

Abstract
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) occur in approximately 200,000 patients per year. Studies indicate that lung endothelium plays a significant role in ALI. The authors’ recent in vitro studies demonstrate a novel mechanism of β-nicotinamide adenine dinucleotide (β-NAD)–induced protection against gram-positive (pneumolysin, PLY) and gram-negative (lipopolysaccharide, LPS) toxin–induced lung endothelial cell (EC) barrier dysfunction. The objective of the current study was to evaluate the protective effect of β-NAD against LPS-induced ALI in mice. C57BL/6J mice were randomly divided into 4 groups: vehicle, β-NAD, LPS, and LPS/β-NAD. After surgery, mice were allowed to recover for 24 hours. Evans blue dye–albumin (EBA) was given through the internal jugular vein 2 hours prior to the termination of the experiments. Upon sacrificing the animals, bronchoalveolar lavage fluid (BALF) was collected and the lungs were harvested. β-NAD treatment significantly attenuated the inflammatory response by means of reducing the accumulation of cells and protein in BALF, blunting the parenchymal neutrophil infiltration, and preventing capillary leak. In addition, the histological examination demonstrated decreased interstitial edema in the LPS/β-NAD specimens, as compared to the LPS-only specimens. The mRNA levels of the anti-inflammatory cytokines were up-regulated in the LPS group treated with β-NAD compared to the LPS-only–treated group. β-NAD treatment down-regulated the mRNA levels of the proinflammatory cytokines. These findings suggest that β-NAD could be investigated as a therapeutic option against bacterial toxin–induced lung inflammation and ALI in mice.
Keywords: acute lung injury, ARDS, cytokines, inflammation, LPS, β-NAD
Respiratory failure due to acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are known causes of hospitalization andmorbidity. ALI is characterized by a disturbance of the pulmonary capillary barrier, followed by increased lung permeability and infiltration of activated neutrophils into the lungs [1-3]. Importantly, ALI also leads to systemic inflammation, which is also associated with endothelial capillary barrier dysfunction, reduced cardiac output, and increased risk of cardiovascular events [4]. In ALI/ARDS, the pulmonary endothelial barrier dysfunction leads to noncardiogenic pulmonary edema, with neutrophil and fluid accumulation in the interstitial spaces and eventually in the alveoli. ARDS incidence in the United States is 79 per 100,000 population per year, with a high mortality rate of 30% to 50% depending on the cause [5, 6]. The most common causes include septic shock and pneumonia, with a lower incidence occurring in trauma, advanced age, and excessive alcohol consumption [7-12]. Pulmonary endothelial barrier dysfunction is a hallmark of these lung diseases for which there is no standard therapy. In recent years, the pulmonary endothelium has been the subject of physiologic and molecular research, with a goal of elucidating pathways that can be targeted for pharmacologic therapy.
Recent studies indicate that β-nicotinamide adenine dinucleotide (β-NAD) is an important vascular mediator [13, 14] that released into the extracellular fluids under proinflammatory conditions [15, 16]. It elicits cellular effects through activation of G protein–coupled purine receptors P2Y1 and P2Y11 [17, 46]. It is known that β-NAD is released into the extracellular fluids under proinflammatory conditions as an important vascular mediator [15, 18]. Since β-NAD is released from the EC [18], we hypothesized that it may participate in the regulation of the endothelial permeability. Our recently published in vitro studies demonstrate a novelmechanism of β-NAD–mediated rapid and dose-dependent increase in transendothelial electrical resistance (TER) of the human pulmonary artery endothelial cell barrier [19]. In the present study, we studied the effect of β-NAD in lipopolysaccharide (LPS)-induced murine model of ALI.
MATERIALS AND METHODS
Chemicals and Reagents
All the chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise indicated. LPS is Escherichia coli serotype 055:B5. Phosphatebuffered saline (PBS) and the Hanks balanced salt solution (HBSS) were obtained from Gibco (Invitrogen, xx, CA). BCA Protein Assay kit (Pierce Chemical, Rockford, IL) was used to measure the total protein. Myeloperoxidase (MPO) assay kit was purchased from Cayman Chemicals (Ann Arbor, MI).
Animals
All research was conducted in compliance with the Animal Welfare Act and was approved by the Institutional Animal Care and Use Committee. Female C57BL/6J mice (8 to 10 weeks old) weighing 20 to 25 g were purchased from Charles River Laboratory (Wilmington, MA). All animals were housed in plastic cages and had access to food and water throughout the experiment. The animals were kept at room temperature and exposed to continuous cycles of 12-hour light and darkness.
Animal Surgical Procedure
Female C57BL/6J (20 to 25 g) mice were anesthetized with intraperitoneal ketamine (150 mg/kg of body weight) and acetylpromazine (15 mg/kg) before the exposure of the trachea via neck and chest incision. LPS dissolved in sterile PBS was instilled intratracheally via a 20-gauge catheter. Fifteen minutes later the mice received either β-NAD (final calculated plasma concentration 50 μM) or sterile PBS through the internal jugular vein. The animals were allowed to recover for 22 hours. Two hours before the end of the experiment, anesthesia was readministered and Evans blue dye–albumin (EBA; 20 mg/kg Evans blue in 4% bovine serum albumin) was administered via the internal jugular vein. After 120 minutes the mouse was fully exsanguinated and the chest cavity opened and lungs were washed free of blood by injecting saline/EDTA via the right ventricle. Bronchoalveolar lavage fluid (BALF) was obtained by the instillation of 1 mL of 10% HBSS, the lungs rinsed once and the fluid was collected and processed for protein and cell count. The lungs were collected and stored at −80°C. In some experiments the right lung was placed in 4% paraformaldehyde for histology and in another experiment the EBA was injected into the right internal jugular vein as described above at 2 hours prior to termination of the experiment and the left lung gross anatomy view was photographed.
