

Summary
Previous studies have shown that growth hormone (GH) recruits the adapter protein SH2B1β to the GH-activated, GH receptor-associated tyrosine kinase JAK2, implicating SH2B1β in GH-dependent actin cytoskeleton remodeling, and suggesting that phosphorylation at serines 161 and 165 in SH2B1β releases SH2B1β from the plasma membrane. Here, we examined the role of SH2B1β in GH regulation of macrophage migration. We show that GH stimulates migration of cultured RAW264.7 macrophages, and primary cultures of peritoneal and bone marrow-derived macrophages. SH2B1β overexpression enhances, whereas SH2B1 knockdown inhibits, GH-dependent motility of RAW macrophages. At least two independent mechanisms regulate the SH2B1β-mediated changes in motility. In response to GH, tyrosines 439 and 494 in SH2B1β are phosphorylated. Mutating these tyrosines in SH2B1β decreases both basal and GH-stimulated macrophage migration. In addition, mutating the polybasic nuclear localization sequence (NLS) in SH2B1β or creating the phosphomimetics SH2B1β(S161E) or SH2B1β(S165E), all of which release SH2B1β from the plasma membrane, enhances macrophage motility. Conversely, SH2B1β(S161/165A) exhibits increased localization at the plasma membrane and decreased macrophage migration. Mutating the NLS or the nearby serine residues does not alter GH-dependent phosphorylation on tyrosines 439 and 494 in SH2B1β. Mutating tyrosines 439 and 494 does not affect localization of SH2B1β at the plasma membrane or movement of SH2B1β into focal adhesions. Taken together, these results suggest that SH2B1β enhances GH-stimulated macrophage motility via mechanisms involving phosphorylation of SH2B1β on tyrosines 439 and 494 and movement of SH2B1β out of the plasma membrane (e.g. as a result of phosphorylation of serines 161 and 165).
Key words: SH2B1, Phosphorylation, Growth hormone, Macrophage, Motility
Introduction
SH2B1 (SH2-B/PSM) is an SH2 domain-containing adaptor protein that is recruited to the cytokine receptor-associated kinase JAK2 in response to multiple ligands, including growth hormone (GH) (Rui et al., 1997), leptin (Li et al., 2007) and most likely prolactin (Rider et al., 2009). It is also recruited to the ligand-activated form of a number of receptor tyrosine kinases, including the receptor for nerve growth factor (NGF) (Qian et al., 1998; Rui et al., 1999). SH2B1 has four isoforms (α, β, γ, and δ), which share 631 amino acids (a.a.) and diverge only in their C-terminal region (Yousaf et al., 2001). All four isoforms share a phenylalanine (Phe) zipper dimerization domain, nuclear localization sequence (NLS), nuclear export sequence (NES), Pleckstrin homology (PH) domain and SH2 domain but exhibit unique C-termini. Among these four isoforms, the ubiquitously expressed beta isoform has been studied most extensively.
SH2B1β has been implicated in a number of cellular functions that involve changes in the actin cytoskeleton. In addition to enhancing NGF-induced neurite outgrowth (Qian et al., 1998; Rui et al., 1999; Zhang et al., 2006), SH2B1 has been implicated in growth factor-induced mitogenesis (Riedel et al., 2000) and GH- and prolactin-induced changes in membrane ruffling and cell motility (Diakonova et al., 2002b; Herrington et al., 2000; Rider and Diakonova, 2011; Rider et al., 2009). In the context of GH, overexpression of SH2B1β has been shown to enhance GH-induced changes in membrane ruffling of 3T3-F442A fibroblasts and movement in a wounding assay of 293T cells ectopically expressing GH receptor (Diakonova et al., 2002b; Herrington et al., 2000). Given this regulation of the actin cytoskeleton by GH, we were curious to determine whether endogenous SH2B1 plays a role in the motility of cells that undergo directed movement.
GH has been previously implicated in the migration of several types of immune cells, including human monocytes (Wiedermann et al., 1993) and both resting and activated human T cells (Taub et al., 1994). In vivo, GH was shown to direct human T cell trafficking from the thymus to peripheral murine lymphoid tissues (Taub et al., 1994). Chemoattraction is an essential step for immune cells, including macrophages, to infiltrate host tissues and respond to infection. In addition, accumulation of macrophages in tissues such as adipose tissue is a central feature of the inflammatory response to obesity, a response that contributes to insulin resistance (Ouchi et al., 2011; Weisberg et al., 2003; Xu et al., 2003). Based on these findings, we hypothesized that GH would promote the migration of macrophages and that the ability of GH to stimulate that migration would require SH2B1.
In this work, we demonstrate that GH enhances the motility of macrophages using several in vitro models and that maximal GH-induced macrophage migration requires SH2B1. We further show that GH induces phosphorylation of Tyr439 and Tyr494 in SH2B1β, and that their phosphorylation is required for SH2B1β to enhance GH-induced macrophage migration. We also provide evidence that the ability of SH2B1β to leave the plasma membrane is required for macrophage motility, movement that is facilitated by phosphorylation of Ser161 and Ser165. Phosphorylation of Tyr439 and Tyr494, and Ser165 appear to work independently to enhance cell migration. Tyrosyl phosphorylation most likely affects interaction of SH2B1β with other critical proteins. Serine phosphorylation most likely enhances the movement of SH2B1β within the plasma membrane or between the plasma membrane, cytoplasm, and focal adhesions.
These results provide important insight into how SH2B1β contributes to GH-activated macrophage motility. Because multiple ligands that bind to immune cells activate JAK2, the results also suggest that SH2B1 may contribute to the motility of immune cells in response to a broader array of cytokines. Our studies also raise the possibility that defects in the migration of macrophages or other cell types may contribute to the phenotype of patients with gene deletions that include SH2B1 (severe early-onset obesity, insulin resistance, developmental delay) (Bachmann-Gagescu et al., 2010; Bochukova et al., 2010; Walters et al., 2010), or point mutations in SH2B1 (severe early-onset childhood obesity, insulin resistance, short stature, maladaptive behavior) (Doche et al., 2012).
Results
GH acts as a chemoattractant for macrophages
GH has been reported to serve as a chemoattractant for human T cells (Taub et al., 1994) and monocytes (Wiedermann et al., 1993). To determine whether GH also has the ability to induce chemotactic recruitment of macrophages, which arise from monocytes, we analyzed the migration of RAW 264.7 macrophages (RAW macrophages) using a transwell migration assay. RAW cells were added to the upper chamber of each transwell, and human GH (hGH) (500 ng/ml) was added to the lower chamber, upper chamber, or both chambers. After 6 hours, cells that had migrated to the bottom of the filter membrane were fixed, stained and counted using a light microscope (Fig. 1A). When GH was present in only the upper chamber or in both chambers, no statistically significant change in the number of migrated cells was observed. In contrast, the number of migrated cells was dramatically increased when GH was present only in the lower chamber. This chemotactic migration of RAW cells was dependent on the concentration of GH, with a statistically significant increase in migration observed at a GH concentration as low as 5 ng/ml (Fig. 1B). These data indicate that GH is able to induce directed macrophage movement (chemotaxis) at concentrations of GH well within the physiological range for rodent plasma GH [pulsatile peak levels of 200–300 ng/ml (Edén, 1979; Steyn et al., 2011) and ghrelin-stimulated levels of GH up to 600 ng/ml (Morozumi et al., 2011)]. Based on these results, GH was added only to the lower chamber of the transwell plate for subsequent migration assays. Because hGH binds to both GH and prolactin receptors in rodent cells, we repeated the experiment using bovine GH (bGH), which binds only to GH receptors. bGH, like hGH, stimulates chemotactic migration of RAW cells (supplementary material Fig. S1), consistent with GH acting through the GH receptor to stimulate migration. In addition, when primary macrophages were isolated from mice and studied, both hGH and bGH stimulated the chemotaxis of primary macrophages derived from both the peritoneum (Fig. 1C) and the bone marrow (Fig. 1D). We also found that the number of migrated cells was dramatically increased when GH was present in either the lower chamber alone or in both chambers when the incubation was extended to 18 hours (supplementary material Fig. S2). This suggests that GH is also able to elicit random cell movement (chemokinesis) over a longer time period.
Fig. 1.
