

Abstract
The maternal skeleton resorbs during lactation to provide calcium to milk and the lost mineral content is restored after weaning. The changes are particularly marked in Ctcgrp null mice, which lose 50% of spine mineral content during lactation but restore it fully. The known calciotropic hormones are not required for skeletal recovery to occur; therefore, unknown factors that stimulate bone formation may be responsible. We hypothesized that the genes responsible for regulating postweaning bone formation are differentially regulated in bone or marrow, and this regulation may be more marked in Ctcgrp null mice. We confirmed that Ctcgrp null mice had twice as many osteoclasts and 30–40% fewer osteoblasts as compared with wild-type mice during lactation but no deficit in osteoblast numbers after weaning. Genome-wide microarray analyses on tibial RNA showed differential expression of 729 genes in wild-type mice at day 7 after weaning vs prepregnancy, whereas the same comparison in Ctcgrp null mice revealed only 283 genes. Down-regulation of Wnt family inhibitors, Sost and Dkk1, and inhibition of Mef2c, a sclerostin stimulator, were observed. Ctsk, a gene expressed during osteoclast differentiation, and Igfbp2, which stimulates bone resorption, were inhibited. Differential regulation of genes involved in energy use was compatible with a net increase in bone formation. The most marked changes occurred in genes not previously associated with bone metabolism. In conclusion, the postlactation skeleton shows dynamic activity with more than 700 genes differentially expressed. Some of these genes are likely to promote bone formation during postweaning by stimulating the proliferation and activity of osteoblasts, inhibiting osteoclasts, and increasing energy use.
In women, bone mineral content (BMC) is reduced by 5%–10% during 3–6 months of exclusive lactation, but this is restored fully within 6–12 months after weaning (1). Despite this transitory loss of skeletal mass and strength, lactation is associated with a long-term neutral or protective effect against the development of low bone mass, fractures, or osteoporosis (2–5). Similarly, rodents typically lose 20%–25% of BMC during 3 weeks of lactation and regain it fully after 2–3 weeks (2, 6).
A brain-breast-bone circuit coordinates the uncoupling of osteoclast and osteoblast activity during lactation to promote bone loss (6). The pituitary releases prolactin and oxytocin in response to suckling; the hypothalamus responds to suckling and prolactin by suppressing estradiol production by the ovaries; mammary tissue releases PTHrP in response to suckling and high prolactin; and osteoclasts proliferate and activate in response to PTHrP, low estradiol, and possibly oxytocin and other factors (1, 7). Clinical and animal data confirm that PTHrP and low estradiol promote bone loss through osteoclast-mediated bone resorption and osteocytic osteolysis (1, 2, 8–11). Ctcgrp null mice, which lack the gene encoding calcitonin and calcitonin gene-related peptide-α, lose twice as much BMC as wild-type (WT) mice during lactation (attributable to the loss of calcitonin and increased mammary expression of PTHrP) but restore the skeleton fully within 18 days after weaning (6).
During postweaning unidentified mechanisms are triggered that reverse the uncoupling to promote bone formation and suppress resorption. The remarkable nature of postweaning bone recovery is that it is rapid and complete, unlike the slow and partial recovery that the adult skeleton achieves after diverse causes of bone loss (2, 12–17). Identifying the factors that stimulate bone formation in the postweaning interval may lead to new treatments for bone loss and skeletal fragility.
Few clinical studies have examined the mechanisms that are invoked after weaning. Ovulatory menses often resume prior to weaning and return of estradiol levels to normal likely facilitates skeletal recovery. However, this restoration of estradiol alone is unlikely to explain the speed and completeness of recovery after lactation because bone loss associated with GnRH analog-induced estradiol deficiency in reproductive-age women is not fully restored after a year of normal ovarian function (2). Furthermore, when estradiol is given to peri- or postmenopausal women who are estrogen deficient, both bone formation and resorption are suppressed (18, 19).
Forced weaning in rodents provokes rapid apoptosis of osteoclasts, whereas osteoblast numbers surge above the high values that were already present during lactation (20–22). This leads to thickening of thinned trabecular plates and cortices, filling of osteocyte lacunae, and restoration of depleted mineral content (6, 21, 23). Bone biomarkers confirm marked suppression of bone resorption in this period compared with lactation, whereas bone formation markers remain at high levels (21, 24). By dual x-ray absorptiometry (DXA), the spine, hindlimb, and whole-body BMC return to normal or above within 14 days for Black Swiss mice and by 21–28 days in CD1 and C57BL6/J (6, 21, 24). Bone microarchitecture, as assessed by microcomputed tomography, returns to normal at differing rates for each skeletal site (25). Studies in genetically altered mice indicate that PTH, PTHrP, calcitonin, and the vitamin D receptor are not required for skeletal recovery after lactation (2, 6, 24, 26, 27); thus, the factors that contribute to skeletal recovery remain unclear.
Our overarching hypothesis is that weaning triggers systemic and local signals that potently suppress bone resorption (by inducing osteoclast apoptosis and inhibiting osteoclast function) and activate bone formation (by stimulating the proliferation and function of mature osteoblasts and osteocytes). The primary signals may be intrinsic to bone or marrow and act locally on bone cells and their precursors. Alternatively, the primary signals may arise extrinsic to bone (in hypothalamus, pituitary, mammary tissue, ovary, etc) and are released into the circulation as hormones or cytokines that act distally on bone cells.
For the present report, we hypothesized that in response to both intrinsic and extrinsic factors, the genes responsible for suppressing bone resorption and stimulating bone formation will show marked changes within bone between prepregnancy and the postweaning recovery phase in WT mice. Moreover, because Ctcgrp null mice lose twice as much BMC during lactation as WT mice but still regain it fully, the changes in gene expression might be more prominent in Ctcgrp null mice. To the best of our knowledge, this is the first report that examines, in a nonbiased manner, genome-wide changes in gene expression during postweaning.
Materials and Methods
Animal husbandry
The originally described Ctcgrp null mice (28) were backcrossed into Black Swiss (Taconic, Germantown, New York) for more than 10 generations (6). Timed matings of WT and Ctcgrp null sisters were done. Mice had ad libitum access to a diet containing 1% calcium and 0.75% phosphorus. The Institutional Animal Care Committee of Memorial University of Newfoundland approved all procedures involving animals.
Reproductive cycles and data collection time points
The animals were 10 weeks of age at the start of the experiments. We established this as the baseline for pregnancy/lactation studies involving Black Swiss mice because BMC of whole body, spine, and hindlimb reaches a stable plateau by this age (6, 29). The full reproductive cycle includes 5–10 days prepregnancy (baseline) interval, 18.5 days of pregnancy, 21 days of lactation, and 21 days of postweaning recovery. In this report the main time points were baseline, midlactation, end of lactation, and day 7 of postweaning.
Histomorphometry
Lumbosacral vertebrae were harvested at midlactation, whereas tibiae were obtained at midlactation and the end of lactation. After removing all soft tissues, the bones were fixed in 10% buffered formalin and embedded in methacrylate. Histomorphometric analysis of 5-μm toluidine blue-stained sections was carried out across the width of the secondary spongiosa, commencing 370 μm below the growth plate and extending for 1.11 mm as previously described using the Osteomeasure system (Osteometrics, Decatur, Georgia) (24, 26, 30, 31).
RNA extraction
Maternal tibiae with marrow intact were harvested at baseline and day 7 of postweaning and then snap frozen in liquid nitrogen. Total RNA was extracted and purified using the RNeasy midikit (QIAGEN, Toronto, Ontario, Canada). RNA quality was confirmed with the Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, California).
Microarray
RNA was analyzed at the Centre for Applied Genomics, Hospital for Sick Children (Toronto, Ontario, Canada) using the Mouse Gene ST 1.0 array (Affymetrix, Santa Clara, California). Primary data analysis was done at the Statistical Analysis Core Facility of the Centre for Applied Genomics. Raw data were normalized using the robust multiarray average algorithm (32), and differentially expressed genes were identified using the local-pooled-error test (33). The false discovery rate (34) was set at 0.01 such that genes with adjusted P < .01 were considered to be statistically significant. Pathways and network analysis of the microarray data were generated using IPA (Ingenuity Systems, Inc, Redwood City, California).