Protein Estimation and Cell Count From the BALF
The BALF was centrifuged (500 × g, 15 minutes, 4°C), supernatant was centrifuged again (16,500 × g, 10 minutes, 4°C), and pure BALF was used to measure total protein. Cell pellets were suspended in Hanks’ solution, and red blood cells were lysed by hypotonic shock (0.2% NaCl) for 5 minutes. Cell suspensions were centrifuged (500 × g, 10 minutes, 4°C). Then formalin (3.7 %) was instilled onto the cell pellet and the cells were then counted on a hemocytometer.
Lung Permeability Measurements Using EBA
Measurement of EBA concentration in the lungs was performed by injection of EBA (20 mg/kg) into the right internal jugular vein 2 hours before the termination of the experiment to assess the vascular leak. Lungs free of blood were weighed and snap frozen in liquid nitrogen. The left lung was weighed and homogenized, then incubated with 2 volumes of formamide (18 hours, 60°C) and centrifuged (5000 × g, 30 minutes, 20°C). The optical density of the supernatant was determined spectrophotometrically at 620 nm. The extravasated EBA concentration (μg/g lung) in the lung homogenate was calculated against a standard curve (micrograms of Evans blue dye per gram lung) as described previously [20]. In a separate experiment, the EBA was injected into the right internal jugular vein as described above at 2 hours prior to termination of the experiment and the left lung gross anatomy view was photographed with a Leica NCL150 Camera.
Lung Histology
Lungs perfused free of blood with EDTA, were immersed in 4% buffered paraformaldehyde for 18 hours at 4°C prior to histological evaluation by hematoxylin and eosin staining (H&E staining) as described previously [20]. The right lung lobes were used for consistency. H&E staining was done by deparafinizing and hydrating the slides to water. The slides were stained in Harris hematoxylin for 15 minutes and eosin for 30 seconds. The slides were dehydrated, cleared, and mounted with cytoseal.
Measurement of Myeloperoxidase (MPO) Activity
MPO is a hemoprotein that is abundantly expressed in polymorphonuclear leukocytes (neutrophils) and secreted during their activation. MPO assay was carried out according to the manufacturer’s assay protocol. Lungs of vehicle-, LPS-, or LPS/β-NAD–treated mice were used for the MPO assay. Lungs (free of blood) were weighed and snap frozen in liquid nitrogen. The right lung was weighed and homogenized in lysis buffer and then centrifuged (12,000 × g, 30 minutes). The protein from the clear supernatant was estimated and normalized from different treatment and analyzed for the MPO levels by enzyme-linked immunosorbent assay (ELISA).
Immunohistochemical Analysis of Myeloperoxidase
Four-micron sections were cut from paraffin blocks andmounted on treated slides (Superfrost plus; VWR Scientific Products, Suwanee, GA). Slides were air dried overnight, then placed in a 60°C oven for 30 minutes. Slides were then deparaffinized in 2 changes of xylene for 7 minutes, then ran through graded alcohols 2 changes of absolute ethanol for 2 minutes each, 2 changes of 95% ethanol for 2 minutes, 80% ethanol for 2 minutes, and 70% ethanol for 2 minutes to distilled water. Slides were pretreated with Target Retrival Solution (Dako, Carpinteria, CA) using a steamer (Black and Decker rice steamer), and slides rinsed in distilled water. Endogenous peroxidase was quenched with 0.3% H2O2 in distilled water for 5 minutes followed by distilled water for 2 minutes, and placed in 1× PBS for 5 minutes. Slides were then incubated with MPO primary antibody (Dako) for 30 minutes at room temperature followed by 2 changes of 1× PBS. Slides were then incubated with secondary antibody peroxidase-conjugated Affinipure F(ab)2 fragment donkey anti-rabbit immunoglobulin G (IgG) (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 hour and rinsed in 2 changes of 1× PBS. Bound antibody was detected with DAB substrate kit (Dako; DAB substrate kit for peroxidase [horseradish peroxidase, HRP]). Slides were then counterstained with hematoxylin (Richard-Allan Scientific, Kalamazoo, MI).
Quantitative Real-Time Polymerase Chain Reaction (qPCR)
Total RNA was prepared from the lungs of mouse tissue using RNeasy mini kit (Qiagen, Valencia, CA). The mRNA was reverse-transcribed into complementary deoxyribonucleic acid (cDNA) using iScript reagents from Bio-Rad on a programmable thermal cycler (PCR-Sprint; Thermo Electron, Milford, MA). Fifty nanograms of cDNA was amplified in each real-time polymerase chain reaction using ABgene reagents (distributed by Fisher Scientific) in Bio-Rad myiQ Cycler. The primers for genes specific to the mice were used from the PrimerBank (http://pga.mgh.harvard.edu/primerbank/index.html) [21, 22] and custom designed primers (Integrated DNA Technologies, Coralville, IA). The forward and reverse primers sequences are shown in Table 1. The reverse transcription reaction was carried out for 25 minutes at 42°C and terminated for 5 minutes at 85°C. qPCR was performed by denaturation for 30 seconds at 94°C and annealing for 30 seconds at 60°C for a total of 40 cycles. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize the expression of the target genes.
TABLE 1.
