

Abstract
Copper-catalyzed borylation of a variety of organic halides with bis(pinacolato)diboron allows the preparation of diverse potassium organotrifluoroborates. The reactions are mild and general, providing access to a variety of interesting, boron-containing building blocks, including those containing piperidine, pyrrole, azetidine, tetrahydropyran and oxetane substructures. Representative Minisci reactions are reported for select examples.
As a consequence of their successful and, thus, increasing use in a variety of reactions, the demand for new boron-containing building blocks is constantly growing.1 Among them, organotrifluoroborates have been shown to be a valuable alternative to boronic acids owing to the increased stability conferred by the tetracoordinated boron atom.2,3 In particular, the efficiency of the organotrifluoroborates in a broad range of reactions has been demonstrated, taking advantage of the reactivity of the boron-carbon bond, as well as reactions where the boron is retained.4,5
Aliphatic potassium organotrifluoroborates can be easily obtained by simply treating appropriate organoboron precursors with potassium bifluoride (KHF2) or KF in the presence of tartaric acid.6 In general, the requisite organoborons can be accessed by different methods: 1) reaction of an organometallic (organomagnesium or organolithium) reagent with an electrophilic boron species BX3 (X=Cl, F, OR);7,8 2) hydroboration of alkenes;9,10 or 3) borylation of alkanes by C–H bond activation.11,12 Recently, much attention has been placed on the use of nucleophilic boron species and, in preference to the highly reactive boryllithiums or NHC-boranes,13,14 activation of dibora compounds by a nucleophile was found to be a more efficient and useful synthetic protocol.15,16 More precisely, copper(I)-catalyzed reactions were particularly successful in the case of 1,4-additions to unsaturated carbonyl compounds,17,18 and after the pioneering work on activated electrophiles by Miyaura,19 the last year has witnessed the emergence of aliphatic borylation of halogenated substrates. Therefore, primary, secondary, and even tertiary alkylboronates have been prepared by Cu-,20,21 Ni-,22,23 or Pd-catalysis,24 providing access to previously problematic targets.25 Herein, we report the use of this method for the preparation of a variety of organotrifluoroborate building blocks of potential use as synthons in the development of drug-like compounds.26
We recently reported the preparation of potassium β-alkoxyethyltrifluoroborates, using an adaptation of Marder and Liu’s conditions,20 wherein a copper(I)-catalyzed borylation of the corresponding primary bromides was followed by treatment with KHF2 to afford the target structures. The resulting organotrifluoroborates were subsequently used in Suzuki-Miyaura cross-coupling reactions (eq 1).27
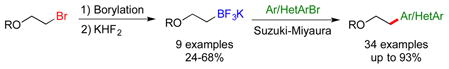 |
(1) |
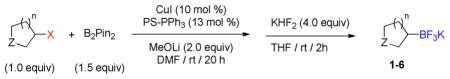
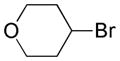
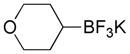
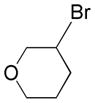
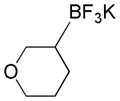
Based on the success of this approach, we explored other alkyl halides as substrates for the copper-promoted borylation, and we were particularly interested in saturated heterocycles bearing a secondary halogen, which would provide access to heterocyclic trifluoroborates that are challenging to access by other means. To this end, N-Boc-4-bromo-piperidine was used as a test substrate under the same conditions used for the β-alkoxyethyl system. The desired transformation proceeded smoothly and afforded, after treatment with potassium bifluoride, the corresponding potassium organotrifluoroborate 1a in 72% yield (Table 1, entry 1). As was reported previously, the use of polystyrene-supported triphenylphosphine (PS-PPh3) was found to be crucial to obtain a pure product, avoiding contamination with triphenylphosphine oxide. Moreover, these conditions proved to be general and have been successfully applied to various substrates. Indeed, different protecting groups on the nitrogen atom are tolerated (Table 1, entries 1 and 2) and six-, five- and even four-membered nitrogen heterocycles provided the desired products in acceptable yields (Table 1, entries 1, 3 and 4, respectively). Additionally, the analogous oxygenated heterocyclic trifluoroborates 4, 5 and 6 (Table 1, entries 5 –8), were obtained under the same conditions in decent yields, thus affording an alternative route to the iridium-catalyzed borylation of cyclic ethers recently reported by Hartwig.28 It should be noted that these new building blocks were obtained in yields that appear to be reflective of the efficiency of the borylation step. The trifluoroborates synthesized were found to be perfectly stable to air and moisture upon storage.
Table 1.
Borylation of Various Heterocyclic Halidesa
| |||
|---|---|---|---|
| entry | alkyl halide | product | yield (%) |
| 1 |
|
1a |
72 |
| 2 |
|
1b |
51 |
| 3 |
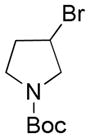
|
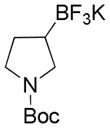 2 |
59 |
| 4 |
|
3 |
54 |
| 5 |
|
 4a |
27 |
| 6 |
|
 4a |
52 |
| 7 |
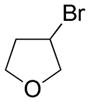
|
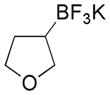 5 |
52 |
| 8 |
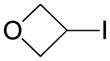
|
6 |
30 |
Reaction conditions: Halide (1.0 equiv), B2pin2 (1.5 equiv), CuI (10 mol %), PS-PPh3 (13 mol %), MeOLi (2.0 equiv), DMF ([halide] = 0.2 M), rt, 20 h then sat. aq KHF2 (4.0 equiv), THF, rt, 2 h.