Real-time quantitative PCR (qPCR)
We used Taqman gene expression assays and Fast Advanced master mix from Applied Biosystems/Life Technologies (Burlington, Ontario, Canada) to analyze the expression of selected genes (Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org). Details of methods have been previously reported (6, 24, 35). Briefly, cDNA was synthesized using the Taqman high-capacity cDNA reverse transcription kit (Applied Biosystems), and single-plex qPCRs were run in triplicate on the ViiA 7 real-time PCR system (Applied Biosystems) (35, 36). Relative expression was determined from the threshold cycle (CT) normalized to the reference gene.
Statistical analysis
Histomorphometry data were analyzed by a 2-way ANOVA using StatPlus:Mac Professional 2009, build 5.8.3.8 (AnalystSoft Inc, Vancouver, British Columbia, Canada). Tukey's test determined which pairs of means differed significantly from each other and are presented as mean ± SE. The qPCR data were analyzed by the comparative CT method (ΔΔCT) (37) and reported as mean ± SD.
Results
Histomorphometry
Ctcgrp null mice have higher bone mass than WT mice as young adults; this difference is more readily discerned by microcomputed tomography and histomorphometry than DXA (6, 28). In our prior studies that used longitudinal DXA measurements, the BMC of the trabecular-rich lumbar spine declined by 51.6 ± 4.6% in Ctcgrp null mice vs 24.4 ± 5.4% in WT mice by the end of lactation (P < .001) (6). In the current study, histomorphometry at midlactation revealed trabecular bone volume (BV/TV) in the vertebrae of 14.33 ± 1.38% in WT mice and 13.49 ± 2.12% in Ctcgrp null mice. At this time point, compared with their WT sisters, Ctcgrp null mice showed 30%–40% lower osteoblast parameters (osteoblast number, osteoblast surface, osteoid surface) and doubled osteoclast number and surface (Figure 1), even though there was no difference in these parameters at baseline (28).
Figure 1.

Histomorphometry of vertebrae at midlactation. Osteoblast parameters were halved in lactating Ctcgrp null vertebrae compared with WT. A, osteoblast number (NOb/BS). B, Osteoblast surface (ObS/BS). C, Osteoid surface (OS/BS). Osteoclast parameters were more than double that of WT in Ctcgrp null vertebrae. D, Osteoclast number (NOc/BS). E, Osteoclast surface (OcS/BS). These findings are consistent with the doubling in BMC loss experienced by Ctcgrp null mice.
In our prior study, the BMC of the hind limb fell below prepregnancy baseline by 28.4 ± 2.9% in Ctcgrp nulls vs 12.9 ± 3.4% in WT mice at the end of lactation (P < .001) (6). In the current study, by histomorphometry at midlactation, the BV/TV of the tibia was 5.17 ± 1.49% in the WT mice vs 2.88 ± 0.87% in the Ctcgrp null mice (P = NS). In this Black Swiss genetic background, the BV/TV is typically 14%–16% in 3- to 4-month old mice (38, 39). Similar to the vertebrae, in Ctcgrp null tibiae, the osteoblast-related parameters were reduced by 30%–40% compared with WT mice (Figure 2, A–C, left panels), and osteoclast number and surface were more than 2-fold higher than in the WT mice (Figure 2, D and E, left panels).
Figure 2.

Histomorphometry of tibiae at midlactation and onset of weaning. Similar to the vertebrae, osteoblast parameters (A–C, left panels) were halved, whereas osteoclast parameters (D and E, left panels) were doubled in lactating Ctcgrp null mice vs WT mice. These findings are consistent with Ctcgrp null mice losing twice as much BMC as WT mice during lactation. At the onset of postweaning, osteoblast parameters (A–C, right panels) increased. The relative increase was greater in Ctcgrp null mice such that osteoblast parameters no longer differed between genotypes. In Ctcgrp null mice, osteoclast parameters (D and E, right panels) remained double the WT values, but osteoclast surface (E, right panel) declined significantly in both genotypes from midlactation. NOb/BS, osteoblast number; NOc/BS, osteoclast number; ObS/BS, osteoblast surface; OcS/BS, osteoclast surface; OS/BS, osteoid surface.
Additional tibiae were examined at the end of natural lactation (21 days), at which point BMC is at a trough value and recovery of bone mass has begun. Osteoblasts more than doubled in number in both genotypes, whereas osteoblast and osteoid surface increased to cover 40%–50% of the trabecular bone surface (Figure 2, A–C, right panels). Osteoclast parameters remained twice normal in Ctcgrp null mice compared with WT mice (Figure 2, D and E, right panels), although osteoclast surface fell in both genotypes to half that of midlactation (Figure 2E, right panel).
Microarray on tibiae
We performed a genome-wide microarray analysis on RNA extracted from tibiae at prepregnancy and day 7 after weaning. At this time point, the bone formation markers osteocalcin and propeptide of type 1 collagen are increased (24, 40), and WT and Ctcgrp null mice have both recovered 40%–50% of the deficit in bone mass (6). In WT mice, 729 genes showed a 1.5-fold or greater differential expression at day 7 after weaning compared with baseline (Supplemental Table 2), whereas 324 genes showed a 2-fold or greater differential expression. The largest fold changes occurred in genes not previously described in bone or marrow, including a 28.6-fold increase in Mela [melanoma associated antigen (MAGE)], and a 6- to 8-fold increases in the 19 different Mup genes [(major urinary proteins (MUPs)]. Changes in known genes that could cause increased bone formation included a 1.65-fold down-regulation of Sost (sclerostin) and a 1.87-fold down-regulation of Dkk1 (Dickkopf-1). In addition, a 1.61-fold down-regulation of Ctsk (cathepsin K) could explain decreased bone resorption or fewer osteoclasts during postweaning. Notably, there was no change in the genes encoding PTHrP or PTH/PTHrP receptor. Several chondrocytic genes were down-regulated including Acan (aggrecan, −2.31-fold), Matn3 (matrilin 3, −3.26-fold), col10a1 (collagen type X α1, −4.62-fold), and col2a1 (collagen type 2 α1, −2.42-fold).
Several genes involved in energy use showed differential expression, including a 3.12-fold increase in Ppargc1a (peroxisome proliferator-activated receptor-γ coactivator-1α or Pgc1α), a 2.37-fold increase in Pdk4 (pyruvate dehydrogenase kinase, isoenzyme 4), a 2.22-fold increase in Ucp3 (uncoupling protein 3), and a 1.84-fold increase in Slc2a4 (glucose transporter type 4, insulin responsive or GLUT-4), which is the key transporter for glucose in osteoblasts.
In Ctcgrp null mice, 283 genes showed differential expression on the genome-wide microarray, with the largest change being a 105-fold increase in expression of Mela (Supplemental Table 3). The genes showing differential expression were largely a subset of the genes that showed significant changes in WT mice. Mup genes increased 1.9-fold. Genes involved in energy use were also represented, including 3.30-fold increase in Ppargc1a, a 2.20-fold increase in Slc2a4, and a 2.15-fold increase in Nr4a1 (nuclear receptor subfamily 4, group A, member 1, which uses Pgc1α as a coactivator).
Comparing Ctcgrp null mice with WT mice at baseline, 15 genes showed differential expression (Table 1). Among these were a 2.32-fold increase in Calcr (calcitonin receptor) and a 2.37-fold increase in Calcb (calcitonin gene related protein-β). Other marked changes included a 9.47-fold decrease in Trim12 (tripartite motif containing 12), a 2.84-fold down-regulation of Npy (neuropeptide Y), and a 4.86-fold up-regulation of Gdpd3 (glycerophosphodiester phosphodiesterase domain containing 3).
Table 1.