Sequences of qPCR Primers and Gene Accession Numbers.
| Name of Gene | Primer sequence | Primer Bank ID | Accession Number |
|---|---|---|---|
| GAPDH | F-CATGGCCTCCAAGGAGTAAGA | Custom made | M32599 |
| R-GAGGGAGATGCTCAGTGTTGG | |||
| IL -1α | F-GCACCTTACACCTACCAGAGT | 6754328a1 | NM 010554.4 |
| R-AAACTTCTGCCTGACGAGCTT | |||
| IL -1β | F-GCAACTGTTCCTGAACTCAACT | 6680415a1 | NM 008361.3 |
| R-ATCTTTTGGGGTCCGTCAACT | |||
| IL-4 | F-GGTCTCAACCCCCAGCTAGT | 10946584a1 | NM 021283.2 |
| R-GCCGATGATCTCTCTCAAGTGAT | |||
| IL-6 | F-TAGTCCTTCCTACCCCAATTTCC | 13624311a1 | NM 031168.1 |
| R-TTGGTCCTTAGCCACTCCTTC | |||
| IL-10 | F-GCTCTTACTGACTGGCATGAG | 6754318a1 | NM 010548.1 |
| R-CGCAGCTCTAGGAGCATGTG | |||
| IL-13 | F-CCTGGCTCTTGCTTGCCTT | 6680403a1 | NM 008355.3 |
| R-GGTCTTGTGTGATGTTGCTCA | |||
| IFN-γ | F-ATGAACGCTACACACTGCATC | 33468859a1 | NM 008337 |
| R-CCATCCTTTTGCCAGTTCCTC | |||
| TNF-α | F-CCCTCACACTCAGATCATCTTCT | 7305585a1 | NM 013693 |
| R-GCTACGACGTGGGCTACAG |
Statistics
Values are expressed as mean ± SEM of 3 to 5 independent experiments. For multiple comparisons, analysis of variance (ANOVA) and post hoc multiple comparison tests were applied. Student’s t test was used for comparisons of 2 sample means. A P value of <.05 was considered statistically significant.
RESULTS
β-NAD Attenuates LPS-Induced BALF Protein Accumulation, Inflammatory Infiltration, and EBA Extravasation in Lungs
Measurement of BALF protein demonstrated that mice challenged with LPS for 24 hours significantly increased the pulmonary BALF protein concentration, indicative of increased alveolar epithelial barrier permeability, as compared to mice given saline or β-NAD alone. This increase in LPS-induced BALF protein accumulation was significantly attenuated when mice were treated with LPS/β-NAD, suggesting a possible protective role of β-NAD (Figure 1A).
FIGURE 1.
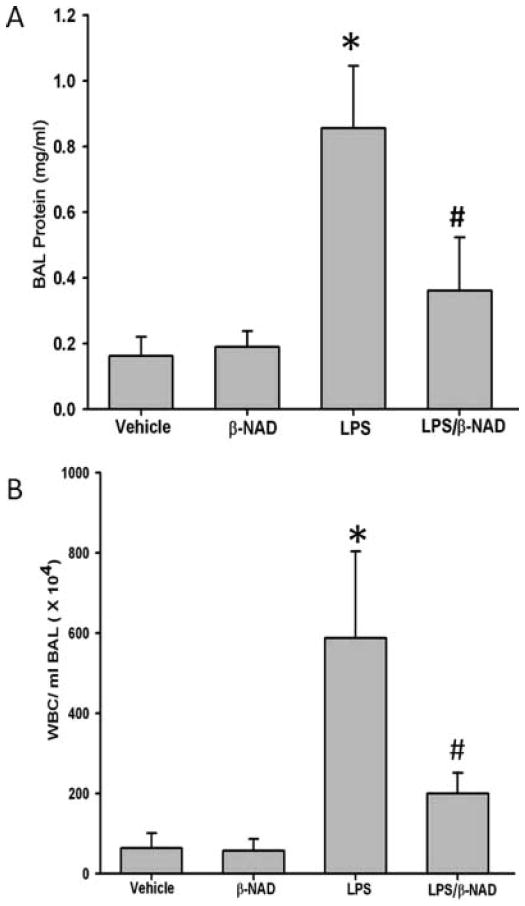
Bronchoalveolar lavage fluid (BALF) protein and cell counts. (A) BALF was collected at 24 hours after treatment, centrifuged, and protein was estimated in the clear supernatant using Bradford protein estimation kit. β-NAD treatment reduced total protein accumulation in the BALF of LPS-induced lung injury. The asterisk (*) indicates that a value significantly (P < .05) differs from the vehicle group and the number sign (#) indicates that a value significantly (P < .05) differs from LPS group (n = 4 for each group). (B) β-NAD reduces WBC accumulation in BALF of LPS-treated mice compared to untreated mice. The BALF was collected at 24 hours after treatment, centrifuged, and the cells were counted in hemocytometer. β-NAD reduced total WBCs in BALF. The asterisk (*) indicates that a value significantly (P < .001) differs from the vehicle group and the number sign (#) indicates that a value significantly (P < .05) differs from LPS group (n = 4 for each group). The error bars represent the standard error of the mean.
The white blood cell (WBC) count was consistent with the protein and EBA results. A quantitative microscopic assessment of the cell count of BALF using a hemocytometer showed that control lungs contained only a few neutrophils. By contrast, LPS treatment led to an increased infiltration of neutrophils, as compared to vehicle-treated animals, which was however significantly reduced in the LPS/β-NAD–treated mice (Figure 1B). Various doses of β-NAD demonstrated similar responses to LPS stimulation (Supplementary Figure 1; available with the online edition of this article).
LPS challenge also induced capillary leak, as evidenced by extravasation of EBA into the lung parenchyma (Figure 2A). The level of EBA was significantly attenuated by β-NAD in the LPS-treated mice, to the level approaching vehicle or β-NAD treatment alone (Figure 2A). A gross view of the LPS challenged left lung photograph indicates the results are consistent with an increase in EBA leakage and a decreased extravasation in the LPS/β-NAD–treated lung (Figure 2B).
FIGURE 2.