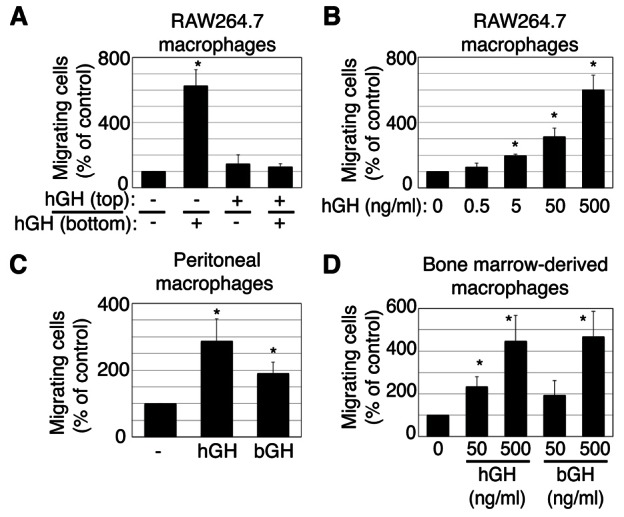
GH acts as a chemoattractant for macrophages. (A–D) Migration capacity of RAW264.7 macrophages (A,B), peritoneal macrophages (C) or bone marrow-derived macrophages (D) was analyzed using a transwell assay. Macrophages (5×105 cells/well) were placed in the upper chamber. (A) hGH (500 ng/ml) was added to the upper chamber, lower chamber or both as indicated for 6 hours. (B) The indicated concentration of hGH was added to the lower chamber for 6 hours. (C) hGH (500 ng/ml) or bGH (500 ng/ml) was added to the lower chamber for 12 hours. (D) The indicated concentration of hGH or bGH was added to the lower chamber for 12 hours. Values were normalized to the number of migrating cells in the absence of GH. The means ± s.e.m. from three independent experiments are shown. *P<0.05 by two-tailed paired Student's t-test compared with non-GH-treated cells.
SH2B1β is required for macrophage migration in response to GH
Our previous studies suggested that the adaptor protein SH2B1β (Fig. 2A) enhances GH-stimulated membrane ruffling and cytoskeleton dynamics (Diakonova et al., 2002b; Herrington et al., 2000), functions associated with cell motility. To examine whether SH2B1β plays a role in GH-mediated macrophage migration, we established pooled RAW cell lines that stably express GFP or GFP-SH2B1β. Immunoblots of cell lysates using an anti-SH2B1 antibody (αSH2B1) revealed similar amounts of endogenous SH2B1 in the two cell lines (Fig. 2B, lanes 1 and 2, lower band) and substantial overexpression of GFP-SH2B1β (Fig. 2B, lanes 1 and 2, upper band). Both cell lines were analyzed using the transwell migration assay for their ability to migrate in response to 0, 25 or 500 ng/ml GH added for 6 hours to the lower chamber (Fig. 2C, left panel). In control GFP-expressing RAW cells, the number of migrated cells doubled in the presence of 25 ng/ml GH and nearly tripled in the presence of 500 ng/ml GH compared to untreated cells. In GFP-SH2B1β-expressing RAW cells, GH-stimulated migration was substantially higher at 25 ng/ml GH than in control GFP-expressing RAW cells. GH-dependent migration was not further enhanced in the presence of 500 ng/ml GH. These results suggest that SH2B1β causes a leftward shift in the GH dose response curve, consistent with the leftward shift in the GH dose response curve reported previously for membrane ruffling in 3T3-F442A fibroblasts overexpressing SH2B1β (Herrington et al., 2000).
Fig. 2.

SH2B1 is required for GH-dependent macrophage migration. (A) Schematic of SH2B1β. Numbers indicate the amino acid position (Y, tyrosine). The P (proline-rich), DD (dimerization domain), NLS, NES, PH and SH2 domains are shown. (B) Cell lysates from RAW cell lines that stably express GFP, GFP-SH2B1β, shControl or shSH2B1 were immunoblotted with αSH2B1 or αvinculin. (C) RAW cell lines described in B were subjected to a transwell migration assay. Cells (5×105 cells/well) were placed in the upper chamber. hGH (0, 25 or 500 ng/ml) was added to the lower chamber for 6 hours. Values were normalized to non-hGH conditions in GFP-expressing control cells (left panel) or to non-hGH conditions in shControl-expressing cells (right panel). The means ± s.e.m. from three independent experiments are shown. P<0.05 by one-tailed paired Student's t-test compared with: non-hGH-treated (*) or hGH (25 ng/ml)-treated (#) GFP control cells; non-hGH-treated cells stably expressing GFP-SH2B1β (&); non-hGH-treated (Δ) or hGH (500 ng/ml)-treated (!) shControl cells; and non-hGH-treated shSH2B1 cells (+).
To confirm a role for endogenous SH2B1 in macrophage migration, we established pooled RAW cells stably expressing a 21-nucleotide long small hairpin shRNA targeted against all isoforms of SH2B1 (shSH2B1 cells). Pooled RAW cells stably expressing a nontargeting shRNA with low sequence similarity to known genes were used as a control (shControl cells). Immunoblotting cell lysates with αSH2B1 showed a reduction of at least 90% in the level of endogenous SH2B1 in the shSH2B1 cells (Fig. 2B, lanes 3 and 4). Immunoblotting cell lysates with αvinculin showed that an equal amount of protein was loaded for both cell types. In shControl cells, after GH treatment, the number of cells that migrated was three times higher than in the absence of GH (Fig. 2C, right panel), similar to the effect of GH on GFP-expressing cells (Fig. 2C, left panel). In contrast, in shSH2B1-expressing RAW cells, the number of cells that migrated in the absence of GH was reduced by 50% compared to shControl cells while the number of migrated cells in the presence of GH was reduced by 2/3. Thus, reduction of endogenous SH2B1 reduces both basal migration and the ability of GH to enhance macrophage migration. Taken together, the results in Fig. 2 provide strong evidence that SH2B1β plays a critical role in both basal and GH-dependent motility of RAW macrophages.
GH stimulates phosphorylation of SH2B1β on Tyr439 and Tyr494
We previously identified tyrosines 439 and 494 as the two major sites in SH2B1β that are phosphorylated by the tyrosine kinase JAK2 using 2-dimensional peptide mapping of in vitro phosphorylated SH2B1β. We also implicated these two phosphotyrosines in SH2B1β enhancement of GH-stimulated membrane ruffling in 3T3-F442A fibroblasts (O’Brien et al., 2003). We therefore investigated whether GH-dependent motility of RAW macrophages requires GH-dependent phosphorylation of Tyr439 and Tyr494. We first verified that GH stimulates phosphorylation of Tyr439 and Tyr494 in endogenous SH2B1. To assess phosphorylation at Tyr439 and Tyr494 of SH2B1, we developed two phosphospecific antibodies, one directed to phosphoY439 (pY439) and the other directed to pY494. To verify that each antibody was specific to the pTyr that it was designed to recognize, myc or myc-tagged SH2B1β constructs (WT, Y439/494F (2YF), Y439F or Y494F) were expressed in 293T cells with FLAG-JAK2. The tagged SH2B1βs were immunoprecipitated using αmyc and immunoblotted with either of these two phosphospecific antibodies. When blotting myc-SH2B1β WT with αpY439 SH2B1, we detected a prominent band that co-migrates with a slower migrating, more highly phosphorylated form of myc-SH2B1β WT. This band was present at similar levels when we blotted myc-SH2B1β Y494F with αpY439 SH2B1 (Fig. 3A, lanes 5 and 8). This band was not observed when JAK2 was absent (Fig. 3A, lanes 1 to 4) or when we blotted SH2B1β Y439F or Y439/494F (2YF), both of which lack the antibody epitope (Fig. 3A, lanes 6 and 7; supplementary material Fig. S3A). Immunoblotting cell lysates with αpY439 SH2B1 gave a pattern similar to immunoblotting the various immunoprecipitated myc-SH2B1β (Fig. 3A). A fainter band that co-migrates with the faster migrating, unphosphorylated form of myc-SH2B1β was present in all lanes (Fig. 3A), suggesting that αpY439 SH2B1 cross-reacts to a modest extent with unphosphorylated myc-SH2B1β or another protein. Immunoblotting with αmyc revealed equal expression and immunoprecipitation of the different myc-tagged SH2B1β constructs while blotting with FLAG antibody (αFLAG) revealed equal expression of FLAG-JAK2.
Fig. 3.
