high incidence for which the role of antibody-mediated autoimmunity is appreciated but not understood. Of these, we selected type 1 diabetes (T1D), multiple sclerosis (MS) and rheumatoid arthritis (RA) for autoantibody repertoire analysis by high-throughput PhIP-Seq screening. Strong genetic linkage to class II HLA alleles in each of these diseases supports the view that there is an important role for antigen presentation and subsequent activation of helper T cells with self-specificity.[3] The role of B cells in these diseases is less clear, but several lines of evidence indicate that a deeper understanding of patient antibody specificities may provide insight into disease pathogenesis. For example, pancreatic beta cell destruction in T1D is thought to be largely a consequence of cytotoxic T cell activity, yet autoantibodies targeting islet-associated antigens are routinely used for diagnosis and risk stratification.[4] In MS, secondary lymphoid tissue with germinal center activity often forms in the meninges of patients with advanced disease[5] and oligoclonal IgG bands of unknown specificity are found in cerebrospinal fluid (CSF; detectable in about 95% of patients compared with 10%-15% of controls)[6]. Patients with RA are classified as seropositive or seronegative depending on the presence of rheumatoid factor (antibodies against the Fc portion of IgG) and/or anti-citrullinated protein antibodies (ACPA). Beneficial clinical response to CD20+ B cell depletion therapy in RA has prompted the adoption of rituximab as a second line therapy for patients with high disease activity and features of a poor prognosis.[7, 8] In the treatment of MS and T1D, several studies have demonstrated a benefit for B cell depletion, but with perhaps more elusive optimal dosing regimens.[9, 10] The inherent pathogenicity of autoantibodies in these diseases is a topic of intense investigation.
Here we report a PhIP-Seq analysis of autoantibody repertoires from a large number of T1D, RA, and MS patients, for comparison to each other and to a set of 73 healthy controls. Our findings describe both known and novel antibody specificities, and methodologically sets the stage for additional large scale PhIP-Seq investigations.
2. Methods
2.1 Patient samples
Specimens originating from patients were collected after informed written consent was obtained and under a protocol approved by the local governing human research protection committee. In some cases, de-identified discarded specimens (synovial fluid) were collected under an exempt protocol approved by the local governing human research protection committee. Type 1 diabetic patient blood samples (n=39, <40 years at diagnosis, male/female ratio = 1.18, average age 18±2 years, range 3–37 years) were obtained within 7 days after initiation of insulin treatment. Age/sex-matched healthy control samples (n=41, male/female ratio = 1.18, average age 18±2 years, range 4–37 years) were obtained from patients undergoing elective minor surgery. Controls were verified to be negative for all known type 1 diabetic autoantibodies. The diabetes autoantibodies were determined by liquid-phase radiobinding assays [11] or indirect immunofluorescence assay (ICA) as described previously.[12] A chimeric ZnT8 protein (gene SEC30A8 is a chimeric construct of two peptides, amino acids 268–369) was used for RIA and contains both CR and CW variants.[13] MS patients’ neurological history, relapse features, neurological examination, MRI and CSF findings were collected. Patients were diagnosed with relapsing-remitting MS according to the McDonald criteria. Viral encephalitis serum samples were provided by the New York State Department of Health. Sera from patients infected with West Nile virus or St. Louis Encephalitis virus were reactive in ELISA tests and were confirmed by cross species plaque reduction neutralization tests with paired acute and convalescent sera. Sera from patients with enteroviral infection were collected on the same day as spinal fluids for which PCR tests for enteroviruses were positive.[14] Synovial fluid from knee joints was obtained during clinically-indicated arthrocentesis from patients with RA or other forms of inflammatory arthritis performed at the Arthritis Center of the Brigham and Women’s Hospital. Medical record review ascertained the diagnosis as assigned by the treating American Board of Internal Medicine-certified rheumatologist, supplemented by review of laboratory and radiologic data. Breast cancer patient serum samples were obtained from the Dana-Farber/Harvard Cancer Center (DF/HCC) Breast SPORE Blood Bank. These samples were originally collected under Protocol #93-085 at the DF/HCC. Healthy control samples were collected at Brigham and Women’s Hospital from subjects self-reported to be free of MS or other autoimmune disease. All serum and CSF samples were stored in aliquots at −80°C.
2.2 Phage Immunoprecipitation and sequencing library preparation
The T7-Pep library was prepared as described previously[1] and stored at −80 °C until used. For all samples, the final amount of IgG added to each 1 ml IP mix was estimated to be 2 μg. Serum/plasma samples were assumed to have 10 μg/μl of Ig. Non serum/plasma samples’ protein content was measured by Bradford assay and the Ig concentration estimated as follows. Several CSF or synovial fluid samples were electrophoretically separated under reducing conditions on a polyacrylamide gel and the Coomassie brilliant blue densitometric signal from the Ig light chain was compared to that from known input concentrations of commercially obtained IgG. We made the simplifying assumption that all Ig was IgG, and assumed a constant fluid-type fractional IgG contribution to the total protein content of each sample. For CSF the IgG fraction was estimated to be 29%, and for synovial fluid was estimated to be 15%. Each 1 ml IP mix contained 5×1010 T7-Pep phage particles and 2 ng of positive control SAPK4 C-19 antibody (Santa Cruz, sc-7585) diluted in M9LB (Novagen) with 100 μg/ml ampicillin. 1 ml IP mixes were placed in each well of a 96 deep well plate. Each patient sample or control was randomly assigned to a position on the IP plate and 2 μg of IgG was added to each well. The plate was then carefully sealed with adhesive optical tape (Applied Biosystems) and placed on a rotator for 20 hours at 4 °C. 40 μl of 1:1 Protein A/Protein G slurry (Invitrogen, 100-02D, 100-04D) was then added to each well. The re-sealed plate was placed back on a rotator for 4 hours at 4 °C. The beads were next subjected to a robotic IP protocol, which was carried out by a BioMek FX liquid handling robot. Briefly, IPs were washed in 440 μl IP Wash Buffer (150 mM NaCl, 50 mM Tris-HCL, 0.1% NP-40, pH 7.5) by pipetting up and down 30 times, for a total of 3 washes. IPs were resuspended in 40 μl of pure water, heated to 95 °C for 10 minutes and then frozen at −80 °C.