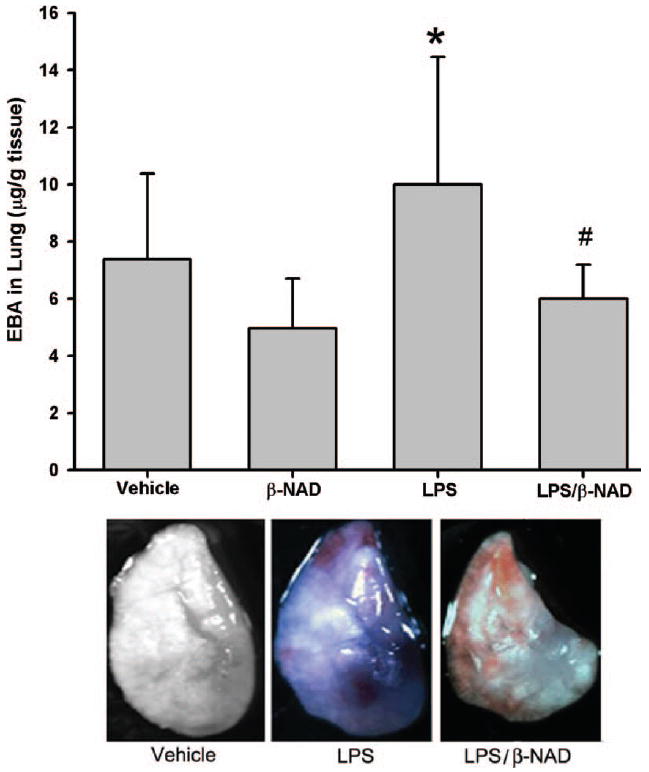
Evans blue dye–albumin (EBA) extravasations in BALF. (A) EBA was injected into the internal jugular vein 2 hours before the termination of the experiment. LPS challenge increased EBA leakage from the vascular space into surrounding lung tissue in the LPS group with notable attenuation in the LPS/β-NAD mice group. Both groups are compared to control and β-NAD-only–treated mice. The asterisk (*) indicates that a value significantly (P < .05) differs from the vehicle group and the number sign (#) indicates that a value significantly (P < .05) differs from LPS group (n = 4 for each group). The error bars represent the standard error of the mean. (B) β-NAD attenuated EBA leakage into the lung parenchyma on gross examination. Mice were grouped and the LPS group received LPS (0.9 mg/kg, i.t.) with PBS (i.v.), LPS/β-NAD group with LPS (0.9 mg/kg, i.t.) and β-NAD (5.46 mg/kg, i.v.), and control group with PBS (12 μL i.t. and 30 μL i.v.). EBA was injected into the right internal jugular vein 2 hours prior to termination of the experiment. The mice were sacrificed at 24 hours and immediately the lungs were flushed with EDTA, harvested, and photographed. Gross observation of the lung at 24 hours showed that the LPS/PBS lung exposure increased penetration of the EBA in the lung parenchyma, with minimal leakage in the LPS/β-NAD–treated specimen and none visible in vehicle.
Histological Assessment of β-NAD Effect on LPS-Induced Lung Inflammation
Mice challenged with LPS for 24 hours demonstrated an inflammatory response typical for ALI/ARDS, as compared to vehicle controls (Figure 3). Histological evaluation of the lung tissue showed an increased interstitial edema and infiltration of neutrophils in the LPS-treated lung, which was much less prominent in the LPS/β-NAD–treated lung. However, the histology was noted to be heterogeneous as typically occurs in ALI. Histological specimens in the control mice displayed normal lung parenchyma (Figure 3) and were indistinguishable from lungs isolated from β-NAD-alone–treated mice (data not shown).
FIGURE 3.
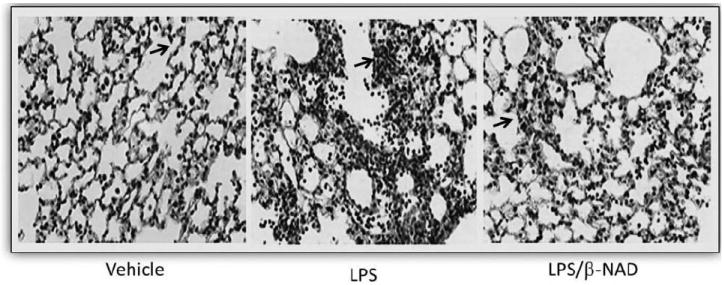
Histopathology. β-NAD inhibits the inflammation in lungs of mice in LPS-induced ALI. Lungs perfused free of blood, were immersed in 4% buffered paraformaldehyde at 4°C for 18 hours prior to histological evaluation by hematoxylin and eosin (H&E) staining. H&E staining was done by deparafinizing and hydrating the slides to water. The slides were stained in Harris hematoxylin for 15 minutes and eosin for 30 seconds. The slides were dehydrated, cleared, and mounted with cytoseal. Histological analysis of the lung tissue obtained from the control mice exposed to PBS showed minimal infiltration of neutrophils. In contrast, mice exposed to LPS for 24 hours produced prominent neutrophil infiltration and that was attenuated in LPS/β-NAD simultaneously.
β-NAD Attenuates LPS-Induced Myeloperoxidase Activity in Lungs
Meloperoxidase (MPO) activity, an index of neutrophil sequestrationin the lungs was measured in snap-frozen right lungs, as described previously [20]. As demonstrated in Figure 4A, MPO activity was significantly increased in LPS-challenged mice, as compared to control animals. However, in LPS/β-NAD–treated mice MPO activity was attenuated (Figure 4A), suggesting a β-NAD–activated signaling–mediated protection. In addition, immunohistochemistry data show a significant amount of neutrophil sequestration that was attenuated in LPS/β-NAD–treated mice (Figure 4B), in accordance with the MPO data in Figure 4A.
FIGURE 4.