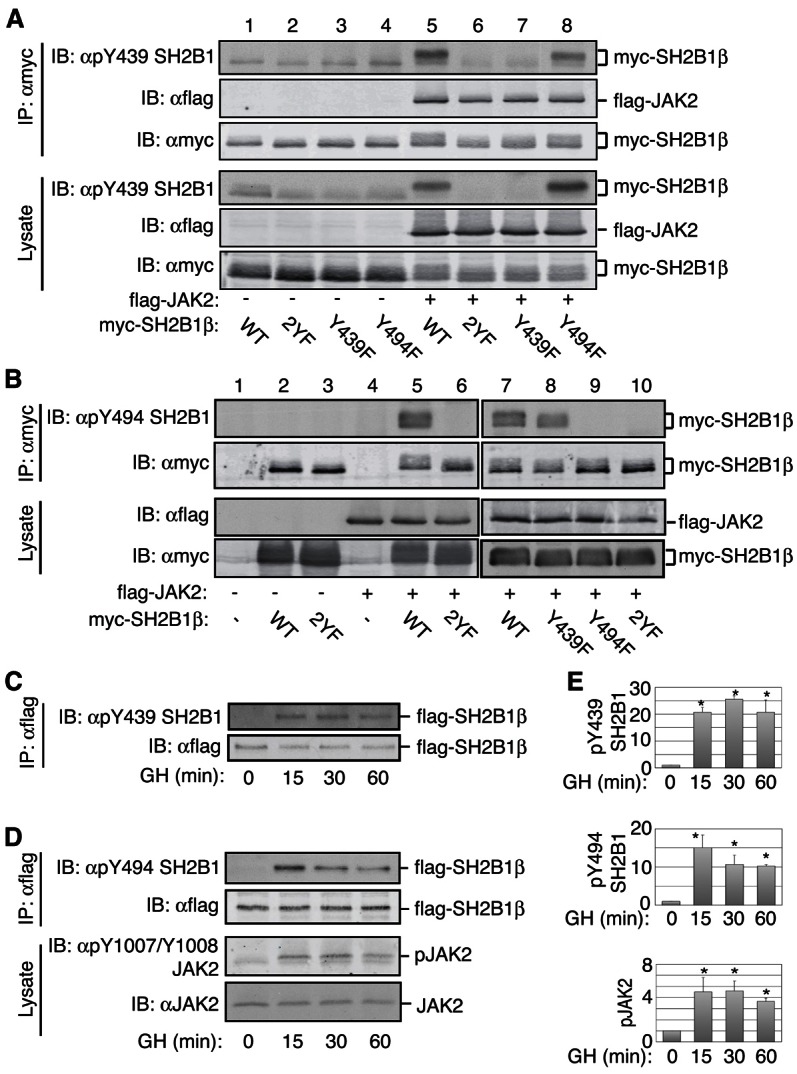
GH stimulates the phosphorylation of SH2B1β at Tyr439 and Tyr494. (A,B) 293T cells expressing myc-SH2B1β [wild-type, Y439/494F (2YF), Y439F or Y494F] or myc vector (−) with or without FLAG-JAK2 were lysed and proteins immunoprecipitated with αmyc. Immunoprecipitated proteins and cell lysates were immunoblotted with the indicated antibodies; n = 3. (C–E) 3T3-F442A fibroblasts expressing FLAG-SH2B1β were treated with hGH (500 ng/ml) for the indicated times. Proteins in cell lysates were immunoprecipitated with αFLAG. Immunoprecipitates and cell lysates were immunoblotted with the indicated antibodies. Level of phosphorylation was normalized for the total FLAG-SH2B1β present. Lanes 1–6 and 7–10 are from separate experiments. Means ± s.e.m. (n = 3) (0 and 15 minutes) or range (n = 2) (30 and 60 minutes) from 2 or 3 independent experiments are shown. *P<0.05 by two-tailed paired Student's t-test compared with GH at 0 minutes.
Similarly, αpY494 SH2B1 recognized myc-SH2B1β WT and Y439F when co-expressed with FLAG-JAK2, but not myc-SH2B1β Y439/494F (2YF) or Y494F, both of which lack the antibody epitope (Fig. 3B; supplementary material Fig. S3B). Immunoblotting the αmyc-SH2B1β immunoprecipitates with αFLAG revealed that FLAG-JAK2 bound equally to the different myc-SH2B1β constructs (Fig. 3A, 2nd panel). Taken together, these results suggest that: 1) the phosphospecific antibodies recognize the appropriate phosphorylated Tyr in SH2B1; 2) both Tyr439 and Tyr494 in SH2B1β are phosphorylated in intact cells by JAK2; 3) phosphorylation of Tyr439 or Tyr494 by JAK2 does not appear to significantly affect the phosphorylation of the other site; and 4) phosphorylation of Tyr439 or Tyr494 does not affect the binding of SH2B1β to JAK2.
These phosphospecific antibodies were then used to examine whether GH induces phosphorylation of SH2B1β on Tyr439 and Tyr494. We transiently expressed FLAG-SH2B1β in the GH-sensitive 3T3-F442A fibroblasts. Cells were treated with GH (500 ng/ml) for various times. FLAG-SH2B1β was immunoprecipitated with αFLAG and immunoblotted with αpY439 SH2B1 (Fig. 3C) or αpY494 SH2B1 (Fig. 3D). Neither Tyr439 nor Tyr494 was phosphorylated in the absence of GH. GH clearly stimulated the phosphorylation of both Tyr439 and Tyr494. Phosphorylation of both Tyr439 and Tyr494 remained elevated for at least 60 minutes. The time course for GH-dependent phosphorylation of SH2B1β on Tyr439 and Tyr494 was consistent with GH-dependent activation of JAK2 assessed by immunoblotting cell lysates with αpY1007/8 JAK2 to monitor the phosphorylation of the activating tyrosines in JAK2 (Fig. 3D). Quantified results from multiple experiments are shown in Fig. 3E. Taken together, our data suggest that GH rapidly induces phosphorylation of SH2B1β on Tyr439 and Tyr494, and that phosphorylation is temporarily consistent with these tyrosines being phosphorylated by JAK2 activated in response to GH.
Phosphorylation of Tyr439 and Tyr494 is required for SH2B1β to enhance GH-dependent macrophage migration
We next examined whether phosphorylation of Tyr439 and Tyr494 is important for SH2B1β-enhancement of GH-dependent macrophage migration. Myc or myc-SH2B1β constructs (WT, Y439/494F, Y439F, Y494F) were transiently expressed in RAW cells. Immunoblotting demonstrated that the various myc-SH2B1β constructs were present at similar levels (Fig. 4A). Both basal and GH-stimulated migration was increased in cells overexpressing myc-SH2B1β WT (Fig. 4B), consistent with the results for the cells stably expressing GFP-SH2B1β WT (Fig. 2C). In contrast, with myc-SH2B1β Y439/494F, both basal and GH-stimulated migration were decreased to a level below that seen in cells expressing vector alone. When Tyr439 or Tyr494 were mutated individually, the decrease in GH-stimulated motility was not quite as profound as when Tyr439 and Tyr494 were mutated in combination. Mutating all nine tyrosines in SH2B1β to phenylalanine did not further suppress SH2B1β-dependent cell migration compared to mutating only Tyr439 and Tyr494 supplementary material Fig. S4). Taken together, these data suggest that phosphorylation of both Tyr439 and Tyr494 in SH2B1β is critical for GH-dependent migration of RAW macrophages and also contributes to basal rates of migration.
Fig. 4.

Mutation of Tyr439 or Tyr494 inhibits SH2B1β-dependent, GH-mediated macrophage migration. (A) RAW cells transiently expressing myc (−) or myc-SH2B1β constructs, as indicated, were lysed and immunoblotted with the indicated antibodies. (B) RAW cells (2×105 cells/well) transiently expressing the indicated constructs were placed in the upper chamber of a transwell and incubated with or without hGH (500 ng/ml) in the lower chamber for 18 hours. Values were normalized to the migration in the absence of GH in myc-expressing control cells. The means ± s.e.m. from three independent experiments are shown. P<0.05 by one-tailed paired Student's t-test compared with non-hGH-treated (*) or hGH-treated (#) myc control cells; and non-hGH-treated (&) or hGH-treated (+) cells expressing wild-type myc-SH2B1β.
Changing the subcellular localization of SH2B1β alters macrophage migration
SH2B1 has both a nuclear localization sequence (NLS) and a nuclear export sequence (NES) that together allow SH2B1 to shuttle between the cytosol and the nucleus (Chen and Carter-Su, 2004; Maures et al., 2009). The polybasic NLS is also required for localization of SH2B1β at the plasma membrane in PC12 and 293T cells, possibly via electrostatic interactions with negatively charged phospholipids in the plasma membrane (Maures et al., 2011). Phosphorylation of serines proximal to the NLS is thought to neutralize the electrostatic interaction and release SH2B1β from the plasma membrane. SH2B1β is then free to migrate and enter the nucleus. Previous results indicate that Ser161 and Ser165 are the most important of the serines proximal to the NLS and that the ability of SH2B1β to shuttle between the plasma membrane, cytosol and nucleus is required for SH2B1β-dependent enhancement of NGF-induced neuronal differentiation of PC12 cells (Maures et al., 2009; Maures et al., 2011). Because the correct cellular localization can greatly impact the function of proteins, we examined how changing the ability of SH2B1β to localize at the plasma membrane, cytosol and nucleus would affect GH-mediated macrophage migration. We first used confocal microscopy of living RAW macrophages to determine the steady-state subcellular distribution of SH2B1β containing mutations found previously to affect the subcellular distribution of SH2B1β in other cell types. As previously observed in PC12 and 293T cells, GFP-SH2B1β WT localizes to the plasma membrane and cytosol of RAW macrophages (Fig. 5B). GFP-SH2B1β mNLS (in which four critical Lys in the NLS are mutated to Ala) fails to localize to the plasma membrane and accumulates in the cytosol of RAW macrophages (Fig. 5C), consistent with the NLS being required for localization of SH2B1β at the plasma membrane. As we reported previously for 293T cells and PC12 cells (Maures et al., 2009), GFP-SH2B1β mNLS also has a greatly reduced rate of nuclear entry compared to GFP-SH2B1β WT, assessed by the accumulation of nuclear SH2B1β when RAW cells are treated with the nuclear export inhibitor, leptomycin B (LMB) (supplementary material Fig. S5). The findings that SH2B1β WT in the presence of LMB and GFP-SH2B1β lacking its nuclear export sequence (ΔNES) (Fig. 5D) accumulate in the nucleus although SH2B1β WT is not seen in the nucleus at steady state indicate that SH2B1β continuously shuttles in and out of the nucleus via its NLS and NES, respectively. GFP-SH2B1β 13SA lacking all 13 Ser/Thr proximal to the NLS (Fig. 5E), SH2B1β S161/165A (supplementary material Fig. S6B), SH2B1β S161A (supplementary material Fig. S6C) and SH2B1β S165A (supplementary material Fig. S6D) were present almost exclusively at the plasma membrane. In contrast, when Ser161 (supplementary material Fig. S6E) or Ser165 (Fig. 5F; supplementary material Fig. S6F) are mutated to the phosphomimetic glutamic acid, SH2B1β is essentially absent from the plasma membrane, consistent with phosphorylation of Ser161 and Ser165 disrupting the interaction of the NLS with the plasma membrane.