Primers used (underlined sequences overlap template, x’s are the indexing barcode specific sequences): PCR1 forward “IS7_HsORF5_2”, 3′-ACA CTC TTT CCC TAC ACG ACT CCA GTC AGG TGT GAT GCT C-5′; PCR1 reverse “IS8_HsORF3_2”, 3′-GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC CGA GCT TAT CGT CGT CAT CC-5′; PCR2 forward “IS4_HsORF5_2”, 3′-AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC TCC AGT-5′; PCR2 reverse “index N” (set of 96), 3′-CAA GCA GAA GAC GGC ATA CGA GAT xxx xxx xGT GAC TGG AGT TCA GAC GTG T-5′; “P5_Primer”, 3′-AAT GAT ACG GCG ACC ACC GA-5′; “P7_Primer_2”, 3′-CAA GCA GAA GAC GGC ATA CGA-5′; “Internal HsORF3′ TaqMan FAM Probe”, 3′-GCC GCA AGC TTG TCG AGC GAT G-5′ (modified with 5′ 6-FAM-ZEN-3′ Iowa Black FQ); T7-Pep Library Sequencing Primer “T7-Pep_96_SP”, 3′-GCT CGG GGA TCC AGG AAT TCC GCT GCG C-5′; Standard Illumina Multiplex Index Sequencing Primer “Index SP”, 3′-GAT CGG AAG AGC ACA CGT CTG AAC TCC AGT CAC-5′. For each 50 μl PCR1 reaction, the following components were mixed with 30 μl from each IP: 8.75 μl water, 10 μl 5x Herculase Buffer, 0.5 μl of 100 mM dNTP, 0.125 μl of 100 μM IS7_HsORF5_2 forward primer, 0.125 μl of 100 μM IS8_HsORF3_2 reverse primer, and 0.5 μl of Herculase II enzyme (Agilent). The reaction was then thermocycled: 1. 95 °C for 2 min; 2. (95 °C, 20s; 58 °C, 30s; 72 °C, 30s) × 30 cycles; 3. 72 °C, 3 min. A set of 96 7-nucleotide barcode-containing primers designed using the method of Meyer et al.[15] was used for PCR2. For each 50 μl PCR2 reaction, the following components were mixed with 5 μl of the appropriate “index N” reverse primer and 1.5 μl of unpurified PCR1 product: 32.38 μl water, 10 μl 5x Herculase Buffer, 0.5 μl of 100 mM dNTP, 0.125 μl of 100 μM IS4_HsORF5_2 forward primer, and 0.5 μl of Herculase II enzyme. The reaction was then thermocycled: 1. 95 °C for 2 min; 2. (95 °C, 20s; 58 °C, 30s; 72 °C, 30s) × 10 cycles; 3. 72 °C, 3 min. Unpurified PCR2 product (diluted 10,000x in water) was quantified using real time quantitative PCR with P5_Primer, P7_Primer_2 and Internal HsORF3′ TaqMan FAM Probe on a 7500 Fast PCR-System (Applied Biosystems). 300 ng of each PCR2 product were combined in a single tube, mixed and the 316 bp product purified on a 2% agarose gel. The pooled 96 samples were sequenced by the Harvard Medical School Biopolymers Facility on 2 or 3 lanes of an Illumina HiSeq 2000 using 93+7 single end cycles (93 cycles from the “T7-Pep_96_SP” primer, and 7 cycles from the “Index SP” primer) to obtain between 300 and 450 million reads per IP plate.
2.3 PhIP-Seq informatics and statistical analysis
We developed an informatics pipeline for processing the single end, 100 nucleotide sequencing data generated from high throughput PhIP-Seq experiments. Unless otherwise noted, scripts were written in python, and are available online for download from: https://github.com/laserson/phip-stat. Note that these commands are for dispatch to the Platform LSF job scheduler. The count data for each IP was analyzed one sample at a time by comparison to the counts obtained by sequencing the un-enriched T7-Pep library. We used our generalized Poisson significance assignment algorithm [1] to compute −log10 P-values for each peptide/sample pair. Briefly, the IP count distribution for each input count was fitted to a generalized Poisson (GP) distribution. The two GP parameters, λ and θ, were then regressed to form a joint distribution between the IP counts and the GP parameters such that each IP count could be evaluated for its likelihood of enrichment. Subsequent computational analysis was performed in MATLAB software (MathWorks). Reproducibility between each replica pair was assessed as follows. Scatter plots of the log10 of the −log10 P-values were generated, and a sliding window of width 0.05 was moved in steps from −2 to 3 across the x-axis. The mean and standard deviation of the values within this window were calculated at each step and plotted as a function of −log10 P-values (see Supplementary Figure 1.A for example). For each replica pair, we determined the −log10 P-value at which the mean was equal to the standard deviation. A histogram plot of these values are given as Supplementary Figure 1.B. Based on this data, we chose a −log10 P-value of 4 to be our cutoff for considering a peptide to be significantly enriched in an IP experiment. Within each 96-well plate screened, several samples were run in duplicate so that the reproducibility of each run’s automated IPs could be assessed. To exclude peptides that precipitated nonspecifically, we ignored the 1,404 peptides that displayed enrichment with −log10 P-values equal to 3 or greater in 2 or more out of 8 negative control IPs (protein A/G beads only).
