

Abstract
Allergic airway inflammation is characterized by increased expression of pro-inflammatory mediators, inflammatory cell infiltration, mucus hypersecretion, and airway hyperresponsiveness, in parallel with oxidative DNA base and strand damage, whose etiological role is not understood. Our goal was to establish the role of 8-oxoguanine (8-oxoG), a common oxidatively damaged base, and its repair by 8-oxoguanine DNA glycosylase 1 (Ogg1) in allergic airway inflammatory processes. Airway inflammation was induced by intranasally administered ragweed (Ambrosia artemisiifolia) pollen grain extract (RWPE) in sensitized BALB/c mice. We utilized siRNA technology to deplete Ogg1 from airway epithelium; 8-oxoG and DNA strand break levels were quantified by Comet assays. Inflammatory cell infiltration and epithelial methaplasia were determined histologically, mucus and cytokines levels biochemically and enhanced pause was used as the main index of airway hyperresponsiveness. Decreased Ogg1 expression and thereby 8-oxoG repair in the airway epithelium conveyed a lower inflammatory response after RWPE challenge of sensitized mice, as determined by expression of Th2 cytokines, eosinophilia, epithelial methaplasia, and airway hyperresponsiveness. In contrast, 8-oxoG repair in Ogg1-proficient airway epithelium was coupled to an increase in DNA single-strand break (SSB) levels and exacerbation of allergen challenge-dependent inflammation. Decreased expression of the Nei-like glycosylases Neil1 and Neil2 that preferentially excise ring-opened purines and 5-hydroxyuracil, respectively, did not alter the above parameters of allergic immune responses to RWPE. These results show that DNA SSBs formed during Ogg1-mediated repair of 8-oxoG augment antigen-driven allergic immune responses. A transient modulation of OGG1 expression/activity in airway epithelial cells could have clinical benefits.
Keywords: allergic inflammation, DNA repair, oxidative DNA damage, oxidative stress, 8-oxoguanine, 8-oxoguanine DNA glycosylase 1
1. Introduction
Exposure to pollen grains is the major cause of seasonal allergic reactions in the skin, eyes, and upper and lower respiratory tracts [1, 2]. Air pollutants can modify the antigenic (allergenic) properties of pollen and generate reactive oxygen species (ROS)4, which promote pro-inflammatory signaling in airway epithelium, thereby increase the severity and complexity of allergic symptoms in atopic subjects [3, 4]. It has been reported that pollen grains, including those of short ragweed (Ambrosia artemisiifolia), have intrinsic NAD(P)H oxidase (NOX) activity [5, 6]. Upon interaction of pollen with mucosal surfaces, pollen NOX becomes activated and generates ROS, leading to the formation of lipid peroxides (4-HNE, MDA) and oxidized glutathione (GSSG) [5], and activation of transcription factors (including NF- κB) [7]; together with antigens this provokes robustallergic inflammation in the airways and conjunctiva in sensitized subjects [5, 8, 9]. Additionally, we previously reported that inhibition/inactivation of the pollens’ NOX activity decreases both T helper 1 (Th1) and Th2 responses [5, 6, 10–12].
The pollen NOX primarily generates superoxide anions (O2•−), which are reduced to H2O2 and then to hydroxyl radicals (•OH), causing damage to macromolecules, including DNA [5, 13]. The primary target of ROS in DNA is guanine, because it has the lowest redox potential of the four nucleobases [14, 15]. Although guanine base lesions vary according to the nature of the oxidants [16], 7,8-dihydro-8-oxoguanine (8-oxoG) is the most frequent oxidation product in both DNA and RNA, and the accumulation of 8-oxoG is considered a biomarker of inflammation [17]. While its repair in RNA is poorly understood [18], 8-oxoG is repaired in DNA via the base excision /single strand break (BER/SSB) repair pathway [19, 20]. In mammalian cells, repair of 8-oxoG is initiated by the 8-oxoguanine DNA glycosylase 1 (OGG1) [21]. The 3′-phospho-α,β-unsaturated aldehyde terminus (apurinic/apyrimidinic [AP] sites) produced by Ogg1 is removed by the 3′-phosphodiesterase activity of the AP endonuclease-1 (APE1) to generate 3′OH for DNA polymerases, which then incorporate the intact base followed by action of DNA ligase to complete the repair process [19, 22]. When 8-oxoG is not removed by OGG1 before DNA replication, MYH (E. coli mutY homolog) removes adenine misincorporated opposite 8-oxoG in the DNA template [23, 24]. The mammalian orthologs of E. coli MutM/Nei are NEIL1 and NEIL2 (Nei endonuclease VIII-like glycosylases), which remove oxidized base lesions, including 8-oxoG, during DNA replication and transcription, respectively [25, 26].
In the present study, we examined the role of 8-oxoG accumulation and its repair in the DNA of airway epithelial cells in allergic immune responses. To do so, we used siRNA to ablate Ogg1 from the airway epithelium of ragweed pollen grain extract (RWPE)-sensitized animals (Ogg1-deficient: Ogg1D mice) before RWPE challenge. Mice treated with control siRNA were used as controls (Ogg1 proficient: Ogg1P mice). We depleted Ogg1 only from the epithelium because environmental oxidative pollutants primarily affect the epithelium, and its constituent cells play a decision-making role in the initiation and manifestation of innate and adaptive inflammatory processes [27–29]. We avoided using Ogg1-defficient mice, as they globally lack Ogg1 activity. For the first time, we show that supra-physiological levels of 8-oxoG have no effect, but DNA repair intermediates, including SSBs generated during the action of Ogg1, increase the expression of Th2 cytokines and allergic immune responses upon challenge of sensitized mice with RWPE. These data link DNA damage repair to allergic immune responses, and also imply that transient modulation of OGG1 activity could have clinical benefits.
2. Experimental procedures
2.1. Cell culture
MLE-12 (American Type Culture Collection), an immortalized type 2 mouse lung epithelial cell line, was grown in RPMI 1640 medium. Ogg1−/− and Ogg1+/+ mouse embryo fibroblast (MEF) cells [30] were kindly provided by Dr. Deborah E. Barnes (Imperial Cancer Research Fund, Clare Hall Labs, United Kingdom) and cultured in DMEM/Ham’s F-12 medium [31]. Mouse airway epithelial cells (AECs) were isolated as described previously [32]. In co-culture studies cells were incubated with neutrophils as described previously [33]. Cell viability was determined by Annexin V assay as we described previously [31].
2.2. Animals, sensitization and challenge
Six- to 8-week-old female BALB/c mice (Harlan Sprague-Dawley; San Diego, CA) were sensitized via intraperitoneal (ip) injections of RWPE (certified LPS-free, Greer Laboratories, Lenoir, North Carolina) on days 0 and 4 [5]. On day 11, mice (n = 7–9) were challenged intra-nasally with 50 μg of RWPE (per challenge) in 60 μL of saline. All experiments were performed according to the NIH Guidelines for the Care and Use of Experimental Animals. The protocol used was approved by the University of Texas Medical Branch Animal Care and Use Committee (#0807044-05).