Owing to the importance of the piperidine moiety in drug discovery, we explored whether this strategy would allow the preparation of other functionalized scaffolds. More precisely, we were particularly interested in the 2-pyridinyl protecting group as it has proven to be efficient in the functionalization of piperidines.29 Consequently, the desired precursor 9 was obtained in four steps from commercially available reagent 8: a PEPPSI ([1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene](3-chloropyridyl)palladium(II) dichloride)-catalyzed Buchwald-Hartwig amination of 2-chloropyridine 7 provided quantitatively the N-arylated intermediate,30 which, after two steps of deprotection and reduction, was successfully converted to the corresponding brominated compound 9 by heating at reflux in aqueous HBr (43% over four steps).31 As in the case of other piperidines 1a and 1b, the borylation of 9 was easily achieved, providing 1c in a yield of 60% (Scheme 1).
Scheme 1.
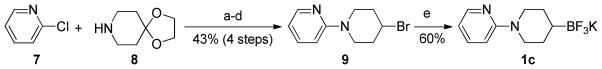
Preparation of potassium N-(2-pyridinyl)-4-trifluoroboratopiperidine 1c
Reactions conditions: a) 7 (1.0 equiv), 8 (1.2 equiv), PEPPSI-IPr (2 mol %), t-BuOK (1.5 equiv), DME, rt, 24 h, quant.; b) TsOH (20 mol %), H2O/acetone:1/3, 150 °C (MW), 1 h, 83%; c) NaBH4 (1.1 equiv), MeOH, rt, 1 h, 90%; d) HBr (48% in H2O, 17 equiv), 100 °C, 14 h, 58% (81% brsm); e) B2pin2 (1.5 equiv); CuI (10 mol %), PS-PPh3 (13 mol %), MeOLi (2.0 equiv), DMF, rt, 20 h then sat. aq KHF2 (4.0 equiv), THF, rt, 2 h, 60%.
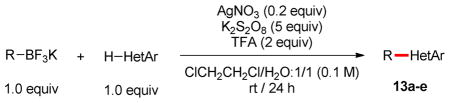
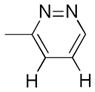
Having synthesized an array of new heterocyclic trifluoroborates, we investigated their behavior in the Minisci reaction with various heteroaromatic compounds. Although conditions previously reported by our laboratory with stoichiometric manganese(III) acetate were only successful in the case of the tetrahydropyran derivative 4a,32 slight modifications of the conditions recently developed by Baran proved to be effective for this transformation.33,34 Indeed, using an increased amount of potassium persulfate and trifluoroacetic acid allowed us to add building blocks 1a, 3 and 4a to lepidine in modest yields (Table 2, compounds 13a–c). Moreover, pyridazine and quinoxaline (Table 2, compounds 13d and 13e–f) were also valuable substrates, opening an alternative route for the preparation of heteroarylazetidines by means of a Minisci reaction.35
Table 2.
Minisci Reaction of Various Heterocyclic Trifluoroboratesa
| ||||
|---|---|---|---|---|
| entry | RBF3K | HetArH | product | yield (%) |
| 1 | 1a |
 10 |
 13a |
54 |
| 2 | 3 | 10 |
 13b |
31 |
| 3 | 4a | 10 |
 13c |
38 |
| 4 | 3 |
 11 |
 13d |
15 |
| 5 | 4a |
 12 |
 13e–f (C4/C5: 40/60) |
37 |
Reaction conditions: Heteroarene (1.0 equiv), potassium organotrifluoroborate (1.1 equiv), AgNO3 (20 mol %), K2S2O8 (5.0 equiv), TFA (2.0 equiv), ClCH2CH2Cl/H2O, 1: 1 ([heteroarene] = 0.1 M), rt, 24 h.
In conclusion, we have prepared various potassium organotrifluoroborate building blocks by copper-catalyzed borylation of the corresponding halides. This method is general and allows the preparation of useful boron-containing synthons of particular interest to medicinal chemistry in useful yields.36 Some of these new trifluoroborates have been successfully tested in the Minisci reaction, affording interesting compounds in a straightforward manner with modest yields.
Experimental Section
General Considerations
All commercially available reagents including anhydrous solvents were used without purification. CuI (99.999%), PEPPSI-IPr, N-Boc-3-bromopyrrolidine, N-Boc-3-iodoazetidine, 3-bromotetrahydropyran, and 3-bromotetrahydrofuran were all contained from commercial sources and used as received. N-Tosyl-4-bromopiperidine was prepared by the procedure of Gong et al.37 Reactions under microwave irradiation were performed in a Biotage Initiator system. Analytical thin-layer chromatography (TLC) was performed on TLC silica gel plates (0.25 mm) precoated with a fluorescent indicator. Flash chromatography (FC) was performed on a Biotage SP4 base system using GRACE Reveleris columns (silica size 40 μm) of various sizes. Visualization was effected with ultraviolet light and ethanolic KMnO4. NMR spectra were recorded on a 400 MHz or a 360 MHz spectrometer. Chemical shifts are given in ppm. 1H NMR chemical shifts were referenced to the residual solvent signal; 13C NMR chemical shifts were referenced to the deuterated solvent signal. Multiplicity was defined by DEPT 135 analysis. The resonance of the carbon center linked to the boron atom was not observed. 19F NMR chemical shifts were referenced to external CFCl3 (0.0 ppm). 11B NMR chemical shifts were referenced to external BF3·OEt2 (0.0 ppm) with a negative sign indicating an upfield shift. Data are presented as follows: chemical shift δ (ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, br = broad), coupling constant J (Hz), integration. High-resolution mass spectra were obtained by electrospray ionization on a TOF instrument.
