

Abstract
The pathogenesis of multiple myeloma (MM) is thought to involve at least two pathways, which generate hyperdiploid (HRD) or nonhyperdiploid (NHRD) tumors, respectively. Apart from chromosome content, the two pathways are distinguished by five primary immunoglobulin heavy chain (IGH) rearrangements (4p16, FGFR3, and MMSET; 6p21, CCND3; 11q13, CCND1; 16q23, MAF; 20q12, MAFB) that are present mainly in NHRD tumors. To determine the prevalence and structures of IGH, immunoglobulin (IG) light chain, and MYC genomic rearrangements in MM, we have done comprehensive metaphase fluorescent in situ hybridization analyses on 48 advanced MM tumors and 47 MM cell lines. As expected, the prevalence of the five primary IGH rearrangements was nearly 70% in NHRD tumors, but only 12% in HRD tumors. However, IGH rearrangements not involving one of the five primary partners, and IG light chain rearrangements, have a similar prevalence in HRD and NHRD tumors. In addition, MYC rearrangements, which are thought to be late progression events that sometimes do not involve an IG heavy or light chain locus, also have a similar prevalence in HRD and NHRD tumors. In contrast to the primary IGH rearrangements, which usually are simple balanced translocations, these other IG rearrangements usually have complex structures, as previously described for MYC rearrangements in MM. We conclude that IG light chain and MYC rearrangements, as well as secondary IGH rearrangements, make similar contributions to the progression of both HRD and NHRD MM tumors.†
INTRODUCTION
It has been hypothesized that the pathogenesis of non-IgM/plasma cell monoclonal gammopathy of undetermined significance (MGUS) and multiple myeloma (MM) involve two distinct pathways that result in a similar prevalence of hyperdiploid (HRD) and nonhyperdiploid (NHRD) tumors (Smadja et al., 1998, 2003; Fonseca et al., 2003b, 2004). HRD tumors have 48–75 chromosomes, typically with extra copies of four to eight odd-numbered chromosomes (3, 5, 7, 9, 11, 15, 19, and 21), or derivatives of these chromosomes. NHRD tumors have <48 and/or >75 chromosomes, but only infrequently have extra copies of these same chromosomes. The two pathways are also distinguished by five recurrent immunoglobulin heavy chain (IGH) rearrangements (4p16, FGFR3, and MMSET; 6p21, CCND3; 11q13, CCND1; 16q23, MAF; 20q12, MAFB) that are present in nearly 70% of NHRD tumors but only in about 10% of HRD tumors (Bergsagel and Kuehl, 2005). The five recurrent IGH rearrangements are thought to be mediated by errors in switch recombination or somatic hypermutation in germinal center B cells (Bergsagel and Kuehl, 2001; Gabrea et al., 2006). These primary translocations mostly appear to be one of the earliest events involved in the pathogenesis of MM.
Dysregulation of a MYC gene, involving complex genomic rearrangements that sometimes but not always include immunoglobulin (IG) sequences, is a very late progression event in MM (Shou et al., 2000; Avet-Loiseau et al., 2001b; Gabrea et al., 2006; Dib et al., in press). Not surprisingly, these rearrangements rarely involve IGH switch or JH regions, since IGH switch recombination and somatic hypermutation are thought to be inactive in normal plasma cells and plasma cell tumors. MYC rearrangements have been proposed to provide a paradigm for secondary genomic rearrangements in MM. Genomic rearrangements involving the IGH locus and other chromosomal partners, or an IG light chain locus, also have been identified in plasma cell MGUS and MM (Fonseca et al., 2002; Kuehl and Bergsagel, 2002). The goal of this study was to do comprehensive metaphase fluorescent in situ hybridization (FISH) analyses to determine the prevalence and characteristics of secondary genomic rearrangements, involving IG or MYC (with or without juxtaposition of IG and MYC sequences), that occur in advanced HRD and NHRD intramedullary MM tumors, and in human myeloma cell lines (HMCL) that reflect an even later stage of tumor progression.
MATERIALS AND METHODS
Tumors and Cell Lines
Bone marrow aspirates were processed for routine chromosome studies as previously described (Shaughnessy et al., 2001). Metaphase spreads were generated from 48 independent MM tumor samples that were obtained from patients at the University of Arkansas for Medical Sciences. All the samples had abundant chromosomal material and an abnormal karyotype as determined by conventional cytogenetic analyses. Most samples that had a t(11;14)(q13;q32) by conventional cytogenetics were not available for this study. Similar to advanced relapsed tumors, 70% of tumors (14 of 20 with gene expression profiling data) in this study had an expression proliferation index >2.0 (Bergsagel et al., 2005; Kuehl and Shaughnessy, unpublished). Metaphase chromosomes from all 48 tumors (Table 1) and 47 HMCL (Table 2) were studied for IG and MYC rearrangements using a variety of FISH probes (later).
TABLE I.
Numeric Chromosomal Abnormalities and Structural Abnormalities of IG or MYC Loci in 48 MM Tumors
| Trisomiesa | IG and MYC Rearrangements | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| Sort# | Chr# | 3 | 5 | 7 | 9 | 11 | 15 | 19 | 21 | Locusb | Target genec | Breakpoints (BP)d | Derivativese |
|
| |||||||||||||
| 1 | 32 | D | d | d | D | D | D | D | D | ||||
|
| |||||||||||||
| 2 | 36 | t | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) | |||||||
|
| |||||||||||||
| 3 | 39 | t | |||||||||||
|
| |||||||||||||
| 4 | 41 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) | ||||||||
| ? | MYC | 1p11~13 ∷ 8q24 | U: der(1) | ||||||||||
|
| |||||||||||||
| 5 | 42 | t | T | T | IGH | MMSET | 4p16 ∷ 14q32 | U*: der(4), mod der(14) see Fig.2C | |||||
| ? | MYC | 1? ∷ 8q24 | U*: mod der(1) see Fig.2E | ||||||||||
|
| |||||||||||||
| 6 | 42 | IGH | CCND1 | 11q13 ∷ 14q32 | B: der(11), der(14) | ||||||||
|
| |||||||||||||
| 7 | 43 | T | IGH | MAF | 14q32 ∷ 16q23 | B: der(14), der(16) | |||||||
| IGL | ? | Xp? ∷ 22q11 | U: der(X) | ||||||||||
| IGK | MYC | 2p11 ∷ 8q24 | U: der(8) | ||||||||||
|
| |||||||||||||
| 8 | 43 | IGH1 | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) | ||||||||
| IGH2 | ? | ins CH @ 17q?21 on 17 | I: 17 (wcp17+, wcp14 -, CH+) | ||||||||||
|
| |||||||||||||
| 9 | 43 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) | ||||||||
|
| |||||||||||||
| 10 | 44 | IGH1 | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) | ||||||||
| IGH2 | NMYC | 2p23 ∷ 14q32 | B: der(2), der(14) | ||||||||||
|
| |||||||||||||
| 11 | 45 | T | IGH1 | ? | 14q32 ∷ 22q13 | B: der(14), der(22) | |||||||
| IGH2 | MYC | ins CH @ 8q24 on 8 | I: 8 (wcp8+,MYC+, wcp14 -, CH+) | ||||||||||
|
| |||||||||||||
| 12 | 45 | IGH1 | CCND1 | 11q13 ∷ 14q32 | B*: der(11), der(14), der(?) see Fig.2C | ||||||||
| IGH2 | ? | ins CH @ ? | I: ? (wcp14 -, CH+) | ||||||||||
|
| |||||||||||||
| 13 | 45 | T | T | t | T | IGL | MYC | 8q24 ∷ 22q11 | B: der(8), der(22) | ||||
|
| |||||||||||||
| 14 | 45 | T | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) | |||||||
| IGL | MYC | ins Cλ @ 8q24 on der(8)t(8;?11)(q24;?p13) | I*: der(8) (wcp8+,MYC+,wcp22-,Cλ+) ×2 | ||||||||||
|
| |||||||||||||
| 15 | 45 | IGH | MAF | 14q32 ∷ 16q23 | B: der(14), der(16) | ||||||||
|
| |||||||||||||
| 16 | 45 | ||||||||||||
|
| |||||||||||||
| 17 | 46 | t | T | T | IGH1 | ? | 7q32 ∷ 14q32 | B: der(7), der(14) | |||||
| IGH2 | MYC | 8q24 ∷ 14q32 | V (U*): mod der(8) (wcp8+, MYC+, wcp14 -, CH+,VH+) | ||||||||||