For analyses of peptide/ORF-disease association, we set all −log10 enrichment P-values less than 4 equal to 0, and −log10 enrichment P-values greater than 4 equal to 1. The data is thus transformed into a binary (0’s and 1’s) peptide enrichment matrix. This allowed us to compute the P-value of association between each peptide’s enrichment and patient disease status using Fisher’s exact test. To construct a null distribution of Fisher’s test P-values, we randomly permuted the sample labels to obtain a corresponding distribution of Fisher’s test P-values. This was repeated 1,000 times and the resulting P-value frequencies were averaged. An “expected” Fisher’s P-value distribution could then be calculated by summing the null distribution from each −log10 P-value to infinity, thus indicating how many peptide/ORF associations with a P-value at least as extreme, would be expected to occur by chance alone. To find the 10% false discovery rate threshold, we compared the expected Fisher P-values to the observed Fisher P-values, summed from each −log10 P-value to infinity. The P-value at which this ratio was found to equal 1:10 was considered to be the 10% false discovery rate threshold. The number of permutations we performed was based in part on computational considerations and in part on standards of practice. Each permutation is computationally intensive, limiting us in practice to a maximum of ~1,000 per hour on Harvard’s High Performance Computing Cluster. This number of permutations is consistent with published methods.[16, 17]
To identify candidate antigens of viral origin, we first constructed a sequence database composed of all proteins from viruses known to have human tropism. The UniProtKB database was queried on March 9, 2012 for these sequences (collapsed onto clusters of 90% identity) by entering the following URL into a web browser: http://www.uniprot.org/uniref/?query=uniprot%3a(host%3a%22Human+%5b9606%5d%22)+identity:0.9 Sequence motifs for alignment were derived from enriched T7-Pep peptide sequences using the Multiple EM for Motif Elicitation (MEME) web tool (http://meme.nbcr.net/meme/cgi-bin/meme.cgi). These motifs were then exported to the Motif Alignment & Search Tool (MAST) web tool (http://meme.nbcr.net/meme/cgi-bin/mast.cgi), where they were aligned against the viral protein sequence database constructed above.
2.4 ELISA testing of CSF samples
High binding capacity streptavidin-coated 96-well ELISA plates (Pierce, USA) were coated with biotin-Krt75_1 or biotin-scrambled peptide at 5 μg/mL in Tris-buffer saline with 0.05% Tween plus 0.1% bovine serum albumin (TBST-BSA), pH 7.2, for 2 hours at room temperature. After three washes with TBST-BSA, CSF samples were normalized to 5 μg/mL IgG and then incubated in the wells with gentle agitation for 1 hour. Wells were washed three times with TBST-BSA. Secondary goat anti-human HRP (Chemicon, USA) was prepared at 1:20,000 and incubated in the wells for one hour with gentle agitation. After three washes with TBST-BSA, 50 μL of One-Step Ultra TMB ELISA developing reagent (Thermo Scientific, USA) was added to each well and allowed to develop for 5 minutes. The reaction was stopped by addition of 50 μl of 1M sulfuric acid. The optical density of each well was measured at 455nm. Data is reported as fold difference from signal from Krt75_1 versus that from scrambled peptide.
3. Results
3.1 Polyautoreactivity and screen sensitivity
We used PhIP-Seq to analyze 298 antibody repertoires. This collection of samples included 39 sera obtained from newly diagnosed T1D patients, 44 synovial fluid samples and 20 sera from RA patients, and 28 CSF samples and 35 sera from MS patients (including 6 matching CSF/serum sets). Additionally, 73 sera from healthy donors, including a set of 41 age/sex-matched controls for the T1D cohort, were analyzed. To control for differences in fluid composition, we screened synovial fluid samples from 19 individuals with gout or osteoarthritis, as well as CSF from 10 patients with non-MS associated meningitis, subacute sclerosing panencephalitis, or paraneoplastic neurological disorder. Finally, we had previously screened a collection of 29 sera from patients with estrogen and progesterone receptor positive breast cancer (BC), and while analysis of the BC dataset is not presented here, it was utilized to increase power of the antigen-disease specificity tests. Table 1 provides a summary of these samples. A more detailed description can be found in Supplementary Table 1. Peptide immuno-enrichments were quantified using massively parallel DNA sequencing. We considered peptides with a P-value < 10−4 (−log10 P-value greater than 4) as scoring positively above background (Methods, Supplementary Figure 1).[1]
Table 1.
Summary of the samples screened by high throughput PhIP-Seq
| Class | Subclass | Fluid | Total |
|---|---|---|---|
| Type 1 Diabetes | serum | 39 | |
|
| |||
| Multiple Sclerosis | (RRMS/SPMS/PPMS) | serum | 35 |
| CSF | 28 | ||
|
| |||
| Rheumatoid Arthritis | Seropositive | serum | 10 |
| synovial fluid | 22 | ||
| Seronegative | serum | 10 | |
| synovial fluid | 22 | ||
|
| |||
| Healthy Controls | serum | 73 | |
|
| |||
| Non MS CSF Controls | SSPE, PND, Meningitis | CSF | 10 |
|
| |||
| Non RA synovial fluid controls | Gout, OA | synovial fluid | 20 |
|
| |||
| Breast Cancer | ER+/PR+ | serum | 29 |
|
| |||
| Total | 298 | ||
RR, relapse remitting MS; SP, secondary progressive MS; PP, primary progressive MS; SSPE, subacute sclerosing panencephalitis; PND, paraneoplastic neurological disorder; OA, osteoarthritis; ER+, estrogen receptor positive; PR+, progesterone receptor positive. Six sets of MS CSF/serum samples are paired.
We first examined sera 73 healthy donors. In total, 14,604 different peptides were enriched by at least one healthy donor. An overwhelming majority (12,727) of these autoreactivities were “personal” in the sense that they were observed to occur in only one individual (Figure 1A). At the other extreme, we observed a smaller number of peptides that were commonly enriched by healthy individuals. For example, we found that serum from 40% of individuals significantly enriched the same peptide from the activin receptor type IIB (ACVR2B), and serum from 44% of individuals was found to harbor reactivity against a peptide from melanoma antigen family E, 1 (MAGEE1). Notably, these two autoreactivities were not significantly correlated with each other, suggesting that they arise independently. We also looked for evidence of multi-epitope targeting within the database. Whereas we did find convincing examples of antigen-driven responses (e.g. the scleroderma antigen CENPC1, Figure 1B), this was not true for the commonly targeted ACVR2B or MAGEE1. We therefore conclude that these commonly occurring anti-peptide antibodies are most likely cross-reactive and because they occur so frequently in the serum of healthy individuals are unlikely to have a pathological consequence.
Figure 1. Enrichment recurrence and multi-epitope targeting.
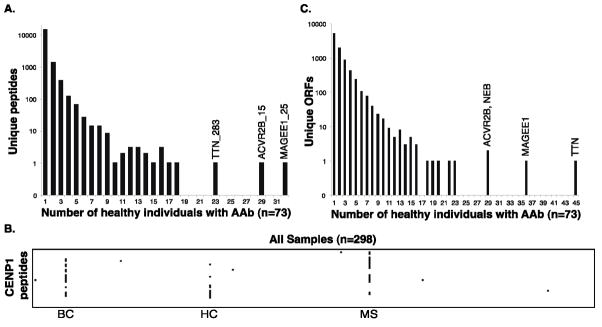
A. Frequency distribution of the 14,604 different peptides enriched above threshold by at least one healthy individual. AAb, autoantibody; TTN, titin; ACVR2B, activin receptor type-2B; MAGEE1, melanoma antigen family E, 1. Number following underscore denotes the 36-residue tile (order is N- to C-terminus).