Fig. 5.

The NLS and its adjacent serine residues regulate the subcellular localization of SH2B1β in RAW macrophages. (A–H) Living RAW cells transiently expressing wild-type GFP-SH2B1β or mutants as indicated were stained with wheat germ agglutinin conjugated with Alexa Fluor 594 (plasma membrane marker) and imaged by confocal microscopy. Top panels show GFP fluorescence. Middle panels show deconvoluted images of an overlay of GFP (green) and plasma membrane marker (red). Bottom panels show the intensity of green and red signal along the yellow arrows in the cells in overlay images. Linescans were determined using the MetaVue linescan tool. Arrows indicate the direction of each linescan. Scale bars: 10 µm.
We next used the transwell migration assay to determine the migration capacity of RAW cells transiently expressing GFP, GFP-SH2B1β WT, or GFP-SH2B1β mNLS. When the NLS was mutated, the number of migrated cells observed in the absence of GH increased substantially, and did not increase further with the addition of GH (Fig. 6A). These results suggest that in contrast to NGF-induced neuronal differentiation of PC12 cells (Maures et al., 2009), nuclear SH2B1 is not required for SH2B1β to stimulate cell migration. The finding that the migration of SH2B1β mNLS is equivalent to the GH-stimulated level raises the possibility that GH stimulates cell motility by releasing SH2B1β from the plasma membrane. In support of this hypothesis, GFP-SH2B1β 13SA and GFP-SH2B1β 2SA (S161/165A), which are localized almost exclusively at the plasma membrane (Fig. 5E; supplementary material Fig. S6B), did not enhance basal or GH-stimulated migration (Fig. 6B). Mutating Ser161 or Ser165 individually to Ala was not as effective at inhibiting motility as mutating both serines together (Fig. 6B). The phosphomimetics GFP-SH2B1β S161E and GFP-SH2B1β S165E, like the mutant NLS, increased cell migration in the absence of GH to the levels seen in GH-stimulated cells expressing GFP-SH2B1β WT. The number of migrating cells was not increased further with the addition of GH (Fig. 6C). We next assessed the effect of GH on the subcellular distribution of GFP-SH2B1β using a Nikon A1 confocal microscope equipped with a Perfect Focus System, ProScan III motorized stage and environmental chamber (37°C, 5% CO2). Images of living RAW macrophages expressing GFP-SH2B1β were taken every 5 minutes for one hour. We did not detect a GH-dependent overall change in subcellular distribution of GFP-SH2B1β. The inability to visualize a change in the amount of GFP-SH2B1β at the plasma membrane may be due to the low levels of GH receptors present in macrophages (Lu et al., 2010). If only a small fraction of the GFP-SH2B1β is phosphorylated in response to GH, it would be difficult to detect a GH-dependent change in the levels of GFP-SH2B1β associated with the plasma membrane.
Fig. 6.
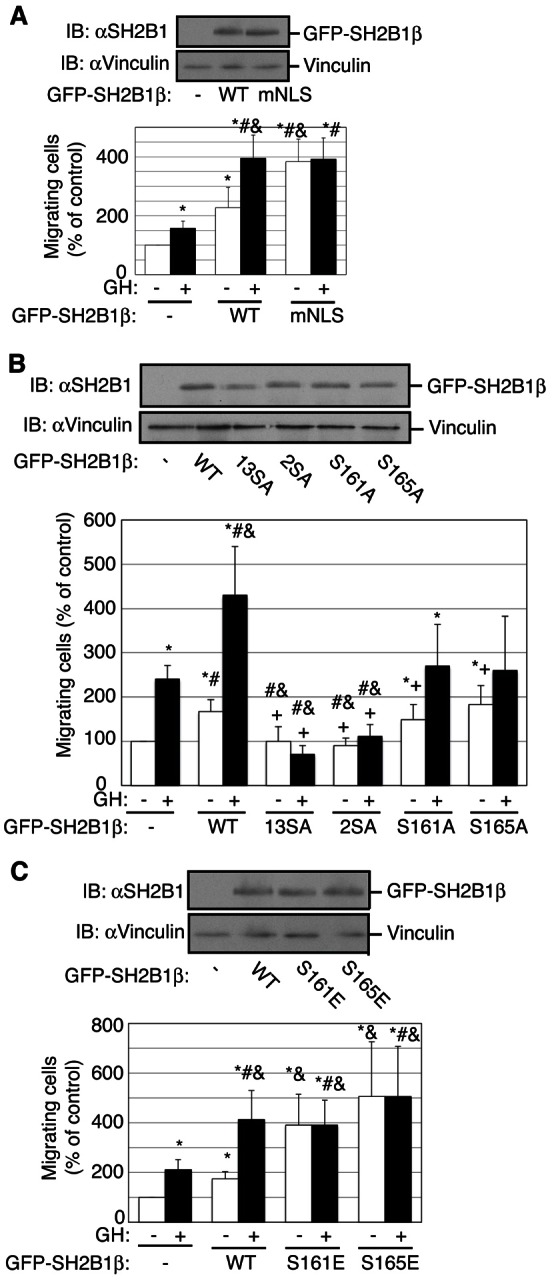
Disruption of the polybasic SH2B1β NLS enhances macrophage migration. Ser161 and Ser165 in SH2B1β play an important role in regulating macrophage migration. (A–C) Cell lysates from RAW cells transiently expressing GFP (−) or the indicated GFP-SH2B1β constructs were immunoblotted with the indicated antibodies. Migration of RAW cells (2×105 cells/well) transiently transfected as indicated was analyzed using a transwell migration assay with or without hGH (500 ng/ml) in the lower chamber for 18 hours. Values were normalized to non-hGH conditions in GFP-expressing control cells. The means ± s.e.m. from three independent experiments are shown. P<0.05 by one-tailed unpaired Student's t-test compared with non-hGH-treated (*) or hGH-treated (#) control cells; and non-hGH-treated (&) or hGH-treated (+) cells expressing wild-type GFP-SH2B1β.
Altering the degree of localization of SH2B1β at the plasma membrane does not affect JAK2-mediated tyrosine phosphorylation on Tyr439 and Tyr494
The results in Figs 3–6 raise the possibility that altering the subcellular localization of SH2B1β affects the ability of SH2B1β to enhance GH-stimulated RAW cell migration because it alters the ability of JAK2 to phosphorylate Tyr439 or Tyr494. To test this, FLAG-JAK2 was co-expressed in 293T cells with either GFP-SH2B1β WT or a GFP-SH2B1β mutant [R555E, 2YF (Y439/494F), mNLS, 2SE (S161/165E), 13SA, 2SA(S161/165A), S161A or S165A]. Equal amounts of the GFP-SH2B1β mutants were immunoprecipitated by αGFP (Fig. 7). As expected, neither αpY439 SH2B1 nor αpY494 SH2B1 bound to GFP-SH2B1β Y439/494F that lacks the antibody epitopes or to GFP-SH2B1β R555E, which contains a mutation in the SH2 domain of SH2B1β that prevents it from binding to and being phosphorylated by JAK2 (Rui and Carter-Su, 1999). However, phosphorylation of Tyr439 and Tyr494 was similar in GFP-SH2B1β WT and the remaining GFP-SH2B1β constructs. These results suggest that the impaired ability of SH2B1β 13SA, 2SA, S161A or S165A to enhance GH-dependent macrophage migration is unlikely to be a consequence of an inability of Tyr439 and Tyr 494 to be phosphorylated by JAK2.
Fig. 7.