Differential Expression of Tibial Genes on Genome-Wide Microarray, Performed at Prepregnancy Baseline (Age 10 Weeks) in Ctcgrp Null vs WT Mice
| Symbol | Gene Name | Identification | Fold Change | Adjusted P Value |
|---|---|---|---|---|
| Gm5574 | Immunoglobulin κ-chain variable 12–47 | ENSMUST00000103364 | 8.57 | <.00001 |
| Gdpd3 | Glycerophosphodiester phosphodiesterase domain containing 3 | NM_024228 | 4.86 | <.00001 |
| Gm10883 | Predicted gene 10883 | ENSMUST00000103378 | 3.26 | <.004 |
| Calcb | Calcitonin-related polypeptide, β | NM_054084 | 2.37 | <.0005 |
| Calcr | Calcitonin receptor | NM_007588 | 2.32 | <.00001 |
| U29423 | cDNA sequence U29423 | ENSMUST00000103376 | 2.15 | <.00001 |
| 2900092E17Rik | RIKEN cDNA 2900092E17 gene | NM_030240 | 2.03 | <.0004 |
| ENSEMBL (mRNA assignment) | Ncrna:snRNA chromosome: NCBIM37:3:35634295:35634399:1 gene: ENSMUSG00000064834 | ENSMUST00000082900 | 1.68 | <.001 |
| Ttn | Titin | ENSMUST00000099981 | 1.20 | <.0001 |
| Gm10877 | Predicted gene 10877 | ENSMUST00000103311 | −1.57 | <.005 |
| LOC100046973 | Similar to (human Ig rearranged γ-chain mRNA, V-J-C region and complete cds.), gene product | ENSMUST00000103366 | −1.80 | <.001 |
| A530023O14Rik | RIKEN cDNA A530023O14 gene | NM_175648 | −2.66 | <.0001 |
| Npy | Neuropeptide Y | NM_023456 | −2.84 | <.0001 |
| AI451617 | Expressed sequence AI451617 | NM_199146 | −6.55 | <.00001 |
| Trim12 | Tripartite motif-containing 12 | NM_023835 | −9.47 | <.00001 |
Results are arranged from greatest to least relative fold change.
Comparing Ctcgrp null mice with WT mice at day 7 after weaning, only 13 genes showed differential regulation (Table 2). These included a 6.49-fold increase in Mela, a 10.92-fold decrease in Trim12, and a 2.67-fold increase in Gdpd3.
Table 2.
Differential Expression of Tibial Genes on Genome-Wide Microarray, Performed at Day 7 After Weaning in Ctcgrp Null vs WT Mice
| Symbol | Name | Identification | Fold Change | Adjusted P Value |
|---|---|---|---|---|
| Mela | Melanoma antigen | BC113756 | 6.49 | <.00001 |
| Mela | Melanoma antigen | D10049 | 4.59 | <.00001 |
| Gdpd3 | Glycerophosphodiester phosphodiesterase domain containing 3 | NM_024228 | 2.67 | <.00001 |
| Fcnb | Ficolin B | NM_010190 | 1.96 | <.006 |
| Igk-V28 | Immunoglobulin κ-chain variable 28 (V28) | DQ078272 | −2.20 | <.006 |
| LOC100046496 | Similar to Ig-κ V-region 24B | ENSMUST00000103322 | −2.42 | <.00001 |
| ENSEMBL (mRNA assignment) | cdna: known chromosome: NCBIM37:6:70166852:70167415:-1 gene: ENSMUSG00000076583 | ENSMUST00000103384 | −2.83 | <.0102 |
| LOC100046973 | Similar to (human Ig rearranged γ-chain mRNA, V-J-C region and complete cds.), gene product | ENSMUST00000103366 | −2.87 | <.00001 |
| AI324046 | Expressed sequence AI324046 | D14625 | −4.38 | <.004 |
| AI451617 | Expressed sequence AI451617 | NM_199146 | −4.43 | <.003 |
| Gm900 | Predicted gene 900 | ENSMUST00000103414 | −4.69 | <.00001 |
| LOC674190 | Similar to Ig heavy chain V region IR2 precursor | ENSMUST00000103484 | −8.41 | <.00001 |
| Trim12 | Tripartite motif-containing 12 | NM_023835 | −10.92 | <.00001 |
Results are arranged from greatest to least relative fold change.
Comparing Ctcgrp null at day 7 after weaning with WT mice at baseline, 300 genes showed differential regulation (Supplemental Table 4), and these were largely a subset of those shown in Supplemental Table 2.
Pathways and networks analysis
The WT microarray results were sorted by pathways and networks analysis (Table 3). Top canonical pathways included calcium signaling (including calcium ATPase, calmodulin, calsequestrin), interferon regulatory factor signaling, glycolysis/gluconeogenesis, and integrin-linked kinase signaling, whereas top upstream regulators included Dmd (dystrophyn), Trim24 (tripartite motif containing 24), IfnA2 (interferon A2), IfnB1 (interferon B1), and Mef2c (myocyte specific enhancer factor 2C, which stimulates Sost). Involved physiological and disease systems included genes associated with skeletal and muscle development; cellular assembly, morphology, function, and death; and carbohydrate metabolism. Similar results were found with the Ctcgrp pathway analysis (not shown). Overall, these results indicate substantial mitogenic responses that may be driving osteoblast proliferation and function, inducing osteoclast apoptosis, inhibiting osteoclast function, and altering pathways to provide the energy needed for the increased cellular activity.
Table 3.
Summary of the Main Categories Revealed From Pathways and Network Analysis of the Tibial Microarray Data
| Top Canonical Pathways | P Value | Ratio |
|---|---|---|
| Calcium signaling | 5.28E-09 | 22/210 (0.105) |
| [Includes Acta1, Atp2a1, Camk2a, Camk2b, Casq1, Casq2, Chrna1, Myh1, Myh2, Myh4, Myh7, Myl1, Myl2, Myl3, Ryr1, Tnnc1, Tnnc2, Tnni2, Tnnt1, Tnnt3, Tpm2, Trdn] | ||
| Activation of interferon regulatory factor by cytosolic pattern recognition receptors | 2.05E-06 | 10/72 (0.139) |
| Interferon signaling | 1.96E-05 | 7/36 (0.194) |
| Glycolysis/gluconeogenesis | 7.49E-05 | 10/130 (0.077) |
| Integrin-linked kinase signaling | 2.09E-04 | 15/192 (0.078) |
| Top Upstream Regulators | P Value of Overlap | Predicted Activation State |
|---|---|---|
| DMD | 4.41E-30 | Activated |
| TRIM24 | 4.03E-23 | Inhibited |
| IFNA2 | 1.72E-22 | Activated |
| IFNB1 (includes EG:15977) | 2.78E-22 | Activated |
| MEF2C | 3.05E-21 | Activated |
| Diseases and Disorders | P Value | Molecules, n |
|---|---|---|
| Skeletal and muscular disorders | 1.99E-20 to 1.61E-02 | 167 |
| Neurological disease | 8.12E-14 to 1.22E-02 | 98 |
| Hereditary disorder | 1.43E-11 to 1.61E-02 | 105 |
| Cardiovascular disease | 3.92E-09 to 1.27E-02 | 66 |
| Developmental disorder | 7.06E-09 to 1.61E-02 | 71 |
| Molecular and Cellular Functions | P Value | Molecules, n |
|---|---|---|
| Cellular assembly and organization | 1.62E-07 to 1.58E-02 | 58 |
| Cellular function and maintenance | 1.62E-07 to 1.37E-02 | 114 |
| Carbohydrate metabolism | 2.34E-07 to 1.58E-02 | 67 |
| Cell morphology | 9.05E-07 to 1.74E-02 | 81 |
| Cell death | 2.08E-06 to 1.61E-02 | 109 |
| Physiological Systems | P Value | Molecules, n |
|---|---|---|
| Organ morphology | 4.18E-32 to 1.74E-02 | 134 |
| Skeletal and muscular system development and function | 4.18E-32 to 1.74E-02 | 153 |
| Embryonic development | 2.62E-12 to 1.74E-02 | 88 |
| Organ development | 2.62E-12 to 1.74E-02 | 98 |
| Organismal development | 2.62E-12 to 1.74E-02 | 118 |
qPCR of selected genes
We used RNA from a subset of the tibiae to confirm expression of selected genes by qPCR. Dkk1 and Sost both inhibit bone formation. By qPCR Dkk1 showed no difference between Ctcgrp null and WT mice at baseline, and in both it suppressed more than 80% during postweaning with more marked suppression observed in Ctcgrp null (Figure 3A). Sost was 1.26-fold higher in Ctcgrp nulls at baseline; during postweaning it was reduced 80% below baseline in both genotypes, achieving a proportionately greater suppression in Ctcgrp null (Figure 3B). The greater reductions in Dkk1 and Sost are consistent with the proportionately greater increase in osteoblast surface in Ctcgrp null mice. Mef2c stimulates Sost, and its expression was reduced 50% in both genotypes during postweaning (Figure 3C). Runx2 inhibits the differentiation of immature osteoblasts into mature osteoblasts, and it too was inhibited during postweaning (Figure 3D).