B. Multi-epitope targeting of the CENPC1 protein. Peptides are organized from top to bottom, N- to C-terminus. Peptide enrichments above threshold are colored black. Three individuals exhibit evidence of multi-epitope (antigen driven) responses. The sample category is denoted below the individual’s data. BC, breast cancer; HC, healthy control; MS, multiple sclerosis.
C. Frequency distribution of the 7,619 different ORFs enriched above threshold by at least one healthy individual. NEB, nebulin.
Patterns of disease-associated autoreactivity may become apparent only in the context of aggregated peptide enrichments, since different individuals may produce antibodies that recognize distinct epitopes of the same protein. We therefore collapsed the peptide enrichment matrix onto an ORF enrichment matrix by taking the most significant value from the set of peptides corresponding to each ORF. Again, if this −log10 P-value was greater than 4, the ORF was considered enriched by the individual. Analysis of ORF enrichments by healthy individuals resulted in a distribution similar to the peptide enrichments, with the majority of significantly enriched ORFs (58%) arising in just one person (Figure 1C). This analysis is biased toward larger proteins being more commonly enriched, and indeed significant reactivity against at least one peptide from titin (TTN, the largest ORF in our library) was observed in 45 of the 73 healthy individuals (Supplementary Discussion).
We next examined a collection of serum samples obtained from 39 newly diagnosed T1D patients. As controls for comparison, we screened sera from 41 healthy donors (matched for age and gender) in the same automated PhIP-Seq run. Titers of clinically utilized autoantibody biomarkers (islet cell cytoplasmic antibody, “ICA”; insulin autoantibody, “IAA”; glutamic acid decarboxylase antibodies, “GADA”; protein tyrosine phosphatase, receptor type, N or insulinoma-associated protein-2 (IA-2) antibodies, “PTPRNA” or “IA-2A”; zinc transporter, member 8 antibodies, “ZnT8A”) were also measured for each of the T1D patients and controls. In order to assess the false negative rate of PhIP-Seq, we compared radioimmunoassay (RIA) measurements for each biomarker in each individual with the corresponding PhIP-Seq ORF enrichment scores. No PhIP-Seq enrichment was observed in any of the patients for insulin or ZnT8A, whereas GAD2 and PTPRN enrichment was observed in some of the T1D patients who had the highest RIA titers for those antigens (Figure 2A and Supplementary Figure 2).
Figure 2. Analysis of T1D and healthy control sera.

A. Sensitivity of PTPRN (IA-2) autoantibody detection by PhIP-Seq compared to RIA in T1D patients (red boxes) and healthy controls (circles). PhIP-Seq values correspond to the most enriched peptide from the PTPRN ORF. A value of >0.5 is considered positive for RIA. None of the HC sera were positive for PTPRN reactivity by RIA or PhIP-Seq. 27 of 39 (69%) T1D patients were positive for PTPRN reactivity by RIA, and 4 of these 27 were positive by PhIP-Seq (15%).
B. Comparison of the total number of unique peptides enriched by individuals of different age groups and disease status. “Youth” individuals are 12 years old or younger; “Adult” individuals are 18 years old or older. Statistical comparisons of the means were performed using the Student’s t test, with one tail.
We reasoned that if the total amount of antibody-self peptide cross-reactivity reflected the complexity of the antibody repertoire, then serum of older individuals should bind a more diverse set of peptides than their younger counterpart. Comparing ages 12 and under (“youth”) with those 18 and older (“adult”), we observed a significant difference in the number of enriched peptides between young and adult healthy controls (P = 0.03; Student’s t test, 1 tail; Figure 2B). However, when we performed the same analysis of the T1D cohort, we found younger T1D patients to be significantly precocious in their development of autoreactive antibodies compared to their age-matched healthy counterpart (P = 0.01).
3.2 Disease-specific autoantibodies
We sought to identify peptide and ORF autoreactivities specifically associated with each autoimmune disease under investigation. For this analysis, each patient sample group was compared to all other samples in the form of a Fisher’s exact test to determine significance of association, considering all enrichments to be as either positive or negative. This analysis was performed for each peptide in the library, and so a distribution of >400,000 Fisher’s P values was obtained. To account for multiple hypothesis testing, we created a null distribution of “expected” Fisher’s P values by randomly permuting the sample labels 1,000 times (Methods). We compared the distribution of expected significance values to that which was actually observed, and then set a threshold for a 10% false discovery rate (FDR). All peptide/ORF autoreactivities that exhibited disease association with this level of confidence are reported in Table 2 (see Supplementary Table 3 for peptide sequences).
Table 2. Peptide/ORF enrichments associated with disease.
All disease-associated autoantigens with a false discovery rate of 10% are listed. ORF-only associations are shown in italics. If the peptide is among a nonrandomly assorted cluster, the name of that cluster is provided. The summary of enrichments provides the total number of individuals from each group that displayed immunoreactivity against the peptide/ORF. T1D, type 1 diabetes; RA, rheumatoid arthritis; MS, multiple sclerosis; HC, healthy controls; BC, breast cancer; OA, osteoarthritis; CSF, cerebrospinal fluid. In parentheses are the total number of individuals from the group. N/A: no longer associated with an expressed sequence. Last column indicates whether protein is predicted to be localized extracellularly: “N”, no; “Y”, yes; “P”, possibly; “L”, likely; “?”, unknown.