2.3. Evaluation of airway inflammation and hyper-responsiveness
Bronchoalveolar lavage fluid (BALF) samples were collected, processed and stained with Wright-Giemsa as we previously described [5, 6, 13]. Lung tissue sections were processed for staining with hematoxylin and eosin or periodic acid-Schiff, photographed, and evaluated as we previously described [5, 6, 13]. In parallel experiments immunofluorescence staining was carried out to identify eosinophils in lung sections. Lung tissue sections were stained with antibodyto the major basic eosinophilic protein, a kind gift form Dr. G Gleich (Division of Allergic Diseases, Department of Internal Medicine, Mayo Clinic and Foundation, Rochester, Minnesota, USA) as we published previously [5]. Airway hyper-responsiveness (AHR) was determined three days after challenge [7, 13]. AHR was expressed as enhanced pause = {(expiratory time/relaxation time)−1} × (peak expiratory flow/peak inspiratory flow). The flow signals and the respiratory parameters were calculated using the Biosystem XA program (Buxco Electronics Inc, Troy N.Y., USA).
2.4. Measurement of ROS levels
ROS levels were assessed using 2′-7′-dihydro-dichlorofluorescein diacetate (H2DCF-DA; Molecular Probes). Dichlorofluorescein (DCF)-mediated fluorescence was determined by fluorescence spectroscopy analysis (FLx800 Bio-Tek Instruments, Winooski, Vermont) [31, 34].
2.5. RNA extraction and real-time (RT)-PCR Analysis
RNA was extracted using an RNeasy kit per the manufacturer’s instructions (Qiagen, Valencia, CA). Total RNA (1 μg) was reverse-transcribed using a SuperScript® III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). qRT-PCR was performed in an ABI7000 thermal cycler [13]. Relative expression levels were calculated by the ΔΔCt method [13, 35].
2.6. Assessment of 8-oxoG and DNA single-strand breaks
8-oxoG in DNA was assessed by determining the levels of OGG1-sensitive sites using an OGG1 FLARE™ Comet assay (Travigen, Gaithersburg, MD) [31, 36, 37]. Briefly, exfoliated AECs were embedded in agarose, cells were lysed, detergent was removed by washing, OGG1 protein was added in the digestion buffer and then the DNA was subjected to alkaline electrophoresis. DNA strand breaks were determined using neutral (double-strand breaks, DSBs) and alkaline (SSBs) electrophoretic conditions [31, 36, 37]. We evaluated 200 cells for each data point, using the Comet Assay IV v4.2 system (Perceptive Instruments, Suffolk, UK).
2.7. Depletion of gene expression
Stealth RNAi™ was used to deplete Ogg1 (Invitrogen Life Technologies). Under mild anesthesia, parallel groups of mice were treated with siRNA to Ogg1 (or control siRNA) intranasally at days 8 and 10 after sensitizing the mice [13]. The mouse orthologs of E. coli endonuclease VIII-like DNA glycosylases were depleted by phosphorothioate-protected oligonucleotides: Neil1 antisense oligonucleotide: 5′-G*C*T* GGC GGG CTG TAG C*T*T* C-3′; sense (5′-G*A*A*GCTACAGCCCGCC*A*G*C-3′ bond) [38], antisense oligonucleotide 5′-C*G*C*CACTCTATCCTCATC*C*C*T-3′, sense: A*G*G*G*-GATGAGGATAGAGTG-G*C*G*8 (Dharmacon; *, indicates a phosphorothioate). Ogg1 levels in cultured cells were decreased by siRNA transfection (Dharmacon) [39]. Decreases in gene expression were determined by qRT-PCR and Western blot analysis.
2.8. Enzyme-Linked Immunosorbent Assays
ELISA was used to determine IL-4, IL-5, and IL-13 levels in BALF samples. The capture antibodies used were ab100710(IL -4), ab114982 (IL-5) and ab100700 (IL-13) (Abcam, Cambridge, MA), as we described [12]. MUC5A/C levels in BALF were also assessed as described [6, 13].
2.9. Assessment of Ogg1’s activity
Ogg1’s repair activity was assayed using a 32P-labeled 31-mer oligonucleotide (5′-GAA GAG AGA AAG AGA X GAA GGA AAG AGA GAA G-3′; Midland Certified Reagent Co., Midland, TX) substrate containing one 8-oxoG (at the X position) as we described previously [39]. The excision reactions were carried out in 40 mM Hepes–KOH (pH 7.6), 5 mM EDTA, 1 mM DTT, 75 mM KCl, 10% glycerol buffer containing 0.2 pmol of 32P-labeled substrate and 20 μg cell extract. In controls 6 pmol recombinant Ogg1 protein per sample was used. The cleaved product was separated from the intact substrate in a 20% polyacrylamide gel containing 8 M urea in Tris-borate-EDTA buffer (pH 8.4). Radioactivity in the separated DNA bands was visualized with a Storm 860 PhosphorImager (Molecular Dynamics) and quantified by densitometry using ImageQuant software.
2.10. Statistical analysis
Statistical analysis was performed by using the Student’s t-test or ANOVA, followed by post hoc tests: Bonferroni’s and Dunnett’s T3 with SPSS 14.0 software. The data are presented as the means ± the standard error of the mean. Differences were considered to be statistically significant at P < 0.05.
3. Results
3.1. RWPE challenge increases 8-oxoG levels in the genome of airway epithelial cells
Sensitized Ogg1P and Ogg1D animals were mock- or RWPE-challenged, and 8-oxoG levels in DNA of exfoliated airway epithelial cells were determined by single-cell DNA gel electrophoresis (Comet assay) combined with OGG1 digestion of DNA [31, 36, 37] (Fig. 1 A). In AECs of Ogg1P mice, RWPE induced 4.1±1.1- and 6.2±1.9-fold increases in 8-oxoG levels at 0.5 h and 1 h after exposure, respectively, compared to pre-challenge levels. By 3 h post-exposure, the 8-oxoG level was decreased significantly (Fig. 1B), in line with the expression of Ogg1 in Ogg1P mice (Fig. 1C) and indicating an effective repair of the oxidized guanine. Nevertheless, time-course studies showed a second-phase increase in 8-oxoG levels between 8 h and 36 h after RWPE challenge (Fig. 1B). This phenomenon is consistent with the kinetics of neutrophil influx into the airways in response to RWPE challenge [11, 40] and ROS generation by activated neutrophils [41]. Next, prior to challenge of mice Ogg1 expression was ablated by Ogg1 siRNA specifically in the airway epithelium (Materials and Methods). Decrease in Ogg1 expression at RNA level was 83±9.1% (Fig. 1C). In Ogg1D mice, 8-oxoG levels steadily increased in the DNA of AECs (Fig. 1B), in line with depletion of Ogg1 expression (Fig. 1C). To provide evidence that the increased 8-oxoG levels in AECs of Ogg1D mice were indeed due to a lack of Ogg1 expression, and not to a change in the airways’ redox capacity, we determined the GSH:GSSG ratio. Administration of Ogg1-specific or control siRNA into the airways of sensitized mice had no effects on the GSH:GSSG ratio in BALF samples (Fig. 1D). Furthermore, we did not observe significant differences in the GSH:GSSG ratio between Ogg1P and Ogg1D mice after RWPE challenge (Fig. 1D). These data indicate that the increased 8-oxoG levels were indeed due to the lack of Ogg1, and not a change in the airways’ redox capacity.
