

Abstract
Linus Pauling proposed that the large rate accelerations for enzymes are due to the high specificity of the protein catalyst for binding the reaction transition state. The observation that stable analogs of the transition states for enzymatic reactions often act as tight-binding binding inhibitors provided early support for this simple and elegant proposal. We review experimental results which support the proposal that Pauling’s model provides a satisfactory explanation for the rate accelerations for many heterolytic enzymatic reactions through high energy reaction intermediates, such as proton transfer and decarboxylation. Specificity in transition state binding is obtained when the total intrinsic binding energy of the substrate is significantly larger than the binding energy observed at the Michaelis complex. The results of recent studies to characterize the specificity in binding of the enolate oxygen at the transition state for the 1,3-isomerization reaction catalyzed by ketosteroid isomerase are reviewed. Interactions between pig heart succinyl-CoA:3-oxoacid coenzyme A transferase (SCOT) and the nonreacting portions of CoA are responsible for a rate increase of 3 × 1012-fold, which is close to the estimated total 5 × 1013-fold enzymatic rate acceleration. Studies that partition the interactions between SCOT and CoA into their contributing parts are reviewed. Interactions of the protein with the substrate phosphodianion group provide a ca. 12 kcal/mol stabilization of the transition state for the reactions catalyzed by triosephosphate isomerase, orotidine 5′-monophosphate decarboxylase and α-glycerol phosphate dehydrogenase. The interactions of these enzymes with the substrate piece phosphite dianion provide a 6 – 8 kcal/mol stabilization of the transition state for reaction of the appropriate truncated substrate. Enzyme activation by phosphite dianion reflects the higher dianion affinity for binding to the enzyme-transition state complex compared with the free enzyme. Evidence is presented that supports a model in which the binding energy of the phosphite dianion piece, or the phosphodianion group of the whole substrate, is utilized to drive an enzyme conformational change from an inactive open form EO to an active closed form EC, by closure of a phosphodianion gripper loop. Members of the enolase and haloalkanoic acid dehalogenase superfamilies use variable capping domains to interact with nonreacting portions of the substrate and sequester the substrate from interaction with bulk solvent. Interactions of this capping domain with the phenyl group of mandelate have been shown to activate mandelate racemase for catalysis of deprotonation of α-carbonyl carbon. We propose that an important function of these capping domains is to utilize the binding interactions with nonreacting portions of the substrate to activate the enzyme for catalysis.
Linus Pauling proposed that the large rate accelerations for enzymatic reactions arise from the high specificity of the protein catalyst for binding the reaction “activated complex”, or transition state, as it is now known.1,2 There is abundant evidence to support this proposal. Stable analogs of the transition states for the reactions catalyzed by triosephosphate isomerase,3–5 proline racemase,6,7 adenosine deaminase,8 and many other enzymes, are tight-binding enzyme inhibitors.9–11 X-ray crystal structures show that enzyme-ligand complexes are stabilized by elaborate networks of protein-ligand interactions that appear sufficiently strong to rationalize enzymatic rate accelerations. Enzymes that operate on small substrates such as ethanol, acetaldehyde or the acetyl group have evolved effective strategies to increase the total substrate binding energy. For example, by recruiting a high molecular weight cofactor such as TPP, NAD/H or PLP, or by attaching a large molecular weight moiety such as coenzyme A, or a charged moiety such as a phosphodianion or pyrophosphotranion group, to the substrate.
The Pauling formalism provides a framework for the development of detailed models for enzyme catalysis that can be tested in experimental and computational studies. It is less useful when transition state theory fails,12,13 but there are no well-documented examples of this for enzyme catalysis. Modern theory has been used to support alternative models that rationalize enzyme catalysis, such as the proposals that interactions between the protein and substrate lower the reaction barrier by modifying the reaction potential energy surface,14 or that enzymatic reactions are accelerated by coupling motion at the protein catalyst to motion along a vibrational mode on the reaction coordinate from substrate to product.14,15 This review highlights experimental results from our laboratory which show that transition state stabilization is central to enzyme catalysis. We do not discuss other models, because the Pauling model provides a satisfactory framework for the rationalization of our experimental observations.
Specificity in Transition State Binding
X-ray crystal structures of liganded enzymes would seem to place in plain view everything that is needed to understand the catalytic rate acceleration. These complexes reveal most, or all, of the important protein-ligand interactions, but this information is less useful than it appears at first glance. The ligand binding energy required to account for an enzymatic rate acceleration is generally so large that it cannot be expressed entirely at the ground state Michaelis complex, because this would result in effectively irreversible ligand binding.16 Consider the decarboxylation of orotidine 5′-monophosphate (OMP) to form uridine 5′-monophosphate (UMP) catalyzed by orotidine 5′-monophosphate decarboxylase (OMPDC, Scheme 1). This enzyme provides a ~31 kcal/mol stabilization of the transition state for the decarboxylation of OMP through a vinyl carbanion intermediate (Figure 1),17,18 which corresponds to KS‡ ≈ 10−23 M for release of the transition state to water.19 If the second-order rate constant for ligand binding is close to the encounter-controlled limit of kd = 108 M−1 s−1,20 then the absolute rate constant for release from OMPDC of a ligand with the same affinity as the transition state would be k−d = KSkd ≈ (10−23 M)(108 M−1 s−1) ≈ 10−15 s−1, and the halftime for ligand release would be on the order of 20 million years! In fact, OMPDC provides a much smaller 8 kcal/mol stabilization of the Michaelis complex with OMP (KS ≈ 1 μM),21 which is formed rapidly and reversibly, so that more than 70% of the intrinsic substrate binding energy is expressed only on moving from the ground state to the transition state (Figure 1).