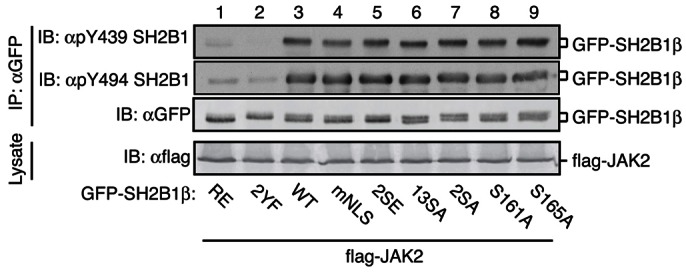
Phosphorylation of Ser161 and Ser165 does not affect JAK2-mediated phosphorylation of SH2B1β at Tyr439 and Tyr494. Cell lysates from 293T cells expressing the indicated GFP-SH2B1β constructs and FLAG-JAK2 were immunoprecipitated with αGFP. Lysates or immunoprecipitation samples were immunoblotted with the indicated antibodies; n = 2.
Tyrosyl phosphorylation of SH2B1β does not affect the localization of SH2B1β at the plasma membrane in RAW macrophages
We next examined whether the impairment of macrophage migration due to mutation of Tyr439 and Tyr494 in SH2B1β could be a consequence of altered subcellular localization of SH2B1β. RAW cells expressing GFP or GFP-SH2B1β constructs (WT, 9YF, or Y439/494F) were labeled with a plasma membrane marker (red) and live cells were imaged by confocal microscopy. Both GFP-SH2B1β 9YF (Fig. 5G) and Y439/494F (Fig. 5H) were present at the plasma membrane and in the cytosol to an extent similar to that seen with GFP-SH2B1β WT (Fig. 5B).
Tyr439 and Tyr494 do not contribute to basal cycling of SH2B1β at focal adhesions
We showed previously that SH2B1β co-localizes with vinculin, FAK, and at the tips of actin filaments at focal adhesions (Lanning et al., 2011). We therefore determined whether mutating Tyr439 and Tyr494 in SH2B1β affects SH2B1β localization to focal adhesions. GFP-SH2B1β Y439/494F localized to focal adhesions at the tips of actin filaments in 3T3-F442A fibroblasts in a manner similar to GFP-SH2B1β WT (Fig. 8A). The dynamics of SH2B1β movement were then measured by fluorescent recovery after photobleaching (FRAP) on individual focal adhesions. GFP-SH2B1β WT and SH2B1β Y439/494F had nearly identical recovery curves, mobile fraction, and t1/2 (time needed to recover to 50% of the mobile fraction) (Fig. 8B,C). This is in contrast to mutating Ser161 and Ser165 to Ala, which substantially decreases dynamic cycling of SH2B1β into and out of focal adhesions (Lanning et al., 2011).
Fig. 8.

Phosphorylation of Tyr439 and Tyr494 does not affect basal cycling of SH2B1β at focal adhesions. (A) 3T3-F442A fibroblasts expressing wild-type GFP-SH2B1β or GFP-SH2B1β Y439/494F were fixed and stained with phalloidin to visualize F-actin. Overlay of the GFP (green) and phalloidin (red) signals is shown in the right-hand panel. All images were obtained by confocal microscopy. Insets in the images are amplifications of the boxed areas. (B) FRAP values were obtained using Olympus Fluoview software. Data were normalized as described in Materials and Methods. (C) The mobile fraction (left panel) and t1/2 (right panel) values were calculated as described in Materials and Methods; n = 13 for wild-type SH2B1β and n = 10 for SH2B1β Y439/494F. Error bars indicate s.e.m.
Discussion
In this study, we document that GH induces both directional (chemotaxis) and random (chemokinesis) migration of RAW264.7 cells (a macrophage-like cell line), primary cultures of peritoneal macrophages and bone marrow-derived macrophages. Thus, macrophages join the growing list of immune cells whose migration is enhanced by GH. The ability of GH to serve as a chemotactic signal for macrophages and stimulate their migration may have several biological implications. Macrophage chemotaxis is important in both inflammatory responses as well as normal homeostatic physiological functions performed by resident macrophages (Chorro and Geissmann, 2010). It is possible that GH mediated activation of macrophage migration into tissues may play a role in both the negative and positive effects of GH replacement therapy.
When we investigated the mechanism by which GH regulates motility of macrophages, we found that SH2B1β plays a key role in both basal and GH-induced macrophage migration. Overexpression of SH2B1β enhances GH-stimulated RAW cell migration, while knockdown of endogenous SH2B1, using shRNA specific to SH2B1, inhibits both basal and GH-stimulated RAW cell migration. A critical role for SH2B1 in GH-induced macrophage migration is consistent with previous studies that used mutated and truncated SH2B1β to implicate SH2B1β in GH-induced membrane ruffling of 3T3-F442A fibroblasts and migration of 293T cells transiently expressing GH receptor into a wound in a wound-healing assay (Diakonova et al., 2002b; Herrington et al., 2000). SH2B1β has also been implicated in platelet-derived growth factor (PDGF)-induced membrane ruffling in 3T3-F442A fibroblasts (Herrington et al., 2000) and prolactin (PRL)-induced membrane ruffling and cell motility (Rider and Diakonova, 2011; Rider et al., 2009).
One possible explanation for how SH2B1β could enhance GH-dependent macrophage motility is if SH2B1β enhances GH-activation of JAK2, with a consequent increase in all GH signaling pathways and cellular responses. However, even though SH2B1β has been shown to activate JAK2 when both are overexpressed in 293T or COS-7 cells (Rui and Carter-Su, 1999), several of our findings argue against the stimulatory effect of SH2B1β on migration being due primarily to a stimulatory effect of SH2B1 on JAK2 activity. While mutating Tyr439 and Tyr494 prevents SH2B1β from enhancing both basal and GH-dependent macrophage motility (Fig. 4), it has no effect on the ability of SH2B1β to activate JAK2 in 293T cells (O’Brien et al., 2003). Furthermore, while the SH2 domain of SH2B1β is sufficient to activate JAK2 (Kurzer et al., 2004), additional regions of SH2B1β, including an N-terminal proline-rich region, are required for SH2B1β to enhance cell migration (Diakonova et al., 2002a).
Our finding that phosphorylation of both Tyr439 and Tyr494 appears to be critical for GH-dependent cell motility is consistent with our previous finding that phosphorylation of Tyr439 and/or Tyr494 is essential for SH2B1β to enhance GH-induced membrane ruffling in 3T3-F442A fibroblasts (O’Brien et al., 2003). Our previous studies used an in vitro kinase assay to demonstrate that JAK2 can phosphorylate Tyr439 and Tyr494 (O’Brien et al., 2003). In this study using phosphospecific antibodies, we were able to monitor the GH-dependent phosphorylation of these two tyrosines. We demonstrated in 3T3-F442A fibroblasts that GH promotes phosphorylation of SH2B1β on both Tyr439 and Tyr494. We think it most likely that Tyr439 and Tyr494, when phosphorylated, serve as binding sites for signaling molecules that mediate or regulate membrane ruffling and cell motility. This would be consistent with other studies suggesting that SH2B1 functions as a scaffold protein that helps stabilize or coordinate complexes that regulate the rearrangement of the actin cytoskeleton by facilitating the interaction of actin with its regulators. For example, SH2B1β has been shown to associate with lamellipodia and focal adhesions via its SH2 domain (Herrington et al., 2000; Lanning et al., 2011), suggesting it is recruited to phosphotyrosines in proteins present in these organelles. An N-terminal proline-rich domain (amino acids 85–106) of SH2B1β has been implicated in cell motility in wound healing studies; this region was also shown to be required for SH2B1β to bind constitutively to Rac (Diakonova et al., 2002a), a critical regulator of cell motility. Regions situated between amino acids 150–200 and 615–670 of SH2B1β have been implicated in actin binding, cross-linking of actin filaments and regulation of actin-based motility of Listeria Monocytogenes (Diakonova et al., 2007). Finally, amino acids 200–260 have been implicated in SH2B1β binding to filamin A (Rider et al., 2009), a protein that binds actin filaments to the plasma membrane (Stossel et al., 2001). Whether pTyr439 and pTyr494 recruit additional actin regulatory proteins to these complexes or phosphorylation of these tyrosines alters the ability of SH2B1 to bind to these or other actin regulating proteins remains to be determined.