General procedure A for the preparation of compounds 1–6
In air, B2pin2 (1.5 equiv), LiOMe (2.0 equiv), PS-PPh3 (13 mol %) and CuI (10 mol %) were weighed in a round-bottomed flask equipped with a stir bar. The flask was closed with a septum, evacuated, and backfilled with N2. DMF (5 mL) and the halide (1.0 mmol, 1.0 equiv) were successively added by syringes (or, if the halide was solid, it was added as a solution in DMF), and the reaction was vigorously stirred at rt for 20 h. Then, the reaction mixture was then diluted with CH2Cl2 (10 mL) and filtered through a pad of Celite, and rinsed with CH2Cl2 (20 mL). The resulting soln was concentrated, poured into sat. aq NH4Cl (20 mL) and extracted three times with Et2O (20 mL). The combined organic layers were washed successively with H2O (50 mL) and brine (50 mL), dried (MgSO4) and concentrated. The residue was solubilized in THF (5 mL) in a round-bottomed flask equipped with a stir bar and sat. aq KHF2 (4.5 M, 0.9 mL, 4.0 equiv) was added. The flask was closed with a septum and the resulting mixture was stirred at rt for 2 h. The reaction mixture was evaporated to dryness, and the resulting salt was extracted several times with hot acetone. The filtrate was concentrated to ~5 mL, and precipitation was achieved by dropwise addition of Et2O (100 mL) at 0 °C. The resulting product was collected by gravity filtration on a fritted funnel and dried to afford the corresponding potassium heterocyclic trifluoroborate as a white solid.
Potassium N-Boc-4-(Trifluoroborato)piperidine 1a
Following general procedure A, the reaction performed with N-Boc-4-bromopiperidine (0.94 g, 3.55 mmol) afforded 741 mg (72%) of the title compound as a white solid. Mp 213–215 °C. 1H NMR (acetone-d6, 400 MHz): δ 3.97 (br, 2H), 2.52 (br, 2H), 1.49 (dd, J = 13, 3 Hz, 2H), 1.41 (s, 9H), 1.26 – 1.10 (m, 2H), 0.28 (m, 1H). 13C NMR (acetone-d6, 100 MHz): δ 155.3 (C), 78.4 (C), 46.6 (2 CH2), 29.1 (2 CH2), 28.8 (3 CH3). 11B NMR (acetone-d6, 128 MHz): δ 4.84 (br). 19F NMR (acetoned6, 377 MHz): δ −148.4. HRMS (ESI) m/z: (M − K)− Calcd. for C10H18BF3NO2 252.1388; Found 252.1380.
Potassium N-Tosyl-4-(trifluoroborato)piperidine 1b
Following general procedure A, the reaction performed with N-tosyl-4-bromopiperidine (596 mg, 1.87 mmol) afforded 330 mg (51%) of the title compound as a white solid. Mp 217–220 °C. 1H NMR (DMSO-d6, 400 MHz): δ 7.56 (d, J = 8 Hz, 2H), 7.41 (d, J = 8 Hz, 2H), 3.51 – 3.49 (m, 2H), 2.40 (s, 3H), 1.99 – 1.93 (m, 2H), 1.46 (dd, J = 13, 2 Hz, 2H), 1.27 – 1.10 (m, 2H), −0.16 (br, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 142.9 (C), 132.9 (C), 129.5 (2 CH), 127.3 (2 CH), 47.9 (2 CH2), 27.4 (2 CH2), 20.9 (CH3). 11B NMR (DMSO-d6, 128 MHz): δ 4.71 (br). 19F NMR (DMSO-d6, 377 MHz): δ −144.6. HRMS (ESI) m/z: (M − K)− Calcd. for C12H16BF3NO2S 306.0952; Found 306.0950.
Potassium N-Boc-3-(Trifluoroborato)pyrrolidine 2
Following general procedure A, the reaction performed with N-Boc-3-bromopyrrolidine (625 mg, 2.50 mmol) afforded 408 mg (59%) of the title compound as a white solid. Mp 195–199 °C. 1H NMR (acetone-d6, 400 MHz): δ 3.35 – 3.22 (m, 2H), 3.05 – 2.91 (m, 2H), 1.69 (dt, J = 12, 6 Hz, 1H), 1.63 – 1.50 (m, 1H), 1.41 (s, 9H), 0.94 (br, 1H). 13C NMR (acetone-d6, 100 MHz): δ 155.2 (C), 77.8 (C), 50.9 (CH2), 50.5 (CH2), 48.3 (CH2), 28.9 (3 CH3). 11B NMR (acetone-d6, 128 MHz): δ 4.55 (br). 19F NMR (acetone-d6, 377 MHz): δ −146.2. HRMS (ESI) m/z: (M − K)− Calcd. for C9H16BF3NO2 238.1232; Found 238.1223.