Figure 1. Changes in 8-oxoG levels in the genomic DNA of airway epithelial cells after RWPE challenge.

Sensitized Ogg1Pand O gg1Dmice (proficient and deficient in O gg1 expression in the airway epithelium, respectively) were challenged with RWPE, and the levels of 8 -oxoG in exfoliated airway epithelial cells were examined at various time points thereafter. (A) Representative images of comet moments with and without digestion of DNA with recombinant hOGG1(n = 200). (B) Quantitation of 8-oxoG levels in the DNA of airway epithelial cells by comet assay (n = 200 comets for each time point) from Ogg1P(n = 7) and O gg1Dmice (n = 6); (C) Ogg1 mRNA levels in airway epithelial cells 48 h after instillation of control or Ogg1-specific Stealth siRNA into the lungs (n = 5–11). (D) GSH:GSSG ratios ± SEM in bronchoalveolar lavage fluid (BALF, n = 7–9). Ogg1P, mice proficient in Ogg1 expression in the airway epithelium, Ogg1D, mice deficient in Ogg1 expression in the airway epithelium; hOGG1: recombinant human OGG1 protein; RWPE, ragweed pollen grain extract. **P< 0.01, *** P< 0.001.
3.2. 8-oxoG repair is associated with DNA strand damage in the RWPE-exposed airway epithelium
In addition to oxidative DNA base lesions, ROS can also cause damage to the sugar moiety, inducing DNA SSBs as well as DSBs [20, 42, 43]. To examine the levels of SSBs and DSBs after RWPE challenge, AECs were subjected to both neutral and alkaline Comet assays [31, 36, 37]. Despite supra-physiological 8-oxoG levels in the genome (Fig. 1B), RWPE challenge did not significantly alter the abundance of SSBs in AECs of either Ogg1P or Ogg1D mice during the first 3-h time period (Fig. 2A). However, during neutrophilia (at 8, 16 and 24 h post-challenge) [5, 11], the abundance of SSBs was significantly increased in the AECs of Ogg1P animals compared to that observed in the AECs of Ogg1D mice (Fig. 2A and 2B, right panels). The levels of SSBs after exposure to RWPE were nearly identical in naïve and sensitized Ogg1P mice (data not shown). The DSB levels did not increase at any time point after RWPE challenge in the AECs of either Ogg1P or Ogg1D mice (Fig. 2B, left panels; microscopic images taken at 16 h time point are shown). Downregulation of Neil1 and Neil2 insignificantly altered the levels of SSBs in AECs (data not shown). These results suggest that the generation of SSBs after RWPE challenge is independent from adaptive immune responses, while it appears to be specifically related to expression of Ogg1 under neutrophilia.
Figure 2. DNA strand damage in the airway epithelium after RWPE challenge of sensitized mice.

Ogg1P and Ogg1Dmice were RWPE -challenged, and exfoliated AECs were subjected to Comet assays. (A) Kinetics of changes in SSB levels in the airway epithelium from Ogg1Pand O gg1D mice after RWPE challenge. For each time points and treatments, >200 comet moments were evaluated. (B) Representative comet moments under neutral (left images) and alkaline (right images) electrophoretic conditions, representing DNA double-strand breaks and single-strand breaks, respectively, 16 h after RWPE challenge. CA, comet assay; Ogg1P, mice proficient in Ogg1 expression in the airway epithelium; O gg1D, mice deficient in Ogg1 expression in the airway epithelium; RWPE, ragweed pollen grain extract; SSBs, single strand breaks;. *P< 0.05, ** P< 0.01, *** P< 0.001.
To confirm these observations, we co-cultured neutrophils with Ogg1P and Ogg1D MLE-12 and MEF cells (at a ratio between neutrophils and MLE-12 or MEF was 1:1). Co-incubation with activated neutrophils [33] for 2 h and 4 h caused a significant increase in SSBs levels in both Ogg1P MLE-12 cells and Ogg1P MEF cells compared to the same Ogg1D cell types (Fig. 3A). To further support these observations, we utilized MEF cells developed from Ogg1−/− mice [30, 31]. Co-culture of activated neutrophils resulted in significantly fewer SSBs in Ogg1−/− compared to Ogg1+/+ MEF cells. Whereas non-activated neutrophils failed to change SSB levels in either MLE-12 or MEF cells (data not shown). On the other hand, RWPE exposure did not significantly affect the levels of SSBs in Ogg1P and Ogg1D cells (Fig. 3B). There were no significant changes in cell viability during co-culture of MLE-12 or MEF cells with activated neutrophils or RWPE as determined by Annexin V assay using flow cytometry (data not shown). The extent of Ogg1 depletion at mRNA and protein levels as well as a decrease in Ogg1’s activity in MLE-12 and MEF Ogg1+/+ cells (MEF Ogg1−/− cells were used as negative control) are shown in Fig. 3E. These results imply that Ogg1 activity is associated with the generation of SSBs during neutrophilia in mice, and in cells cultured with activated neutrophils.
Figure 3. The levels of SSBs and intracellular ROS in cultured cells proficient or deficient in Ogg1 expression after RWPE challenge or co-culture with neutrophils.

(A) SSB levels in cultured cells. MLE-12 cells and mouse embryonic fibroblast (MEF) cells were Ogg1-depleted using siRNA, and co-incubated with activated neutrophils. In parallel experiments Ogg1−/− MEF cells were co-cultured with activated neutrophils. SSB levels were determined by alkaline Comet assays. For each time points and treatments, >200 comet moments were evaluated. **P < 0.01, ***P < 0.001. (B) RWPE challenge did not induce a detectable increase in SSB levels. MLE-12 and Ogg1+/+MEF cells transfected with control siRNA or O gg1 siRNA, as well as Ogg1−/− MEF cells were treated with RWPE for 2 h. Levels of SSBs were determined by alkaline Comet assays. In controls, MLE-12 cells were co-incubated with activated neutrophils for 2 h. For each treatment, >200 comet moments were evaluated. (C) Changes in ROS levels in MLE-12 cells after addition of RWPE or activated neutrophils. ROS levels in MLE -12 cells were determined using 2′-7′-dihydro-dichlorofluorescein diacetate (n = 3–4) as described in Materials and Methods. (D) Ogg1 depletion had no effect on the neutrophil-induced increase in cellular ROS levels. Ogg1 was depleted via siRNA as in (A). Ogg1−/− MEF cells were used as a control (n = 2–4). (E) Ogg1 protein (upper panel) and mRNA (lower panel) levels in control siRNA and Ogg1 siRNA-transfected MLE-12 and Ogg1+/+MEF cells. Right panel: 8 -oxoG excision by cell extracts from control and Ogg1 siRNA-transfected cells. MEF, mouse embryonic fibroblast; RWPE, ragweed pollen grain extract; SSBs, single strand breaks. **P< 0.01, *** P< 0.001.