Our data suggest that GH-dependent macrophage motility also requires the release of SH2B1β from the plasma membrane. Thus, mutations in SH2B1β that increase the cytosolic to plasma membrane ratio of SH2B1β (mNLS, S161E, S165E) result in increased motility whereas mutations that decrease the cytosolic to plasma membrane ratio (S161A, S165A) result in decreased GH-dependent motility. Phosphorylation at Ser161 and Ser165 has previously been implicated in the regulation of other aspects of SH2B1β signaling, including GH-dependent increases in the dynamic cycling of SH2B1β into focal adhesions. Dynamic cycling or turnover rates of multiple focal adhesion proteins (e.g. paxillin) are known to be highly associated with cell movement (Sero et al., 2011; Zaidel-Bar et al., 2007). Phosphorylation at Ser161 and Ser165 has also been reported to decrease the size of focal adhesions and increase the number of focal adhesions per cell (Lanning et al., 2011), changes that are associated with cell motility. These findings suggest that GH-regulated movement of SH2B1β from the plasma membrane to different cellular compartments, including the cytosol and focal adhesions, may be critical for GH-dependent cell motility. Consistent with GH affecting the subcellular localization of SH2B1β by increasing phosphorylation of Ser161 and Ser165, GH has been shown to stimulate the activity of protein kinase C (PKC) (reviewed by Argetsinger and Carter-Su, 1996). Ser161 and Ser165 fit the consensus sequence for PKC and activators of PKC have been shown to stimulate the release of SH2B1β from the plasma membrane (Maures et al., 2011). The fact that we did not find GH to change the overall subcellular distribution of SH2B1β in the cell within the 60-minute timeframe examined may reflect that in these cells too small a percentage of SH2B1β undergo a change in subcellular localization to be detected using our approach, perhaps due to the small number of GH receptors in these cells or because the changes in subcellular localization take longer than 60 minutes to occur. Alternatively, GH-dependent modifications of SH2B1β may not have a substantial impact on the steady state distribution of SH2B1β. At the very least, the ability of SH2B1β to freely move between different subcellular compartments, which is enhanced by serine phosphorylation of SH2B1β, appears to be required for SH2B1β to facilitate cell motility. For example, a form of SH2B1β that has been post-translationally modified in response to GH may need to move to a different subcellular location to facilitate cell motility.
Our findings also suggest that the signaling pathways regulated by phosphorylation at Ser161 and Ser165 and by phosphorylation of Tyr439 and Tyr494 are independent of each other. Phosphorylation at Ser161 and Ser165 does not appear to influence the ability of Tyr439 and Tyr494 to be phosphorylated by JAK2. Conversely, mutating Tyr439 and Tyr494 does not affect the steady state distribution of SH2B1β between the plasma membrane and cytosol, nor the Ser161, Ser165-dependent dynamic cycling of SH2B1β at the focal adhesions.
In conclusion, this is the first study to suggest that SH2B1 has a key role in regulating GH-dependent macrophage migration. Further, our data suggest that SH2B1β has the ability to enhance GH-mediated macrophage migration through two independent mechanisms: one dependent upon JAK2-mediated phosphorylation of SH2B1β at Tyr439 and Tyr494, and the other dependent upon the ability of SH2B1β to move from the plasma membrane to the cytosol. Our findings provide a foundation for future studies concerning a role for SH2B1 in macrophage function in vivo, the mechanism by which SH2B1β movement between different cellular compartments affects cell motility, and how SH2B1β integrates the activities associated with various SH2B1-associated proteins to regulate cell motility and other downstream functions. Numerous ligands involved in immune function activate JAK2 (Ghoreschi et al., 2009). Because SH2B1β appears to be expressed ubiquitously, it seems likely that SH2B1β will be found to influence aspects of signaling downstream of these JAK2-activating ligands, including cell motility. Mutations in SH2B1 have been detected in humans. These mutations prevent SH2B1β-enhancement of GH stimulated motility of macrophages (Doche et al., 2012). Therefore, our findings also raise the possibility that defects in the motility of macrophages as well as the motility of other cell types (e.g. during development) may contribute to early onset childhood obesity, insulin resistance, short stature, and/or maladaptive behaviors observed in the humans with mutations in the SH2B1 gene.
Materials and Methods
Antibodies
Anti-rat SH2B1 (αSH2B1) (1∶1000) (kind gift from L. Rui, University of Michigan, Ann Arbor, MI) was raised against an SH2B1β-GST fusion protein (Duan et al., 2004). Anti-vinculin (V9131) (αvinculin) (1∶1000), anti-FLAG (M2) (αFLAG) (1∶4000) and αFLAG (M2) conjugated agarose beads were from Sigma–Aldrich (St. Louis, MO). Anti-myc (9E10) (αmyc) (1∶100 for immunoprecipitation; 1∶2000 for immunoblotting) was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). αGFP-IRDye800 (1∶4000), αmyc-IRDye700 (1∶1000) and IRDye700- and IRDye800-conjugated anti-mouse (1∶20,000) and anti-rabbit IgG (1∶20,000) were from Rockland Immunochemicals (Gilbertville, PA). αGFP (Cat no. 632460) (1∶100 for immunoprecipitation) was from Clontech (Mountain View, CA). αphospho-JAK2 (Tyr1007/1008) (Cat no. 07-606) (1∶1000), αJAK2 (8E10.2) (1∶1000) and rabbit anti-mouse IgG (1∶100 for immunoprecipitation) were from Millipore (Billerica, MA). Antibodies against peptides containing phosphorylated tyrosine 439 of SH2B1 (Cat no. 1960) (1∶1000) or phosphorylated tyrosine 494 of SH2B1 (Cat no. 1956) (1∶1000) were made in collaboration with Millipore (Billerica, MA). Horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgG (1∶5000) were from Cell Signaling Technology (Danvers, MA).
Reagents
Recombinant 22,000-Da human GH was a kind gift from Eli Lilly & Co. (Indianapolis, IN). Bovine GH was obtained from A. Parlow (National Hormone and Peptide Program). Fetal bovine serum (FBS) was from Atlanta Biologicals (Lawrenceville, GA). Calf serum was from Invitrogen (Grand Island, NY). BSA (fatty acid free) was from Proliant (Ankeny, IA). High range prestained SDS-PAGE standards were from BioRad. Aprotinin and leupeptin were from Roche. Recombinant protein A-agarose was from Repligen (Wattham, MA). Hybond-C Extra nitrocellulose was from Amersham Biosciences (Pittsburg, PA).
Plasmids
The cDNA for murine JAK2 was provided by J. Ihle (St. Jude Children's Research Hospital, Memphis, TN) (Silvennoinen et al., 1993). FLAG-JAK2, and the various rat SH2B1β constructs, FLAG-SH2B1β, GFP-SH2B1β WT, GFP-SH2B1β Δ198–268 (ΔNES), GFP-SH2B1β point mutations (9YF, Y439/494F, S161A, S165A, 2SA(S161/165A), 13SA, S161E, S165E, S161/165E, mNLS), myc-SH2B1β WT, and myc-SH2B1β point mutations (Y439/494F, Y439F, Y494F) have been described previously (Chen and Carter-Su, 2004; Kurzer et al., 2004; Maures et al., 2009; Maures et al., 2011; O’Brien et al., 2003). When expressed in 293T and RAW cells, all mutant SH2B1β proteins migrated in SDS-PAGE gels at the predicted size (data not shown). Control shRNA and shRNA for SH2B1 knockdown have been described previously (Maures et al., 2009).
Cell culture and transfection
Murine 3T3-F442A fibroblasts (stock from Howard Green, Harvard University, Cambridge, MA) and 293T cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 1 mM L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, 0.25 µg/ml amphotericin (DMEM culture medium) and 8% calf serum (growth medium). RAW264.7 macrophages were kindly provided by Joel Swanson (University of Michigan, Ann Arbor, MI). RAW macrophages were grown in DMEM culture medium containing 8% heat-inactivated fetal bovine serum (FBS) (complete medium). Cells were incubated in serum-free medium containing 1% BSA overnight prior to treatment with GH. 293T cells were transiently transfected using calcium phosphate precipitation (Chen and Okayama, 1987). 3T3-F442A fibroblasts and RAW cells were transiently transfected using Amaxa Nucleofector Technology (Lonza, Basal, Switzerland) with solution V and setting U24 or D32, respectively. After 6 hours, fresh growth medium was added. Cells were used 18–24 hours later.
To make RAW cells stably overexpressing GFP or GFP-SH2B1β, RAW cells were transfected with cDNA encoding GFP or GFP-SH2B1β using Fugene HD and then grown in selection medium containing 1 mg/ml G418 (Invitrogen) for at least 30 days. Cells resistant to G418 were subjected to fluorescence cell sorting (FACS); the population with the highest GFP expression level (top 5%) was pooled to avoid clonal variation. Stable pooled cell lines were maintained in complete medium plus 0.5 mg/ml G418 and placed in fresh complete medium the day before each experiment.
To make cells that stably expressed low levels of endogenous SH2B1, SH2B1 siRNA vector described previously (Maures et al., 2009) was introduced into subconfluent RAW cells using Amaxa Nucleofector Technology with solution V and setting T20. The pSuper vector containing a non-targeting shRNA with a low sequence similarity to known genes was used as a control. After 6 hours, fresh complete medium was added. Twenty-four hours later, complete medium containing 5 µg/ml puromycin was added. Selection for pSuper positive RAW cells was carried out for 30 days. Cells were placed in fresh complete medium the day before each experiment.