Scheme 1.
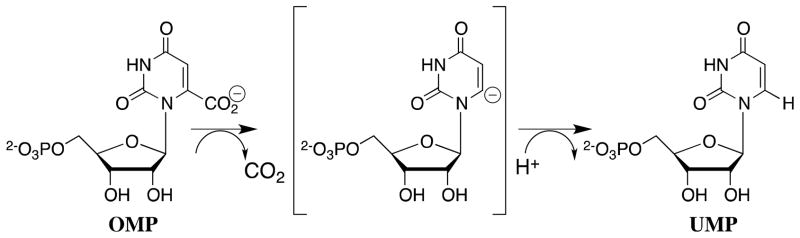
Figure 1.

Reaction coordinate profiles for the nonenzymatic and the OMPDC-catalyzed decarboxylation of OMP to form UMP through a vinyl carbanion-like transition state. The total stabilization of the transition state by interactions with the protein catalyst is estimated to be 31 kcal/mol,19 which is much larger than the observed binding energy for OMP in the ground state Michaelis complex of 8 kcal/mol.
There are no X-ray crystal structures of the short-lived transition state complexes for enzymatic reactions for comparison with the structure of the corresponding Michaelis complex. The structures of complexes with stable ligands, including transition state analogs, provide limited insight into the explanation for the large differences in the enzymatic stabilization of ground state and transition state complexes. For example, structures of OMPDC complexed with the product UMP or tight-binding inhibitors reveal an extensive network of enzyme-ligand interactions that may be sufficient to account for the 31 kcal/mol transition state binding energy.22–30 These structures have generated a great deal of discussion, but they were not sufficient to define the mechanism for the decarboxylation reaction, or to explain the specificity of OMPDC in transition state binding.31
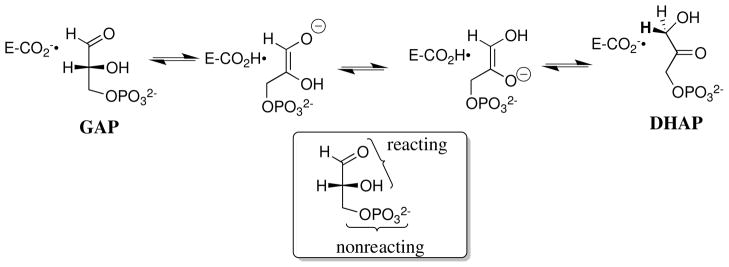
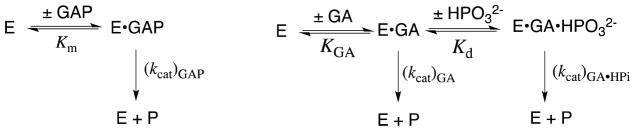
Our discussion of specificity in transition state binding is divided into two sections, as can be illustrated for the isomerization of glyceraldehyde 3-phosphate (GAP) to form dihydroxyacetone phosphate (DHAP) through a pair of enediolate reaction intermediates,32,33 catalyzed by triosephosphate isomerase (TIM, Scheme 2).34,35 The first section describes experiments that characterize the specificity in transition state binding resulting from interactions of the enzyme with the reacting portion of the substrate that undergoes substantial changes in charge, geometry or polarity on moving from the reactant to transition state. For example, the unit increase in negative charge at GAP upon conversion of the substrate to the high-energy enediolate intermediate at TIM (Scheme 2).34,36 The second section discusses experiments that characterize the specificity in transition state binding resulting from interactions of the enzyme with nonreacting substrate fragments, such as the phosphodianion group of GAP (Scheme 2).
Scheme 2.