Next we explored the kinetics and extent of changes in cellular ROS levels mediated by RWPE and neutrophils. Co-culturing Ogg1P MLE-12 cells with activated neutrophils (at a ratio of 1:1) resulted in a sustained increase in ROS levels, while RWPE exposure caused a transient increase (Fig. 3C). Increases in intracellular ROS levels were similar between 0.5 and 1 after addition of RPWE or neutrophils. However, activated neutrophils resulted in sustained increases in intracellular ROS levels. There were statistically significant differences between RWPE and neutrophil challenge-induced levels of ROS at 2 h and 4 h time points in MLE-12 cells (Fig. 3C). Changes in ROS levels were independent from Ogg1 expression (the 4 h time point is shown in Fig. 3D). These data suggest that sustained oxidative stress appears to be associated with formation of SSBs in Ogg1P cells.
3.3. Deficiency in 8-oxoG repair decreases RWPE-induced allergic inflammatory responses
To examine the implication of 8-oxoG repair and SSBs associated with Ogg1 expression in allergic immune responses, sensitized Ogg1D and Ogg1P mice were challenged with RWPE and the extent of airway inflammation was examined thereafter. Surprisingly, despite a steady accumulation of 8-oxoG in the AECs of Ogg1D mice (Fig. 1B), there were significantly fewer eosinophils in the BALF (Fig. 4A) and the subepithelial area (Fig. 4B middle panel; confirmed by immunofluorescence staining; data not shown [11]) lower MUC5A/C levels in BALF (Fig. 4C), and lower epithelial cell metaplasia (Fig. 4D, middle panel) and Phen index (Fig. 4E) compared to the same parameters in the Ogg1P mice (Fig. 4A, right panel of B, C, right panel of D and E). These observations appear to be specific to Ogg1, as depletion of Neil1 glycosylase [25] did not affect the number of eosinophils accumulated into BALF and subepithelium (Fig. 4G).
Figure 4. Depletion of Ogg1 in the airway epithelium decreases allergen-driven inflammation and the expression of Th2 cytokines in the lungs of sensitized mice after RWPE challenge.
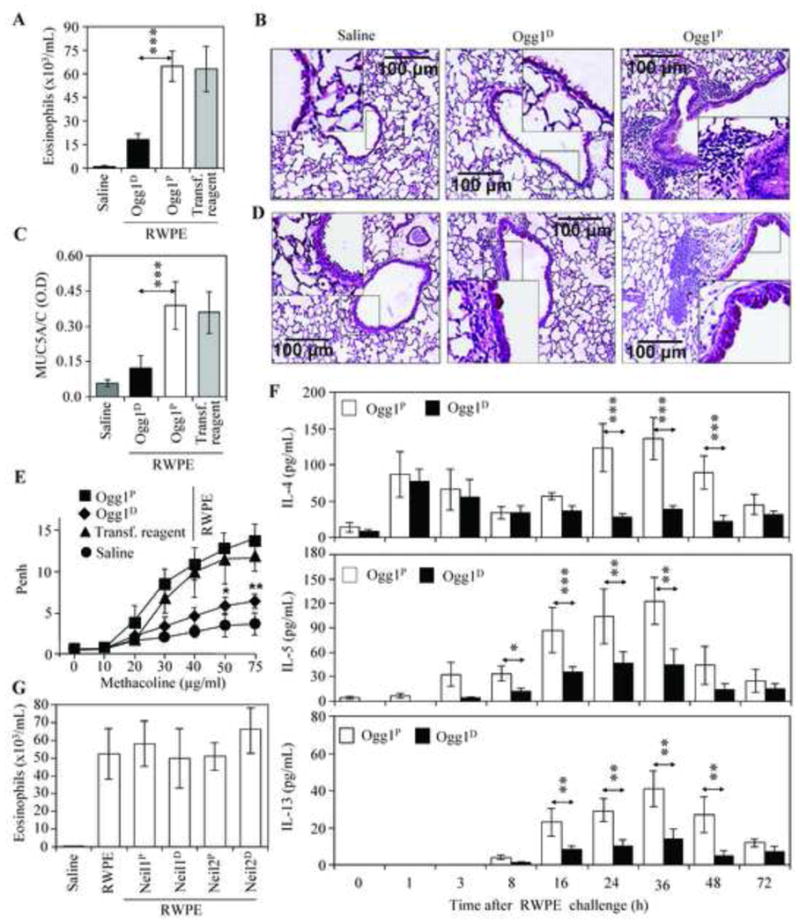
Sensitized Ogg1P and Ogg1Dmice were challenged with saline or RWPE, and BALF and the lungs were then analyzed. (A) The number of eosinophils infiltrated into the BALF (n=9). (B) Inflammatory cell infiltration into the peribronchial area (n= 7–9 per group). Magnification: x96. Scale bars represent 100 μm. (C) MUC5A/C levels in BALF as determined by ELISA (n = 6–9 per group). (D), Airway epithelial hyperplasia (PAS staining; magnification: x96). Representative images are shown out of 6–9 mice per group. (E) Airway hyperresponsiveness was assessed at 60 h post-challenge (n = 6–9 per group). *P< 0.05, **P < 0.01.(F) Changes in levels of IL-4 (upper panel), IL-5 (middle panel) and IL-13 (lower panel) in BALF as a function of time after RPWE challenge (n = 6–8 per group). (G) Depletion of Neil1 and Neil2 DNA glycosylases had no effect on the eosinophil numbers in BALF (n = 5–7 per group). Neil1 and Neil2 were depleted as described in Methods. Neil1P: mice proficient in Neil1 expression in the airway epithelium;Neil1D: mice deficient in Neil1 expression in the airway epithelium; Neil2P: mice proficient in Neil2 expression in the airway epithelium; Neil2D: mice deficient in Neil2 expression in the airway epithelium; Ogg1P, mice proficient in Ogg1 expression in the airway epithelium; Ogg1D, mice deficient in Ogg1 expression in the airway epithelium; RWPE, ragweed pollen grain extract; BALF, bronchoalveolar lavage fluid. *P< 0.05, **P< 0.01, *** P< 0.001.
T helper 2 (Th2) cytokines, including IL-4, IL-5 and IL-13, are thought to play major roles in the accumulation of eosinophils, and in airway metaplasia and mucus production [44]. In the BALF of both Ogg1Pand O gg1Dmice, RWPE challenge induced an immediate increase in IL-4 (1 h and 3 h) (Fig. 4F, upper panel). However, at later time points (8 h, 16 h, 24 h, 36 h and 48 h), its level further increased only in the BALF of Ogg1Pmice. The levels of IL -5 and IL-13 were significantly lower in the BALF of Ogg1Dmice compared to those in BALF of O gg1P animals (Fig. 4F, middle, lower panels). These data imply that O gg1-generated SSBs are associated with the expression of Th2 cytokines and exacerbation of allergic airway inflammation.