| IGH3 | ? | ? ∷ 14q32 | U: der(14) | ||||||||||
|
| |||||||||||||
| 18 | 47 | T | T | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) | ||||||
| IGL | MYC | ins Cλ @ 8q24 on 8 | I: 8 (wcp8+,MYC+, wcp22-, Cλ+) | ||||||||||
|
| |||||||||||||
| 19 | 76 | t | |||||||||||
|
| |||||||||||||
| 20 | 76 | t | tt | t | IGH | MAFB | 14q32 ∷ 20q11 | B*: der(14), der(20) see Fig.2C | |||||
|
| |||||||||||||
| 21 | 77 | t | TT | T | IGH1 | ? | ? ∷ 14q32 | V (U): der(?) (CH+,VH+) | |||||
| IGH2 | ? | ins CH @ ? | I: ? (wcp14 -, CH+) | ||||||||||
|
| |||||||||||||
| 22 | 78 | IGH | MYC | 8q24 ∷ 14q32 | B: der(8), der(14) | ||||||||
|
| |||||||||||||
| 23 | 70 | T | T | t | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) | |||||
|
| |||||||||||||
| 24 | 70 | T | T | T | Tt | T | |||||||
|
| |||||||||||||
| 25 | 60 | T | T | T | T | TTt | TTT | T | TT | ||||
|
| |||||||||||||
| 26 | 56 | T | t | T | t | tt | T | ? | MYC | 8q10 ∷ 8q10 | Amplification MYC: i(8)(q10) ×3 | ||
|
| |||||||||||||
| 27 | 55 | t | t | T | T | TT | T | ||||||
|
| |||||||||||||
| 28 | 55 | t | T | t | t | TT | T | T | |||||
|
| |||||||||||||
| 29 | 55 | T | T | T | TT | T | TT | T | TT | ? | MYC | ins MYC @ ? | I: ? (wcp8-,MYC+) |
|
| |||||||||||||
| 30 | 54 | T | t | T | Tt | t | T | t | |||||
|
| |||||||||||||
| 31 | 54 | T | t | T | t | T | t | T | IGH | ? | 1q11~12 ∷ 14q32 | U: der(14) | |
|
| |||||||||||||
| 32 | 54 | T | T | T | T | t | T | T | T | IGL | CCND3 | 6p21 ∷ 22q11 | U: der(6) |
|
| |||||||||||||
| 33 | 53 | T | t | T | T | T | T | T | T | ||||
|
| |||||||||||||
| 34 | 53 | t | Tt | T | T | T | t | IGH1 | ? | 3q?27 ∷ 14q32 | V (B): der(14) (wcp14+,CH-), der(3) (wcp3+,CH+,VH+,wcp14-) | ||
| IGH2 | MYC | ins CH @ 8q24 on 8 | I: 8 (wcp8+,MYC+, wcp14 -, CH+) | ||||||||||
|
| |||||||||||||
| 35 | 52 | T | T | T | T | T | T | T | |||||
|
| |||||||||||||
| 36 | 52 | T | T | T | T | T | T | IGL | MYC | ins Cλ @ 8q24 on 8 | I: 8 (wcp8+,MYC+,wcp22-, Cλ+) | ||
|
| |||||||||||||
| 37 | 51 | t | T | TT | TT | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) | ||||
| ? | MYC | 6? ∷ 8q24 | U*: mod der(6) | ||||||||||
| ? | MYC | 8q24 ∷ 15? | U: der(15) (×2) | ||||||||||
| ? | MYC | 8q24 ∷ 16? | U: der(16) | ||||||||||
|
| |||||||||||||
| 38 | 51 | t | t | t | T | T | t | T | ? | MYC | ins MYC @ ? | I: ? (wcp8-,MYC+) | |
|
| |||||||||||||
| 39 | 51 | T | T | T | T | T | |||||||
|
| |||||||||||||
| 40 | 51 | T | T | t | T | T | t | IGL1 | ? | ? ∷ 22q11 | U*: der(?) see Fig.2D | ||
| IGL2 | MYC | 8q24 ∷ 22q11 | V (U*): der(22) (Vλ+,Cλ+,wcp22+,MYC+,wcp8-) | ||||||||||
|
| |||||||||||||
| 41 | 51 | T | T | T | T | T | t | T | |||||
|
| |||||||||||||
| 42 | 51 | T | T | t | T | T | T | IGH | MYC | ins CH @ 8q24 on 8 | I: 8 (wcp8+,MYC+, wcp14 -, CH+) | ||
|
| |||||||||||||
| 43 | 51 | T | T | T | T | T | T | ? | MYC | 1p13 ∷ 8q24 | B: der(1) ×2, der(8) ×2 (MYC+) | ||
|
| |||||||||||||
| 44 | 51 | T | T | T | TT | T | IGH | ? | ? ∷ 14q32 | U: der(14) | |||
| IGL | MYC | ins Cλ @ 8q24 on der(8)t(8;11)(q24;q23) | I*: der(8) (wcp8+,MYC+,wcp22-,Cλ+,wcp11+) see Fig.2D | ||||||||||
|
| |||||||||||||
| 45 | 50 | Tt | T | T | TT | T | |||||||
|
| |||||||||||||
| 46 | 50 | T | T | T | Tt | T | T | IGH1 | ? | I?q42 ∷ 14q32 | B: der(1), der(14) | ||
| IGH2 | MYC | 8q24 ∷ 14q32 & ins CH @ 8q24 on mod der(8) | V (U*), I: mod der(8) (wep8+,MYC+,wcp14 -,CH+) & (wcp14 -,CH+,VH+,wcp8-,MYC+) see Fig.2C | ||||||||||
|
| |||||||||||||
| 47 | 49 | T | T | T | T | IGH1 | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) | ||||
| IGH2 | CCND1 | ins CH @ 11q13 on 11 | I: 11 (wcp11+,CCND1+,wcp14-,CH+) | ||||||||||
| ? | MYC | 6q21 ∷ 8q24 | B: der(6) (MYC+), der(8) (MYC+) | ||||||||||
|
| |||||||||||||
| 48 | 48 | T | Tt | IGH | ? | 3q23 ∷ 14q32 | B: der(3), der(14) | ||||||
Extra copies of the odd number chromosomes are shown, where T or D represents a full copy and t or d represents a rearranged copy of that chromosome.
Loci containing IG or unknown (?) regulatory elements that dysregulate the target genes.
Target genes found by FISH to colocalize with IG or unknown (?) loci sequences; for t(4;14), MMSET is presumed on der(4) for all tumors and HMCL with the exception of tumor #5 and XG-7 where MMSET was confirmed by FISH. In contrast to cell lines (Table 2), IRF4 was not a target in any of the 48 advanced tumors.
Breakpoint information is restricted to the particular area on the chromosome that affects an IG or MYC locus; full karyotype is provided in the Supplemental Tables.
Types of rearrangements for loci of interest are given (B, UB, V, I) followed by the derivatives detected (hybridization signals not shown for most balanced and unbalanced translocations); for complex (*) rearrangements, the hybridization signals detected are specified for each derivative; detailed descriptions of complex chromosomes are given in Supplemental Table 2.
Chr#, average number of metaphase chromosomes.
Rearrangements: B: simple balanced translocations or inversions with respect to the breakpoint of interest: IGH with der(14)(wcp14+, CH+, VH−,target gene+), der(other)(wcp14 -,CH−,VH+); IGL with der(22)(wcp22+, Vλ+, Cλ−), der(other)(wcp22+, Vλ−,Cλ+, target gene+). U: unbalanced translocations with respect to the breakpoint of interest, with only one of the derivatives present. V: variant translocations: IGH with der(14)(wcp14+, CH−,VH−), der(other)(wcp14 -,CH+, VH+, target gene+); IGL with der(22)(wcp22+, Vλ+, Cλ+, target gene+), der(other)(wcp22+, Vλ−,Cλ−). I: insertion of IG sequences into another chromosome juxtaposed to target genes (with or without associated wcp). I(tg): insertion of target gene sequences into an IG locus.
Note: bold der indicates that additional rearrangements occurred in addition to the translocation of interest, without affecting the centromere; mod der indicates that additional rearrangements occurred in addition to the translocation of interest, with the modification also replacing the centromere.
TABLE 2.
Structural Abnormalities of IG or MYC Loci in 47 HMCL
| IG and MYC Rearrangements | |||||
|---|---|---|---|---|---|
|
| |||||
| HMCL | Chr# | Locus | Target Gene | Breakpoints (BP) | Derivatives |
|
| |||||
| 8226 | 60 | IGH | ? | 1q12 ∷ 14q32 | U: der(14) ×2 |
| IGL | MAF, MYC(§) | ins MYC @ BP on der(16)t(16;22)(q23;q11) | I(tg) & U*: der(16) (wcp16+,MAF+,wcp8-,MYC+,wcp22+,Cλ+) see Fig.4B | ||
|
| |||||
| ANBL6 | 82 | IGH | MAF | 14q32 ∷ 16q23 | B*: mod der(14) ×3, der(16) ×2 |
|
| |||||
| ARK | 46 | IGH | MYC (§) | 8q24 ∷ 14q32 | B: der(8), der(14) |
|
| |||||
| ARP-1/ARP-1Ca | 69/43 | IGH | MAF | 14q32 ∷ 16q23 | B: der(14) ×2, der(16) ×2 |
| ? | MYC | ? ∷ 8q24 | U: der(8) | ||
|
| |||||
| Delta47 | 45 | IGL1 | ? | 11?p14 ∷ 22q11 | B: der(11) (Vλ-, Cλ+), der(22) (Vλ+, Cλ+) |
| IGL2 | ? | ins Cλ @ 9p24 on 9 | I: 9 (wcp9+, wcp22-, Cλ+) | ||
| IGL3 | MYC (§) | ins Cλ @ 8q24 on der(19)t(8;19)(q13;q13) | I*: der(19) (wcp19+, wcp22-, Cλ+, wcp8+, MYC+) | ||
|
| |||||
| DP6-DJ | 47 | IGH | MYC | ins CH @ 8q24 on 8 | I: 8 (wcp8+, MYC+, wcp14 -, CH+) |
|
| |||||
| EJM | 58 | IGH1 | MAFB | 14q32 ∷ 20q11 | B*: mod der(14), der(20) |
| IGH2 | ? | ins CH @ BP on der(18)t(7;18)(?;q23) | I: der(18) (wcp18+, wcp14 -, CH+, wcp7+) | ||
|
| |||||
| Flam76 | 42 | IGH | CCND1 | 11q13 ∷ 14q32 | B*: der(11), der(14), der(14) see Fig.4A |
| ? | MYC (§,‡) | ins MYC @ BP on der(10)t(10;13) | I: der(10) (wcp10+, wcp8-, MYC+, wcp13+) see Fig.4D | ||