| Dz | Gene Symbol | Gene name associated with peptide or ORF | −log10 Fisher P val | Cluster | Summary of positives
|
Extra-cellular | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T1D (39) | RA (64) | MS (57) | HC (73) | BC (29) | OA/Gout (20) | CSF Ctrl (10) | ||||||
| RA | BCOR | BCL6 corepressor | 5.9 | RA1 | 0 | 11 | 0 | 1 | 0 | 1 | 0 | N |
| LOC645453 | ring finger protein, LIM domain interacting; similar to ring finger protein (C3H2C3 type) 6 | 5.3 | RA1 | 0 | 9 | 1 | 0 | 0 | 0 | 0 | N | |
| ATAD5 | ATPase family, AAA domain containing 5 | 4.1 | RA1 | 0 | 6 | 0 | 0 | 0 | 0 | 0 | N | |
| Hcn3 | hyperpolarization activated cyclic nucleotide-gated potassium channel 3 | 4.0 | 0 | 7 | 0 | 0 | 0 | 1 | 0 | P | ||
| FAM135A | family with sequence similarity 135, member A | 3.4 | 0 | 7 | 1 | 0 | 0 | 1 | 0 | ? | ||
| HRNR | hornerin | 3.4 | 0 | 7 | 0 | 0 | 1 | 0 | 0 | N | ||
| ADAM33 | ADAM metallopeptidase domain 33 | 3.4 | RA2 | 0 | 7 | 0 | 0 | 0 | 1 | 0 | L | |
| PTK2 | PTK2 protein tyrosine kinase 2 | 3.4 | RA2 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | N | |
| SNRPB | small nuclear ribonucleoprotein polypeptides B and B1 | 3.1 | RA2 | 0 | 8 | 1 | 1 | 1 | 1 | 0 | N | |
| KRT33B | keratin 33B | 2.7 | RA2 | 0 | 5 | 0 | 0 | 0 | 1 | 0 | N | |
| ATXN2 | ataxin 2 | 2.7 | RA2 | 0 | 5 | 0 | 0 | 0 | 1 | 0 | N | |
| S100A11 | S100 calcium binding protein A11; S100 calcium binding protein A11 pseudogene | 2.7 | 0 | 5 | 0 | 0 | 0 | 1 | 0 | N | ||
| Lrba | LPS-responsive vesicle trafficking, beach and anchor containing | 2.7 | RA2 | 0 | 5 | 0 | 0 | 0 | 1 | 0 | N | |
| CREB3L1 | cAMP responsive element binding protein 3-like 1 | 2.7 | RA2 | 0 | 5 | 0 | 0 | 0 | 1 | 0 | N | |
| SEPT8 | septin 8 | 2.7 | RA2 | 0 | 5 | 0 | 0 | 0 | 1 | 0 | N | |
|
| ||||||||||||
| MS | Krt75 | keratin 75 | 6.7 | MS1 | 0 | 0 | 9 | 0 | 0 | 0 | 0 | N |
| TRIO | triple functional domain (PTPRF interacting) | 5.9 | MS1 | 0 | 0 | 8 | 0 | 0 | 0 | 0 | N | |
| Sox17 | SRY (sex determining region Y)-box 17 | 5.4 | 0 | 1 | 13 | 5 | 1 | 0 | 0 | N | ||
| LOC388182 | LOC388182 | 5.1 | MS1 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | ? | |
| METTL23 | methyltransferase like 23 | 5.1 | MS1 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | ? | |
| DENND4C | DENN/MADD domain containing 4C | 5.1 | MS1 | 0 | 1 | 9 | 0 | 0 | 0 | 1 | N | |
| PPARGC1A | peroxisome proliferator-activated receptor gamma, coactivator 1 alpha | 5.0 | 0 | 0 | 8 | 0 | 1 | 0 | 0 | N | ||
| SFRS16 | splicing factor, arginine/serine-rich 16 | 4.4 | MS1 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | N | |
| KIAA1045 | KIAA1045 | 4.4 | MS1 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | ? | |
| FRMD4B | FERM domain-containing protein 4B | 4.4 | MS1 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | N | |
| N/A | N/A | 4.4 | MS1 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | ? | |
| RIMS2 | regulating synaptic membrane exocytosis 2 | 4.3 | MS1 | 0 | 1 | 7 | 0 | 0 | 0 | 0 | Y | |
| PPP1R10 | protein phosphatase 1, regulatory (inhibitor) subunit 10 | 3.8 | 1 | 2 | 15 | 9 | 4 | 1 | 0 | N | ||
| Baz2a | bromodomain adjacent to zinc finger domain, 2A | 3.6 | MS1 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | N | |
| tes | testis derived transcript (3 LIM domains) | 3.6 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | P | ||
| USP11 | ubiquitin specific peptidase 11 | 3.1 | 0 | 0 | 6 | 0 | 2 | 0 | 0 | N | ||
We first examined peptide/ORF autoreactivities specifically associated with RA (Table 2, Figure 3). Of the 16 peptides with an FDR <10%, 11 assorted with patients non-randomly as two peptide clusters, “RA1” and “RA2”, composed of 3 and 8 peptides, respectively. Interestingly, none of the RA-associated enrichments appeared to correlate with seropositivity (reactivity against rheumatoid factor and/or ACPA; Figure 3.B). Despite attempts to uncover a shared sequence motif among RA1- or RA2-clustered peptides using blastp and MEME algorithms, none was identified.[18]
Figure 3. RA associated peptides and their clusters.

A. Permutation analysis of peptide enrichments associated with RA. “Observed” bars indicate the number of peptides associated with RA at a given P-value by Fisher’s exact test. “Expected” bars show the number of peptides expected to have a −log10 Fisher P-value at least that extreme due to chance alone (as determined by permuting sample labels).
B. RA1 and RA2 peptide enrichment heat map illustrating nonrandom segregation of above-threshold peptide enrichments and RA patients. −Log10 P-values less than 4 are white and greater than 4 are black. Patients are organized by their seropositivity, and by their RA1/RA2 status.
We next examined the set of 15 peptides that were enriched specifically by MS patients with a disease association FDR of <10% (Figure 4A, Table 2). Eleven of these peptides assorted non-randomly among a subset of MS patients, and motif discovery revealed a 7 amino acid sequence motif contained in all of them (“MS1”, Figures 4B–D). Notably, a motif nearly identical to MS1 was previously identified by Cepok et al. in a phage screen of MS CSF samples,[19] and they reported an alignment with the BRRF2 protein of the Epstein-Barr virus, a pathogen repeatedly implicated in MS pathogenesis. We performed an alignment of our MS1 motif against the UniProt database of all proteins from viruses with human tropism, collapsed onto 90% identity clusters (7,546 UniRef sequences; 656 unique taxa), and also found the best alignment to be with the EBV BRRF2 protein (E value = 1.2; sequence: PAASRSK).