Figure 3.

qPCR of osteoblast and osteoclast genes at day 7 postweaning vs baseline. Reduced expression of Dkk1 (A), Sost (B), and Mef2c (C) predict increased bone formation and reduced Runx2 expression (D) favors differentiation to mature osteoblasts. Ctcgrp null mice had proportionately greater suppression of Dkk1 and Sost, which is consistent with their proportionately greater increase in osteoblast parameters during postweaning. Reduced Ctsk (E) and Igfbp2 (F) expression are consistent with reduced bone resorption. Oxt (G) down-regulated during postweaning, whereas Oxtr (H) showed no significant change. Braces indicate P < .01.
Ctsk expression reflects osteoclast number and activity; by qPCR Ctsk had 30% higher expression in Ctcgrp nulls at baseline (Figure 3E). During postweaning, Ctsk expression decreased below the reference by 80% in WT mice and 70% in Ctcgrp null mice but remained significantly higher in the Ctcgrp null mice (Figure 3E). IGF binding protein-2 (IGFBP2) stimulates osteoclast differentiation and skeletal resorption, and its circulating levels are increased during lactation (41, 42). By qPCR it fell 70% below baseline in both genotypes during postweaning recovery (Figure 3F).
Oxt (oxytocin) and Oxtr (oxytocin receptor) are both expressed in bone cells, but despite recent claims that oxytocin stimulates bone formation after weaning (43), neither showed a significant differential change in the microarray, and so both genes were examined by qPCR. Ctcgrp null mice showed 1.9-fold higher Oxt at baseline, and during postweaning Oxt expression decreased about 90% below baseline, achieving the lowest value in Ctcgrp null mice (Figure 3G). Oxtr showed no significant differences between genotypes or time points (Figure 3H).
The microarray findings prompted us to confirm the regulation of several genes known to be important for energy use. Ppargc1a (Pgc1α) is expressed by osteoblasts, and by qPCR its expression rose 2.3-fold in WT mice and 1.9-fold in Ctcgrp null mice during postweaning recovery (Figure 4A). The transcription factor Ucp3 was 1.2-fold higher in Ctcgrp null mice vs WT mice at baseline, and it increased during postweaning recovery to 2.2-fold in WT mice and 1.9-fold in Ctcgrp null mice (Figure 4B). Foxc2 encodes the transcription factor forkhead box protein C2 (Foxc2), and its expression in Ctcgrp null mice was 1.2-fold higher than WT mice at baseline. In both genotypes it was suppressed to about 80% below reference during postweaning recovery (Figure 4C).
Figure 4.
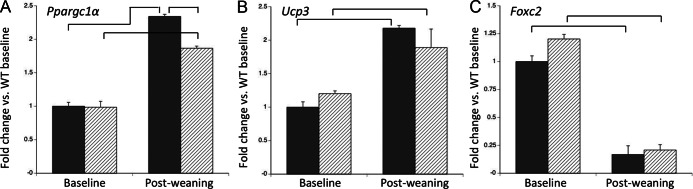
qPCR of genes relevant to energy metabolism at day 7 postweaning vs baseline. Increased expression of Ppargc1α (A) and Ucp3 (B) predict increased osteoblast activity and energy use, whereas decreased expression of Foxc2 (C) is consistent with the use of fatty acids as an energy source for osteoblast activity during postweaning. Braces indicate P < .01.
Mela and Mup showed the largest differential expression on the microarray. At baseline by qPCR, Mela expression was 2-fold higher in Ctcgrp null mice compared with WT mice, and during postweaning it rose to 4.8-fold in WT mice and 1500-fold in Ctcgrp null mice (Figure 5A). This was not due to small changes in a gene with low-level expression; during postweaning the expression of Mela exceeded that of the housekeeping gene in Ctcgrp nulls. Mup showed 2.5-fold higher values in Ctcgrp null mice vs WT mice at baseline, whereas during postweaning its expression rose 2.2-fold in WT mice and decreased slightly in Ctcgrp null mice (Figure 5B).
Figure 5.
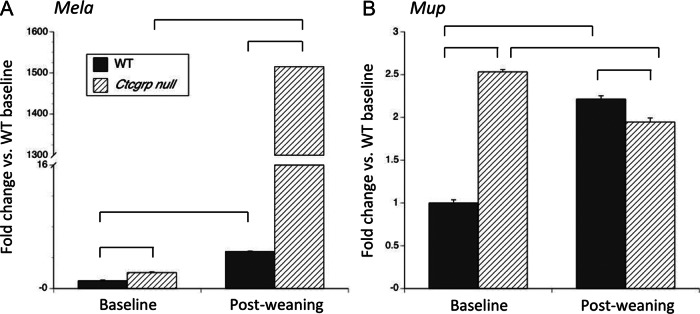
qPCR of Mela and Mup at day 7 postweaning vs baseline. The largest differential expression on the microarray occurred in Mela and Mup, neither of which has previously been shown to be expressed in bone or marrow. In A, qPCR confirmed the microarray findings of up-regulation of Mela during postweaning in both genotypes, with the greater increase achieved in Ctcgrp null mice. On the microarray, each of the 19 different Mup genes had 1.9-fold increased expression in Ctcgrp null mice during postweaning as compared with baseline (Supplemental Table 3) or 5-fold higher expression in Ctcgrp null mice during postweaning as compared with WT baseline (Supplemental Table 4). Shown in B is the result of a single qPCR primer set common to all Mup genes, which suggests that Mup gene expression was 2.5-fold higher in Ctcgrp null mice than WT mice at baseline, whereas during postweaning it decreased slightly in Ctgrp null mice but increased 2-fold in WT mice. It is unknown whether Mela or Mup plays any role in regulating skeletal metabolism during postweaning. Braces indicate P < .01.
Discussion
Lactation causes a loss of bone mass and strength that is normally completely reversed after weaning. We previously reported that Ctcgrp null mice lose double the normal amount of BMC during lactation but regain it fully after weaning (6). Ctcgrp null mice have 5-fold higher deoxypyridinoline levels than WT mice during lactation, confirming increased bone resorption that may be attributable to up-regulated mammary tissue expression of PTHrP and loss of actions of calcitonin in osteoclasts and mammary epithelial cells (6). The present study revealed that compared with WT mice, lactating Ctcgrp null mice have twice the number of osteoclasts and eroded surface and significantly reduced osteoblast-related parameters. Therefore, the greater loss of bone mass in Ctcgrp null mice likely results not only from increased bone resorption but also reduced bone formation. Why osteoblast numbers are reduced during lactation is unknown. Exogenous calcitonin increases the expression of Sost by osteocytes and impairs the anabolic response to PTH injections (44), but that result predicts that lactating Ctcgrp null mice should have reduced expression of Sost and increased bone formation compared with WT mice. Alternatively, loss of calcitonin gene-related peptide (CGRP)-α may contribute to reduced osteoblast numbers because CGRP stimulates differentiation and proliferation of osteoblast-like cells in vitro (45–49), and mice lacking CGRPα display low bone mass with reduced bone formation (50).
At the time of natural weaning when histomorphometry was performed, osteoclast numbers had not yet decreased but osteoclast surface had declined, in keeping with reduced osteoclast activity. In contrast, when weaning is forced by the early removal of pups, osteoclast numbers decrease substantially within 24–48 hours due to apoptosis (20, 22). The early postweaning increase in osteoblast numbers of WT and Ctcgrp null mice implies that a significant stimulus for bone formation has been triggered and is consistent with the rapid restoration of bone mass that both genotypes subsequently achieve. A similar surge in osteoblast parameters has been observed after granulocyte colony-stimulating factor (G-CSF) treatment depletes the marrow of endosteal osteoblasts and suppresses bone formation (51). Beginning several days after G-CSF treatment, osteoblast surface increases from less than 5% to more than 40% and the bone formation rate normalizes. In both lactation and after G-CSF treatment, bone cells may be responding to a systemic trigger.