|
| |||||
| FR4 | 100 | IGH1 | IRTA | ins CH @ Iq21 on der(1)t(1;?)(p?;?) | I*: der(1) (wcp1+, wcp14 -, CH+) |
| IGH2 | IRF4 | 6p25 ∷ 14q32 | V (U): der(6) (wcp14 -, CH+, VH+, wcp6+, IRF4+) ×4 | ||
| IGH3 | MYC (§) | 8q24 ∷ 14q32 | B: der(8) ×2, der(14) ×2 | ||
|
| |||||
| H1112 | 46 | IGH | CCND1 | 11q13 ∷ 14q32 | B: der(11), der(14) |
|
| |||||
| H929 | 45 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) |
| ? | MYC (§,‡) | 8q24 ∷ 20q11 | U: der(8) ×2 | ||
|
| |||||
| INA-6 | 82 | IGH | CCND1 | 11q13 ∷ 14q32 | U: der(14) ×2 |
| ? | MYC | ins MYC @ ? | I: ? (wcp8-, MYC+) ×3 | ||
|
| |||||
| JIM-3 | 60 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) |
| ? | MYC | 6q?21 ∷ 8q24 | U*: mod der(8) ×2 | ||
| ? | MYC | ? ∷ 8q24 | U*: mod der(8) | ||
|
| |||||
| JJN3 | 60 | IGH1 | MAF | 14q32 ∷ 16q23 | B*: der(14), der(14), der(16) ×2 see Fig.4A |
| IGH2 | MYC (§) | ins CH @ 8q24 on 8 & der(14) | I*: 8 (wcp8+, MYC+, wcp14 -, CH+) ×2 & der(14) (wcp8+, MYC+, wcp14+, CH+) see Fig.4A | ||
|
| |||||
| JK-6L | 50 | IGH | MYC | 8q24 ∷ 14q32 | B: der(14) (complex der(14) in 40% of metaphases) (wcp14+, CH+,VH-), der(8) (wcp14 -, CH+, VH+) |
|
| |||||
| Karpas 620 | 68 | IGH1 | CCND1,MYC (§) | ins CH @ 8q24 on der(8)t(8;11)(q24;q13) | I*: der(8) (wcp8+, MYC+, wcp14 -, CH+, wcp11+, CCND1+) see Fig.4A |
| IGH2 | MFC (§) | 8q24 ∷ 14q32 | U: der(14) | ||
| IGH3 | ? | 1q12 ∷ 14q32 | U: der(14) | ||
|
| |||||
| Kas-6 | 65 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B*: der(4), der(4) ×2, mod der(14) ×2 |
| ? | MYC | 1? ∷ 8q24 | U: der(8)dup(8)(q22q24) (MYC+) see Fig.4D | ||
|
| |||||
| KHM-1B | 59 | IGL | MYC | ins Cλ @ 8q24 on 8 | I: 8 (wcp8+, MYC+, wcp22-, Cλ+) |
|
| |||||
| KHM-11 | 75 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) |
| ? | MYC (‡)c | 6?p24 ∷ 8q24 | B: der(6), mod der(8) ×2 (MYC+) | ||
|
| |||||
| KMM1 | 80 | IGH1 | CCND3 | 6p21 ∷ 14q32 | U*: der(14) (wcp14+, CH+, wcp6+, CCND3+) ×2 see Fig.4A |
| IGH2 | MYC (§) | ins MYC @ 14q32 on der(12) & der(4) | I(tg)*: der(12) (wcp14+, CH+, VH+, wcp8-, MYC+) & der(4) (wcp14+, CH+, VH+, wcp8-, MYC+) | ||
|
| |||||
| KMS-11 | 70 | IGH1 | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) |
| IGH2 | MAF | 14q32 ∷ 16q23 | B: der(14), der(16) | ||
| IGH3 | MYC (§) | ins CH @ 8q24 on der(?) & dup(8)(q24.3q24) | I*: der(?) (wcp14 -, CH+, wcp8+, MYC+; wcp8+, MYC+, wcp14 -, CH+) see Fig.4A | ||
|
| |||||
| KMS-12 PE/BM b | 47/75 | IGH1 | CCND1 | 11q13 ∷ 14q32 | B*: der(11), der(14), mod der(14) |
| IGH2 | MYC (§) | ins CH @ 8q24 on 8 | I: 8 (wcp8+, MYC+, wcp14 -, CH+) ×2 | ||
|
| |||||
| KMS-18 | 74 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B*: der(4), mod der(14) ×2 |
| ? | MYC (‡) | 6? ∷ 8q24 | U: der(6) ×2 | ||
| ? | MYC (‡) | 8q24 ∷ 21? | U: der(21) ×2 | ||
|
| |||||
| KMS-20 | 41 | IGH | MYC | 8q24 ∷ 14q32 | B*: der(8), mod der(14) |
|
| |||||
| KMS-26 | 71 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B*: mod der(4), mod der(14) |
| IGL | MYC | 8q24 ∷ 22q11 | U*: mod der(8) (wcp8+, MYC+, wcp22+, Cλ+) ×3 see Fig.4B | ||
|
| |||||
| KMS-28 PE/BMb | 42/42 | IGH1 | FGFR3, MMSET | 4p16 ∷ 14q32 | B*: der(4), mod der(14) |
| IGH2 | MYC | 8q24 ∷ 14q32 | B: der(8), der(14) | ||
|
| |||||
| KMS-34 | 76 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B*: der(4), der(14), mod der(14) |
| ? | MYC | ? ∷ 8q24 | U: der(8) | ||
|
| |||||
| KP6-DJ | 47-89 | IGH | ? | 14q32 ∷ 17?p | V, B: der(14) (wcp14+,CH-,VH-) ×3, der(17) (wcp14 -,CH+,VH+,wcp17+) ×3 |
|
| |||||
| L363 | 46 | IGL | NIK | 17q21 ∷ 22q11 | B: der(17) ×3, der(22) ×2 |
| ? | MYC (§, ‡) | 5? ∷ 8q24 | B: der(5), der(8) ×2 (MYC+) | ||
|
| |||||
| LP1 | 80 | IGH1 | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) |
| IGH2 | ? | ins CH @ ? | I: ?(wcp14 -, CH+) | ||
| IGH3 | ? | ? ∷ 14q32 | V (U)*: mod der(8) (wcp14 -, CH+, VH+) ×2 | ||
| IGH4 | MYC (§) | 8q24 ∷ 14q32 | V (U)*: der(8) (wcp8+, MYC+, wcp14 -, CH+, VH+) ×3 & mod der(8) (wcp8+, MYC+, wcp14 -, CH+, VH+) ×2 | ||
|
| |||||
| MM.1 | 44 | IGH1 | MAF | 14q32 ∷ 16q23 | B: der(14), der(16) |
| IGH2 | MYC (§) | ins CH @ BP on der(3)t(3;8)(?;q24) | I*: der(3) (wcp3+, wcp14 -, CH+, wcp8+, MYC+) | ||
|
| |||||
| MM-S1 | 47 | IGH1 | CCND1 | 11q13 ∷ 14q32 | B: der(11), der(14) |
| IGH2 | MYC (§) | ins CH @ 8q24 on 8 & i(8q) | I*: 8 & i(8q) (wcp8+, MYC+, wcp14 -, CH+) | ||
|
| |||||
| OCI-MY1 | 49 | IGHI | ? | ins CH @ ? | I: ? (wcp14-, CH+) |
| IGH2 | ?? | ins CH @ ?? | I: ?? (wcp14-, CH+) | ||
| IGL | MYC | 8q24 ∷ 22q11 | U: der(8) | ||
|
| |||||
| OCI-MY5 | 46 | IGH1 | MAF | 14q32 ∷ 16q23 | U: der(14) |
| IGH2 | MYC (§) | 8q24 ∷ 14q32 & dup(14)(q32q?22) | U*: der(?) (wcp8+,MYC+,wcpl4+,CH+;wcp14+,CH+,wcp8+,MYC+) see Fig.4A | ||
|
| |||||
| OCI-MY7 | 78 | IGH1 | CCND1 | 11q13 ∷ 14q32 | U: der(14) ×2 |
| IGH2 | ? | ?X?q ∷ 14q32 | B: der(?X), der(14) | ||
| ? | MYC(‡) | 3? ∷ 8q24 | U: der(8) ×2 | ||
|
| |||||
| OPM-1/2 b | 75/75 | IGH1 | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) |
| IGH2 | MYC (§) | ins CH & wcp14 @ 8q24 on der(8)t(8; 14)(q24;q32)t(1;14)(?q12;q?) | I*: der(8) (wcp8+, MYC+, wcp14+, CH+) | ||
|
| |||||
| PE-1 | 83 | IGH | CCND1 | 11q13 ∷ 14q32 | U*: der(14) ×2, mod der(14) |
|
| |||||
| PE-2 | 72 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4), der(14) |
| IGK | NMYC | 2p12 ∷ 2p25 | B: der(2)inv(2)(p25p12)(wcp2+, Cκ+, NMYC+) see Fig.4C | ||
|
| |||||
| Sachi | 69 | IGH1 | MAFB | 14q32 ∷ 20q11 | B*: der(14) ×2, der(20) ×2 |
| IGH2 | MYC | 8q24 ∷ 14q32 | B: der(8), der(14) | ||
|
| |||||
| SKMM1 | 79 | IGH1 | MAFB | 14q32 ∷ 20q11 | B: der(14), der(20) |
| IGH2 | IRF4 | 6p25 ∷ 14q32 | U*: der(8) (wcp14+, CH+, wcp6-, IRF4+) | ||
| IGH3 | MYC (§) | 8q24 ∷ 14q32 | U*: der(8) (wcp8+, MYC+, wcp14+, CH+) | ||
|
| |||||
| SKMM2 | 37 | IGH | CCND1 | 11q13 ∷ 14q32 | U: der(14) |
| ? | MYC (‡) | ins MYC @ ?9 | I: ?9 (wcp8-, MYC+) | ||
|
| |||||
| U266 | 39 | IGH | CCND1 | ins CH @ 11q13 on der(11)t(11;?)(q14;7) | I: der(11) (wcp11+, CCND1+, wcp14 -, CH+) |
| ? | LMYC (§) | 1p13 ∷ 1p34 | U: der(1)inv(p34p13) (LMYC @ p13) | ||
|
| |||||
| UTMC-2 | 77 | IGH | FGFR3, MMSET | 4p16 ∷ 14q32 | B: der(4) ×2, der(14) ×2 |
| ? | MYC (‡) | del(8q24) | U: del (.5-1 Mb tel of MYC) | ||
|
| |||||
| XG-1 | 44 | IGH1 | CCND1 | 11q13 ∷ 14q32 | U: der(14) ×2 |
| IGH2 | MYC | 8q24 ∷ 14q32 | V (U)*: der(8) (wcp8+, MYC+, wcp14 -, CH+, VH+) | ||
|
| |||||
| XG-2 | 49 | IGH | ? | 12q24.31 ∷ 14q32 | B: der(12) (wcp14 -, CH+, VH+), der(14) (wcp14+, CH+, VH-) |
| IGL1 | MAFB | ins Cλ @ 20q11 on 20 | I: 20 (wcp20+, MAFB+, wcp22-, Cλ+) | ||
| IGL2 | CD40 | ins CD40 @ 22q11 on 22 | I(tg): 22 (wcp22+, Vλ+, Cλ+, wcp20-, CD40+) ×2 | ||
| ? | MYC (‡) | 2q? ∷ 8q24 | U: der(2) | ||
|
| |||||
| XG-6 | 77 | IGL | 7 | 16q23 ∷ 22q11 | B: der(16) (wcp16+, MAF+, wcp22-, Cλ+) ×2, der(22) |
|
| |||||
| XG-7 | 43 | IGH1 | MMSET | 4p16 ∷ 14q32 | U: der(4) (VH+, MMSET+, FGFR3-) |
| IGH2 | IRF4 | ins IRF4 @ 14q32 on der(14)t(4;14)(p16;q32) | I(tg)*: der(14) (wcp14+, CH+, FGFR3-, wcp6-, IRF4+, 4ptel+) | ||
| ? | MYC (‡) | ins MYC @ ? on 17 | I: 17 (wcp17+, wcp8-, MYC+) | ||
| ? | MYC (‡) | ins MYC @? on 19 | I: 19 (wcp19+, wcp8-, MYC+) | ||
See Table I legend.
Two sublines; ARP-IC subline was incorrectly identified as CAG in Annunziata et al., 2007.
Two independent lines from the same patient.
MYC rearrangements previously described (Shou et al., 2000).
MYC rearrangements recently described (Dib et al., 2008).