Potassium N-Boc-3-(Trifluoroborato)azetidine 3
Following general procedure A, the reaction performed with N-Boc-3-iodoazetidine (1.63 g, 5.75 mmol) afforded 826 mg (54%) of the title compound as a white solid. Mp 182–185 °C. 1H NMR (DMSO-d6, 400 MHz): δ 3.57 (br, 4H), 1.34 (s, 9H), 1.31 (br, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 155.6 (C), 77.1 (C), 51.9 (CH2), 50.7 (CH2), 28.2 (3 CH3). 11B NMR (DMSO-d6, 128 MHz): δ 4.11 (br). 19F NMR (DMSO-d6, 377 MHz): δ −145.2. HRMS (ESI) m/z: (M − K)− Calcd. for C8H14BF3NO2 224.1075; Found 224.1067.
Potassium 4-(Trifluoroborato)tetrahydropyran 4a
Following general procedure A, the reaction performed with 4-bromotetrahydropyran (0.5 g, 3.0 mmol) afforded 155 mg (27%) of the title compound as a white solid. Mp >250 °C. 1H NMR (DMSO-d6, 400 MHz,): δ 3.74 (d, J = 10 Hz, 2H), 3.12 (td, J = 10, 3 Hz, 2H), 1.32 – 1.13 (m, 4H), 0.21 – 0.19 (m, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 69.3 (2 CH2), 28.9 (2 CH2). 11B NMR (DMSO-d6, 128 MHz): δ 4.39 (q, J = 58 Hz). 19F NMR (DMSO-d6, 377 MHz): δ −145.0. HRMS (ESI) m/z: (M − K)− Calcd. for C5H9BF3O 153.0704; Found 153.0697. Analytical data are consistent with that previously reported.32
Potassium 3-(Trifluoroborato)tetrahydropyran 4b
Following general procedure A, the reaction performed with 3-bromotetrahydropyran (0.5 g, 3.0 mmol) afforded 304 mg (52%) of the title compound as a white solid. Mp 204–207 °C. 1H NMR (DMSO-d6, 400 MHz,): δ 3.73 – 3.64 (m, 2H), 3.03 – 3.17 (m, 2H), 1.57 – 1.53 (m, 1H), 1.37 – 1.31 (m, 2H), 1.20 – 1.07 (m, 1H), 0.40 (br, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 72.3 (CH2), 67.7 (CH2), 28.0 (CH2), 25.7 (CH2). 11B NMR (DMSO-d6, 128 MHz): δ 3.80 (br). 19F NMR (DMSO-d6, 377 MHz): δ −143.2. HRMS (ESI) m/z: (M − K)− Calcd. for C5H9BF3O 153.0704; Found 153.0697.
Potassium 3-(Trifluoroborato)tetrahydrofuran 5
Following general procedure A, the reaction performed with 3-bromotetrahydrofuran (0.5 g, 3.3 mmol) afforded 307 mg (52%) of the title compound as a white solid. Mp 169–171 °C. 1H NMR (DMSO-d6, 400 MHz,): δ 3.63 (t, J = 8 Hz, 1H), 3.53 (td, J = 8, 3 Hz, 1H), 3.41 – 3.35 (m, 1H), 3.23 (dd, J = 10, 8 Hz, 1H), 1.68 – 1.57 (m, 1H), 1.46 (t, J = 10 Hz, 1H), 0.78 (br, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 71.8 (CH2), 67.6 (CH2), 29.5 (CH2). 11B NMR (DMSO-d6, 128 MHz): δ 4.32 (q, J = 59 Hz). 19F NMR (DMSO-d6, 377 MHz): δ −141.0. HRMS (ESI) m/z: (M − K)− Calcd. for C4H7BF3O 139.0548; Found 139.0538. Analytical data are consistent with that previously reported.28
Potassium 3-(Trifluoroborato)oxetane 6
Following general procedure A, the reaction performed with 3-iodooxetane (2.14 g, 11.63 mmol) afforded 580 mg (30%) of the title compound as a white solid. Mp 211–213 °C. 1H NMR (DMSO-d6, 400 MHz,): δ 4.44 – 4.42 (br, 4H), 1.93 (br, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 74.7 (2 CH2). 11B NMR (DMSO-d6, 128 MHz): δ 3.97 (q, J = 59 Hz). 19F NMR (DMSO-d6, 377 MHz): δ −144.2. HRMS (ESI) m/z: (M − K)− Calcd. for C3H5BF3O 125.0391; Found 125.0381.