Our primary interest was to identify the key genes that stimulate bone formation and inhibit bone resorption during postweaning recovery. We hypothesized that postweaning bone would show marked differential regulation of genes responsible for stimulating bone formation and down-regulating bone resorption and that such genes might show more marked changes in Ctcgrp null mice. We studied intact bone because genes in the marrow are intimately involved in regulating bone metabolism.
That 729 genes in WT mice had a 1.5-fold or greater change during postweaning and underscores how active bone and marrow are during the postweaning interval. The microarray revealed changes in genes that should increase osteoblast and decrease osteoclast function and cell number, alter calcium signaling, and increase energy use. Analyzing Ctcgrp null mice reduced the number of 1.5-fold or greater differentially regulated candidate genes by more than half, but this reduction may not have been useful because there were false negatives among the excluded genes. For example, qPCR showed significant differential regulation in Dkk1, Sost, Ctsk, and Igfbp2 that was not apparent in the Ctcgrp null microarray. Discordance between results of microarray and qPCR arise because qPCR has 10-fold greater sensitivity, better ability to discriminate less than 2-fold differences in expression, and probe placement that enables the detection of multiple splice variants, whereas a single probe may represent 1 or more genes on the microarray (52, 53). A comparative study of WT and Ctcgrp null mice found only 13 differentially regulated genes during postweaning (Table 2), but those remaining genes seem unlikely candidates to explain increased bone formation.
Pathways and network analysis confirmed differential regulation of genes involved in skeletal and muscle development; cellular morphology, proliferation, function, and death; and carbohydrate and energy metabolism. We were guided by these results and recent literature in selecting genes for confirmative study by qPCR.
We expected to see differential regulation of key genes that stimulate osteoblast number or function. Pathway analysis indicated the involvement of Mef2c, a key upstream enhancer of Sost (54, 55), and we confirmed the down-regulation of Mef2c by qPCR. Dkk1 and Sost each decreased by 80% during postweaning recovery, with greater suppression in Ctcgrp null mice, consistent with the up-regulation of the Wnt/β-catenin signaling pathway and its role in stimulating bone formation and regeneration (56). DKK1 and sclerostin both potently inhibit Wnt signaling (57). Their physiological importance has been confirmed by loss-of-function mutations in Sost, which cause high bone mass in humans (58, 59), whereas ablation of Sost or heterozygous deletion of Dkk1 causes high bone mass in mice (57, 60–62). Conversely, the power of this signaling pathway to suppress bone formation has been confirmed by overexpression of Dkk1 or Sost in mice, which causes low bone mass due to reduced bone formation (57, 61, 63, 64). Monoclonal antibodies to DKK1 and sclerostin stimulate bone formation in animal models (57); a sclerostin antibody has reached phase III clinical trials. Overall, the up-regulation in Wnt signaling may explain the marked stimulation in osteoblast number and function that occurs during postweaning.
Runx2 stimulates the commitment of mesenchymal precursors to become immature osteoblasts but inhibits their differentiation into mature osteoblasts; its expression must be inhibited in immature osteoblasts for fully mature osteoblasts to develop under the influence of Wnt signaling and other factors (65, 66). Conversely, inhibition of Runx2 in mature osteoblasts does not reduce their activity or expression of col1a1 and osteocalcin (65). Therefore, decreased Runx2 expression is compatible with increased osteoblast maturation and sustained osteoblast function.
The pathway analysis revealed significant alteration in other upstream regulators that control cellular proliferation, differentiation, and apoptosis, including Trim24, Ifna2, and Ifnb1. These genes may contribute to the divergent changes in osteoblast and osteoclast cell numbers, which in turn cause a relative uncoupling of bone formation and resorption during postweaning. More specifically to osteoclasts, reduced expression of Ctsk and Igfb2 may drive or reflect the lower osteoclast numbers and function. Ctsk is highly expressed by osteoclasts and encodes cathepsin K, a cysteine protease that degrades type I collagen (67, 68). The 80% reduction in Ctsk expression during postweaning is consistent with lower osteoclast numbers and reduced bone resorption. The importance of Ctsk has been demonstrated by loss-of-function mutations, which lead to absent or impaired osteoclasts and osteopetrotic phenotypes in humans and mice (68–72). Conversely, overexpression of Ctsk leads to excessive osteoclast-mediated bone resorption and osteoporosis (73). These observations prompted the development of cathepsin K inhibitors, which are now in phase III clinical trials as treatments for osteoporosis (72). Reduced expression of Igfbp2 may also reduce osteoclast numbers during postweaning because IGFBP2 stimulates osteoclast differentiation, fusion, and resorption, whereas Igfbp2 null mice have fewer osteoclasts and reduced bone resorption (74).
The reduced expression of several genes that regulate chondrocyte development (Acan, Matn3, col10a1, and col2a1) imply preferential stimulation of osteoblastogenesis over chondrocyte differentiation from the same precursor pool. Redd et al (75) previously reported that cartilaginous growth up-regulates during pregnancy but down-regulates during lactation; if this persists during postweaning, it could explain why we found reduced expression of chondrocyte genes. Alternatively, the down-regulation may be attributable to the older age of the mice at postweaning as compared with prepregnancy baseline when cartilage is still growing.
Oxytocin reaches the maternal circulation from the pituitary in response to suckling (76), and its main role is to contract mammary myoepithelial cells to cause milk ejection. If milk cannot be ejected, mammary cells undergo apoptosis and lactation ceases. But oxytocin conceivably regulates bone metabolism because it is also produced in bone marrow (77), and osteoclasts and osteoblasts both express the oxytocin receptor (78). Oxytocin stimulates osteoblast differentiation and function, promotes osteoclast formation, but inhibits osteoclast function and skeletal resorption (79, 80). Oxt and Oxtr null mice have low bone mass with low bone formation rates (79). Recent work has shown that estradiol stimulates expression of oxytocin in bone marrow (77) and that bone-anabolic actions of estradiol (at pharmacological doses) are blunted by loss of Oxtr (43). Because estradiol levels increase as lactation wanes, the authors of that recent work deduced that osteoblast-derived oxytocin stimulates postweaning bone recovery (43). Oxt null mice cannot lactate and therefore cannot be used to assess whether oxytocin regulates bone formation during lactation or postweaning (81). However, in our studies Oxtr showed no change in expression between time points or genotypes, whereas Oxt was down-regulated 90% during postweaning recovery and achieved even greater suppression in Ctcgrp null mice. Therefore, our results do not support an essential role for oxytocin and its receptor in stimulating bone formation after weaning.
Increased substrate use is required for active bone formation and cellular proliferation that occurs in bone and marrow during postweaning. The pathway analysis indicated up-regulation in glycolysis/gluconeogenesis, which may support the precept that the energy needs of bone cells are provided through gluconeogenesis and fatty acid use during the recovery phase. Consistent with this, our finding that Ppargc1a expression is increased may provide insight into energy use by osteoblasts. Pgc1α is a coactivator that also modulates the expression of UCP1, which is essential for nonshivering thermogenesis in brown fat, regulates mitochondrial biogenesis and oxidative metabolism in diverse cell types, and is up-regulated in muscle in response to exercise (82). Pgc1α also stimulates osteoblast function and gene expression, and its expression in primary mouse osteoblasts and calvariae is induced by PTH (83), whereas overexpression of Ppargc1a in skeletal muscle protects against age-related bone loss (84). Pdk4 is an immediate downstream target of Ppargc1a and acts to inhibit the pyruvate dehydrogenase complex, thereby shifting preferred energy use from glucose to fatty acids. Its 2-fold up-regulation during recovery from lactation may indicate that fatty acids become a preferred energy source when osteoblasts are synthesizing collagen.
Similarly, altered Ucp3 and Foxc2 expression support that osteoblasts may change the energy sources used during postweaning. Ucp3 is expressed in skeletal muscle and brown adipose tissue in which it is important for energy use (85). Ucp3 and Ucp1 expression are normally suppressed during lactation, consistent with the sparing of free fatty acids as a fuel source (86), whereas we found that Ucp3 expression increased during postweaning recovery, consistent with metabolism of fatty acids to provide energy for osteoblast work. Foxc2 encodes a transcription factor that is expressed in diverse tissues, including brown adipose tissue and bone. Overexpression of Foxc2 in adipose tissue results only in enhanced brown-like adipocytes and increased bone mass (B. Lecka-Czernik, personal communication to C. J. Rosen), whereas Foxc2 null mice have multiple defects including undermineralized skeletons (87, 88). Foxc2 is up-regulated by PTH or bone morphogenetic protein 2 and promotes proliferation and differentiation of osteoblasts, possibly through interactions with the Wnt signaling pathway (87, 89). However, we found Foxc2 was significantly down-regulated during recovery, which may also reflect the preferential use of fatty acids for ATP generation in mesenchymal cells destined to become osteoblasts rather than for thermogenesis in adipocytes.