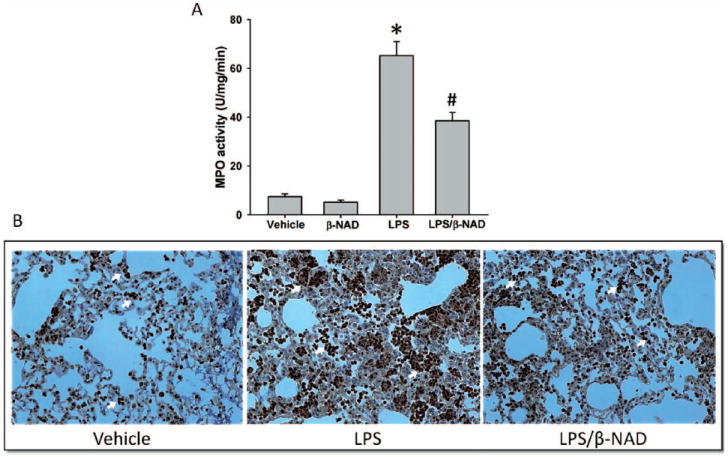
Myeloperoxidase (MPO) activity and staining. (A) Myeloperoxidaselevels in lung tissues of mice treated with or without β-NAD in LPS challenge. Neutrophil infiltration was analyzed by quantifying MPO levels in lungs tissues. β-NAD treatment significantly attenuates the MPO activity in lungs. (B) Myeloperoxidase staining was performed in lung tissues and the markedly increased infiltration of neutrophils (arrows) was observed in the lungs of mice from the LPS group, which was significantly attenuated by β-NAD treatment. The asterisk (*) indicates that a value significantly (P < .001) differs from the vehicle group and the number sign (#) indicates that a value significantly (P < .05) differs from LPS group (n = 4 for each group). The error bars represent the standard error of the mean.
β-NAD Inhibits Gene Expression of Proinflammatory, and Up-regulates Expression of Anti-inflammatory, Cytokines in LPS-Treated Lungs
It is well known that proinflammatory gene expression is up-regulated in response to endotoxin leading to sequestration and degranulation of neutrophils in the lungs. To identify if β-NAD affects LPS-induced expression of pro- or anti-inflammatory genes, lung tissues were harvested from the mice treated with or without β-NAD immediately after the LPS insult. The mRNA was prepared and the qPCRwas performed with specific primers for proinflammatory (interleukin [IL]-1α, IL-1β, interferon [IFN]-γ, and tumor necrosis factor [TNF]-α) and anti-inflammatory (IL-4, IL-10, and IL-13) cytokines to quantify the expression levels. GAPDH was used as internal control. As shown in Figure 5 (upper panel), gene expression of all 3 tested proinflammatory cytokines was up-regulated in LPS-stimulated mice groups, but their levels were significantly attenuated in LPS/β-NAD–treated mice. This suggests the involvement of β-NAD in the attenuation of proinflammatory cytokine gene expression levels. In addition, gene expression levels of anti-inflammatory cytokines (IL-4, IL-10, and IL-13) were low in the LPS-treated groups, but, were significantly up-regulated in the LPS/β-NAD–treated mice group, implying initiation of the resolution of LPS-induced inflammation in the presence of β-NAD treatment.
FIGURE 5.
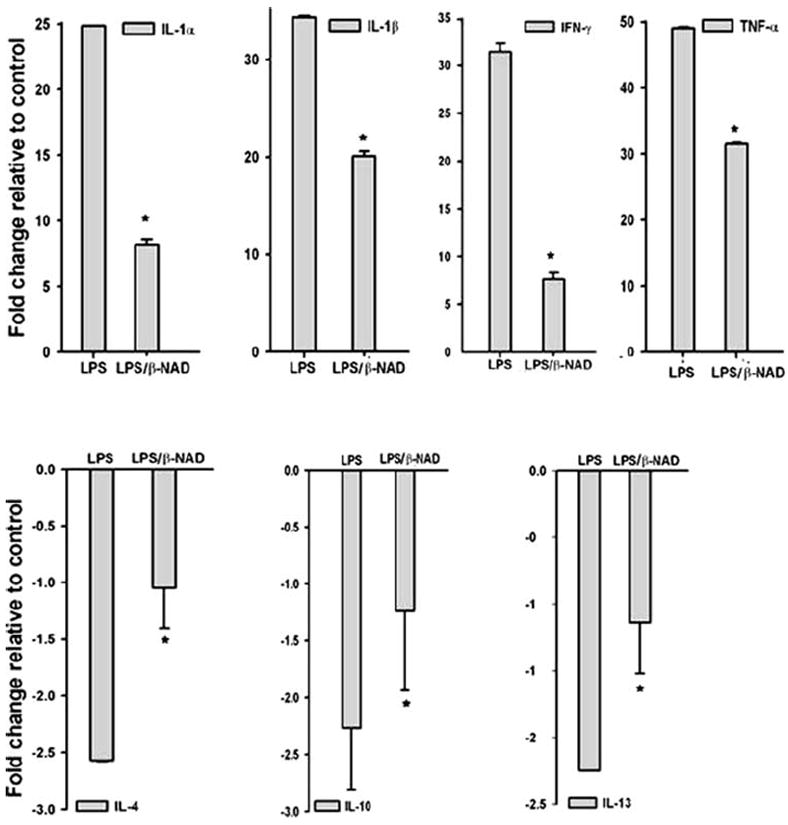
Quantitative real-time PCR (qPCR). qPCR analysis of proinflammatory (upper panel) and anti-inflammatory (lower panel) cytokine gene expression from lungs of mice challenged with vehicle, LPS, and LPS/β-NAD. The bar represents the average fold change compared with vehicle and the expression levels were normalized to the value of housekeeping gene GAPDH mRNA. Data are shown as the mean ± SEM (n = 4 for each group). *P < .05 versus LPS group.