4. Discussion
Exposure to environmental oxidative pollutants exacerbates airborne allergen-mediated innate and adaptive immune cascades. Under inflammatory conditions, increased levels of oxidative DNA base and strand damage have also been documented, and considered as biomarkers of inflammation [17]. Although the role of DNA repair intermediates in exacerbating inflammation has been proposed, their etiological role has not been investigated. In the present study, we document for the first time that oxidative damage to guanine in DNA has no direct effect, while SSBs generated during Ogg1-initiated base excision repair of 8-oxoG in the airway epithelium are etiologically associated with Th2-type responses, including cytokine expression, inflammatory cell infiltration, mucus production and AHR in a mouse model of allergic airway inflammation. Exacerbation of the inflammatory response was specific to Ogg1, as depletion of the DNA glycosylases Neil1 and Neil2 had no significant effects on RWPE-induced allergic airway inflammation in our experimental model. These unexpected observations suggest that it is not 8-oxoG accumulation in the genome but the Ogg1-initiated repair of 8-oxoG that helps to exacerbate antigen-induced allergic inflammation.
Among DNA bases, guanine is the primary target of both radical and non-radical oxidants due to its low redox potential [15, 45]. Therefore, it was not surprising to observe that 8-oxoG levels in the DNA of AECs were increased due to the elevated oxidative stress induced by the intrinsic NOX activity of RWPE and the ROS generated by infiltrating neutrophils. In Ogg1D animals8 -oxoG levels were continuously increased due to a lack of repair by O gg1, as there were no differences in the redox balance of the airways between Ogg1Pand O gg1Dmice as measured by the GSH:GSSG ratio, a gauge of oxidative stress in airway inflammation [46]. That the inflammatory parameters were significantly higher in Ogg1Pmice led us to conclude that the repair of 8-oxoG rather than its genomic accumulation is associated with the allergic inflammatory processes.
Ogg1 excises 8-oxoG (and also FapyG) from DNA with high efficiency [19], generates AP sites (5′ phosphate and 3′ blocking phospho-α,β unsaturated aldehyde ends), and via its glycosylase/AP lyase activity also cleaves the DNA backbone at these sites by transiently generating single-strand gaps [42, 43, 47]. Under physiological conditions Ogg1 forms complexes with downstream canonical repair and non-repair proteins, and single-strand gaps are fully corrected in consecutive steps of BER/SSBR [19, 20]. Indeed, this appears to be the case immediately after RWPE challenge, as 8-oxoG was repaired in the epithelium of Ogg1P mice without a significant increase in SSBs.
Activated neutrophils inhibit nucleotide excision repair via release of ROS and MPO-generated hypochlorous acid in lungs and human pulmonary epithelial cells [48]. In contrast, sustained oxidative stress by activated neutrophils did not alter 8-oxoG excision activity of Ogg1 in lysates of the lungs or in airway epithelial cell cultures (A549, MLE-12; Bacsi et al., unpublished observation). Thus an intriguing question remains – how Ogg1-initiatedbase excision repair of 8-oxoG is involved in the formation of DNA strand breaks. It has been proposed that during oxidative stress, when the 8-oxoG level in DNA is elevated, a concerted glycosylase/AP lyase reaction by Ogg1 would create numerous SSBs in the genome that could have pathophysiological consequences [47]. Thus one possible explanation could be that during sustained oxidative stress, there is an imbalance in glycosylase and AP lyase activities of Ogg1, generating SSBs in the genome. This action of Ogg1 could result from improper protein-protein interactions with canonical BER enzymes, such as APE1, and ligases or non -canonical proteins in the BER/SSBR pathways [42, 47, 49]. It has also been suggested that oxidative stress may compromise Ogg1’s binding to AP-sites, and thereby decrease its protective effects on 5′ phosphate and 3′ blocking phospho-α,β unsaturated aldehyde ends against ROS and AP site-cleaving enzymes, such as Nth1 and DNA topoisomerases [42, 47, 50]leading to SSBs. While the exact molecular mechanism is the subject of our future studies, it appears that Ogg1′ activity could lead to a transient increase in SSBs in the airway epithelium during RWPE-induced neutrophilia. SSBs are significantly increased in Ogg1P animals; therefore, it is reasonable to propose that the allergic inflammation is augmented by DNA damage response signaling. In support of this idea, SSB-and DSB -initiated signaling activates transcription factors (e.g. NF -κB), and consequently the expression of proinflammatory chemokines/cytokines [51–57]. For example, DNA strand breaks induce expression of IL-4, IL-10, and TGF-β in an ataxia telangiectasia-mutated deficient colitis model [58]. Recent studies also showed that DNA damage signaling is etiologically associated with Th2 responses, including IgE and IgG1 production during OVA sensitization/immunization [59].
Moreover, we found that the levels of SSBs were similarly increased in the epithelium of both sensitized and naïve Ogg1P animals at 8 h, 16 h, and 24 h after RWPE challenge. Importantly, there was no eosinophilia observed in naïve mice as a consequence of DNA strand damage induced by neutrophils in response to RWPE. In contrast, in sensitized Ogg1Panimals there was robust accumulation of eosinophils, and increased epithelial hyperplasia and AHR, when compared to those in sensitized Ogg1Dmice. It is thus feasible to propose that perturbed Ogg1-initiated repair of 8-oxoG leads to a transient increase in SSBs, which promotes Th2 cytokine expression and exacerbates allergic inflammation. In support of this idea, previous studies demonstrated that atopic subjects have more DNA breaks in their nasal epithelial cells than do non asthmatics upon oxidative environmental exposures [60].
In controls, we investigated the role of the Nei endonuclease VIII-like Neil1 and Neil2 glycosylases [25, 61, 62]. Neil1 preferentially initiates base excision repair of ring-fragmented purines (e.g., the purine-derived lesions 2,6-diamino-4-hydroxy-5-formamidopyrimidine and 4,6-diamino-5-formamidopyrimidine) and some saturated pyrimidines [26]. Neil1 knockout and heterozygous mice develop obesity, dyslipidemia, fatty liver disease and hyperinsulinemia [61]. Inflammatory processes are highly integrated into these conditions associated with Neil1 deficiency [63]. Unexpectedly, depletion of Neil1 in the airway epithelium had no significant effect on levels SSBs and eosinophil counts after RWPR challenge. On the other hand, depletion of the Neil2 (which preferentially repairs the cytosine oxidation product 5-hydroxyuracil [25, 62]) in the airway epithelium caused insignificant changes in SSB levels, while it somewhat but not significantly increased eosinophil count in BALF.