Two surprising findings are the marked up-regulation in Mup and Mela, neither of which have been previously reported in bone or marrow. Nineteen different MUPs are secreted by the liver, bind to volatile pheromones, and are excreted into urine (90, 91). They are best known to establish a characteristic signature scent for the animal that secretes them and to trigger adaptive behavioral or developmental responses in the recipient that senses them. But MUPs in the systemic circulation regulate lipid metabolism, suppress hepatic gluconeogenesis, promote energy expenditure in skeletal muscle, show GH-like effects, and may have other roles that have yet to be defined (90). Mela encodes MAGE, which is best known for a wide expression in a variety of malignant tumors and in germ cells of the testis; Mela expression is silent in most normal adult tissues (92, 93). MAGE regulates cell cycle progression and apoptosis during germ cell development (92, 93). It remains to be seen whether the marked up-regulation of Mup and Mela in bone or marrow implicates a role in regulating osteoblasts, osteoclasts, or energy use during this time.
Overall, these findings within bone and marrow confirm substantial changes in numerous genes that may be stimulating osteoblast proliferation and function, inducing osteoclast apoptosis, inhibiting osteoclast function, and stimulating fat metabolism as a fuel source. Ctcgrp null mice differ from WT mice mainly in their marked loss of bone during lactation (increased osteoclast and decreased osteoblast parameters) and not in the ability to stimulate bone formation parameters during postweaning. The observed changes in gene regulation and osteoblast/osteoclast parameters during postweaning may reflect responses to intrinsic and extrinsic factors and do not necessarily point to any 1 of the factors as being the key signal that promotes bone formation after weaning. Inactivation of Mef2c to down-regulates Sost and activate Wnt signaling, for example, might prove sufficient to explain bone recovery after lactation, but the key question will remain as to what triggers suppression of Mef2c after weaning.
Limitations of this work include that we compared postweaning (17–18 weeks old) to prepregnancy baseline (10 weeks old) to match the experimental design of our longitudinal studies; consequently, some of the observed differences in gene expression may be due to age. The steady plateau in BMC at prepregnancy baseline indicates a stable balance of bone formation and resorption in mice from the Black Swiss strain; conversely, age-matched controls are already undergoing gradual bone loss by 17–18 weeks of age. Some changes in gene expression may be due to altered cell numbers or programmed responses to postweaning that have nothing to do with altering bone turnover. Inclusion of bone marrow may drown out smaller signals from bone cells, so focused study of RNA from bone alone may also be needed.
In summary, our novel findings of enhanced Wnt signaling, impaired Ctsk and Igfbp2 expression, and altered expression of genes regulating apoptosis and cell proliferation, may explain the uncoupling of bone formation and resorption in tibiae during postweaning. Altered regulation of genes involved in substrate use may indicate a switch from carbohydrate to fat as an energy source for the osteoblastic work required during the recovery phase. Numerous other genes are differentially expressed, including many that have not been previously described in bone or marrow. Some of these genes (including Mela and Mup, which achieve proportionately higher expression during postweaning in Ctcgrp null mice) may have as-yet-undefined roles in regulating skeletal metabolism. Although Ctcgrp null mice have increased bone resorption during lactation, they have a normal osteoblastic response during postweaning and greater suppression of Dkk1 and Sost, which may boost bone recovery. It remains to be determined whether a primary signal(s) in bone triggers the stimulation of bone formation after lactation or whether the observed alterations in tibial gene expression are initiated in response to systemic signals. For this reason we are separately pursuing studies to identify circulating proteins during postweaning recovery and to determine whether any are required to stimulate bone formation after lactation.
Supplementary Material
Acknowledgments
We thank Dr T. J. Martin for his critical review of the manuscript.
This work was supported by an operating grant from the Canadian Institutes of Health Research (to C.S.K.).
Disclosure Summary: The authors have nothing to declare.
Footnotes
- BMC
- bone mineral content
- BV/TV
- trabecular bone volume
- CT
- threshold cycle
- CGRP
- calcitonin gene-related peptide
- Ctsk
- cathepsin K
- Dkk1
- Dickkopf-1
- DXA
- dual x-ray absorptiometry
- Foxc2
- forkhead box protein C2
- G-CSF
- granulocyte colony-stimulating factor
- Gdpd3
- glycerophosphodiester phosphodiesterase domain containing 3
- IfnA2
- interferon A2
- IfnB1
- interferon B1
- IGFBP2
- IGF binding protein-2
- MAGE
- melanoma associated antigen
- Mef2c
- myocyte specific enhancer factor 2C
- MUP
- major urinary protein
- Oxt
- oxytocin
- Oxtr
- oxytocin receptor
- Ppargc1a
- peroxisome proliferator-activated receptor-γ coactivator-1α or Pgc1α
- qPCR
- quantitative PCR
- Sost
- sclerostin
- Trim12
- tripartite motif containing 12
- Trim24
- tripartite motif containing 24
- Ucp3
- uncoupling protein 3
- WT
- wild type.
References
- 1. Kovacs CS. Calcium and bone metabolism disorders during pregnancy and lactation. Endocrinol Metab Clin N America. 2011;40:795–826 [DOI] [PubMed] [Google Scholar]
- 2. Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium and lactation. Endocr Rev. 1997;18:832–872 [DOI] [PubMed] [Google Scholar]
- 3. Sowers M. Pregnancy and lactation as risk factors for subsequent bone loss and osteoporosis. J Bone Miner Res. 1996;11:1052–1060 [DOI] [PubMed] [Google Scholar]
- 4. Polatti F, Capuzzo E, Viazzo F, Colleoni R, Klersy C. Bone mineral changes during and after lactation. Obstet Gynecol. 1999;94:52–56 [DOI] [PubMed] [Google Scholar]
- 5. Chantry CJ, Auinger P, Byrd RS. Lactation among adolescent mothers and subsequent bone mineral density. Arch Pediatr Adolesc Med. 2004;158:650–656 [DOI] [PubMed] [Google Scholar]
- 6. Woodrow JP, Sharpe CJ, Fudge NJ, Hoff AO, Gagel RF, Kovacs CS. Calcitonin plays a critical role in regulating skeletal mineral metabolism during lactation. Endocrinology. 2006;147:4010–4021 [DOI] [PubMed] [Google Scholar]
- 7. Kovacs CS. Control of skeletal homeostasis during pregnancy and lactation—lessons from physiological models. In: Thakker RV, Whyte MP, Eisman JA, Igarashi T, eds. Genetics of Bone Biology and Skeletal Disease. San Diego: Academic Press/Elsevier; 2012:221–240 [Google Scholar]
- 8. Teti A, Zallone A. Do osteocytes contribute to bone mineral homeostasis? Osteocytic osteolysis revisited. Bone. 2009;44:11–16 [DOI] [PubMed] [Google Scholar]
- 9. Qing H, Ardeshirpour L, Pajevic PD, et al. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J Bone Miner Res. 2012;27:1018–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. VanHouten JN, Dann P, Stewart AF, et al. Mammary-specific deletion of parathyroid hormone-related protein preserves bone mass during lactation. J Clin Invest. 2003;112:1429–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. VanHouten JN, Wysolmerski JJ. Low estrogen and high parathyroid hormone-related peptide levels contribute to accelerated bone resorption and bone loss in lactating mice. Endocrinology. 2003;144:5521–5529 [DOI] [PubMed] [Google Scholar]