DISCUSSION
LPS, a major component of the outer membrane of gram-negative bacteria, is an endotoxin that induces a strong immune response in mammals. As such, it promotes the secretion of proinflammatory cytokines in many cell types [23, 24]. In the present study, we have evaluated the effect of β-NAD– on LPS-induced lung inflammation—and vascular leak. It is well known that endothelial permeability leads to a noncardiogenic pulmonary edema in ALI/ARDS [25-28]. ALI is characterized by pulmonary microvascular endothelial gaps and disarray with cellular breakdown and subsequent endothelial permeability and interstitial edema [29-31]. Recent studies suggest that in proinflammatory conditions, β-NAD released from cellular sources, including the endothelium into the extracellular fluid, is an important vascular mediator [18, 32]. Our experimental data demonstrate that β-NAD administration significantly attenuates the accumulation of protein in LPS-induced murine models of ALI (Figure 1), suggesting an improvement of barrier function via β-NAD–induced signaling. In addition, measurement of EBA extravasation into the lung parenchyma showed that the LPS-induced increase in albumin was also attenuated in the β-NAD–treated mice (Figure 2), implying that there is attenuation of capillary vascular leak in the β-NAD–treated mice, as compared to the LPS-only–treated mice. Our recently published in vitro studies [19] demonstrated that in transendothelial resistance (TER) measurement assays, LPS caused significant endothelial barrier disruption and the addition of β-NAD to the cells attenuated the barrier disruption. Our current in vivo results are thus consistent with the in vitro findings [19].
LPS-induced murine lung injury is a model that has been shown to be consistent with sepsis-induced ALI [33-36]. The injury is characterized by neutrophil infiltration into the lungs within 24 hours, with an associated increase in inflammatory mediators, interstitial edema and early mortality. These factors contribute to the oxidative stress and inflammatory response of the host. Histological evaluation of the lung tissue isolated from LPS-treated animals demonstrated extensive interstitial edema and increased neutrophil contents. This illustrates the EC barrier disruption, characteristic of ALI/ARDS, as well as the histologic heterogeneity known to occur during complicating ventilator strategies in animal and human models of ALI/ARDS [37].
The histological examination of LPS-challenged lung tissue showed morphological changes that were attenuated in the lungs from the LPS/β-NAD–treated groups. In addition, LPS/β-NAD–treated specimens showed significant reduction in edema formation, as compared to the LPS-only specimens.
The pathogenesis of ALI is thought to be mediated by proinflammatory cytokines, such as tumor necrosis factor (TNF) and interleukins (IL-1 and IL-6), which are released from macrophages and neutrophils [38]. On the cellular level, β-NAD has been demonstrated to represent a proinflammatory cytokine mediator in human granulocytes [16]. In this study, we analyzed the effect of β-NAD treatment on the gene expression of proinflammatory and anti-inflammatory cytokines. Our data indicate that LPS/β-NAD–treated mice had lower expression levels of the proinflammatory cytokines, but an increased gene expression of anti-inflammatory cytokines in their lungs, as compared to the LPS-only–treated mice. Cytokines are synthesized as a host response to infection or disease [39]. On a cellular level, they activate mitogen-activated protein kinases (MAPKs), which phosphroylate transcription factors for gene expression [39]. It is known that an important transcription factor regulating the expression of IL-1, IL-6, IL-8, and TNF-α genes is nuclear factor kappa B (NF-κB). It is likely that the regulation of NF-κB in alveolar macrophages after exposure to LPS is critical to the inflammatory response that occurs in ARDS [39, 40]. Endothelial damage is associated with numerous inflammatory events. During sepsis, proinflammatory cytokines are first necessary for initiating an effective inflammatory process against infection, but anti-inflammatory cytokines can lead to a down-regulation of the inflammatory response, leading to a depression of the immune system [41, 42]. Within the alveolar space, the balance between proinflammatory and anti-inflammatory mediators favors ongoing inflammation [43]. IL-1 and TNF-α induce endothelial adhesion molecules that are essential precursors to chemotaxis of neutrophils, which ultimately results in the generation of toxic factor. Further analysis of gene expression profiling in the future would help us to better understand the role of β-NAD in anti-inflammatory mechanisms.
Extracellular β-NAD is an important vascular mediator that elicits cellular effects on endothelial cells [44, 45] and interacts with 2 purinergic receptors, namely, P2Y1 and P2Y11 [17, 46]. Our published in vitro studies have demonstrated that extracellular β-NAD mediates protection against LPS-induced lung endothelial cell barrier dysfunction via both P2Y1 and P2Y11 receptors [19]. In this study, we demonstrated that extracellular β-NAD protects LPS-induced lung injury. However, the mechanisms underlying this protective effect remain unknown. Their elucidation may not only unravel novel mechanisms of lung endothelial barrier protection, but also establish a basis for potential applications of β-NAD for treating acute lung injury. Previous studies have indicated that β-NAD plays significant roles in multiple biological functions, including energy metabolism, mitochondrial functions, aging, gene expression, calcium homeostasis, and immunological functions [47, 48]. Another study also indicated that administration of β-NAD decreases ischemic brain damage partially by blocking autophagy in a mouse model of brain ischemia [47].
It is known that in various pathophysiological conditions, reactive oxidants cause DNA strand breakage and subsequent activation of the nuclear enzyme poly(ADP ribose) polymerase (PARP). Activation of PARP results in cellular dysfunction. Activated PARP-1 cleaves off nicotinamide from NAD+ (β-NAD) and polymerizes the remaining ADP-ribose units into long, branching PAR polymer covalently attached to glutamate or aspartate residues of suitable acceptor proteins [49]. This causes depletion of the cellular stores of its substrate β-NAD. Resynthesis of β-NAD consumes adenosine triphosphate (ATP), causing cell death by energy depletion. Previous studies have shown that extracellular β-NAD treatment decreases PARP-mediated cell death of primary neurons and astrocytes [50, 51]. Therefore, extracellular β-NAD therapy might be an effective way to treat acute lung injury and possibly ARDS.