Studies utilizing Ogg1−/− mice showed an increased resistance to endotoxin-induced organ dysfunction, diabetes induced by streptozotocin and contact hypersensitivity induced by oxazolone [64]. Moreover, a meticulous study by Li and colleagues documented a decrease in ovalbumin (OVA)-induced airway inflammation in sensitizedOgg1 −/− mice, and concluded that Ogg1 expression is associated with oxidative stress after OVA challenge, leading toSTAT6 and NF-κB activation, as well as enhanced IL-4, IL-6, IL-10, and IL-17 expression in wild-type (WT) mice [65]. They supported their observation by showing that depletion of Ogg1 by siRNA markedly decreased ROS levels in MLE-12 cells after exposure to dust mite extract [65]. In our model, depletion of Ogg1 did not alter the redox balance (GSH:GSSG ratio) in the airways of experimental mice or the intracellular ROS levels of Ogg1-expressing vs. non-expressing AECs and MEF cells mediated by RWPE or activated neutrophils. Results by Li and colleagues using OVA-sensitized Ogg1 deficient mice support the validity of our observations, although they did not consider exacerbation of allergic immune responses due to 8-oxoG repair and generation of DNA strand breaks. Together these results suggest that DNA glycosylases could modulate inflammatory processes via a variety of mechanisms, including generation of DNA SSBs, which appears to play a critical role in exacerbation of antigen-induced inflammation in sensitized animals, and possibly in atopic human subjects.
In our model, Ogg1 was depleted only in the airway epithelium, so our results suggest that DNA strand damage associated with Ogg1’s expression/activity in AECs induces discrete signaling pathways to enhance the expression of Th2 cytokines, recruitment of eosinophils, induction of epithelial hyperplasia and AHR. These observations are consistent with those showing that oxidative environmental pollutants provoke and exacerbate airway inflammatory processes, and with the view that airway inflammation is primarily driven by airway epithelial disorder(s), rather than being linked purely to immunological pathways [27–29].
In conclusion, ROS are known to increase inflammatory chemokine/cytokine expression and exacerbate antigen-mediated allergic immune responses. In our experimental model, ROS levels were identical in both naïve and sensitized Ogg1-proficient and -deficient airways, while RWPE challenge induced severe allergic immune responses only in sensitized Ogg1-proficient animals. These results imply that DNA repair associated with Ogg1 generates DNA SSBs that are etiologically linked to allergic immune responses. From our results it is also plausible that the level of Ogg1 expression/activity in atopic subjects could be etiologically linked to clinical manifestations of allergic responses provoked by various allergens. Our results also indicate that the transient modulation of Ogg1’s activity in the airway epithelium could decrease the severity of inflammation and so have clinical benefits.
Highlights.
The role of 8-oxoG repair by OGG1 in allergic airway inflammation was established.
Inflammation was induced by intranasal ragweed pollen challenge of sensitized mice.
Pollen exposure and infiltrating neutrophils increase 8-oxoG levels in the genome.
Repair of 8-oxoG only under neutrophilia resulted in DNA single-strand breaks.
Down-regulation of OGG1 in the airways decreased allergic immune responses.
Acknowledgments
We thank Dr. David Konkel (Department of Biochemistry and Molecular Biology, UTMB) for critically reading and editing the manuscript. This work was supported by grants from National Institutes of Environmental Health and Sciences (RO1-ES018948 to I.B.), National Institute of Allergic and Infectious Diseases (P01 AI062885 to I.B, Director A. AR Brasier) and NHLBI Proteomic Center (HV2005 Director A. Kurosky). The work was also supported by the TAMOP 4.2.1/B-09/1/KONV-2010-0007 project (to A.B.). The project is co-financed by the European Union and the European Social Fund. A.B. was also supported by the Janos Bolyai Fellowship from the Hungarian Academy of Sciences. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Abbreviations: 8-oxoG: 7,8-dihydro-8-oxoguanine; AECs: Airway epithelial cells; AHR: Airway hyperresponsiveness; APE1: AP-endonuclease 1; AP sites: Apurinic/apyrimidinic site; BER: Base excision repair; BALF: Bronchoalveolar lavage fluid; CA: Comet assay; DSB: Double-strand break; FapyG: 2,6-diamino-4-hydroxy-5-formamidopyrimidine; GSH, GSSG: reduced and oxidized glutathione, respectively; H2DCF-DA: 2′-7′-dihydro-dichlorofluorescein diacetate; MEF: Murine embryonic fibroblast; Neil1D/Neil2D: Deficient in Neil1/Neil2 expression; Neil1P/Neil2P: Proficient in Neil1/Neil2 expression; NOX: NAD(P)H oxidase; Ogg1: 8-oxoguanine DNA glycosylase 1; Ogg1D: Deficient in Ogg1 expression; Ogg1P: Proficient in Ogg1 expression; ROS: Reactive oxygen species; RWPE: Ragweed pollen grain extract; SSBs: Single-strand breaks
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Attila Bacsi, Email: bacsi.attila@gmail.com.
Leopoldo Aguilera-Aguirre, Email: leaguile@utmb.edu.
Bartosz Szczesny, Email: baszczes@utmb.edu.
Zsolt Radak, Email: radak@tf.hu.
Tapas K. Hazra, Email: tkhazra@utmb.edu.
Sanjiv Sur, Email: sasur@utmb.edu.
Xueqing Ba, Email: baxq755@nenu.edu.cn.
Istvan Boldogh, Email: sboldogh@utmb.edu.