Bone marrow-derived macrophages and thioglycolate-elicited peritoneal macrophages
C57BL/6J mice were purchased from Jackson Laboratory, Bar Harbor, ME. Mice were fed normal diets consisting of 4.5% calories from fat (Lab Diet 5002; PMI Nutrition International, St Louis, MO), housed in a specific pathogen-free facility on a 12-hour light/12-hour dark cycle and given free access to food and water. All animal use was in compliance with the Institute of Laboratory Animal Research Guide for the Care and Use of Laboratory Animals. Animal procedures were approved by the University Committee on Use and Care of Animals (UCUCA) at the University of Michigan. For bone marrow-derived macrophages, bone marrow was isolated from femurs and tibias of the mice and cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum and 20% L929 conditioned medium for 7 days. Peritoneal macrophages were elicited by intraperitoneal (i.p.) injection of aged 30% thioglycollate broth (3 ml/mouse; Sigma) and recovered 4 days later by lavage with ice-cold PBS followed by enrichment by adhesion to plastic.
Cell migration assay
Parental, stably transfected, and transiently transfected RAW cells were incubated in serum-free medium containing 1% BSA (deprivation medium) overnight. Murine bone marrow-derived macrophages, parental and stably transfected RAW cells (5×105 cells), or transiently transfected RAW (2×105 cells) were suspended in deprivation medium and added to the upper chamber of transwell inserts (5-µm pore size) (Costar, Lowell, MA). For murine peritoneal macrophages, 5×105 cells were suspended in complete medium and added to the upper chamber of transwell inserts. After 2 hours (macrophages had attached to the upper side of the transwell insert), medium in the upper chamber was replaced by deprivation medium. Inserts were placed in the lower chamber, which contained 10 µg of type I collagen and the indicated concentration of GH in deprivation medium. Transwell units were incubated at 37°C for the time indicated, fixed in methanol and then air-dried. Cells in the upper chamber of the transwell unit were removed with a cotton swab. Filter membranes were stained with hematoxylin and eosin stain (1∶10 dilution) for one hour and destained with double distilled water. Migrated cells on the lower side of the filter membrane were counted using a light microscope. For each experiment, at least three independent fields were counted for each condition. Each experiment was performed three times, on separate days. The number of migrated cells counted per field per condition ranged from ten to over 800. For the control (-GH) cells used for normalization, the number of counted cells per field averaged 26; the calculated total number of control (-GH) cells that migrated ranged between 0.1% and 0.3% (500–1500 cells) of the total number of cells plated.
Immunoprecipitation and immunoblotting
Cells were lysed in RIPA buffer (25 mM Tris pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 10 mM NaF, 1 mM Na3VO4, protease inhibitors (1 mM PMSF, 10 µg/ml aprotinin, 10 µg/ml leupeptin), centrifuged (16,000×g at 4°C for 10 minutes) and the supernatant was collected (cell lysate). For immunoprecipitations, cell lysates were pre-cleared by incubating with agarose beads (Sigma) for one hour and then incubated overnight with either 1) αFLAG-agarose beads or 2) αGFP or αmyc plus rabbit αmouse IgG followed by one hour incubation with protein A-agarose beads (Amersham Biosciences). Bound proteins were eluted and resolved by SDS-PAGE, transferred to nitrocellulose, immunoblotted with the indicated antibodies, and visualized using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE) or enhanced chemiluminescence (Pierce, Rockford, IL).
Cell imaging
Cell imaging was performed using an Olympus FluoView 500 laser scanning confocal microscope and FluoView version 5.0 software. RAW cells were plated on 35-mm glass bottom dishes (MatTek, Ashland, MA). To label the plasma membrane, cells were incubated for 45 seconds with wheat germ agglutinin-conjugated Alexa Fluor 594 (Invitrogen), washed, and resuspended in Ringer's Buffer (10 mM HEPES, 155 mM NaCl, 2 mM CaCl2, 5 mM KCl, 1 mM MgCl2, 10 mM NaH2PO4 and 10 mM glucose, pH 7.2). All transfected cells were viewed (≥20 cells/experiment). Two or more independent experiments were conducted with each SH2B1β mutant. Generally, for each GFP-SH2B1β construct, 90% of the cells showed the same subcellular distribution, independent of expression level. Image planes were chosen to allow maximal visualization of the plasma membrane and nucleus. Deconvoluted images were obtained using AutoQuant X2 software from three merged z section confocal images (each 0.6 µm distance apart). Linescan profiles were obtained using MetaVue Software (Universal Imaging, Sunnyvale, CA). Each value represents an average of 15 pixels (∼1.5 µm) perpendicular to and centered around the scanned line.
Fluorescence recovery after photobleaching
FRAP experiments were performed and analyzed as described previously (Lanning et al., 2011). Briefly, GFP-SH2B1β WT or GFP-SH2B1β (Y439/494F) were transiently expressed in 3T3-F442A fibroblasts. The GFP signal in individual focal adhesions was bleached by three iterations of 100% laser power. Fluorescent intensity measurements were taken every 6 seconds from 30 individual focal adhesions in ten individual cells for each condition. Data were normalized to unbleached sections of cytosol after background subtraction. SigmaPlot 11.0 (San Jose, CA) was used to fit curves to FRAP data by applying the nonlinear regression of exponential rise to maximum and the double fours best fit equation, y = a(1−e−bx)+c(1−e−dx). From this equation, the mobile fraction of SH2B1β (the maximum percentage fluorescence recovery) and t1/2 (time needed to recover to 50% of the mobile fraction) were determined.
Supplementary Material
Acknowledgments
We thank Dr Liangyou Rui for αSH2B1 antibody; Dr Joel Swanson for RAW264.7 macrophages; Dr Travis Maures and Joel Cline for help with the constructs; Dr Stephen Lentz for assistance with confocal microscopy and FRAP; Dr Edward Stuenkel for assistance with FRAP; Heekon Cha and Aaron Taylor for help with curve fitting; Barbara Hawkins for help with the manuscript; and Drs Jessica Schwartz, Ram Menon, Bridgette Ray and Melissa Dobson for helpful discussions. Confocal microscopy was performed in the Morphology and Imaging Core of the Michigan Diabetes Research and Training Center (NIH P60-DK20572). cDNAs were sequenced by the University of Michigan DNA Sequencing Core with support from the University of Michigan Comprehensive Cancer Center (NIH P30-CA46592).
Footnotes
Author contributions
H.-W.S. designed and performed experiments, interpreted data, and wrote the manuscript, N.J.L and D.L.M. designed and performed experiments and interpreted data, L.S.A. interpreted data, and edited the manuscript, C.N.L. contributed essential reagents, interpreted data and edited the manuscript, C.C.-S. designed experiments, interpreted data, and wrote the manuscript.