Preparation of Potassium N-(2-Pyridinyl)-4-trifluoroboratopiperidine 1c. N-(2-Pyridinyl)-4-piperidinone Ethylene Ketal
In air, KOt-Bu (9.0 g, 79.86 mmol, 1.5 equiv) and PEPPSI-IPr (702 mg, 1.06 mmol, 2 mol %) were weighed in a 250 mL round bottom flask equipped with a stir bar. The flask was closed with a septum, evacuated, and backfilled with N2. DME (50 mL), 2-chloropyridine (6.04 g, 53.24 mmol, 1.0 equiv) and 4-piperidinone ethylene ketal (9.16 g, 63.89 mmol, 1.2 equiv) were successively added by syringes. The reaction was stirred at rt for 3 d. Then, the reaction mixture was filtered through a pad of Celite (rinsed with 100 mL of EtOAc) and the filtrate was evaporated to afford the crude product. Purification by flash chromatography (SiO2, CH2Cl2 to CH2Cl2/EtOAc, 1: 1) afforded 11.8 g (quant) of the title compound as a pale yellow oil. 1H NMR (CDCl3, 360 MHz,): δ 8.22 – 8.12 (m, 1H), 7.52 – 7.38 (m, 1H), 6.75 – 6.63 (m, 1H), 6.63 – 6.50 (m, 1H), 3.99 (s, 4H), 3.74 – 3.63 (m, 4H) 1.81 – 1.72 (m, 4H). 13C NMR (CDCl3, 90 MHz): δ 158.8 (C), 147.9 (CH), 137.4 (CH), 112.7 (CH), 107.5 (C), 107.0 (CH), 64.3 (2 CH2), 43.4 (2 CH2), 34.3 (2 CH2 HRMS (ESI) m/z: (M + H)+ Calcd. for C12H17N2O2 221.1290; Found 221.1279. Analytical data are consistent with that previously reported.29
N-(2-Pyridinyl)-4-piperidinone
In air, a solution of N-(2-pyridinyl)-4-piperidinone ethylene ketal (9.8 g, 44.4 mmol, 1.0 equiv) and p-TsOH monohydrate (846 mg, 4.45 mmol, 10 mol %) in acetone (36 mL) and H2O (12 mL) was split equally in four 10–20 mL microwave vials equipped with a stir bar. The tubes were sealed and each of them was irradiated at 150 °C for 30 min. The tops were removed and another portion of p-TsOH (10 mol %) was added. The tubes were sealed and each of them was irradiated at 150 °C for 30 min. The tops were removed and the reaction mixtures were combined and concentrated to afford the crude product. Purification by flash chromatography (SiO2, CH2Cl2 to CH2Cl2/MeOH, 98:2) afforded 6.5 g (83%) of the title compound as a yellow oil. 1H NMR (CDCl3, 360 MHz,): δ 8.22 – 8.15 (m, 1H), 7.56 – 7.46 (m, 1H), 6.73 (d, J = 8 Hz, 1H), 6.65 (dd, J = 7, 5 Hz, 1H), 3.89 (d, J = 12 Hz, 4H), 2.48 (d, J = 12 Hz, 4H). 13C NMR (CDCl3, 90 MHz): δ 208.4 (C), 157.7 (C), 148.1 (CH), 137.7 (CH), 113.5 (CH), 106.8 (CH), 44.6 (2 CH2), 40.5 (2 CH2). HRMS (ESI) m/z: (M + H)+ Calcd. for C10H13N2O 177.1028; Found 177.1008.
N-(2-Pyridinyl)-4-hydroxypiperidine
In air, N-(2-pyridinyl)-4-piperidinone (6.2 g, 35.3 mmol, 1.0 equiv) was solubilized in MeOH (150 mL) in a 250 mL round bottom flask equipped with a stir bar. The flask was closed with a septum and cooled to 0 °C. NaBH4 (1.5 g, 38.8 mmol, 1.1 equiv) was added portionwise. The reaction was stirred at rt for 1 h. Then, the reaction mixture was poured into sat aq NH4Cl (200 mL) and the resulting solution was extracted with EtOAc (2×100 mL). The combined organic layers were washed with brine (100 mL), dried (MgSO4) and evaporated to afford the crude product. Purification by flash chromatography (SiO2, CH2Cl2 to CH2Cl2/MeOH, 95:5) afforded 5.7 g (90%) of the title compound as a pale yellow oil. 1H NMR (CDCl3, 360 MHz,): δ 8.16 (dd, J = 5, 1 Hz, 1H), 7.51 – 7.40 (m, 1H), 6.73 – 6.62 (m, 1H), 6.58 (dd, J = 7, 5 Hz, 1H), 4.05 (ddd, J = 9, 8, 4 Hz, 2H), 3.90 (tt, J = 9, 4 Hz, 1H), 3.20 – 3.04 (m, 2H), 2.13 (br, 1H), 2.03 – 1.87 (m, 2H) 1.57 (dtd, J = 13, 9, 4 Hz, 2H). 13C NMR (CDCl3, 90 MHz): δ 159.2 (C), 147.9 (CH), 137.5 (CH), 112.8 (CH), 107.2 (CH), 68.1 (CH), 43.1 (2 CH2), 33.8 (2 CH2). HRMS (ESI) m/z: (M + H)+ Calcd. for C10H15N2O 179.1184; Found 179.1155.
N-(2-Pyridinyl)-4-bromopiperidine 9
In air, N-(2-pyridinyl)-4-hydroxypiperidine (4.5 g, 25.5 mmol, 1.0 equiv) was solubilized in HBr, 48% in H2O (50 mL, 17 equiv) in a 250 mL round bottom flask equipped with a stir bar, and the flask was equipped with a condenser. The reaction was heated at 120 °C (oil bath temperature) for 14 h. Then, the reaction mixture was cooled to rt and 1 M aq NaOH was added until pH > 12. The resulting solution was extracted with CH2Cl2 (3×100 mL) and the combined organic layers were dried (MgSO4) and evaporated to afford the crude product. Purification by flash chromatography (SiO2, CH2Cl2 to CH2Cl2/MeOH, 95:5) afforded 3.6 g (58%) of the title compound as a yellow oil (and 1.24 g of recovered starting material). 1H NMR (CDCl3, 360 MHz,): δ 8.24 – 8.12 (m, 1H), 7.55 – 7.42 (m, 1H), 6.74 – 6.53 (m, 2H), 4.42 (dt, J = 8, 4 Hz, 1H), 4.02 – 3.84 (m, 2H), 3.52 – 3.33 (m, 2H), 2.26 – 2.17 (m, 2H), 2.14 – 1.95 (m, 2H). 13C NMR (CDCl3, 90 MHz): δ 159.0 (C), 148.0 (CH), 137.5 (CH), 113.1 (CH), 107.1 (CH), 50.2 (CH), 44.1 (2 CH2), 35.4 (2 CH2). HRMS (ESI) m/z: (M + H)+ Calcd. for C10H14BrN2 241.0340; Found 241.0369.