Figure 4. MS associated peptides share a sequence motif.
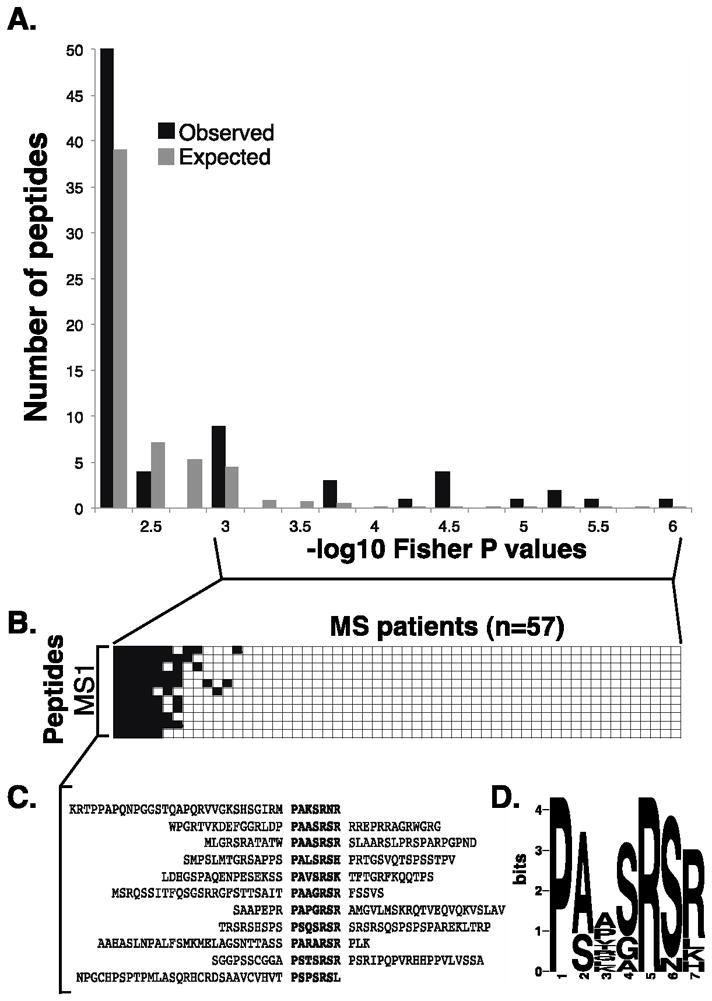
A. Permutation analysis of peptide enrichments associated with MS. “Observed” bars indicate the number of peptides associated with MS at a given P-value by Fisher’s exact test. “Expected” bars show the number of peptides expected to have a −log10 Fisher P-value at least that extreme due to chance alone (as determined by permuting sample labels).
B. Peptide enrichment heat map (as in Figure 1) illustrating nonrandom segregation of MS1 peptide enrichments (rows) and MS patients (columns). −Log10 P-values of enrichment less than 4 are white and greater than 4 are black. Patients are organized by their MS1 status.
C. Alignment of the co-segregated peptides reveals a shared epitope.
D. MS associated epitope (MS1) motif logo, calculated from the peptides in C (MEME software).
We considered the possibility that a peptide containing the MS1 motif might have clinical utility in the form of an ELISA assay. To this end, we immobilized the peptide which performed best in our PhIP-Seq screen, Krt75_1 (9 positives of 57 MS samples, versus 0 positive of 239 non MS samples). Of 25 MS CSF samples tested by ELISA, 3 were positive, compared to 0 of 19 CSF samples from individuals with other inflammatory neurological diseases (Supplementary Figure 3A). Eight of the ELISA-tested MS samples had also been screened using PhIP-Seq, and we found the latter method to have a greater sensitivity (Supplementary Figure 3B).
3.3 Analysis of matched MS samples
We obtained six sets of individually matched MS CSF-serum samples. Each of these samples was screened in duplicate, and we considered peptides that were reproducibly enriched with a −log10 P-value greater than 3 in both replicates from either compartment. For each of these MS patient pairs, we plotted the average −log10 P-value for each peptide’s CSF enrichment against the average serum enrichment (Figure 5). In all cases we observed a strong correlation in the enrichment profiles between these two fluid compartments. A majority of the enrichments were found in both serum and CSF, with a trend toward stronger enrichment in the serum. In several cases, however, we did find peptides that were more highly enriched in the CSF. For example, CSF from patient 9292 enriched two homologous peptides from interferon alpha 5 and 14 much more significantly than serum from the same patient (Figure 5A; Supplementary Table 2). This is unlikely to reflect cross-reactivity of inhibitor antibodies to therapeutic interferon beta, however, as the homologous peptide from interferon beta was not enriched in either compartment.
Figure 5. Analysis of MS patient CSF/serum pairs.

Scatter plots of matched samples from the same individuals. Each sample was analyzed in duplicate and the average −log10 P-value is plotted for peptides enriched by more than −log10 P value of 3 in both duplicates.
A. Patient 9292 peptide enrichments in CSF versus serum, and enrichment of nearly identical peptides from IFN-α5/14 specifically in the CSF.
B. Patient 10894 peptide enrichments in CSF versus serum, and CSF-specific enrichment of the JCV motif is illustrated.
C. Patient 8911 peptide enrichments in CSF versus serum. D. PhIP-Seq serum profile in patient 8911 taken at two time points.
We systematically examined all the CSF-specifically enriched peptides (enriched by CSF antibodies with −log10 P-value of at least 3 greater than the corresponding serum enrichment) that were identified in the six patients (Supplementary Table 2). Motif discovery was performed on each set of CSF-specifically enriched peptides, and one motif was uncovered for patient 10894 (Figure 5B and Supplementary Table 2). This motif was searched into the database of human viruses, and a significant alignment was found with the major capsid protein VP1 of the JC polyomavirus (JCV; E value = 0.03; sequence: RRVKNP). Similar to EBV, JCV infection is highly prevalent, infecting 70 to 90 percent of humans. Also of note, JCV can cross the blood-brain barrier into the central nervous system, where it infects oligodendrocytes and astrocytes.[20]
Some MS patients exhibited little or no CSF-specific autoreactivity, an example of which is shown in Figure 5C (patient 8911). This patient, however, did have serum samples drawn on two separate occasions within one year, which allowed us to examine the persistence of PhIP-Seq enrichments over this length of time. The scatterplot (Figure 5D) reveals minimal time-dependent changes.