- 12. Collet P, Uebelhart D, Vico L, et al. Effects of 1- and 6-month spaceflight on bone mass and biochemistry in two humans. Bone. 1997;20:547–551 [DOI] [PubMed] [Google Scholar]
- 13. Holick MF. Perspective on the impact of weightlessness on calcium and bone metabolism. Bone. 1998;22:105S–111S [DOI] [PubMed] [Google Scholar]
- 14. Tilton FE, Degioanni JJ, Schneider VS. Long-term follow-up of Skylab bone demineralization. Aviat Space Environ Med. 1980;51:1209–1213 [PubMed] [Google Scholar]
- 15. Hermus AR, Smals AG, Swinkels LM, et al. Bone mineral density and bone turnover before and after surgical cure of Cushing's syndrome. J Clin Endocrinol Metab. 1995;80:2859–2865 [DOI] [PubMed] [Google Scholar]
- 16. Lufkin EG, Wahner HW, O'Fallon WM, et al. Treatment of postmenopausal osteoporosis with transdermal estrogen. Ann Intern Med. 1992;117:1–9 [DOI] [PubMed] [Google Scholar]
- 17. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Use of oral corticosteroids and risk of fractures. J Bone Miner Res. 2000;15:993–1000 [DOI] [PubMed] [Google Scholar]
- 18. Reid IR. Menopause. In: Favus MJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 5th ed Washington, DC: ASBMR Press; 2003:86–89 [Google Scholar]
- 19. Gallagher JC. Effect of estrogen on bone. In: Favus MJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 5th ed Washington, DC: ASBMR Press; 2003:327–330 [Google Scholar]
- 20. Miller SC, Bowman BM. Rapid inactivation and apoptosis of osteoclasts in the maternal skeleton during the bone remodeling reversal at the end of lactation. Anat Rec (Hoboken). 2007;290:65–73 [DOI] [PubMed] [Google Scholar]
- 21. Ardeshirpour L, Dann P, Adams DJ, et al. Weaning triggers a decrease in receptor activator of nuclear factor-κB ligand expression, widespread osteoclast apoptosis, and rapid recovery of bone mass after lactation in mice. Endocrinology. 2007;148:3875–3886 [DOI] [PubMed] [Google Scholar]
- 22. Miller SC, Anderson BL, Bowman BM. Weaning initiates a rapid and powerful anabolic phase in the rat maternal skeleton. Biol Reprod. 2005;73:156–162 [DOI] [PubMed] [Google Scholar]
- 23. Qing H, Bonewald LF. Osteocyte remodeling of the perilacunar and pericanalicular matrix. Int J Oral Sci. 2009;1:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirby BJ, Ardeshirpour L, Woodrow JP, et al. Skeletal recovery after weaning does not require PTHrP. J Bone Miner Res. 2011;26:1242–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu XS, Ardeshirpour L, VanHouten JN, Shane E, Wysolmerski JJ. Site-specific changes in bone microarchitecture, mineralization, and stiffness during lactation and after weaning in mice. J Bone Miner Res. 2012;27:865–875 [DOI] [PubMed] [Google Scholar]
- 26. Fudge NJ, Kovacs CS. Pregnancy up-regulates intestinal calcium absorption and skeletal mineralization independently of the vitamin D receptor. Endocrinology. 2010;151:886–895 [DOI] [PubMed] [Google Scholar]
- 27. Kirby BJ, Karaplis AC, Kovacs CS. Calcitriol increases during pregnancy and bone formation upregulates post-lactation without parathyroid hormone. Bone. 2011;48:S101–S102 [Google Scholar]
- 28. Hoff AO, Catala-Lehnen P, Thomas PM, et al. Increased bone mass is an unexpected phenotype associated with deletion of the calcitonin gene. J Clin Invest. 2002;110:1849–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sharpe CJ, Fudge NJ, Kovacs CS. A rapid 35% flux in bone mass occurs during pregnancy and lactation cycles in mice [abstract]. International Bone and Mineral Society Meeting, Osaka, Japan, June 3–7, 2003 Bone. 2003;32 (suppl):S227 [Google Scholar]
- 30. Parfitt AM, Drezner MK, Glorieux FH, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610 [DOI] [PubMed] [Google Scholar]
- 31. Sims NA, Brennan K, Spaliviero J, Handelsman DJ, Seibel MJ. Perinatal testosterone surge is required for normal adult bone size but not for normal bone remodeling. Am J Physiol Endocrinol Metab. 2006;290:E456–E462 [DOI] [PubMed] [Google Scholar]
- 32. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264 [DOI] [PubMed] [Google Scholar]
- 33. Jain N, Thatte J, Braciale T, Ley K, O'Connell M, Lee JK. Local-pooled-error test for identifying differentially expressed genes with a small number of replicated microarrays. Bioinformatics. 2003;19:1945–1951 [DOI] [PubMed] [Google Scholar]
- 34. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B (Methodological). 1995;57:289–300 [Google Scholar]
- 35. Simmonds CS, Karsenty G, Karaplis AC, Kovacs CS. Parathyroid hormone regulates fetal-placental mineral homeostasis. J Bone Miner Res. 2010;25:594–605 [DOI] [PubMed] [Google Scholar]
- 36. Kovacs CS, Woodland ML, Fudge NJ, Friel JK. The vitamin D receptor is not required for fetal mineral homeostasis or for the regulation of placental calcium transfer. Am J Physiol Endocrinol Metab. 2005;289:E133–E144 [DOI] [PubMed] [Google Scholar]
- 37. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-ΔΔC(T)] Method. Methods. 2001;25:402–408 [DOI] [PubMed] [Google Scholar]
- 38. Montero A, Okada Y, Tomita M, et al. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Invest. 2000;105:1085–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garner SC, Pi M, Tu Q, Quarles LD. Rickets in cation-sensing receptor-deficient mice: an unexpected skeletal phenotype. Endocrinology. 2001;142:3996–4005 [DOI] [PubMed] [Google Scholar]
- 40. Kirby BJ, Karaplis AC, Kovacs CS. 2010 Stimulation of bone formation and mineralization post-weaning without parathyroid hormone. J Bone Miner Res 2010(suppl 1). Available at http://www.asbmr.org/Meetings/AnnualMeeting/AbstractDetail.aspx?aid=59f8e1fc-623d-4826-a568-29e32e11f525 Accessed February 19, 2013
- 41. Breier BH, Milsom SR, Blum WF, Schwander J, Gallaher BW, Gluckman PD. Insulin-like growth factors and their binding proteins in plasma and milk after growth hormone-stimulated galactopoiesis in normally lactating women. Acta Endocrinol (Copenh). 1993;129:427–435 [DOI] [PubMed] [Google Scholar]
- 42. Vicini JL, Buonomo FC, Veenhuizen JJ, Miller MA, Clemmons DR, Collier RJ. Nutrient balance and stage of lactation affect responses of insulin, insulin-like growth factors I and II, and insulin-like growth factor-binding protein 2 to somatotropin administration in dairy cows. J Nutr. 1991;121:1656–1664 [DOI] [PubMed] [Google Scholar]
- 43. Colaianni G, Sun L, Di Benedetto A, et al. Bone marrow oxytocin mediates the anabolic action of estrogen on the skeleton. J Biol Chem. 2012;287:29159–29167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gooi JH, Pompolo S, Karsdal MA, et al. Calcitonin impairs the anabolic effect of PTH in young rats and stimulates expression of sclerostin by osteocytes. Bone. 2010;46:1486–1497 [DOI] [PubMed] [Google Scholar]
- 45. Villa I, Melzi R, Pagani F, Ravasi F, Rubinacci A, Guidobono F. Effects of calcitonin gene-related peptide and amylin on human osteoblast-like cells proliferation. Eur J Pharmacol. 2000;409:273–278 [DOI] [PubMed] [Google Scholar]
- 46. Cornish J, Callon KE, Lin CQ, et al. Comparison of the effects of calcitonin gene-related peptide and amylin on osteoblasts. J Bone Miner Res. 1999;14:1302–1309 [DOI] [PubMed] [Google Scholar]
- 47. Wang YS, Wang YH, Zhao GQ, Li YB. Osteogenic potential of human calcitonin gene-related peptide alpha gene-modified bone marrow mesenchymal stem cells. Chin Med J (Engl). 2011;124:3976–3981 [PubMed] [Google Scholar]
- 48. Han N, Jiang BG, Wang TB, Zhang PX, Kou YH, Zhang DY. [Effect of calcitonin gene-related peptide on RUNX2 expression in primary rat osteoblasts]. J Peking Univ Health Sci. 2011;43:652–656 [PubMed] [Google Scholar]