Potassium N-(2-Pyridinyl)-4-(trifluoroborato)piperidine 1c
Following general procedure A, the reaction performed with N-(2-pyridinyl)-4-bromopiperidine 9 (3.4 g, 14.1 mmol) afforded 2.27 g (60%) of the title compound as a white solid. Mp 215–217 °C 1H NMR (DMSO-d6, 400 MHz): δ 8.06 – 7.98 (m, 1H) 7.41 (ddd, J = 9, 7, 2 Hz, 1H), 6.67 (d, J = 9 Hz, 1H), 6.51 – 6.42 (m, 1H), 4.19 – 4.16 (m, 2H), 2.61 – 2.56 (m, 2H), 1.49 – 1.45 (m, 2H), 1.20 – 1.09 (m, 2H) 0.23 – 0.18 (m, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 159.3 (C), 147.4 (CH), 137.1 (CH), 111.2 (CH), 106.6 (CH), 47.2 (2 CH2), 27.6 (2 CH2). 11B NMR (DMSO-d6, 128 MHz): δ 4.47 (br). 19F NMR (DMSO-d6, 377 MHz): δ −144.5. HRMS (ESI) m/z: (M − K)− Calcd. for C10H13BF3N2 229.1129; Found 229.1122.
General Procedure B for the Minisci Reactions
In air, potassium organotrifluoroborate (1.1 equiv), AgNO3 (0.2 equiv) and K2S2O8 (5.0 equiv) were weighed in a reaction tube equipped with a stir bar. ClCH2CH2Cl (2.5 mL), H2O (2.5 mL), heteroarene (0.5 mmol, 1.0 equiv) and TFA (2.0 equiv) were successively added, and the tube was sealed. The reaction was vigorously stirred at rt for 24 h. Then, the reaction mixture was poured into 20 mL of a 1/1:v/v mixture of sat. aq NaHCO3 and 5 % aq NaS2O3, and the resulting solution was extracted three times with CH2Cl2 (30 mL). The combined organic layers were dried (MgSO4) and evaporated to afford the crude product. Purification by flash chromatography (SiO2, CH2Cl2 to CH2Cl2/MeOH, 95:5) afforded the desired product.
2-(N-Boc-4-Piperidinyl)lepidine 13a
Following general procedure B, the reaction performed with 1a (160 mg, 0.55 mmol) and 10 (72 mg, 0.50 mmol) afforded 88 mg (54%) of the title compound as a pale yellow oil. 1H NMR (CDCl3, 360 MHz): δ 8.04 (d, J = 8 Hz, 1H), 7.96 (d, J = 8 Hz, 1H), 7.73 – 7.64 (m, 1H), 7.57 – 7.47 (m, 1H), 7.15 (s, 1H), 4.29 (br, 2H), 3.09 – 2.96 (m, 1 H), 2.89 (br, 2H), 2.69 (s, 3H), 2.04 – 1.92 (m, 2H), 1.92 – 1.75 (m, 2H), 1.50 (s, 9H). 13C NMR (CDCl3, 90 MHz): δ 164.3 (C), 154.8 (C), 147.6 (C), 144.6 (C), 129.5 (CH), 129.1 (CH), 127.1 (C), 125.6 (CH), 123.6 (CH), 120.0 (CH), 79.4 (C), 45.5 (CH), 43.6 (2 CH2), 31.6 (2 CH2), 28.5 (3 CH3), 18.8 (CH3). HRMS (ESI) m/z: (M + H)+ Calcd. for C20H27N2O2 327.2072; Found 327.2059.