In summary, our studies have shown that β-NAD attenuates the LPS-induced lung inflammation and permeability increase in vivo. Gross observation of the lung and histological evaluation is consistent with these results. Whether or not the β-NAD therapy will become a viable strategy for the treatment of acute lung injury will largely depend on safety issues, although β-NAD is a natural signaling molecule.
Supplementary Material
Acknowledgments
This work was supported, in part, by grants, Biomedical Research Grant from the American Lung Association (Southeast) to N.S.U. and the National Institute of Health (HL094609 to R.L. and HL083327 and HL101902 to A.D.V. and N.S.U.).
Footnotes
Supplementary Material
Supplementary Figure 1 is available with the online edition of this article.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31(4 Suppl):S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 2.Razavi HM, Wang le F, Weicker S, Rohan M, Law C, McCormack DG, et al. Pulmonary neutrophil infiltration in murine sepsis: role of inducible nitric oxide synthase. Am J Respir Crit Care Med. 2004;170:227–233. doi: 10.1164/rccm.200306-846OC. [DOI] [PubMed] [Google Scholar]
- 3.Wang le F, Patel M, Razavi HM, Weicker S, Joseph MG, McCormack DG, et al. Role of inducible nitric oxide synthase in pulmonary microvascular protein leak in murine sepsis. Am J Respir Crit Care Med. 2002;165:1634–1639. doi: 10.1164/rccm.2110017. [DOI] [PubMed] [Google Scholar]
- 4.Suda K, Tsuruta M, Eom J, Or C, Mui T, Jaw JE, et al. Acute lung injury induced cardiovascular dysfunction: effects of IL-6 and budesonide/formoterol. Am J Respir Cell Mol Biol. 2011 Jan 21;45:510–516. doi: 10.1165/rcmb.2010-0169OC. [DOI] [PubMed] [Google Scholar]
- 5.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 6.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 7.Seitz DH, Froba JS, Niesler U, Palmer A, Veltkamp HA, Braumuller ST, et al. Inhaled hydrogen sulfide induces suspended animation, but does not alter the inflammatory response after blunt chest trauma. Shock. 2011 Nov 15; doi: 10.1097/SHK.0b013e31823f19a0. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, et al. Protective effect of suberoylanilide hydroxamic acid against LPS-induced septic shock in rodents. Shock. 2009;32:517–523. doi: 10.1097/SHK.0b013e3181a44c79. [DOI] [PubMed] [Google Scholar]
- 9.Liu JC, Wang HT, Wang W. Protective effects of alanylglutamine on acute lung injury induced by lipopolysaccharide in rats. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2008;33:1095–1100. [PubMed] [Google Scholar]
- 10.Boe DM, Richens TR, Horstmann SA, Burnham EL, Janssen WJ, Henson PM, et al. Acute and chronic alcohol exposure impair the phagocytosis of apoptotic cells and enhance the pulmonary inflammatory response. Alcohol Clin Exp Res. 2010;34:1723–1732. doi: 10.1111/j.1530-0277.2010.01259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang SM, Gabelaia L, Gauthier TW, Brown LA. N-acetylcysteine improves group B streptococcus clearance in a rat model of chronic ethanol ingestion. Alcohol Clin Exp Res. 2009;33:1197–1201. doi: 10.1111/j.1530-0277.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmickl CN, Shahjehan K, Li G, Dhokarh R, Kashyap R, Janish C, et al. Decision support tool for early differential diagnosis of acute lung injury and cardiogenic pulmonary edema in medical critically-ill patients. Chest. 2011 Oct 26; doi: 10.1378/chest.11-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMPK pathway. J Biol Chem. 2009 Nov 24; doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imai SI. “Clocks” in the NAD World: NAD as a metabolic oscillator for the regulation of metabolism and aging. Biochim Biophys Acta. 2009 Nov 6; doi: 10.1016/j.bbapap.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Bruzzone S, Moreschi I, Guida L, Usai C, Zocchi E, De Flora A. Extracellular NAD+ regulates intracellular calcium levels and induces activation of human granulocytes. Biochem J. 2006;393(Pt 3):697–704. doi: 10.1042/BJ20051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mutafova-Yambolieva VN, Hwang SJ, Hao X, Chen H, Zhu MX, Wood JD, et al. Beta-nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc Natl Acad Sci U S A. 2007;104:16359–16364. doi: 10.1073/pnas.0705510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smyth LM, Bobalova J, Mendoza MG, Lew C, Mutafova-Yambolieva VN. Release of beta-nicotinamide adenine dinucleotide upon stimulation of postganglionic nerve terminals in blood vessels and urinary bladder. J Biol Chem. 2004;279:48893–48903. doi: 10.1074/jbc.M407266200. [DOI] [PubMed] [Google Scholar]
- 19.Umapathy NS, Zemskov EA, Gonzales J, Gorshkov BA, Sridhar S, Chakraborty T, et al. Extracellular beta-nicotinamide adenine dinucleotide (beta-NAD) promotes the endothelial cell barrier integrity via PKA- and EPAC1/Rac1-dependent actin cytoskeleton rearrangement. J Cell Physiol. 2010;223:215–223. doi: 10.1002/jcp.22029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolosova IA, Mirzapoiazova T, Moreno-Vinasco L, Sammani S, Garcia JG, Verin AD. Protective effect of purinergic agonist ATPgammaS against acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;294:L319–L324. doi: 10.1152/ajplung.00283.2007. [DOI] [PubMed] [Google Scholar]