- 49. Wang L, Shi X, Zhao R, et al. Calcitonin-gene-related peptide stimulates stromal cell osteogenic differentiation and inhibits RANKL induced NF-κB activation, osteoclastogenesis and bone resorption. Bone. 2010;46:1369–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schinke T, Liese S, Priemel M, et al. Decreased bone formation and osteopenia in mice lacking α-calcitonin gene-related peptide. J Bone Miner Res. 2004;19:2049–2056 [DOI] [PubMed] [Google Scholar]
- 51. Winkler IG, Sims NA, Pettit AR, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116:4815–4828 [DOI] [PubMed] [Google Scholar]
- 52. Etienne W, Meyer MH, Peppers J, Meyer RA., Jr Comparison of mRNA gene expression by RT-PCR and DNA microarray. Biotechniques. 2004;36:618–620, 622,, 624–616 [DOI] [PubMed] [Google Scholar]
- 53. Morey JS, Ryan JC, Van Dolah FM. Microarray validation: factors influencing correlation between oligonucleotide microarrays and real-time PCR. Biol Proc Online. 2006;8:175–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kramer I, Baertschi S, Halleux C, Keller H, Kneissel M. Mef2c deletion in osteocytes results in increased bone mass. J Bone Miner Res. 2012;27:360–373 [DOI] [PubMed] [Google Scholar]
- 55. Bonewald LF. Mef2c does more than regulate Sost in osteocytes: distinct gender effects. IBMS Bone Key. 2012;9 [Google Scholar]
- 56. Baron R, Rawadi G. Targeting the Wnt/β-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007;148:2635–2643 [DOI] [PubMed] [Google Scholar]
- 57. Ke HZ, Richards WG, Li X, Ominsky MS. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr Rev. 2012;33(5):747–783 [DOI] [PubMed] [Google Scholar]
- 58. Balemans W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10:537–543 [DOI] [PubMed] [Google Scholar]
- 59. Brunkow ME, Gardner JC, Van Ness J, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–869 [DOI] [PubMed] [Google Scholar]
- 61. Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. 2010;25:178–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Morvan F, Boulukos K, Clement-Lacroix P, et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21:934–945 [DOI] [PubMed] [Google Scholar]
- 63. Winkler DG, Sutherland MK, Geoghegan JC, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. MacDonald BT, Joiner DM, Oyserman SM, Sharma P, Goldstein SA, He X, Hauschka PV. Bone mass is inversely proportional to Dkk1 levels in mice. Bone. 2007;41:331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Komori T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010;339:189–195 [DOI] [PubMed] [Google Scholar]
- 66. Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol. 2012;13:27–38 [DOI] [PubMed] [Google Scholar]
- 67. Costa AG, Cusano NE, Silva BC, Cremers S, Bilezikian JP. Cathepsin K: its skeletal actions and role as a therapeutic target in osteoporosis. Nat Rev Rheumatol. 2011;7:447–456 [DOI] [PubMed] [Google Scholar]
- 68. Boskey AL, Gelb BD, Pourmand E, et al. Ablation of cathepsin K activity in the young mouse causes hypermineralization of long bone and growth plates. Calcif Tissue Int. 2009;84:229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fujita Y, Nakata K, Yasui N, et al. Novel mutations of the cathepsin K gene in patients with pycnodysostosis and their characterization. J Clin Endocrinol Metab. 2000;85:425–431 [DOI] [PubMed] [Google Scholar]
- 70. Saftig P, Hunziker E, Wehmeyer O, et al. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci USA. 1998;95:13453–13458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gowen M, Lazner F, Dodds R, et al. Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J Bone Miner Res. 1999;14:1654–1663 [DOI] [PubMed] [Google Scholar]
- 72. Boonen S, Rosenberg E, Claessens F, Vanderschueren D, Papapoulos S. Inhibition of cathepsin K for treatment of osteoporosis. Curr Osteoporos Rep. 2012;10:73–79 [DOI] [PubMed] [Google Scholar]
- 73. Kiviranta R, Morko J, Uusitalo H, Aro HT, Vuorio E, Rantakokko J. Accelerated turnover of metaphyseal trabecular bone in mice overexpressing cathepsin K. J Bone Miner Res. 2001;16:1444–1452 [DOI] [PubMed] [Google Scholar]
- 74. DeMambro VE, Maile L, Wai C, et al. Insulin-like growth factor-binding protein-2 is required for osteoclast differentiation. J Bone Miner Res. 2012;27:390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Redd EH, Miller SC, Jee WS. Changes in endochondral bone elongation rates during pregnancy and lactation in rats. Calcif Tissue Int. 1984;36:697–701 [DOI] [PubMed] [Google Scholar]
- 76. Dawood MY, Khan-Dawood FS, Wahi RS, Fuchs F. Oxytocin release and plasma anterior pituitary and gonadal hormones in women during lactation. J Clin Endocrinol Metab. 1981;52:678–683 [DOI] [PubMed] [Google Scholar]
- 77. Colaianni G, Di Benedetto A, Zhu LL, et al. Regulated production of the pituitary hormone oxytocin from murine and human osteoblasts. Biochem Biophys Res Commun. 2011;411:512–515 [DOI] [PubMed] [Google Scholar]
- 78. Colucci S, Colaianni G, Mori G, Grano M, Zallone A. Human osteoclasts express oxytocin receptor. Biochem Biophys Res Commun. 2002;297:442–445 [DOI] [PubMed] [Google Scholar]
- 79. Tamma R, Colaianni G, Zhu LL, et al. Oxytocin is an anabolic bone hormone. Proc Natl Acad Sci USA. 2009;106:7149–7154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu X, Shimono K, Zhu LL, et al. Oxytocin deficiency impairs maternal skeletal remodeling. Biochem Biophys Res Commun. 2009;388:161–166 [DOI] [PubMed] [Google Scholar]
- 81. Nishimori K, Young LJ, Guo Q, Wang Z, Insel TR, Matzuk MM. Oxytocin is required for nursing but is not essential for parturition or reproductive behavior. Proc Natl Acad Sci USA. 1996;93:11699–11704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bostrom P, Wu J, Jedrychowski MP, et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nervina JM, Magyar CE, Pirih FQ, Tetradis S. PGC-1α is induced by parathyroid hormone and coactivates Nurr1-mediated promoter activity in osteoblasts. Bone. 2006;39:1018–1025 [DOI] [PubMed] [Google Scholar]
- 84. Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA. 2009;106:20405–20410 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85. Vidal-Puig A, Solanes G, Grujic D, Flier JS, Lowell BB. UCP3: an uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem Biophys Res Commun. 1997;235:79–82 [DOI] [PubMed] [Google Scholar]
- 86. Xiao XQ, Grove KL, Grayson BE, Smith MS. Inhibition of uncoupling protein expression during lactation: role of leptin. Endocrinology. 2004;145:830–838 [DOI] [PubMed] [Google Scholar]
- 87. Park SJ, Gadi J, Cho KW, et al. The forkhead transcription factor Foxc2 promotes osteoblastogenesis via up-regulation of integrin beta1 expression. Bone. 2011;49:428–438 [DOI] [PubMed] [Google Scholar]
- 88. Iida K, Koseki H, Kakinuma H, et al. Essential roles of the winged helix transcription factor MFH-1 in aortic arch patterning and skeletogenesis. Development. 1997;124:4627–4638 [DOI] [PubMed] [Google Scholar]
- 89. Kim SH, Cho KW, Choi HS, et al. The forkhead transcription factor Foxc2 stimulates osteoblast differentiation. Biochem Biophys Res Commun. 2009;386:532–536 [DOI] [PubMed] [Google Scholar]
- 90. Zhou Y, Rui L. Major urinary protein regulation of chemical communication and nutrient metabolism. Vitam Horm. 2010;83:151–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Beynon RJ, Hurst JL. Multiple roles of major urinary proteins in the house mouse, Mus domesticus. Biochem Soc Trans. 2003;31:142–146 [DOI] [PubMed] [Google Scholar]
- 92. Sang M, Wang L, Ding C, et al. Melanoma-associated antigen genes—an update. Cancer Lett. 2011;302:85–90 [DOI] [PubMed] [Google Scholar]
- 93. Ohman Forslund K, Nordqvist K. The melanoma antigen genes—any clues to their functions in normal tissues? Exp Cell Res. 2001;265:185–194 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.