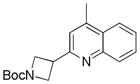
2-(N-Boc-3-Azetidinyl)lepidine 13b
Following general procedure B, the reaction performed with 3 (145 mg, 0.55 mmol) and 10 (72 mg, 0.50 mmol) afforded 47 mg (31%) of the title compound as a brown oil. 1H NMR (CDCl3, 400 MHz): δ 8.05 (dd, J = 9, 1 Hz, 1H), 7.96 (dd, J = 8, 1.0 Hz, 1H), 7.69 (ddd, J = 8, 7, 1 Hz, 1H), 7.53 (ddd, J = 8, 7, 1 Hz, 1H), 7.25 (s, 1H), 4.43 – 4.34 (m, 2H), 4.32 – 4.24 (m, 2H), 4.07 – 3.95 (m, 1H), 2.70 (d, J = 1 Hz, 3H), 1.48 (s, 9H). 13C NMR (CDCl3, 100 MHz): δ 160.8 (C), 156.5 (C), 147.4 (C), 145.1 (C), 129.6 (CH), 129.3 (CH), 127.1 (C), 126.0 (CH), 123.6 (CH), 119.9 (CH), 79.4 (C), 54.5 (2 CH2), 35.7 (CH), 28.4 (3 CH3), 18.7 (CH3). HRMS (ESI) m/z: (M + H)+ Calcd. for C18H23N2O2 299.1759; Found 299.1789. Analytical data are consistent with that previously reported.35
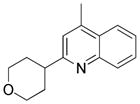
2-(4-Tetrahydropyranyl)lepidine 13c
Following general procedure B, the reaction performed with 4a (106 mg, 0.55 mmol) and 10 (72 mg, 0.50 mmol) afforded 44 mg (38%) of the title compound as a white solid. Mp 108 °C 1H NMR (CDCl3, 400 MHz): δ 8.04 (dd, J = 8, 1 Hz, 1H), 7.93 (dd, J = 8, 1 Hz, 1H), 7.66 (ddd, J = 8, 7, 1 Hz, 1H), 7.49 (ddd, J = 8, 7, 1 Hz, 1H), 7.16 (s, 1H), 4.16 – 4.07 (m, 2 H), 3.58 (td, J = 12, 2 Hz, 2H), 3.11 (tt, J = 11.9, 4.0 Hz, 1H), 2.65 – 2.70 (m, 3H), 2.09 – 1.95 (m, 2H), 1.95 – 1.86 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 164.1 (C), 147.5 (C), 144.8 (C), 129.4 (CH), 129.0 (CH), 127.0 (C), 125.5 (CH), 123.5 (CH), 119.8 (CH), 68.0 (2 CH2), 44.2 (CH), 32.2 (2 CH2), 18.7 (CH3). HRMS (ESI) m/z: (M + H)+ Calcd. for C15H18NO 228.1388; Found 228.1386. Analytical data are consistent with that previously reported.32
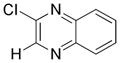
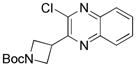
2-Chloro-3-(N-Boc-3-azetidinyl)quinoxaline 13d
Following general procedure B, the reaction performed with 3 (66 mg, 0.25 mmol) and 11 (41 mg, 0.25 mmol) afforded 12 mg (15%) of the title compound as a colorless oil. 1H NMR (CDCl3, 500 MHz): δ, 8.14–8.12 (m, 1H), 8.03–8.01 (m, 1H), 7.82–7.78 (m, 2H), 4,46 (br, 2H), 4,41 (t, J = 8.0 Hz, 2H), 4.36–4.33 (m, 1H), 1.47 (s, 9H). 13C NMR (CDCl3, 125.8 MHz): δ 156.3 (C), 153.1 (C), 146.8 (C), 141.0 (C), 140.5 (C), 130.6 (CH), 130.3 (CH), 128.9 (CH), 128.0 (CH), 79.6 (C), 38.8 (2 CH2), 32.9 (CH), 28.3 (3 CH3). HRMS (ESI) m/z: (M2 + H)+ Calcd. for C32H37N6O4Cl2 639.2253; Found 639.2231.
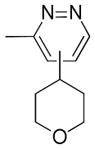
3-Methyl-4-(4-tetrahydropyranyl)pyridazine 13e and 3-Methyl-5-(4-tetrahydropyranyl)pyridazine 13f
Following general procedure B, the reaction performed with 4a (106 mg, 0.55 mmol) and 12 (47 mg, 0.50 mmol) afforded 33 mg (37%) of 13e and 13f (C4 and C5 regioisomers, respectively) as a 40:60 mixture as a brown oil. 1H NMR (CDCl3, 400 MHz): δ 8.95 (d, J = 5 Hz, 1H), 8.89 (d, J = 2 Hz, 1H), 7.21 (d, J = 5 Hz, 1H), 7.11 (d, J = 2 Hz, 1H), 4.12 – 4.01 (m, 4H), 3.58 – 3.44 (m, 4H), 2.73 – 2.92 (m, 2H), 2.72 (s, 3H), 2.66 (s, 3H), 1.68 – 1.79 (m, 8H). 13C NMR (CDCl3, 100 MHz): δ 159.7 (C), 158.8 (C), 150.0 (CH), 149.2 (CH), 144.1 (C), 143.0 (C), 124.3 (CH), 122.9 (CH), 67.8 (2 CH2), 67.6 (2 CH2), 38.3 (CH), 36.4 (CH), 32.2 (2 CH2), 31.7 (2 CH2), 22.2 (CH3), 19.8 (CH3). HRMS (ESI) m/z: (M + H)+ Calcd. for C10H15N2O 179.1184; Found 179.1202.
Supplementary Material
Acknowledgments
This research was supported by NIH (NIGMS R01 GM35249) and the Neuroscience Medicinal Chemistry Department of Janssen. Michel Carpentier (Janssen) and Dr. Rakesh Kohli (University of Pennsylvania) are acknowledged for obtaining HRMS data, and we thank DaWeon Ryu (University of Pennsylvania) for providing some characterization data.
Footnotes
Copies of NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org
Contributor Information
Frederik Rombouts, Email: frombout@its.jnj.com.
Gary A. Molander, Email: gmolandr@sas.upenn.edu.