FISH and SKY Probes
The following probes were described elsewhere: CH BAC, Cλ BAC, Cκ cos, all three including strong 3′ enhancers, VH telomeric cos, LMYC genomic fragment, MYC BAC (GS-93F05), MYC plasmid, CCND1 (6.22 cosmid), CCND3 (ccnd3cy13 cosmid), FGFR3 (L184d6 cosmid), and MMSET (L190b4 cosmid) (Chesi et al., 1998b; Gabrea et al., 1999; Shou et al., 2000; Shaughnessy et al., 2001; Dib et al., in press). NMYC (MYCN) (AC010145), MAFB (AL035665), NIK (MAP3K14) (AC003963) are identified by their GenBank accession numbers. MAF and IRF4/MUM1 were obtained by screening Genome Systems BAC libraries. Vλ and Vκ cosmids, each containing the most centromeric V sequences, were prepared by subcloning Vλ and Vκ BAC fragments (Genome Systems) into Supercos. SKY probes were obtained from Applied Spectral Imaging (Vista, CA). FISH probes were labeled by nick translation with either biotin-16-dUTP (Roche, Indianapolis, IN) followed by avidin-FITC (Sigma, St. Louis, MO) detection or digoxigenin-11-dUTP (Roche, Indianapolis, IN) followed by TRITC antibody (Sigma, St. Louis, MO) detection. Whole chromosome painting (wcp) probes were generated in our laboratory by direct PCR labeling with Cy5-5-dUTP of chromosome-specific template DNA, generously provided by Dr. Thomas Ried (Center for Cancer Research, National Cancer Institute, Bethesda, Maryland).
FISH and SKY Hybridization
FISH hybridizations of metaphase chromosomes have been described in detail elsewhere (Shou et al., 2000). SKY analyses were carried out according to the manufacturer’s protocol ASI. For sequential hybridizations, slides were analyzed and the position on the slide of 5–10 metaphases was recorded. The slides were then prepared for rehybridization by shaking twice in 4×SSC/Tween (15 min each time), rinsing briefly with water, dehydrating in ethanol, and stripping of the old probes in 70% formamide denaturing solution (72°C for 2 min).
Strategy for Detection of Structural Chromosome Abnormalities
An initial screen was conducted on all MM tumor samples and HMCL for each IG locus and the MYC locus. For the IG loci, three-color FISH hybridizations were performed using constant (C) region probes that include strong enhancer sequences, variable (V) region probes, and whole chromosome painting probes. For the MYC locus, two-color FISH hybridizations were performed using the MYC and wcp8 probes. For translocation partners easily identified by their morphology, a second round of FISH hybridization was performed using a wcp probe and a specific target gene probe. For complex rearrangements, sequential SKY and FISH hybridizations were performed on the same metaphase chromosomes.
RESULTS
Hyperdiploid and Nonhyperdiploid Tumors
The 48 advanced tumors studied were subdivided into two groups based on the metaphase chromosome number (Table 1). The first 22 tumors, which have <48 and/or >75 chromosomes, were classified as NHRD. The remaining 26 tumors, which have 48–75 chromosomes, were classified as HRD. Consistent with published studies, extra copies or partial copies of eight odd-numbered chromosomes (3, 5, 7, 9, 11, 15, 19, and 21) occur frequently in HRD tumors, but only infrequently in NHRD tumors. Extra copies of at least four of these chromosomes are present in 24 of 26 HRD tumors, with the two exceptions having the minimum (#48, 48 chromosomes) or close to the maximum (#23, 70 chromosomes) number of chromosomes that define this group. By contrast, only two (#1, 13) of 22 NHRD tumors had extra copies, or partial copies, of at least four of these chromosomes. Curiously, one NHRD tumor (#1) has only 32 chromosomes, with two copies of each of the eight odd chromosomes and only one copy of most other chromosomes (see Discussion).
Four Types of IGH Rearrangements in MM Tumors
Nine of the 48 tumors (#6, 7, 10, 11, 15, 31, 34, 46, and 48) had IGH rearrangements characterized previously by conventional cytogenetic and SKY analyses, and four other tumors were shown to have an add(14)(q32) (#12, 17, 20, and 22) (Supplemental Table 1) (Sawyer et al., 1998, 2001). For a comprehensive analysis, we subjected all 48 tumor samples to specific IGH FISH analyses. An initial screen, using a CH probe, which hybridizes with Eα1 and Eα2 enhancer sequences, a distal VH probe located within 100 kb of the 14q telomere, and a wcp14 probe revealed 36 IGH rearrangements in 26 tumors (Table 1). There were four kinds of rearrangements: (1) classical balanced IGH translocations; (2) variant IGH translocations; (3) unbalanced (but nonvariant) IGH translocations; and (4) IGH insertions.
Classical balanced (B) IGH translocations are distinguished by a FISH hybridization pattern of dissociated CH and VH signals, with CH remaining on der(14) and VH relocating to the telomere of the other derivative (Fig. 1B and D). Tumors #2, 4, 5, 8, 9, 10, 14, 18, 23, 37, and 47 have the karyotypically cryptic t(4;14)(p16;q32) with the FGFR3 gene colocalizing with CH on der(14), while VH is on der(4) (Fig. 1D). Other balanced classical translocations, which were confirmed by FISH analyses, were found in tumors #6 and 12 (CCND1), #7 and 15 (MAF), #10 (NMYC), #22 (MYC) and #11, 17, 46, and 48 (unidentified target genes).
Figure 1.
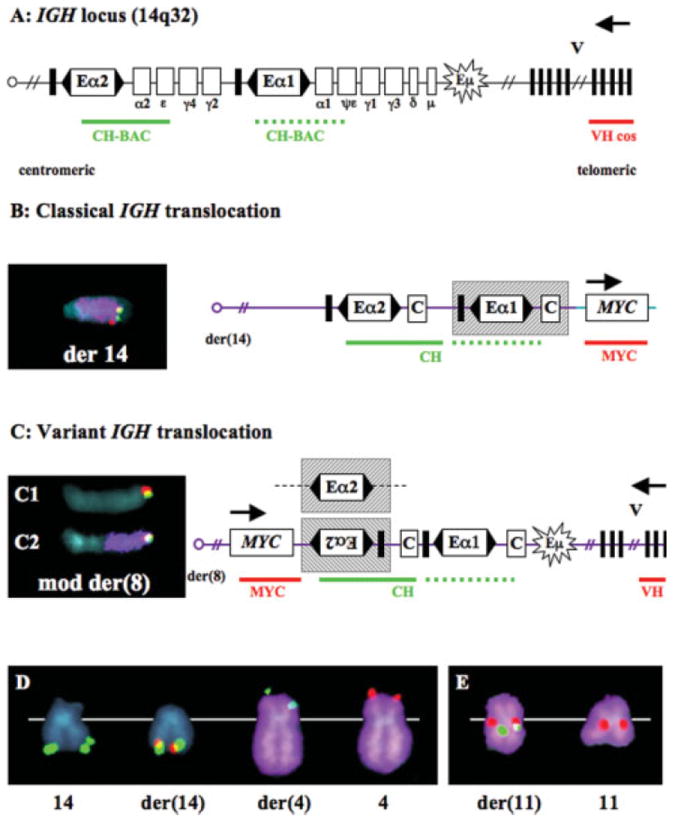
Structures of IGH translocations in MM tumors. (A) The 1 Mb IGH locus is depicted with boxed coding regions, enhancer elements, and direction of transcription (arrow). Eα1 and Eα2 enhancers control gene expression bidirectionally (flanking arrowheads), but insulator sequences (thick vertical lines) may block centromeric effects. Eμ is the bidirectional intronic enhancer. The VH cosmid (red line) located 100 kb from the telomere is essentially a 14q tel probe. The CH BAC (green line) detects Eα1 and Eα2 sequences. (B) FISH analysis detected a classical balanced t(8;14) in the ARK HMCL. CH (green) and MYC (red) colocalize on der(14) (purple). The diagram depicts a breakpoint centromeric to MYC (8q24) and telomeric to the Eα1 enhancer (shaded box) that presumably dysregulates MYC. (C) FISH analysis detected a variant unbalanced t(8;14) in tumor #17. CH (green) and VH (red) colocalize on mod der(8) (C1). MYC (red) is juxtaposed to CH (green) on mod der(8) (purple) (C2). The diagram depicts a breakpoint telomeric to MYC and centromeric to Eα2; MYC may be juxtaposed and thus dysregulated either by an inverted Eα2 enhancer or an Eα2 enhancer with deleted insulator sequences (alternative shaded boxes). (D, E) FISH detection of classical t(4;14) and CH insertion at 11q13 in tumor #47. (D) One copy of CH (green) colocalizes with VH (green) on normal chromosome 14, a second copy of CH colocalizes with FGFR3 (red) on der(14), one copy of VH is on der(4) (purple), and one copy of FGFR3 is on chromosome 4 (purple) (E). A third copy of CH (green) is shown to colocalize with CCND1 (red) on der(11) (purple), while a second copy of CCND1 is on normal chromosome 11 (purple).
Variant (V) IGH translocations are distinguished by a FISH hybridization pattern in which the VH and CH probes, without wcp14, colocalize at the telomere of another chromosome, and der(14) has no CH signal. One tumor (#34) had a balanced t(3;14)(q?27;q32) variant IGH translocation, with der(3) having CH and VH signals colocalizing at the telomere, and der(14) lacking CH or VH signals. Three other tumors (#17, 21, and 46) had unbalanced variant IGH translocations, with no der(14), but VH and CH probes colocalizing at the telomeric end of another chromosome (adjacent to MYC at 8q24.1 in tumors #17 and 46) (Fig. 1C). None of the variant IGH translocations would be detected by interphase FISH analyses designed to identify dissociation of the VH and CH probes. In addition, it should be noted that it is only the balanced variant IGH translocation that might be suspected by conventional cytogenetic or SKY analyses.
Excluding variant translocations, unbalanced (UB) IGH translocations are distinguished by a FISH hybidization pattern in which there is either a der(14) having a CH signal or another chromosome having a telomeric VH signal. This kind of unbalanced translocation was found in four tumors. Tumor #31 had a CH signal on der(14)t(1;14)(q11~12;q32), but no der(1) with VH. Tumors #17 and #44 had CH signals at the telomere of der(14) but no detectable chromosome sequences telomeric to CH, and no derivative with VH, which limits our ability to identify the partner chromosomes or target genes. Tumor #5 had no der(14) with CH, but two copies of VH colocalize with the MMSET gene, one copy on der(4) and the other on a more complex derivative chromosome.