4. Discussion
In this study we report the first large scale PhIP-Seq screen of individuals with different autoimmune diseases for direct comparison to healthy controls and to each other. These data provide an unbiased, proteomic-scale assessment of precise autoreactivities found within 298 independent antibody repertoires. The vast majority of autoreactivities were individually unique, lending support to the notion that each person possesses a unique autoantibody fingerprint, of which the impact on phenotype remains to be explored. It is interesting to note that as our database of enriched peptides grows, so will the number of peptides recurrently enriched by a small fraction of the population - a situation analogous to the ongoing identification of progressively less common alleles in sequenced human genomes. Screening large numbers of genotyped individuals may additionally reveal correlations between autoreactivities and HLA haplotypes, antibody variable domain alleles, and other immunogenetic modifiers.
Our unbiased method revealed a large number of novel peptide autoreactivities, but when compared to RIA-determined titers of known autoantibodies, appears to suffer from relatively low sensitivity. We detected no anti-insulin antibodies in the T1D patients, with the important caveat that we did not charcoal-extract insulin from the serum prior to performing PhIP-Seq, which is standard protocol for the RIA assay. It is therefore possible that the anti-insulin antibodies were occupied by endogenous or injected insulin and therefore not accessible for peptide binding. Additionally, ZnT8 RIA titers were obtained using a fusion protein consisting of two allelic variants of the immunodominant epitope, and so the single sequence present in T7-Pep (the “CR” variant) may have contributed to the low sensitivity. The most important source of the high false negative rate, however, is most likely the limited amount of conformational structure inherent to 36 amino acid peptide tiles. The findings presented here thus highlight the need for improved display libraries that include more complex epitopes. Despite this limitation, we were able to observe a significantly accelerated polyautoreactivity in younger T1D patients compared with their matched controls. To our knowledge, this finding has not been explicitly reported previously.
Our RA screens uncovered a set of novel disease-associated anti-peptide antibodies. Fifteen novel autoantigen reactivities were identified, which can be clustered into two antigen groups, called here RA1 and RA2. Thirteen out of the 64 RA patients (20%) exhibited immunoreactivity against at least one RA1 peptide, compared to 3 of 232 non-RA individuals (1.3%). In addition, 16 of the RA patients (25%) exhibited immunoreactivity against at least one RA2 peptide, compared to 6 non-RA individuals (2.6%). Taken together, 26 of the RA patients (41%) exhibited immunoreactivity against at least one RA1 or RA2 peptide, compared to 9 non-RA individuals (3.9%; P = 7.8×10−13, Fisher’s exact test, one tail); 16 RA patient samples enriched at least two peptides from RA1 or RA2. To our knowledge, none of these peptides have been previously implicated by prior serological studies in RA. It is important to note that the T7-displayed peptides are not subject to post-translational modification, such as citrullination; therefore we would not have expected to detect antibodies against known RA antigens such as citrullinated fibrinogen and citrullinated alpha-enolase.
Much effort has been invested in the search for antibody specificities in the CSF of MS patients. Cortese et al. used a library of constrained nonamers to find mimitopes for CSF antibodies in 2 MS patients.[21] One of the sequences (KPPNP) is contained within several of the T7-Pep library peptides. Of them, one peptide from XP_499190.1 (SQQWRENPRTQNQSAVERKPPNPEPVSSGEKTPEPR), was enriched by 6 of 57 MS patients and 9 of 235 non-MS individuals, and so was significantly associated with MS (Fisher’s P value = 0.05). Rand et al. used a small collection of CSF samples from MS patients to screen a phage library of random hexamers.[22] They uncovered an enriched sequence (RRPFF) in several individuals with MS, and reported alignment with the heat shock protein αB crystallin and the Epstein-Barr virus nuclear antigen (EBNA-1). In our study, the most commonly enriched peptide by healthy individuals, MAGEE1_25, contains this precise sequence (RAFAEGWQALPHFRRPFFEEAAAEVPSPDSEVSSYS; Figure 1A). MAGEE1_25 was enriched with similar frequency by serum of MS patients and healthy controls (17/29 MS and 32/73 HC). Of the six MS patients for which we had matching CSF and serum samples, two had MAGEE1_25 antibodies. Both of these patients exhibited stronger enrichment in their serum than in their CSF. Taken together, we believe the PhIP-Seq data are consistent with a scenario in which RRPFF antibodies occur with equal frequency in the serum of MS and healthy individuals, and suggest that they are unlikely to be produced specifically within the CNS. In contrast, the BRRF2 epitope (MS1) was targeted specificity by patients with MS. Importantly, the MS1 antibodies exhibited a notable degree of polyspecificity for self peptides (Figure 4B). While this manuscript was in preparation, Srivastava et. el. reported that serum autoantibodies recognizing the extracellular loop of the potassium channel KIR4.1 (residues 83–120) could be detected in 46.9 percent of patients with multiple sclerosis.[23] A KIR4.1 peptide spanning residues 88–123 is present in T7-Pep, but was not significantly enriched by any patients in our study.
The findings presented here point to the accumulating value of high throughput, low cost PhIP-Seq screening. As the sample size of our database grows, so will the power to detect rare, yet significantly disease-associated autoantibodies. Quantitative elucidation of these diverse autoreactivities will be particularly important for understanding complex, heterogeneous autoimmune disease pathogeneses. In the future, methods that query linear and conformational epitopes, post translational modifications, and T cell epitopes from both human and pathogen proteomes will eventually provide us with a more comprehensive characterization of the adaptive immune system and its role in disease.