- 21.Spandidos A, Wang X, Wang H, Dragnev S, Thurber T, Seed B. A comprehensive collection of experimentally validated primers for polymerase chain reaction quantitation of murine transcript abundance. BMC Genomics. 2008;9:633. doi: 10.1186/1471-2164-9-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003;31:154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Li HY, Li H, Zhao J, Su L, Zhang Y, et al. Lipopolysaccharide activated phosphatidylcholine-specific phospholipase C and induced IL-8 and MCP-1 production in vascular endothelial cells. J Cell Physiol. 2011;226:1694–1701. doi: 10.1002/jcp.22500. [DOI] [PubMed] [Google Scholar]
- 24.Mako V, Czucz J, Weiszhar Z, Herczenik E, Matko J, Prohaszka Z, et al. Proinflammatory activation pattern of human umbilical vein endothelial cells induced by IL-1beta, TNF-alpha, and LPS. Cytometry A. 2010;77:962–970. doi: 10.1002/cyto.a.20952. [DOI] [PubMed] [Google Scholar]
- 25.Bersten A, Sibbald WJ. Acute lung injury in septic shock. Crit Care Clin. 1989;5:49–79. [PubMed] [Google Scholar]
- 26.Luce JM. Acute lung injury and the acute respiratory distress syndrome. Crit Care Med. 1998;26:369–376. doi: 10.1097/00003246-199802000-00043. [DOI] [PubMed] [Google Scholar]
- 27.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006;27:337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 28.Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol. 2009;77:1763–1772. doi: 10.1016/j.bcp.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta D, Bhattacharya J, Matthay MA, Malik AB. Integrated control of lung fluid balance. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1081–L1090. doi: 10.1152/ajplung.00268.2004. [DOI] [PubMed] [Google Scholar]
- 30.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- 31.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 32.Han X, Uchiyama T, Sappington PL, Yaguchi A, Yang R, Fink MP, et al. NAD+ ameliorates inflammation-induced epithelial barrier dysfunction in cultured enterocytes and mouse ileal mucosa. J Pharmacol Exp Ther. 2003;307:443–449. doi: 10.1124/jpet.103.056556. [DOI] [PubMed] [Google Scholar]
- 33.Rojas M, Woods CR, Mora AL, Xu J, Brigham KL. Endotoxin-induced lung injury in mice: structural, functional, and biochemical responses. Am J Physiol Lung Cell Mol Physiol. 2005;288:L333–L341. doi: 10.1152/ajplung.00334.2004. [DOI] [PubMed] [Google Scholar]
- 34.Rudkowski JC, Barreiro E, Harfouche R, Goldberg P, Kishta O, D’Orleans-Juste P, et al. Roles of iNOS and nNOS in sepsis-induced pulmonary apoptosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L793–L800. doi: 10.1152/ajplung.00266.2003. [DOI] [PubMed] [Google Scholar]
- 35.Mirzapoiazova T, Kolosova IA, Moreno L, Sammani S, Garcia JG, Verin AD. Suppression of endotoxin-induced inflammation by taxol. Eur Respir J. 2007;30:429–435. doi: 10.1183/09031936.00154206. [DOI] [PubMed] [Google Scholar]
- 36.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L379–L399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bastarache JA, Blackwell TS. Development of animal models for the acute respiratory distress syndrome. Dis Model Mech. 2009;2:218–223. doi: 10.1242/dmm.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JY, Park JS, Strassheim D, Douglas I, Diaz del Valle F, Asehnoune K, et al. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol. 2005;288:L958–L965. doi: 10.1152/ajplung.00359.2004. [DOI] [PubMed] [Google Scholar]
- 39.Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–508. doi: 10.1378/chest.118.2.503. [DOI] [PubMed] [Google Scholar]
- 40.Carter AB, Monick MM, Hunninghake GW. Lipopolysaccharide-induced NF-kappaB activation and cytokine release in human alveolar macrophages is PKC-independent and TK- and PC-PLC-dependent. Am J Respir Cell Mol Biol. 1998;18:384–391. doi: 10.1165/ajrcmb.18.3.2972. [DOI] [PubMed] [Google Scholar]
- 41.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 42.Bone RC, Grodzin CJ, Balk RA. Sepsis: a new hypothesis for pathogenesis of the disease process [review] Chest. 1997;112:235–243. doi: 10.1378/chest.112.1.235. [DOI] [PubMed] [Google Scholar]
- 43.Adhikari N, Burns KE, Meade MO. Pharmacologic therapies for adults with acute lung injury and acute respiratory distress syndrome. Cochrane Database Syst Rev. 2004;(4) doi: 10.1002/14651858.CD004477.pub2. CD004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, et al. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. 2010;285:3133–3144. doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imai S. “Clocks” in the NAD World: NAD as a metabolic oscillator for the regulation of metabolism and aging. Biochim Biophys Acta. 2010;1804:1584–1590. doi: 10.1016/j.bbapap.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreschi I, Bruzzone S, Nicholas RA, Fruscione F, Sturla L, Benvenuto F, et al. Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J Biol Chem. 2006;281:31419–31429. doi: 10.1074/jbc.M606625200. [DOI] [PubMed] [Google Scholar]
- 47.Zheng C, Han J, Xia W, Shi S, Liu J, Ying W. NAD(+) administration decreases ischemic brain damage partially by blocking autophagy in a mouse model of brain ischemia. Neurosci Lett. 2012 Jan 12; doi: 10.1016/j.neulet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 48.Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- 49.Virag L. Poly(ADP-ribosyl)ation in asthma and other lung diseases. Pharmacol Res. 2005;52:83–92. doi: 10.1016/j.phrs.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 50.Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–18902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.