The final group of IGH rearrangements involves simple insertions (I). The CH signals, which are dissociated from telomeric VH signals, are found internally on recipient chromosomes, usually without a detectable wcp14 signal. In half of the tumors that have a simple CH insertion (#11, 34, 42, and 46), the inserted CH probe colocalized with the MYC gene at 8q24. In tumor #8 the recipient chromosome is 17, but a target gene has not been identified. In tumors #12 and 21 our FISH and SKY analyses failed to classify the recipient chromosome because of poor metaphase quality, although we were able to eliminate MYC as the target gene. In tumor #47, which also has a balanced t(4;14) (see above), inserted CH sequences colocalized with CCND1 at 11q13 (Fig. 1E). In this tumor we therefore have identified two independent, karyotypically silent IGH rearrangements, neither of which was detected by conventional cytogenetic or SKY analyses.
Remodeling of Chromosomes Carrying IGH Rearrangements
In each of the four kinds of IGH rearrangements described above, we have found tumors with complex derivative chromosomes that result from further rearrangements of translocated chromosomes. New breakpoints may occur far from the IGH rearrangement, and thus should not affect the juxtaposed target gene and CH sequences (e.g., tumor #17). Alternatively, new breakpoints may occur in very close proximity to the IGH rearrangement, thereby masking the target gene juxtaposed to CH sequences (tumors #5, 12, 20, and 46) (Fig. 2C). Tumor #20 has a complex, balanced translocation, which the conventional cytogenetic analysis detected as an add(14)(q32). Our initial FISH screen revealed one CH signal on der(14)t(4;14) (q22;q32), and one VH signal on an unknown small chromosome (not shown). A subsequent hybridization confirmed a der(14)t(4;14) but no colocalizing CH and FGFR3 signals (Fig. 2A). Sequential SKY analysis was carried out on the same metaphase chromosomes, which revealed both that the VH sequences are on der(20) and that there is a thin strip of chromosome 20 sequences at the t(4;14) breakpoint (Fig. 2B). A final hybridization confirmed colocalization on der(14)t(14;20)(q32;q11) t(4;20)(q22;q?) of CH and MAFB (not shown). It appears that an initial t(14;20)(q32;q11) juxtaposed CH and MAFB sequences, resulting in the presumptive dysregulation of MAFB, and that der(14) subsequently underwent a second translocation with chromosome 4.
Figure 2.
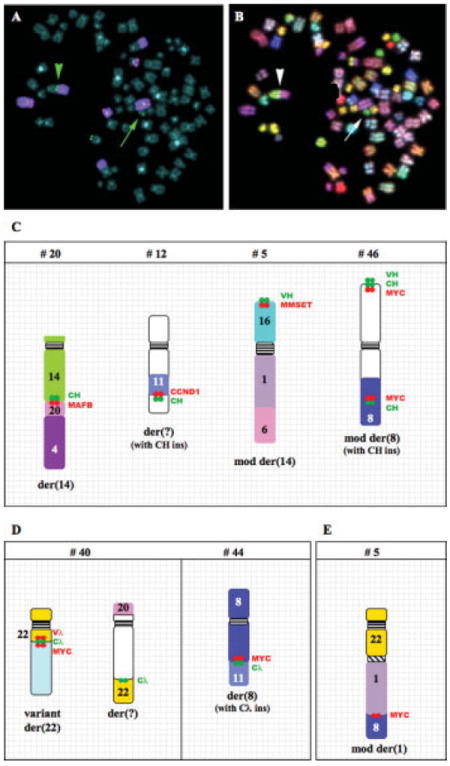
Complex chromosome rearrangements in MM tumors. Sequential FISH (A) and SKY (B) analysis of the complex IGH translocation t(14;20)t(4;20) in tumor #20. Metaphase chromosomes were hybridized with CH (green), VH (green), FGFR3 (red), and wcp4 (purple). The arrowhead shows a copy of CH at the t(4;14) breakpoint, while the arrow indicates a copy of VH on a small chromosome. The same metaphase chromosomes were stripped and rehybridized with SKY probes to show that the VH is on der(20) (arrow) and that chromosome 20 sequences are inserted at the t(4;14) breakpoint (arrowhead). Examples of chromosomes with rearrangements of the IGH (C), IGL (D), and MYC (E) loci are shown for selected MM tumors. Relative positions of CH, VH, Cλ, and target gene sequences (MAFB, CCND1, MMSET, MYC) are indicated.
IGH Rearrangements in HMCL
FISH analyses showed that 43 of 47 HMCL had 68 different IGH rearrangements, whereas the other four HMCL had IG light chain rearrangements (Table 2 and below). The same four kinds of structural abnormalities identified in MM tumors also were identified for the HMCL.
Classical balanced translocations (B) juxtapose CH sequences mostly to recurrent target genes, FGFR3/MMSET (13 cases), CCND1 (four cases), MAF (five cases), MAFB (three cases), and MYC (seven cases); but also to unidentified genes in XG-2 and in OCI-MY7, which also had an unbalanced translocation involving CCND1. XG-7 has a complex, apparently balanced classical t(4;14), with der(4) having colocalizing telomeric MMSET and VH sequences, but with der(14) having colocalizing CH, IRF4 and 4p telomeric sequences, but no FGFR3 sequences. It seems likely that the original t(4;14) in XG-7 generated a der(14) that was modified by an insertion of IRF4 (6p25) and deletion of FGFR3.
Five variant IGH translocations (V) were found in HMCL, four of which were unbalanced (IRF4 in FR4; MYC in LP1 and XG-1; unknown target gene in LP1), and one of which was balanced (t(14;17) in Kp6). The balanced translocation in XG-2 is similar to a balanced variant translocation since it has both CH and VH signals on der(12). However, it does not meet our definition of a variant IGH translocation since it has a CH signal on der(14). In any case, 3′ IGH enhancer sequences potentially could dysregulate a gene on der(14), der(12), or both in XG-2.
Excluding variant IGH translocations, approximately one fourth of IGH translocations are unbalanced (U). Target genes include MMSET (one case), CCND1 (four cases), CCND3 (one case), MAF (one case), IRF4 (two cases), and MYC (two cases). In addition, both 8226 and Karpas-620 have CH signals on der(14)t(1;14)(q12;q32), but a target gene has not been identified. Of the unbalanced translocations, about half are highly complex, resulting in rearranged derivative chromosomes, in which the CH sequences can be juxtaposed to more than one target gene. For example, Karpas-620 has a der(14)t(8;14) with CH and MYC sequences, and a der(8)t(8;11) with CH, MYC, and CCND1 sequences (Fig. 4A). More complete FISH mapping analyses (not shown) suggest that der(14)t (11;14) underwent a subsequent translocation involving the MYC locus that generated the two chromosomes described above. See Figure 4A for other examples of complex unbalanced IGH translocations.
Figure 4.
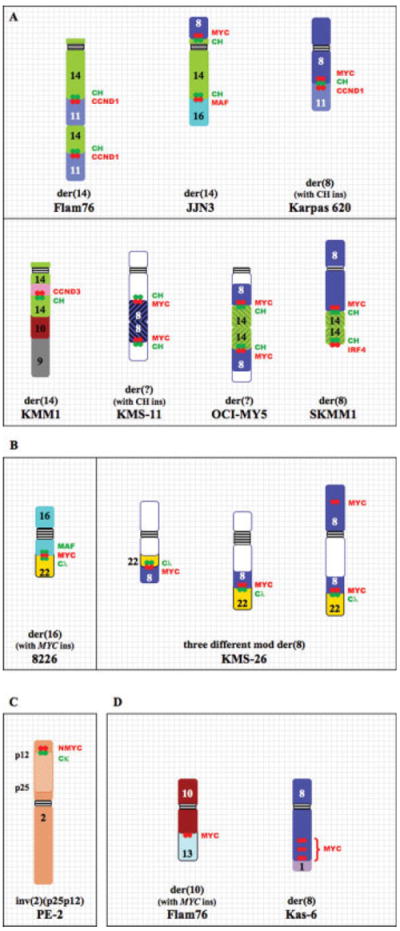
Complex chromosome rearrangements in HMCL. Examples of chromosomes with complex rearrangements of the IGH (A), IGL (B), and IGK (C) loci are shown for selected HMCL. Relative positions of CH, VH, Cλ, Cκ, and target gene sequences (CCND1, MAF, MYC, and NMYC) are indicated. Karyotypic abnormalities for the MYC locus that do not involve an IG locus are shown (D).
There are two kinds of IGH insertions (I) in the HMCL. First, CH sequences without associated wcp14 are inserted at a single site in an apparently normal chromosome region, e.g., FR4, JJN-3, KMS-12PE, MM-S1, and U266. Second, CH sequences with or without associated wcp14 are inserted at the breakpoint of two chromosomes, e.g., EJM, MM.1, OPM-1, and KMS-11 (Fig. 4A). For JJN-3, the telomeric end of chromosome 8 moves onto the p arm of chromosome 14, with CH and MYC juxtaposed at the breakpoint (Fig. 4A). For KMM-1, FISH analyses and cloning results show that chromosome 21 (without wcp21) and MYC (without wcp8) sequences are inserted into the IGH locus at 14q32 (Shou et al., 2000).
IGL Rearrangements in MM Tumors
Conventional cytogenetic and SKY analyses detected IGL rearrangements in 5 of 48 tumors (#7, 13, 14, 32, and 40) (Supplemental Table 1). FISH analyses of metaphase chromosomes from all 48 tumor samples revealed nine IGL rearrangements in eight tumors (Table 1), so that three tumors had karyotypically silent rearrangements.
Similar to IGH rearrangements, there are four kinds of IGL rearrangements (Fig. 3). Classical balanced IGL translocations (B) are distinguished by a FISH hybridization pattern that identifies dissociation of Vλ and Cλ signals, with Vλ remaining on der(22) and Cλ relocating to the breakpoint of the other derivative chromosome. Only one tumor sample (#13) had a balanced (B) translocation, and this involved MYC. A variant IGL translocation (V) is distinguished by a FISH hybridization pattern in which der(22) retains both Vλ and Cλ sequences. Only one tumor (#40) had a variant IGL translocation, and this was an unbalanced variant translocation that involved MYC. Excluding the tumor with the variant translocation, five tumors have unbalanced translocations (U) in which there was no der(22), but only another chromosome derivative with a Cλ signal. The target genes in these tumors included MYC (tumors #14 and 44), CCND3 (tumor #32), and unknown genes (tumors #7 and 40). Finally, two tumors (#18 and 36) had insertions (I) of Cλ sequences without a detectable wcp22 signal within chromosome 8, and juxtaposed to MYC at 8q24.1.
Figure 3.