References
- 1.Dharajiya N, Boldogh I, Cardenas V, Sur S. Role of pollen NAD(P)H oxidase in allergic inflammation. Curr Opin Allergy Clin Immunol. 2008;8:57–62. doi: 10.1097/ACI.0b013e3282f3b5dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vrtala S, Grote M, Duchene M, van Ree R, Kraft D, Scheiner O, Valenta R. Properties of tree and grass pollen allergens: reinvestigation of the linkage between solubility and allergenicity. Int Arch Allergy Immunol. 1993;102:160–169. doi: 10.1159/000236567. [DOI] [PubMed] [Google Scholar]
- 3.D’Amato G, Liccardi G, D’Amato M, Cazzola M. Outdoor air pollution, climatic changes and allergic bronchial asthma. Eur Respir J. 2002;20:763–776. doi: 10.1183/09031936.02.00401402. [DOI] [PubMed] [Google Scholar]
- 4.Annesi-Maesano I, Rouve S, Desqueyroux H, Jankovski R, Klossek JM, Thibaudon M, Demoly P, Didier A. Grass Pollen Counts, Air Pollution Levels and Allergic Rhinitis Severity. Int Arch Allergy Immunol. 158:397–404. doi: 10.1159/000332964. [DOI] [PubMed] [Google Scholar]
- 5.Boldogh I, Bacsi A, Choudhury BK, Dharajiya N, Alam R, Hazra TK, Mitra S, Goldblum RM, Sur S. ROS generated by pollen NADPH oxidase provide a signal that augments antigen-induced allergic airway inflammation. J Clin Invest. 2005;115:2169–2179. doi: 10.1172/JCI24422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bacsi A, Choudhury BK, Dharajiya N, Sur S, Boldogh I. Subpollen particles: carriers of allergenic proteins and oxidases. J Allergy Clin Immunol. 2006;118:844–850. doi: 10.1016/j.jaci.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yadav UC, Naura AS, Aguilera-Aguirre L, Ramana KV, Boldogh I, Sur S, Boulares HA, Srivastava SK. Aldose reductase inhibition suppresses the expression of Th2 cytokines and airway inflammation in ovalbumin-induced asthma in mice. J Immunol. 2009;183:4723–4732. doi: 10.4049/jimmunol.0901177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yadav UC, Aguilera-Aguirre L, Ramana KV, Boldogh I, Srivastava SK. Aldose reductase inhibition prevents metaplasia of airway epithelial cells. PLoS One. 2010;5:e14440. doi: 10.1371/journal.pone.0014440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bacsi A, Dharajiya N, Choudhury BK, Sur S, Boldogh I. Effect of pollen-mediated oxidative stress on immediate hypersensitivity reactions and late-phase inflammation in allergic conjunctivitis. J Allergy Clin Immunol. 2005;116:836–843. doi: 10.1016/j.jaci.2005.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Csillag A, Boldogh I, Pazmandi K, Magyarics Z, Gogolak P, Sur S, Rajnavolgyi E, Bacsi A. Pollen-induced oxidative stress influences both innate and adaptive immune responses via altering dendritic cell functions. J Immunol. 2010;184:2377–2385. doi: 10.4049/jimmunol.0803938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dharajiya N, Choudhury BK, Bacsi A, Boldogh I, Alam R, Sur S. Inhibiting pollen reduced nicotinamide adenine dinucleotide phosphate oxidase-induced signal by intrapulmonary administration of antioxidants blocks allergic airway inflammation. J Allergy Clin Immunol. 2007;119:646–653. doi: 10.1016/j.jaci.2006.11.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dharajiya N, Vaidya S, Sinha M, Luxon B, Boldogh I, Sur S. Allergen challenge induces Ifng dependent GTPases in the lungs as part of a Th1 transcriptome response in a murine model of allergic asthma. PLoS One. 2009;4:e8172. doi: 10.1371/journal.pone.0008172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aguilera-Aguirre L, Bacsi A, Saavedra-Molina A, Kurosky A, Sur S, Boldogh I. Mitochondrial dysfunction increases allergic airway inflammation. Journal of Immunology. 2009;183:5379–5387. doi: 10.4049/jimmunol.0900228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dizdaroglu M. Oxidative damage to DNA in mammalian chromatin. Mutat Res. 1992;275:331–342. doi: 10.1016/0921-8734(92)90036-o. [DOI] [PubMed] [Google Scholar]
- 15.Steenken S, Jovanovic SV. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 16.Chan SW, Dedon PC. The biological and metabolic fates of endogenous DNA damage products. J Nucleic Acids. 2010;2010:929047. doi: 10.4061/2010/929047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dedon PC, DeMott MS, Elmquist CE, Prestwich EG, McFaline JL, Pang B. Challenges in developing DNA and RNA biomarkers of inflammation. Biomark Med. 2007;1:293–312. doi: 10.2217/17520363.1.2.293. [DOI] [PubMed] [Google Scholar]
- 18.Radak Z, Boldogh I. 8-Oxo-7,8-dihydroguanine: links to gene expression, aging, and defense against oxidative stress. Free Radic Biol Med. 2010;49:587–596. doi: 10.1016/j.freeradbiomed.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitra S, Hazra TK, Roy R, Ikeda S, Biswas T, Lock J, Boldogh I, Izumi T. Complexities of DNA base excision repair in mammalian cells. Mol Cells. 1997;7:305–312. [PubMed] [Google Scholar]
- 20.HP, Hegde ML, Rao KS, Mitra S. Oxidative genome damage and its repair in neurodegenerative diseases: function of transition metals as a double-edged sword. J Alzheimers Dis. 2011;24:183–198. doi: 10.3233/JAD-2011-110281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimura S. Involvement of mammalian OGG1(MMH) in excision of the 8-hydroxyguanine residue in DNA. Free Radic Biol Med. 2002;32:813–821. doi: 10.1016/s0891-5849(02)00778-5. [DOI] [PubMed] [Google Scholar]
- 22.Izumi T, Wiederhold LR, Roy G, Roy R, Jaiswal A, Bhakat KK, Mitra S, Hazra TK. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology. 2003;193:43–65. doi: 10.1016/s0300-483x(03)00289-0. [DOI] [PubMed] [Google Scholar]
- 23.McCullough AK, Dodson ML, Lloyd RS. Initiation of base excision repair: glycosylase mechanisms and structures. Annu Rev Biochem. 1999;68:255–285. doi: 10.1146/annurev.biochem.68.1.255. [DOI] [PubMed] [Google Scholar]
- 24.Dodson ML, Michaels ML, Lloyd RS. Unified catalytic mechanism for DNA glycosylases. J Biol Chem. 1994;269:32709–32712. [PubMed] [Google Scholar]
- 25.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, Mitra S, Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem. 2002;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- 27.Holgate ST. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev. 2011;242:205–219. doi: 10.1111/j.1600-065X.2011.01030.x. [DOI] [PubMed] [Google Scholar]
- 28.Swamy M, Jamora C, Havran W, Hayday A. Epithelial decision makers: in search of the ‘epimmunome’. Nat Immunol. 2010;11:656–665. doi: 10.1038/ni.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locksley RM. Asthma and allergic inflammation. Cell. 2010;140:777–783. doi: 10.1016/j.cell.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci U S A. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bacsi A, Chodaczek G, Hazra TK, Konkel D, Boldogh I. Increased ROS generation in subsets of OGG1 knockout fibroblast cells. Mech Ageing Dev. 2007;128:637–649. doi: 10.1016/j.mad.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson DJ, Gray MA, Kilanowski FM, Tarran R, Randell SH, Sheppard DN, Argent BE, Dorin JR. Murine epithelial cells: isolation and culture. J Cyst Fibros. 2004;3(Suppl 2):59–62. doi: 10.1016/j.jcf.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 33.Knaapen AM, Seiler F, Schilderman PA, Nehls P, Bruch J, Schins RP, Borm PJ. Neutrophils cause oxidative DNA damage in alveolar epithelial cells. Free Radic Biol Med. 1999;27:234–240. doi: 10.1016/s0891-5849(98)00285-8. [DOI] [PubMed] [Google Scholar]