Supplementary Material
Acknowledgments
We would like to thank Stewart Rudnicki at the Institute of Chemistry and Cell Biology (ICCB) at Harvard Medical School for helping to automate PhIP-Seq. Paul I W de Bakker provided valuable statistical direction. We also thank the Dana-Farber/Harvard Cancer Center (DF/HCC) Specialized Programs of Research Excellence (SPORE) in breast cancer for providing the breast cancer patient sera, the Human Brain and Spinal Fluid Resource Centre (VA Greater Los Angeles Healthcare System, West Los Angeles Healthcare Center) for providing CSF, and the members of the Belgian Diabetes Registry who participated in the recruitment of diabetic patients for the current study. This work was supported in part by grants from the United States Department of Defense (W81XWH-10-1-0994 and W81XWH-04-1-0197), and a HITI/Helmsley Trust Pilot Grant in Type 1 Diabetes to S.J.E. N.L.S. is a fellow of the Susan G. Komen for the Cure Foundation. L.Q. receives support from Fondo de Investigaciones Sanitarias (CM 09/00017), Carlos III Institute of Health, Spain. K.C.O. receives support from the Nancy Davis Foundation. P.A.N. was supported in part by the Cogan Family Foundation. I.W and G.M acknowledge the Belgian Fund for Scientific Research (FWO Vlaanderen; senior clinical research fellowship). S.J.E. is an investigator with the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Larman HB, Zhao Z, Laserson U, Li MZ, Ciccia A, Gakidis MA, Church GM, Kesari S, Leproust EM, Solimini NL, Elledge SJ. Autoantigen discovery with a synthetic human peptidome. Nat Biotechnol. 2011;29:535–541. doi: 10.1038/nbt.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larman MSHB, Nazareno R, Lam T, Sauld J, Steen H, Kong SW, Pinkus JL, Amato AA, Elledge SJ, Greenberg SA. Cytosolic 5′-Nucleotidase 1A Autoimmunity in Sporadic Inclusion Body Myositis. Annals of neurology. 2012 doi: 10.1002/ana.23840. In press. [DOI] [PubMed] [Google Scholar]
- 3.Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L, Eisenbarth GS. Prediction and prevention of Type 1 diabetes mellitus. Journal of diabetes. 2011;3:48–57. doi: 10.1111/j.1753-0407.2010.00102.x. [DOI] [PubMed] [Google Scholar]
- 5.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164–174. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Link H, Huang YM. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. J Neuroimmunol. 2006;180:17–28. doi: 10.1016/j.jneuroim.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 8.Saag KG, Teng GG, Patkar NM, Anuntiyo J, Finney C, Curtis JR, Paulus HE, Mudano A, Pisu M, Elkins-Melton M, Outman R, Allison JJ, Suarez Almazor M, Bridges SL, Jr, Chatham WW, Hochberg M, MacLean C, Mikuls T, Moreland LW, O’Dell J, Turkiewicz AM, Furst DE. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum. 2008;59:762–784. doi: 10.1002/art.23721. [DOI] [PubMed] [Google Scholar]
- 9.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM, Raskin P, Rodriguez H, Schatz DA, Wherrett D, Wilson DM, Lachin JM, Skyler JS. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 11.De Grijse J, Asanghanwa M, Nouthe B, Albrecher N, Goubert P, Vermeulen I, Van Der Meeren S, Decochez K, Weets I, Keymeulen B, Lampasona V, Wenzlau J, Hutton JC, Pipeleers D, Gorus FK. Predictive power of screening for antibodies against insulinoma-associated protein 2 beta (IA-2beta) and zinc transporter-8 to select first-degree relatives of type 1 diabetic patients with risk of rapid progression to clinical onset of the disease: implications for prevention trials. Diabetologia. 2010;53:517–524. doi: 10.1007/s00125-009-1618-y. [DOI] [PubMed] [Google Scholar]
- 12.Vermeulen I, Weets I, Asanghanwa M, Ruige J, Van Gaal L, Mathieu C, Keymeulen B, Lampasona V, Wenzlau JM, Hutton JC, Pipeleers DG, Gorus FK. Contribution of antibodies against IA-2beta and zinc transporter 8 to classification of diabetes diagnosed under 40 years of age. Diabetes Care. 2011;34:1760–1765. doi: 10.2337/dc10-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wenzlau JM, Liu Y, Yu L, Moua O, Fowler KT, Rangasamy S, Walters J, Eisenbarth GS, Davidson HW, Hutton JC. A common nonsynonymous single nucleotide polymorphism in the SLC30A8 gene determines ZnT8 autoantibody specificity in type 1 diabetes. Diabetes. 2008;57:2693–2697. doi: 10.2337/db08-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’connor KC, Mclaughlin KA, De Jager PL, Chitnis T, Bettelli E, Xu C, Robinson WH, Cherry SV, Bar-Or A, Banwell B, Fukaura H, Fukazawa T, Tenembaum S, Wong SJ, Tavakoli NP, Idrissova Z, Viglietta V, Rostasy K, Pohl D, Dale RC, Freedman M, Steinman L, Buckle GJ, Kuchroo VK, Hafler DA, Wucherpfennig KW. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med. 2007;13:211–217. doi: 10.1038/nm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor protocols. 2010;2010 doi: 10.1101/pdb.prot5448. pdb prot5448. [DOI] [PubMed] [Google Scholar]
- 16.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Efron B, Tibshirani R. An introduction to the bootstrap. Chapman & Hall; New York: 1993. [Google Scholar]
- 18.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Bio. 1994;2:28–36. [PubMed] [Google Scholar]
- 19.Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Bussow K, Sommer N, Hemmer B. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest. 2005;115:1352–1360. doi: 10.1172/JCI23661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, Dugan A, Stanifer M, Bhatnagar A, Kroeze WK, Roth BL, Atwood WJ. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306:1380–1383. doi: 10.1126/science.1103492. [DOI] [PubMed] [Google Scholar]
- 21.Cortese I, Tafi R, Grimaldi LM, Martino G, Nicosia A, Cortese R. Identification of peptides specific for cerebrospinal fluid antibodies in multiple sclerosis by using phage libraries. Proc Natl Acad Sci USA. 1996;93:11063–11067. doi: 10.1073/pnas.93.20.11063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rand KH, Houck H, Denslow ND, Heilman KM. Molecular approach to find target(s) for oligoclonal bands in multiple sclerosis. J Neuro Neurosurg Psychiatr. 1998;65:48–55. doi: 10.1136/jnnp.65.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Srivastava R, Aslam M, Kalluri SR, Schirmer L, Buck D, Tackenberg B, Rothhammer V, Chan A, Gold R, Berthele A, Bennett JL, Korn T, Hemmer B. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med. 2012;367:115–123. doi: 10.1056/NEJMoa1110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.