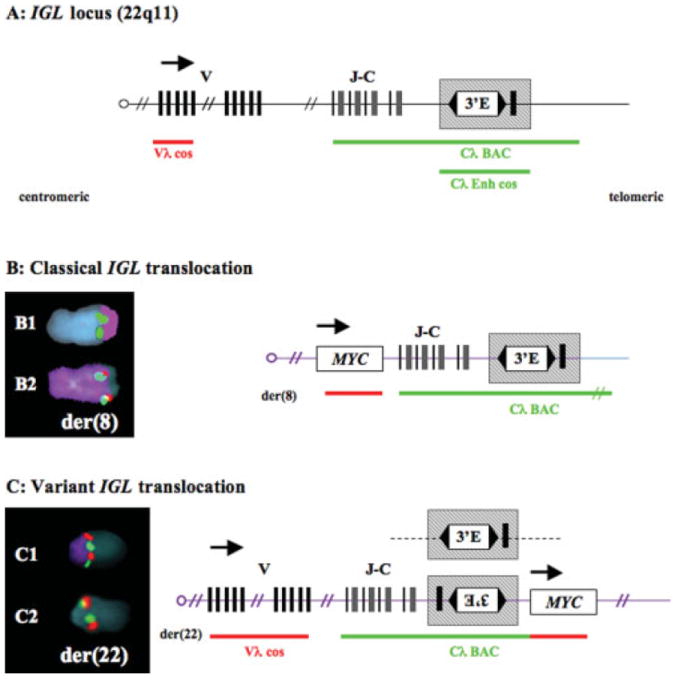
Structures of Igλ translocations in MM tumors. (A) The 1 Mb IGL locus is depicted with boxed coding regions and direction of transcription (arrow). The 3′ enhancer (E) is depicted as controlling gene expression bidirectionally, except for putative telomeric insulator sequences (thick vertical line). The Vλ cosmid (red line) detects centromeric sequences, the Cλ-BAC detects JC and 3′E sequences (green line) and Cλ-cos detects only the 3′E. (B) FISH analysis detected a classical balanced t(8;22) in tumor #13. Cλ (green) and telomeric 22 sequences (purple) translocate to der(8) (B1). Cλ (green) colocalizes with MYC (red) on der(8) (purple) (B2). (C) FISH analysis detected a variant unbalanced and complex t(8;22) in tumor #40. Cλ (green) and Vλ (red) colocalize on der(22) (purple) (C1). MYC (red) is juxtaposed to Cλ (green) on der(22) (C2).
IGL Rearrangements in HMCL
Using the same hybridization techniques and IGL probes described earlier, we found that 8 of 47 HMCL analyzed had 12 distinct IGL rearrangements (Table 2, Fig. 4B). Four HMCL have both light and heavy chain rearrangements: 8226, KMS-26, OCI-MY1, and XG-2, but four HMCL have only light chain rearrangements: Delta-47, KHM-1B, L363, and XG-6.
Delta-47, KMS-26, and L363 have balanced translocations with breakpoints at 11?p14, 8q24 (MYC), and 17q21 (NIK), respectively. The rearrangement in L363 has a cloned breakpoint 6.8 kb upstream of Cλ1, ~48 kb from the 3′Cλ enhancer (Annunziata et al., 2007). OCI-MY1, 8226, and XG-6 had unbalanced translocations, with the der(22) and associated Vλ signal being absent in all three lines. In OCI-MY1, Cλ and MYC were juxtaposed at 8q24 on der(8). In 8226, Cλ and MAF were juxtaposed at 16q23 on der(16), and there was associated over-expression of MAF (Chesi et al., 1998a). In XG-6, Cλ is juxtaposed to BAC sequences that include MAF at 16q23 on der(16). We did not, however, find over-expression of MAF in XG-6, suggesting that a different unidentified gene may be the target for dysregulation (Kuehl, unpublished).
Delta-47, KHM-1B, and XG-2 have simple insertions that are detected by Cλ probe signals, without associated wcp22 probe signals. In Delta-47 and KHM-1B, Cλ signals were juxtaposed near MYC at 8q24, with an additional Cλ insertion at 9p24 also detected in Delta-47. Finally, in XG-2, a Cλ signal was inserted near MAFB at 20q11.
In addition to insertions of Cλ sequences into other chromosomes, two HMCL have insertions of other genes near a Cλ sequences. In 8226, MYC sequences without an associated wcp8 signal were found at the breakpoint of the der(16)t(16;22) unbalanced translocation (Fig. 4B). This suggests that the t(16;22) that dysregulates MAF occurred initially, but was followed by insertion of a MYC gene so that both MAF and MYC are dysregulated by Cλ 3′ enhancer sequences on der(16)t(16;22). In XG-2, the over-expression of CD40 was reported to be associated with insertion of the CD40 gene near Cλ and Vλ sequences on chromosome 22 (Keats et al., 2007). Importantly, the detection of both of these insertions required an assay that used target gene specific probes, and could not have been detected by assays that used only IGL probes (similar to MYC insertion into IGH locus in KMM-1 or IRF4 insertion replacing FGFR3 on der(14) in XG-7).
MYC Rearrangements in MM Tumors
We confirmed MYC abnormalities in 7 of 48 tumors (#4, 7, 13, 14, 26, 43, and 47) and clarified or corrected cytogenetic and SKY findings in three other tumors (#22, 37, and 44) (Supplemental Table 1). We also identified new MYC rearrangements in 11 other tumors (#5, 11, 17, 18, 29, 34, 36, 38, 40, 42, and 46). For tumor #10, we confirmed a classical balanced t(2;14)(p23;q32) that results in the juxtaposition of CH and NMYC sequences.
The MYC rearrangements, which are found in 22 of 47 MM tumors, include involvement of the following: (1) an IGH locus (#10, 11, 17, 22, 34, 42, and 46); (2) an IGL locus (#13, 14, 18, 36, 40, and 44); (3) an IGK locus (#7); or (4) no detectable IG sequences (#4, 5, 26, 29, 37, 38, 43, and 47) (Table 1). The eight tumors without apparent involvement of an IG locus deserve further comment. First, tumors #4, 5, 43, and 47 have simple unbalanced translocations, three of which involve chromosome 1. Second, tumors #29 and 38 have MYC sequences without a wcp8 signal inserted into un-identified chromosomes. Third, tumor #37 had three highly complex rearrangements that involve MYC, suggesting that sequential breaks and reunions involving the MYC locus occurred, giving rise to these complex derivative chromosomes. Finally, tumor #26 has three copies of an i(8)(q10) and thus a MYC amplification without any evidence for a rearrangement near the MYC gene.
MYC Rearrangements in HMCL
Previously, we have characterized in detail the MYC rearrangements found in 30 HMCL (Table 2 MYC) (Shou et al., 2000; Dib et al., in press). We extended our FISH analyses to encompass the remaining 17 HMCL available to us, and identified partner chromosomes that were previously reported as unknown. For example, we found here that Delta47 has a Cλ insertion into der(19)t(8;19). Overall, FISH hybridizations reveal MYC rearrangements in 39 of 47 HMCL, plus one LMYC rearrangement (U266) and one NMYC rearrangement (PE-2). Six HMCL (ANBL6, EJM, H1112, KP-6, PE-1, and XG-6) have no karyotypic abnormality of a MYC gene, but array CGH analyses detected rearrangements near the MYC gene in two HMCL (EJM and XG-6) (Dib et al., in press).
Rearrangements that involve the IGH locus (19 HMCL), the IGL locus (5 HMCL), and the IGK locus (inversion to NMYC in PE-2) have been described earlier or in Table 2 (see also Fig. 4). For 14 HMCL that have MYC rearrangements not involving an IG locus, we find balanced and unbalanced translocations as well as simple and complex insertions (Table 2 and Fig. 4B), with some of these results having been presented in detail elsewhere (Shou et al., 2000; Dib et al., in press). One HMCL (UTMC-2) has two different chromosomes: i(8)(q10) and der(5)t(5;8)(?;q11), both of which have an ~500 kb deletion immediately telomeric to MYC. Finally, in the U266 HMCL, which expresses LMYC but not MYC, LMYC was found to have been relocated from 1p34 close to the centromere at 1p13 as the result of a translocation, with an associated inversion.
DISCUSSION
Identification of IG and MYC Genomic Rearrangements in MM Tumors and Cell Lines
A comprehensive analysis of IG and MYC rearrangements was performed on 48 advanced MM tumors and 47 independent HMCL. Selection of tumors was dependent on the availability of an abnormal karyotype and enough additional metaphase chromosome material to enable multiple metaphase FISH analyses. Unfortunately, most tumors with a t(11;14) were not included in the samples available for analysis. The initial strategy was to do separate three-color metaphase FISH analyses for the three IG loci and the MYC locus. Additional FISH and SKY analyses—sometimes done sequentially on marked metaphase spreads—were used to characterize more fully rearrangements detected in the initial analyses, as well as to identify potential target genes for dysregulation by strong enhancer elements found in IG loci. In all HMCL and the few tumors where expression microarray data were available, we confirmed over-expression of target genes identified by FISH (Kuehl and Shaughnessy, unpublished). Apart from insertion of unknown sequences, inversions, and small (less than 5 Mb) deletions, or amplifications, most nonphysiological IG and MYC rearrangements should be detected by this strategy.
Defining Hyperdiploid and Nonhyperdiploid Tumors
The classification of tumors strictly by chromosome number instead of chromosome content is problematic (Table 1, Results), particularly since we do not understand the molecular basis for this classification (Chng et al., 2006, 2007). Perhaps this is illustrated best by tumor #1, which is markedly hypodiploid (32 chromosomes), and thus classified as a NHRD tumor. However, this tumor has single copies of most chromosomes, but extra copies of all eight odd-numbered chromosomes, with the chromosomal imbalance suggesting that this tumor might have a “HRD” phenotype. This extreme hypodiploid phenotype is associated with a similar kind of chromosome imbalance in other tumors: one tumor included cells with 31 or 61~63 chromosomes, both associated with extra copies of chromosomes 3, 7, 9, 11, 15, and 19 (Sawyer, unpublished); a second tumor (#129) included cells with 30 or 57~58 chromosomes, both associated with extra copies of at least five of the eight odd-number chromosomes (Smadja et al., 2001); and a third tumor (#21) had 30 chromosomes, again with extra copies of at least five of the eight odd-numbered chromosomes (Avet-Loiseau et al., 2001a). As summarized elsewhere, HMCL appear to be selectively generated from NHRD tumors, and it is possible that some “HRD” HMCL are not generated from HRD tumors (Bergsagel et al., 2005).