Funding
This work was supported by the National Institutes of Health (NIH) [grant numbers R01-DK54222 to C.C.S. and R01-DK090262 to C.N.L.]. D.L.M. was supported by an NIH National Research Service Award (NRSA) award [grant number DK091976]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.113050/-/DC1
References
- Argetsinger L. S., Carter-Su C. (1996). Mechanism of signaling by growth hormone receptor. Physiol. Rev. 76, 1089–1107 [DOI] [PubMed] [Google Scholar]
- Bachmann-Gagescu R., Mefford H. C., Cowan C., Glew G. M., Hing A. V., Wallace S., Bader P. I., Hamati A., Reitnauer P. J., Smith R. et al. (2010). Recurrent 200-kb deletions of 16p11.2 that include the SH2B1 gene are associated with developmental delay and obesity. Genet. Med. 12, 641–647 10.1097/GIM.0b013e3181ef4286 [DOI] [PubMed] [Google Scholar]
- Bochukova E. G., Huang N., Keogh J., Henning E., Purmann C., Blaszczyk K., Saeed S., Hamilton-Shield J., Clayton-Smith J., O’Rahilly S. et al. (2010). Large, rare chromosomal deletions associated with severe early-onset obesity. Nature 463, 666–670 10.1038/nature08689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Carter-Su C. (2004). Adapter protein SH2-B β undergoes nucleocytoplasmic shuttling: implications for nerve growth factor induction of neuronal differentiation. Mol. Cell. Biol. 24, 3633–3647 10.1128/MCB.24.9.3633-3647.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Okayama H. (1987). High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7, 2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorro L., Geissmann F. (2010). Development and homeostasis of ‘resident’ myeloid cells: the case of the Langerhans cell. Trends Immunol. 31, 438–445 10.1016/j.it.2010.09.003 [DOI] [PubMed] [Google Scholar]
- Diakonova M., Bokoch G., Swanson J. A. (2002a). Dynamics of cytoskeletal proteins during Fcgamma receptor-mediated phagocytosis in macrophages. Mol. Biol. Cell 13, 402–411 10.1091/mbc.01-05-0273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diakonova M., Gunter D. R., Herrington J., Carter-Su C. (2002b). SH2-Bbeta is a Rac-binding protein that regulates cell motility. J. Biol. Chem. 277, 10669–10677 10.1074/jbc.M111138200 [DOI] [PubMed] [Google Scholar]
- Diakonova M., Helfer E., Seveau S., Swanson J. A., Kocks C., Rui L., Carlier M. F., Carter-Su C. (2007). Adapter protein SH2-Bbeta stimulates actin-based motility of Listeria monocytogenes in a vasodilator-stimulated phosphoprotein (VASP)-dependent fashion. Infect. Immun. 75, 3581–3593 10.1128/IAI.00214-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doche M. E., Bochukova E. G., Su H. W., Pearce L. R., Keogh J. M., Henning E., Cline J. M., Saeed S., Dale A., Cheetham T. et al. (2012). Human SH2B1 mutations are associated with maladaptive behaviors and obesity. J. Clin. Invest. 122, 4732–4736 10.1172/JCI62696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan C., Li M., Rui L. (2004). SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. J. Biol. Chem. 279, 43684–43691 10.1074/jbc.M408495200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edén S. (1979). Age- and sex-related differences in episodic growth hormone secretion in the rat. Endocrinology 105, 555–560 10.1210/endo-105-2-555 [DOI] [PubMed] [Google Scholar]
- Ghoreschi K., Laurence A., O’Shea J. J. (2009). Janus kinases in immune cell signaling. Immunol. Rev. 228, 273–287 10.1111/j.1600-065X.2008.00754.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J., Diakonova M., Rui L., Gunter D. R., Carter-Su C. (2000). SH2-B is required for growth hormone-induced actin reorganization. J. Biol. Chem. 275, 13126–13133 10.1074/jbc.275.17.13126 [DOI] [PubMed] [Google Scholar]
- Kurzer J. H., Argetsinger L. S., Zhou Y-J., Kouadio J-L., O’Shea J. J., Carter-Su C. (2004). Tyrosine 813 is a site of JAK2 autophosphorylation critical for activation of JAK2 by SH2-B β. Mol. Cell. Biol. 24, 4557–4570 10.1128/MCB.24.10.4557-4570.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanning N. J., Su H. W., Argetsinger L. S., Carter-Su C. (2011). Identification of SH2B1β as a focal adhesion protein that regulates focal adhesion size and number. J. Cell Sci. 124, 3095–3105 10.1242/jcs.081547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Zhou Y., Carter-Su C., Myers M. G., Jr, Rui L. (2007). SH2B1 enhances leptin signaling by both Janus kinase 2 Tyr813 phosphorylation-dependent and -independent mechanisms. Mol. Endocrinol. 21, 2270–2281 10.1210/me.2007-0111 [DOI] [PubMed] [Google Scholar]
- Lu C., Kumar P. A., Fan Y., Sperling M. A., Menon R. K. (2010). A novel effect of growth hormone on macrophage modulates macrophage-dependent adipocyte differentiation. Endocrinology 151, 2189–2199 10.1210/en.2009-1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maures T. J., Chen L., Carter-Su C. (2009). Nucleocytoplasmic shuttling of the adapter protein SH2B1β (SH2-Bbeta) is required for nerve growth factor (NGF)-dependent neurite outgrowth and enhancement of expression of a subset of NGF-responsive genes. Mol. Endocrinol. 23, 1077–1091 10.1210/me.2009-0011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maures T. J., Su H-W., Argetsinger L. A., Grinstein S., Carter-Su C. (2011). Phosphorylation controls a dual-function polybasic nuclear localization sequence in the adapter protein SH2B1β to regulate its cellular function and distribution. J. Cell Sci. 124, 1542–1552 10.1242/jcs.078949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozumi N., Hanada T., Habara H., Yamaki A., Furuya M., Nakatsuka T., Inomata N., Minamitake Y., Ohsuye K., Kangawa K. (2011). The role of C-terminal part of ghrelin in pharmacokinetic profile and biological activity in rats. Peptides 32, 1001–1007 10.1016/j.peptides.2011.01.021 [DOI] [PubMed] [Google Scholar]
- O’Brien K. B., Argetsinger L. S., Diakonova M., Carter-Su C. (2003). YXXL motifs in SH2-Bbeta are phosphorylated by JAK2, JAK1, and platelet-derived growth factor receptor and are required for membrane ruffling. J. Biol. Chem. 278, 11970–11978 10.1074/jbc.M210765200 [DOI] [PubMed] [Google Scholar]
- Ouchi N., Parker J. L., Lugus J. J., Walsh K. (2011). Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 11, 85–97 10.1038/nri2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X., Vass W. C., Papageorge A. G., Anborgh P. H., Lowy D. R. (1998). N terminus of Sos1 Ras exchange factor: critical roles for the Dbl and pleckstrin homology domains. Mol. Cell. Biol. 18, 771–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider L., Diakonova M. (2011). Adapter protein SH2B1β binds filamin A to regulate prolactin-dependent cytoskeletal reorganization and cell motility. Mol. Endocrinol. 25, 1231–1243 10.1210/me.2011-0056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider L., Tao J., Snyder S., Brinley B., Lu J., Diakonova M. (2009). Adapter protein SH2B1β cross-links actin filaments and regulates actin cytoskeleton. Mol. Endocrinol. 23, 1065–1076 10.1210/me.2008-0428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel H., Yousaf N., Zhao Y., Dai H., Deng Y., Wang J. (2000). PSM, a mediator of PDGF-BB-, IGF-I-, and insulin-stimulated mitogenesis. Oncogene 19, 39–50 10.1038/sj.onc.1203253 [DOI] [PubMed] [Google Scholar]
- Rui L., Carter-Su C. (1999). Identification of SH2-bbeta as a potent cytoplasmic activator of the tyrosine kinase Janus kinase 2. Proc. Natl. Acad. Sci. USA 96, 7172–7177 10.1073/pnas.96.13.7172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L., Mathews L. S., Hotta K., Gustafson T. A., Carter-Su C. (1997). Identification of SH2-Bbeta as a substrate of the tyrosine kinase JAK2 involved in growth hormone signaling. Mol. Cell. Biol. 17, 6633–6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L., Herrington J., Carter-Su C. (1999). SH2-B is required for nerve growth factor-induced neuronal differentiation. J. Biol. Chem. 274, 10590–10594 10.1074/jbc.274.15.10590 [DOI] [PubMed] [Google Scholar]
- Sero J. E., Thodeti C. K., Mammoto A., Bakal C., Thomas S., Ingber D. E. (2011). Paxillin mediates sensing of physical cues and regulates directional cell motility by controlling lamellipodia positioning. PLoS ONE 6, e28303 10.1371/journal.pone.0028303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvennoinen O., Witthuhn B. A., Quelle F. W., Cleveland J. L., Yi T., Ihle J. N. (1993). Structure of the murine Jak2 protein-tyrosine kinase and its role in interleukin 3 signal transduction. Proc. Natl. Acad. Sci. USA 90, 8429–8433 10.1073/pnas.90.18.8429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyn F. J., Huang L., Ngo S. T., Leong J. W., Tan H. Y., Xie T. Y., Parlow A. F., Veldhuis J. D., Waters M. J., Chen C. (2011). Development of a method for the determination of pulsatile growth hormone secretion in mice. Endocrinology 152, 3165–3171 10.1210/en.2011-0253 [DOI] [PubMed] [Google Scholar]
- Stossel T. P., Condeelis J., Cooley L., Hartwig J. H., Noegel A., Schleicher M., Shapiro S. S. (2001). Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2, 138–145 10.1038/35052082 [DOI] [PubMed] [Google Scholar]
- Taub D. D., Tsarfaty G., Lloyd A. R., Durum S. K., Longo D. L., Murphy W. J. (1994). Growth hormone promotes human T cell adhesion and migration to both human and murine matrix proteins in vitro and directly promotes xenogeneic engraftment. J. Clin. Invest. 94, 293–300 10.1172/JCI117320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters R. G., Jacquemont S., Valsesia A., de Smith A. J., Martinet D., Andersson J., Falchi M., Chen F., Andrieux J., Lobbens S. et al. (2010). A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature 463, 671–675 10.1038/nature08727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg S. P., McCann D., Desai M., Rosenbaum M., Leibel R. L., Ferrante A. W., Jr (2003). Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedermann C. J., Reinisch N., Braunsteiner H. (1993). Stimulation of monocyte chemotaxis by human growth hormone and its deactivation by somatostatin. Blood 82, 954–960 [PubMed] [Google Scholar]
- Xu H., Barnes G. T., Yang Q., Tan G., Yang D., Chou C. J., Sole J., Nichols A., Ross J. S., Tartaglia L. A. et al. (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousaf N., Deng Y., Kang Y., Riedel H. (2001). Four PSM/SH2-B alternative splice variants and their differential roles in mitogenesis. J. Biol. Chem. 276, 40940–40948 10.1074/jbc.M104191200 [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar R., Milo R., Kam Z., Geiger B. (2007). A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J. Cell Sci. 120, 137–148 10.1242/jcs.03314 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Zhu W., Wang Y. G., Liu X. J., Jiao L., Liu X., Zhang Z. H., Lu C. L., He C. (2006). Interaction of SH2-Bbeta with RET is involved in signaling of GDNF-induced neurite outgrowth. J. Cell Sci. 119, 1666–1676 10.1242/jcs.02845 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.