- 34.Das A, Hazra TK, Boldogh I, Mitra S, Bhakat KK. Induction of the human oxidized base-specific DNA glycosylase NEIL1 by reactive oxygen species. J Biol Chem. 2005;280:35272–35280. doi: 10.1074/jbc.M505526200. [DOI] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Olive PL, Wlodek D, Banath JP. DNA double-strand breaks measured in individual cells subjected to gel electrophoresis. Cancer Res. 1991;51:4671–4676. [PubMed] [Google Scholar]
- 37.Bacsi A, Kannan S, Lee MS, Hazra TK, Boldogh I. Modulation of DNA-dependent protein kinase activity in chlorambucil-treated cells. Free Radic Biol Med. 2005;39:1650–1659. doi: 10.1016/j.freeradbiomed.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 38.Maiti AK, Boldogh I, Spratt H, Mitra S, Hazra TK. Mutator phenotype of mammalian cells due to deficiency of NEIL1 DNA glycosylase, an oxidized base-specific repair enzyme. DNA Repair (Amst) 2008;7:1213–1220. doi: 10.1016/j.dnarep.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhakat KK, Mokkapati SK, Boldogh I, Hazra TK, Mitra S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol Cell Biol. 2006;26:1654–1665. doi: 10.1128/MCB.26.5.1654-1665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taube C, Dakhama A, Rha YH, Takeda K, Joetham A, Park JW, Balhorn A, Takai T, Poch KR, Nick JA, Gelfand EW. Transient neutrophil infiltration after allergen challenge is dependent on specific antibodies and Fc gamma III receptors. J Immunol. 2003;170:4301–4309. doi: 10.4049/jimmunol.170.8.4301. [DOI] [PubMed] [Google Scholar]
- 41.Knaapen AM, Gungor N, Schins RP, Borm PJ, Van Schooten FJ. Neutrophils and respiratory tract DNA damage and mutagenesis: a review. Mutagenesis. 2006;21:225–236. doi: 10.1093/mutage/gel032. [DOI] [PubMed] [Google Scholar]
- 42.Izumi T, Schein CH, Oezguen N, Feng Y, Braun W. Effects of backbone contacts 3′ to the abasic site on the cleavage and the product binding by human apurinic/apyrimidinic endonuclease (APE1) Biochemistry. 2004;43:684–689. doi: 10.1021/bi0346190. [DOI] [PubMed] [Google Scholar]
- 43.Mitra S, Izumi T, Boldogh I, Bhakat KK, Hill JW, Hazra TK. Choreography of oxidative damage repair in mammalian genomes. Free Radic Biol Med. 2002;33:15–28. doi: 10.1016/s0891-5849(02)00819-5. [DOI] [PubMed] [Google Scholar]
- 44.Alam R, Gorska MM. Mitogen-activated protein kinase signalling and ERK1/2 bistability in asthma. Clin Exp Allergy. 41:149–159. doi: 10.1111/j.1365-2222.2010.03658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cadet J, Douki T, Ravanat JL. One-electron oxidation of DNA and inflammation processes. Nat Chem Biol. 2006;2:348–349. doi: 10.1038/nchembio0706-348. [DOI] [PubMed] [Google Scholar]
- 46.Reynaert NL. Glutathione biochemistry in asthma. Biochim Biophys Acta. 1810:1045–1051. doi: 10.1016/j.bbagen.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 47.Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–438. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gungor N, Haegens A, Knaapen AM, Godschalk RW, Chiu RK, Wouters EF, van Schooten FJ. Lung inflammation is associated with reduced pulmonary nucleotide excision repair in vivo. Mutagenesis. 2010;25:77–82. doi: 10.1093/mutage/gep049. [DOI] [PubMed] [Google Scholar]
- 49.Izumi T, Hazra TK, Boldogh I, Tomkinson AE, Park MS, Ikeda S, Mitra S. Requirement for human AP endonuclease 1 for repair of 3′-blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis. 2000;21:1329–1334. [PubMed] [Google Scholar]
- 50.Sidorenko VS, Nevinsky GA, Zharkov DO. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair (Amst) 2007;6:317–328. doi: 10.1016/j.dnarep.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 51.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. Embo J. 2011;30:1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jamaluddin M, Wang S, Boldogh I, Tian B, Brasier AR. TNF-alpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal. 2007;19:1419–1433. doi: 10.1016/j.cellsig.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 53.Yang Y, Xia F, Hermance N, Mabb A, Simonson S, Morrissey S, Gandhi P, Munson M, Miyamoto S, Kelliher MA. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-kappaB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol Cell Biol. 2011;31:2774–2786. doi: 10.1128/MCB.01139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell. 2011;145:92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 55.Hadian K, Krappmann D. Signals from the nucleus: activation of NF-kappaB by cytosolic ATM in the DNA damage response. Sci Signal. 2011;4:pe2. doi: 10.1126/scisignal.2001712. [DOI] [PubMed] [Google Scholar]
- 56.Oumouna M, Datta R, Oumouna-Benachour K, Suzuki Y, Hans C, Matthews K, Fallon K, Boulares H. Poly(ADP-ribose) polymerase-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5. J Immunol. 2006;177:6489–6496. doi: 10.4049/jimmunol.177.9.6489. [DOI] [PubMed] [Google Scholar]
- 57.Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 58.Westbrook AM, Schiestl RH. Atm-deficient mice exhibit increased sensitivity to dextran sulfate sodium-induced colitis characterized by elevated DNA damage and persistent immune activation. Cancer Res. 2010;70:1875–1884. doi: 10.1158/0008-5472.CAN-09-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marichal T, Ohata K, Bedoret D, Mesnil C, Sabatel C, Kobiyama K, Lekeux P, Coban C, Akira S, Ishii KJ, Bureau F, Desmet CJ. DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med. 2011;17:996–1002. doi: 10.1038/nm.2403. [DOI] [PubMed] [Google Scholar]
- 60.Fortoul TI, Valverde M, del Lopez CM, Bizarro P, Lopez I, Sanchez I, Colin-Barenque L, Avila-Costa MR, Rojas E, Ostrosky-Shejet P. Single-cell gel electrophoresis assay of nasal epithelium and leukocytes from asthmatic and nonasthmatic subjects in Mexico City. Arch Environ Health. 2003;58:348–352. [PubMed] [Google Scholar]
- 61.Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, Ballinger SW, Corless CL, McCullough AK, Lloyd RS. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc Natl Acad Sci U S A. 2006;103:1864–1869. doi: 10.1073/pnas.0507444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Banerjee D, Mandal SM, Das A, Hegde ML, Das S, Bhakat KK, Boldogh I, Sarkar PS, Mitra S, Hazra TK. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J Biol Chem. 2011;286:6006–6016. doi: 10.1074/jbc.M110.198796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18:363–374. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- 64.Mabley JG, Pacher P, Deb A, Wallace R, Elder RH, Szabo C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. Faseb J. 2005;19:290–292. doi: 10.1096/fj.04-2278fje. [DOI] [PubMed] [Google Scholar]
- 65.Li G, Yuan K, Yan C, Fox J, 3rd, Gaid M, Breitwieser W, Bansal AK, Zeng H, Gao H, Wu M. 8-Oxoguanine-DNA glycosylase 1 deficiency modifies allergic airway inflammation by regulating STAT6 and IL-4 in cells and in mice. Free Radic Biol Med. 2012;52:392–401. doi: 10.1016/j.freeradbiomed.2011.10.490. [DOI] [PMC free article] [PubMed] [Google Scholar]