Prevalence of IGH Rearrangements in MM Tumors and Cell Lines
Similar to results published by others, IGH rearrangements were identified in 26 (54%) of 48 tumors (Table 1) (Avet-Loiseau et al., 2002; Fonseca et al., 2003a, 2004). As expected, there was a marked difference in the prevalence of IGH rearrangements in NHRD versus HRD tumors (Fonseca et al., 2003b; Smadja et al., 2003). IGH rearrangements were present in 17 (77%) of 22 NHRD tumors, but only nine (35%) of 26 HRD tumors. More strikingly, four (4p16, FGFR3/MMSET; 11q13, CCND1; 16q23, MAF; 20q12, MAFB) of the five recurrent IGH rearrangements were present in 13 (59%) of the 22 NHRD tumors [18(67%) of 27 assuming sample selection excluded the analysis of five tumors with a t(11;14)], but only three (12%) of 25 HRD tumors. In contrast, IGH rearrangements were identified in 43 (91%) of 47 HMCL, and the five recurrent rearrangements were identified in 34 (72%) of 47 HMCL (Table 2), consistent with the hypothesis that most HMCL are generated from NHRD tumors (Bergsagel et al., 2005).
Excluding the five recurrent IGH rearrangements, there are 19 additional IGH rearrangements in 13 (27%) of the 48 tumors. Seven (37%) of these 19 rearrangements involve a MYC gene. However, unlike the recurrent IGH rearrangements, these IGH rearrangements have a similar prevalence in NHRD and HRD tumors, with 11 in seven (32%) of 22 tumors (0.50/tumor) and 8 in six (23%) of 26 tumors (0.32/tumor), respectively. Significantly, reexamination of SKY analyses on 150 MM tumors identified apparent IGH rearrangements not involving the five recurrent partners with the same prevalence (0.28 per tumor) in HRD and NHRD tumors (Sawyer et al., 1998, 2001). In addition to the 35 IGH rearrangements that affect the five recurrent partners, there are 33 additional IGH rearrangements in 27 of the 47 HMCL (0.72/HMCL). Nineteen (56%) of these 33 rearrangements involve a MYC gene. The higher prevalence of IGH rearrangements that do not involve one of the five recurrent partners in HMCL compared to primary tumors is consistent with the hypothesis that they represent secondary IGH rearrangements that occur during tumor progression.
Prevalence of IG Light Chain Rearrangements in MM Tumors and Cell Lines
We detected 10 IG light chain (nine immunoglobulin lambda IGL) rearrangements in eight (17%) of the 48 MM tumors (0.21/tumor), with five each in NHRD and HRD tumors. Seven rearrangements involved the MYC gene, and one involved CCND3, a recurrent IGH translocation partner. In addition, we detected 13 IG light chain (12 IGL) rearrangements in nine (19%) of the 47 HMCL (0.28/HMCL). Five IG light chain rearrangements involved MYC, one involved NMYC, and two (MAF and MAFB) are recurrent IGH translocation partners. The IG light chain rearrangements are three- to fivefold less prevalent than IGH rearrangements. Also, immunoglobulin kappa (IGK) rearrangements appear to be ~10-fold less frequent than IGL rearrangements. Although our estimate of IGK rearrangements might be somewhat low since we only did FISH analyses for IGK rearrangements in 20 tumors and 20 HMCL, SKY analyses on 150 MM tumors identified apparent rearrangements of IGL in 24 tumors (15 involving MYC) and IGK in two tumors (both involving MYC) (Sawyer et al., 1998, 2001).
Prevalence of MYC Rearrangements in MM
MYC rearrangements were identified in 22 (46%) of 48 MM tumors, and were equally distributed between the NHRD (43%) and HRD (48%) groups, a result that is consistent with reported SKY analyses on 150 MM tumors (Sawyer et al., 1998, 2001). Among the 22 MYC rearrangements (21 MYC and one NMYC), seven involved an IGH locus (32%), six involved an IGL locus (27%), one involved an IGK locus (2%), and eight did not appear to involve an IG locus (32%). The recurrent involvement of 1p11~13 (tumors #4, 5, and 43; plus six published tumors), and to a lesser extent 6q21 (tumors #37 and 47; plus five published tumors) (Lewis and MacKenzie, 1984; Sawyer et al., 1998, 2001; Smadja et al., 1998), are the only apparent recurrent non-IG chromosomal loci involved in MYC rearrangements. MYC rearrangements were identified in 41 (87%) of 47 HMCL, although two additional HMCL (EJM, XG-6) had MYC rearrangements detected by array CGH (Dib et al., in press). Among the 41 MYC rearrangements (39 MYC, one LMYC, and one NMYC), 19 rearrangements involved the IGH locus (46%), five involved the IGL locus (12%), one involved the IGK locus (2%), and 16 had no apparent involvement of an IG locus (39%). These MYC rearrangements have additional complexities, often with involvement of three chromosomes and sometimes with associated amplification, duplication, or inversion (Shou et al., 2000) (also Tables 1 and 2). More than a third of MYC abnormalities detected in the MM tumors and HMCL would not have been detected by either conventional cytogenetic or SKY analyses, and only half of the rearrangements not involving an IG locus would have been detected by interphase FISH analyses using probes that flank the MYC locus.
Anatomy of IG Rearrangements
As described earlier, we have classified nonphysiological rearrangements of the IG loci into four groups: classical balanced rearrangements (B); variant rearrangements (V); unbalanced but nonvariant rearrangements (U); and insertions (I). Balanced rearrangements can be either reciprocal translocations or inversions (e.g., IGK/NMYC inversion in PE-2, Table 2). We use the term “variant” IGH rearrangement when there is no dissociation of V and C probes on either derivative chromosome, as depicted in Figure 1. This variant configuration has not been described for primary IG translocations in other kinds of B cell tumors, and has not been found among the 52 IGH rearrangements that involve the five recurrent partners in MM tumors and HMCL (Tables 3 and 4). However, for rearrangements not involving the five recurrent partners, we find that eight of 53 IGH rearrangements have the variant configuration.
TABLE 3.
Anatomy of IG and MYC Rearrangements in 48 MM Tumors
| Rearrangements | # | B | U | V | I | %B | %V+I |
|---|---|---|---|---|---|---|---|
| IGH.5 recurrent | 17 | 15 | 1 | 0 | 1 | 88 | 6 |
| IGH.MYC | 7 | 2 | 0 | 1 | 4 | 29 | 71 |
| IGH. Other | 12 | 4 | 3 | 2 | 3 | 33 | 42 |
| IGL/IGK | 10 | 1 | 4 | 1 | 4 | 10 | 50 |
| IGL/IGK.MYC | 7 | 1 | 1 | 1 | 4 | 14 | 71 |
#Number of rearrangements. As described in the Results, the different kinds of rearrangements are: B, balanced; U, unbalanced; V, variant; and I, insertion.
TABLE 4.
Anatomy of IG and MYC Rearrangements in 47 HMCL
| Rearrangements | # | B | U | V | I | %B | %V+I |
|---|---|---|---|---|---|---|---|
| IGH.5 recurrent | 35 | 26 | 7 | 0 | 2 | 74 | 6 |
| IGH.MYC | 19 | 6 | 3 | 2 | 8 | 32 | 53 |
| IGH. Other | 15 | 2 | 4 | 3 | 6 | 13 | 60 |
| IGL/IGK | 13 | 4 | 3 | 0 | 6 | 31 | 46 |
| IGL/IGK.MYC | 6 | 1 | 2 | 0 | 3 | 17 | 50 |
#Number of rearrangements. As described in the Results, the different kinds of rearrangements are: B, balanced; U, unbalanced; V, variant; and I, insertion.
There is a striking difference in the distribution of the four kinds of rearrangements for IGH translocations involving the five recurrent partners compared to all other IGH and IG light chain rearrangements (Tables 3 and 4). The former have a much higher proportion of balanced translocations, and a much lower proportion of rearrangements having the variant or insertion configuration.
Multiple Independent Aberrant IG Rearrangements in a Tumor Cell
Two or more independent IGH rearrangements have been thought to be rare in MM tumors (Avet-Loiseau et al., 2002). However, we found nine of 48 advanced MM tumors and 19 of 47 HMCL with two or more IGH rearrangements, and five HMCL with three or more different IGH rearrangements. Coincident IGH and IG light chain rearrangements in the same tumor cell also occur, with one tumor (#7) having rearrangements that involve each of the three IG loci.
Although our finding that nearly 20% of advanced MM tumors have two IGH rearrangements is unexpected, we note that in one study, two of 59 MGUS/SMM tumors were found to have two different IGH rearrangements (Fonseca et al., 2002). For most tumors with two different IGH rearrangements, there is involvement of one of the five recurrent IGH partners plus a MYC gene (Tables 1 and 2). Rarely, however, two of the five different recurrent IGH partners are involved in IGH rearrangements in the same tumor cell, i.e., t(4;14) plus insertion of 3′Eα near CCND1 in tumor #47 (Table 1); and t(4;14) plus t(14;16) in KMS-11 HMCL (Table 2). Other published examples include two MGUS/SMM tumors with t(4;14) plus t(14;16) or t(4;14) plus a fraction of cells with t(11;14) (Fonseca et al., 2002); and t(14;16) plus t(6;14) in one of 150 MM tumors analyzed by SKY (Sawyer et al., 2001). A sixth MM tumor most likely has t(11;14) plus t(14;20) based on the marked over-expression of CCND1 and MAFB by microarray analysis (Shaughnessy, unpublished). Thus, all combinations of the three types of recurrent rearrangements (4p + a MAF gene or a CCND gene, and a CCND gene plus a MAF gene) have been seen, suggesting that there can be at least partial complementation between these different translocation partners.
Timing of IG and MYC Rearrangements in MM
IGH rearrangements that involve the five recurrent partners in MM are thought to represent primary translocations that are mediated by errors in IGH switching or somatic hypermutation as B cells pass through germinal centers (Bergsagel and Kuehl, 2001; Gabrea et al., 2006). We have summarized detailed evidence supporting the hypothesis that MYC (MYC ≫ NMYC > LMYC) rearrangements in MM, either with or without juxtaposition to an IG locus, are very late progression events that occur at a time when tumors are becoming more proliferative and less stromal cell dependent (Dib et al., in press). The findings reported here support this hypothesis, since MYC rearrangements were identified in nearly 90% of HMCL and about 45% of advanced, intramedullary MM tumors, most of which have a high proliferation index as determined from expression arrays. By contrast, it has been reported that MYC rearrangements are rare in MGUS or smoldering MM (SMM), but are present in about 15% of MM tumors at the time of diagnosis (Avet-Loiseau et al., 2001b). Therefore, MYC rearrangements provide a paradigm for secondary genomic rearrangements not involving the mechanisms responsible for B-cell-specific DNA modifications.