References
- 1.Hall DG, editor. Boronic Acids. Wiley-VCH; Weinheim: 2011. [Google Scholar]
- 2.Molander GA, Figueroa R. Aldrichimica Acta. 2005;38:49. [Google Scholar]
- 3.Darses S, Genet JP. Chem Rev. 2008;108:288. doi: 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- 4.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- 5.Molander GA, Canturk B. Angew Chem, Int Ed. 2009;48:9240. doi: 10.1002/anie.200904306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lennox AJJ, Lloyd-Jones GC. Angew Chem, Int Ed. 2012;51:9385. doi: 10.1002/anie.201203930. [DOI] [PubMed] [Google Scholar]
- 7.Pintaric C, Olivero S, Gimbert Y, Chavant PY, Dunach E. J Am Chem Soc. 2010;132:11825. doi: 10.1021/ja1052973. [DOI] [PubMed] [Google Scholar]
- 8.Brown HC, Cole TE. Organometallics. 1983;2:1316. [Google Scholar]
- 9.Brown HC, Subba Rao BCJ. Am Chem Soc. 1956;78:2582. [Google Scholar]
- 10.Burgess K, Ohlmeyer MJ. Chem Rev. 1991;91:1179. [Google Scholar]
- 11.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 12.Ishiyama T, Miyaura N. J Organomet Chem. 2003;680:3. [Google Scholar]
- 13.Segawa Y, Yamashita M, Nozaki K. Science. 2006;314:113. doi: 10.1126/science.1131914. [DOI] [PubMed] [Google Scholar]
- 14.Curran DP, Solovyev A, Makhlouf Brahmi M, Fensterbank L, Malacria M, Lacôte E. Angew Chem, Int Ed. 2011;50:10294. doi: 10.1002/anie.201102717. [DOI] [PubMed] [Google Scholar]
- 15.Lee K, Zhugralin AR, Hoveyda AH. J Am Chem Soc. 2009;131:7253. doi: 10.1021/ja902889s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonet A, Gulyas H, Fernandez E. Angew Chem, Int Ed. 2010;49:5130. doi: 10.1002/anie.201001198. [DOI] [PubMed] [Google Scholar]
- 17.Ito H, Yamanaka H, Tateiwa J, Hosomi A. Tetrahedron Lett. 2000;41:6821. [Google Scholar]
- 18.Takahashi K, Ishiyama T, Miyaura N. Chem Lett. 2000:982. [Google Scholar]
- 19.Ishiyama T, Oohashi Z, Ahiko T, Miyaura N. Chem Lett. 2002:780. [Google Scholar]
- 20.Yang CT, Zhang ZQ, Tajuddin H, Wu CC, Liang J, Liu JH, Fu Y, Czyzewska M, Steel PG, Marder TB, Liu L. Angew Chem, Int Ed. 2012;51:528. doi: 10.1002/anie.201106299. [DOI] [PubMed] [Google Scholar]
- 21.Ito H, Kubota K. Org Lett. 2012;14:890. doi: 10.1021/ol203413w. [DOI] [PubMed] [Google Scholar]
- 22.Yi J, Liu JH, Liang J, Dai JJ, Yang CT, Fu Y, Liu L. Adv Synth Catal. 2012;354:1685. [Google Scholar]
- 23.Dudnik AS, Fu GC. J Am Chem Soc. 2012;134:10693. doi: 10.1021/ja304068t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joshi-Pangu A, Ma X, Diane M, Iqbal S, Kribs RJ, Huang R, Wang CY, Biscoe MR. J Org Chem. 2012;77:6629. doi: 10.1021/jo301156e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Georgiou I, Whiting A. Eur J Org Chem. 2012:4110. [Google Scholar]
- 26.Lovering F, Bikker J, Humblet C. J Med Chem. 2009;52:6752. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 27.Fleury-Brégeot N, Presset M, Beaumard F, Colombel V, Oehlrich D, Rombouts F, Molander GAJ. Org Chem. 2012;77:10399– 10408. doi: 10.1021/jo3021665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liskey CW, Hartwig JF. J Am Chem Soc. 2012;134:12422. doi: 10.1021/ja305596v. [DOI] [PubMed] [Google Scholar]
- 29.Prokopcová H, Bergman SD, Aelvoet K, Smout V, Herrebout W, Van der Veken B, Meerpoel L, Maes BUW. Chem Eur J. 2010;16:13063. doi: 10.1002/chem.201001887. [DOI] [PubMed] [Google Scholar]
- 30.Organ MG, Abdel-Hadi M, Avola S, Dubovyk I, Hadei N, Kantchev EAB, O’Brien CJ, Sayah M, Valente C. Chem Eur J. 2008;14:2443. doi: 10.1002/chem.200701621. [DOI] [PubMed] [Google Scholar]
- 31.Bailey JM, Booth H, Al-Shirayda ARY, Trimble ML. J Chem Soc, Perkin Trans 2. 1984:737. [Google Scholar]
- 32.Molander GA, Colombel V, Braz VA. Org Lett. 2011;13:1852. doi: 10.1021/ol2003572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seiple IB, Su S, Rodriguez RA, Gianatassio R, Fujiwara Y, Sobel AL, Baran PS. J Am Chem Soc. 2010;132:13194. doi: 10.1021/ja1066459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lockner JW, Dixon DD, Risgaard R, Baran PS. Org Lett. 2011;13:5628. doi: 10.1021/ol2023505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duncton MA, Estiarte MA, Johnson RJ, Cox M, O’Mahony DJR, Edwards WT, Kelly MG. J Org Chem. 2009;74:6354. doi: 10.1021/jo9010624. [DOI] [PubMed] [Google Scholar]
- 36.Burkhard JA, Guérot C, Knust H, Rogers-Evans M, Carreira EM. Org Lett. 2010;12:1944. doi: 10.1021/ol1003302. [DOI] [PubMed] [Google Scholar]
- 37.Yu X, Yang T, Wang S, Xu H, Gong H. Org Lett. 2011;13:2138. doi: 10.1021/ol200617f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.