We have shown here that the five recurrent IGH rearrangements are mostly simple balanced translocations, whereas all other IGH, IG light chain, and MYC rearrangements are mostly insertions, and variant or unbalanced translocations. Moreover, in contrast to the five recurrent IGH rearrangements that occur predominantly in NHRD tumors, all other IGH, IG light chain, and MYC rearrangements occur with a similar frequency in NHRD and HRD tumors. Unlike MYC rearrangements, IG light chain rearrangements and IGH rearrangements not involving one of the five recurrent partners occur with a significant frequency in MGUS (Fonseca et al., 2002). Therefore, we suggest that IG light chain rearrangements, IGH rearrangements not involving one of the five recurrent partners, and perhaps some IGH rearrangements involving the five recurrent partners (especially tumors with two recurrent IGH translocations, see above) represent secondary rearrangements that can occur at all stages of pathogenesis, including MGUS tumors (Fig. 5).
Figure 5.
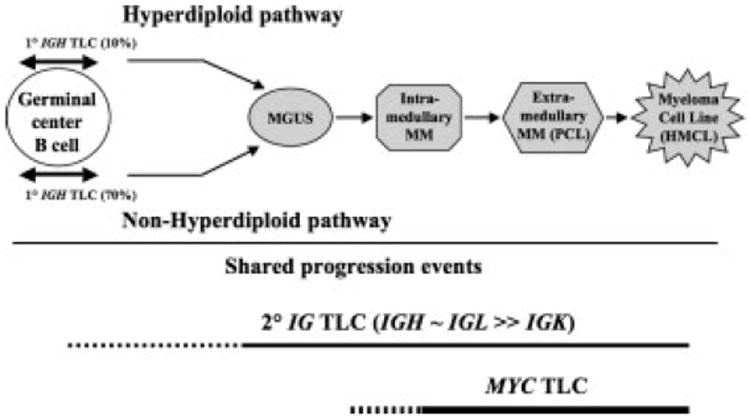
Primary and secondary genomic rearrangements during MM pathogenesis. There are two proposed pathways of pathogenesis for MM: hyperdiploid (HRD) and nonhyperdiploid (NHRD). Primary translocations that are mediated mainly by errors in IGH switch recombination or somatic hypermutation in germinal center B cells occur mostly in NHRD tumors. Secondary IG genomic rearrangements (TLC) that can occur at all stages of pathogenesis and MYC genomic rearrangements that occur at late stages of pathogenesis have a have a similar prevalence in HRD and NHRD tumors.
Supplementary Material
Acknowledgments
Supported by: National Cancer Institute; Grant numbers: CA55819, CA97513; Center for Cancer Research (Intramural Research Program of the NIH, National Cancer Institute); Lebow Fund to Cure Myeloma, Nancy and Stephen Grand Philanthropic Fund.
Footnotes
This article is a US Government work and, as such, is in the public domain in the United States of America.
This article contains Supplementary Material available at www.interscience.wiley.com/jpages/1045-2257/suppmat.
References
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, Dave S, Hurt EM, Tan B, Zhao H, Stephens O, Santra M, Williams DR, Dang L, Barlogie B, Shaughnessy JD, Jr, Kuehl WM, Staudt LM. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avet-Loiseau H, Daviet A, Brigaudeau C, Callet-Bauchu E, Terre C, Lafage-Pochitaloff M, Desangles F, Ramond S, Talmant P, Bataille R. Cytogenetic, interphase, and multicolor fluorescence in situ hybridization analyses in primary plasma cell leukemia: A study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myelome and the Groupe Francais de Cytogenetique Hematologique. Blood. 2001a;97:822–825. doi: 10.1182/blood.v97.3.822. [DOI] [PubMed] [Google Scholar]
- Avet-Loiseau H, Gerson F, Margrangeas F, Minvielle S, Harousseau J-L, Bataille R. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood. 2001b;98:3082–3086. doi: 10.1182/blood.v98.10.3082. [DOI] [PubMed] [Google Scholar]
- Avet-Loiseau H, Facon T, Grosbois B, Magrangeas F, Rapp MJ, Harousseau JL, Minvielle S, Bataille R. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation. Blood. 2002;99:2185–2191. doi: 10.1182/blood.v99.6.2185. [DOI] [PubMed] [Google Scholar]
- Bergsagel PL, Kuehl WM. Chromosomal translocations in multiple myeloma. Oncogene. 2001;20:5611–5622. doi: 10.1038/sj.onc.1204641. [DOI] [PubMed] [Google Scholar]
- Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J., Jr Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106:296–303. doi: 10.1182/blood-2005-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Bergsagel PL, Shonukan OO, Martelli ML, Brents LA, Chen T, Schrock E, Ried T, Kuehl WM. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood. 1998a;91:4457–4463. [PubMed] [Google Scholar]
- Chesi M, Nardini E, Lim RSC, Smith KD, Kuehl WM, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998b;92:3025–3034. [PubMed] [Google Scholar]
- Chng WJ, Ketterling RP, Fonseca R. Analysis of genetic abnormalities provides insights into genetic evolution of hyperdiploid myeloma. Genes Chromosomes Cancer. 2006;45:1111–1120. doi: 10.1002/gcc.20375. [DOI] [PubMed] [Google Scholar]
- Chng WJ, Kumar S, Vanwier S, Ahmann G, Price-Troska T, Henderson K, Chung TH, Kim S, Mulligan G, Bryant B, Carpten J, Gertz M, Rajkumar SV, Lacy M, Dispenzieri A, Kyle R, Greipp P, Bergsagel PL, Fonseca R. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007;67:2982–2989. doi: 10.1158/0008-5472.CAN-06-4046. [DOI] [PubMed] [Google Scholar]
- Dib A, Gabrea A, Glebov O, Bergsagel PL, Kuehl WM. Characterization of MYC translocations in multiple myeloma cell lines. J Natl Cancer Inst. doi: 10.1093/jncimonographs/lgn011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca R, Bailey RJ, Ahmann GJ, Rajkumar SV, Hoyer JD, Lust JA, Kyle RA, Gertz MA, Greipp PR, Dewald GW. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood. 2002;100:1417–1424. [PubMed] [Google Scholar]
- Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA, Dewald GW, Van Ness B, Van Wier SA, Henderson KJ, Bailey RJ, Greipp PR. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003a;101:4569–4575. doi: 10.1182/blood-2002-10-3017. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Debes-Marun CS, Picken EB, Dewald GW, Bryant SC, Winkler JM, Blood E, Oken MM, Santana-Davila R, Gonzalez-Paz N, Kyle RA, Gertz MA, Dispenzieri A, Lacy MQ, Greipp PR. The recurrent IgH translocations are highly associated with non-hyperdiploid variant multiple myeloma. Blood. 2003b;102:2562–2567. doi: 10.1182/blood-2003-02-0493. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel PL, Chesi M, Davies FE, Drach J, Greipp PR, Kirsch IR, Kuehl WM, Hernandez JM, Minvielle S, Pilarski LM, Shaughnessy JD, Jr, Stewart AK, Avet-Loiseau H. Genetics and cytogenetics of multiple myeloma: A workshop report. Cancer Res. 2004;64:1546–1558. doi: 10.1158/0008-5472.can-03-2876. [DOI] [PubMed] [Google Scholar]
- Gabrea A, Bergsagel PL, Chesi M, Shou Y, Kuehl WM. Insertion of excised IgH switch sequences causes overexpression of cyclin D1 in a myeloma tumor cell. Mol Cell. 1999;3:119–123. doi: 10.1016/s1097-2765(00)80180-x. [DOI] [PubMed] [Google Scholar]
- Gabrea A, Leif Bergsagel P, Michael Kuehl W. Distinguishing primary and secondary translocations in multiple myeloma. DNA Repair. 2006;5:1225–1233. doi: 10.1016/j.dnarep.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX, Fogle H, Price-Troska T, Ahmann G, Mancini C, Brents LA, Kumar S, Greipp P, Dispenzieri A, Bryant B, Mulligan G, Bruhn L, Barrett M, Valdez R, Trent J, Stewart AK, Carpten J, Bergsagel PL. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehl WM, Bergsagel PL. Multiple myeloma: Evolving genetic events and host interactions. Nat Rev Cancer. 2002;2:175–187. doi: 10.1038/nrc746. [DOI] [PubMed] [Google Scholar]
- Lewis JP, MacKenzie MR. Non-random chromosomal aberrations associated with multiple myeloma. Hematol Oncol. 1984;2:307–317. doi: 10.1002/hon.2900020402. [DOI] [PubMed] [Google Scholar]
- Kuehl M, Shaughnessy JD, Sawyer JR, Lukacs JL, Munshi N, Desikan KR, Singhal S, Mehta J, Siegel D, Shaughnessy J, Barlogie B. Identification of new nonrandom translocations in multiple myeloma with multicolor spectral karyotyping. Blood. 1998;92:4269–4278. [PubMed] [Google Scholar]
- Sawyer JR, Lukacs JL, Thomas EL, Swanson CM, Goosen LS, Sammartino GJCG, Munshi NC, Tricot G, Shaughnessy JD, Jr, Barlogie B. Multicolour spectral karyotyping identifies new translocations and a recurring pathway for chromosome loss in multiple myeloma. Brit J Haematol. 2001;112:167–174. doi: 10.1046/j.1365-2141.2001.02546.x. [DOI] [PubMed] [Google Scholar]
- Shaughnessy J, Gabrea A, Qi Y, Brents LA, Zhan F, Tian E, Sawyer J, Barlogie B, Bergsagel PL, Kuehl WM. Cyclin D3 at 6p21 is dysregulated by recurrent Ig translocations in multiple myeloma. Blood. 2001;98:217–223. doi: 10.1182/blood.v98.1.217. [DOI] [PubMed] [Google Scholar]
- Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, Dewald G, Kirsch IR, Bergsagel PL, Kuehl WM. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci USA. 2000;97:228–233. doi: 10.1073/pnas.97.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smadja NV, Bastard C, Brigaudeau C, Leroux D, Fruchart C. Hypodiploidy is a major prognostic factor in multiple myeloma. Blood. 2001;98:2229–2238. doi: 10.1182/blood.v98.7.2229. [DOI] [PubMed] [Google Scholar]
- Smadja NV, Fruchart C, Isnard F, Louvet C, Dutel JL, Cheron N, Grange MJ, Monconduit M, Bastard C. Chromosomal analysis in multiple myeloma: Cytogenetic evidence of two different diseases. Leukemia. 1998;12:960–969. doi: 10.1038/sj.leu.2401041. [DOI] [PubMed] [Google Scholar]
- Smadja NV, Leroux D, Soulier J, Dumont S, Arnould C, Taviaux S, Taillemite JL, Bastard C. Further cytogenetic characterization of multiple myeloma confirms that 14q32 translocations are a very rare event in hyperdiploid cases. Genes Chromosomes Cancer. 2003;38:234–239. doi: 10.1002/gcc.10275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.