Specificity in Transition State Binding: Interactions with the Reacting Center
Heterolytic bond cleavage/formation results in a redistribution of charge at the reactant on proceeding to the transition state. Large catalytic rate accelerations for both nonenzymatic and enzymatic reactions are obtained by the placement of groups that provide stabilizing electrostatic, hydrogen bonding or other polar interactions at the site of changing charge.37 For example, Zn2+ provides modest catalysis of the deprotonation of α-carbonyl carbon, by stabilizing the negative change that develops at the enolate oxygen of the delocalized carbanion product.38 Transition state stabilization is largest, and most easily characterized, when the interacting centers carry multiple charges, such as for a metal dication and a phosphodianion group.37
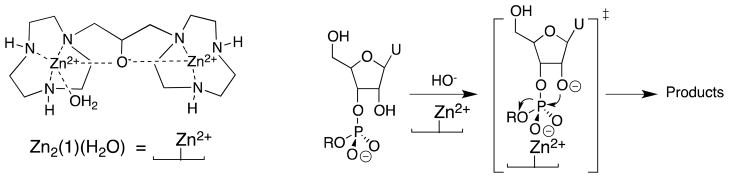
The enzyme-like 9.3 kcal/mol stabilization of the anionic transition state for the base-catalyzed cleavage of the dinucleotide uridylyl-(3′-5′)-uridine and related phosphate diesters by interactions with the cationic dinuclear Zn2+ catalyst Zn2(1)(H2O) (Scheme 3) corresponds to an impressive 107-fold catalytic rate acceleration.39–41 The large 105-fold effect of the K12G mutation at yeast TIM on kcat/Km for the isomerization of GAP shows that interactions between an amine monocation and the transition state for formation of the enediolate phosphate trianion intermediate (Scheme 2) makes a large contribution to the overall enzymatic rate acceleration.42,43
Scheme 3.
It is difficult to characterize the smaller transition state stabilization from hydrogen bonding interactions between enzymes and a carbonyl oxygen that is transformed to an oxyanion by nucleophilic addition to the carbonyl group, or by deprotonation of α-carbonyl carbon. The X-ray crystal structures of enzyme-ligand complexes of trypsin and chymotrypsin suggest that two backbone amide NH groups form hydrogen bonds that stabilize a tetrahedral oxyanion intermediate.44,45 This arrangement of hydrogen bond donors, commonly known as an oxyanion hole, has been observed for enzymes that catalyze the hydrolysis of amides46 and esters,47 and also the deprotonation of α-carbonyl carbon.48–50 There is no simple protocol for the excision of interactions between a protein backbone amide NH and the transition state for these enzyme-catalyzed reactions. However, the amide side chain of Asn-155 provides one of the hydrogen-bonding groups for the oxyanion intermediate of the reaction catalyzed by subtilisin, and this side chain has been varied by site directed mutagenesis.51,52
Oxyanion holes might appear to provide little stabilization of the transition state for an uncatalyzed reaction in water, because hydrogen bonds to the weakly acidic backbone amide NH groups are not expected to be stronger than hydrogen bonds to water. However, Jencks argued that there is a substantial entropic advantage to oxyanion formation at preorganized enzyme active sites, because this eliminates the loss in the translational and rotational entropy of three solvent molecules that occurs when the oxyanion is generated in aqueous solution.16 Warshel emphasized that the water dipoles, which stabilize the developing oxyanion at a transition state for reaction in water, are randomly oriented compared with the preorganized dipoles at an oxyanion hole.53 He has proposed that the lower barrier for the enzymatic reaction represents the elimination of the energetic cost for dipole reorganization that is needed for the nonenzymatic reaction.53,54
3-Oxo-Δ5-steroid isomerase (KSI) catalyzes the reaction shown in Scheme 4, proceeding through a dienolate intermediate that is stabilized by hydrogen bonding to the phenolic side chain of Tyr-16 and the carboxylic acid side chain of Asp-103 (numbering for the enzyme from Pseudomonas putida).55 This hydrogen-bonding network resembles the oxyanion hole of proteases and esterases, but the overall stabilizing interactions should be larger, because of the greater acidity of the amino acid side chains at KSI compared with that of a backbone amide NH group.
Scheme 4.
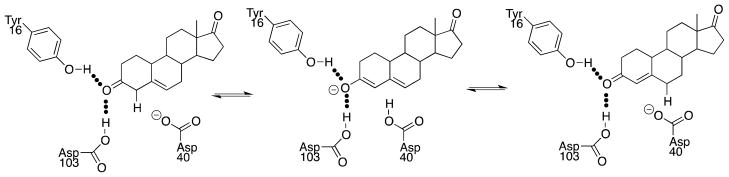
The intramolecular proton transfer catalyzed by KSI is mediated by the carboxylate side chain of Asp-40. The second-order rate constant kcat/Km = 3 × 108 M−1 s−1 for KSI-catalyzed isomerization (Scheme 4)56 is 5 × 1011-fold larger than kAcO = 6 × 10−4 M−1 s−1 for the corresponding deprotonation of the substrate by acetate ion in aqueous solution.57 This shows that the catalytic base at KSI is strongly activated for catalysis by interactions between the substrate and enzyme. The binding interactions of the nonreacting rings of the steroid substrate contribute 3 × 104-fold to this total effect.58 The remaining rate acceleration of ca. 2 × 107-fold is presumably due to either the enhancement of Brønsted base catalysis by Asp-40,59 or to the stabilization of negative charge at the enolate oxygen anion by interactions with the amino acid side chains at the oxyanion hole.
The contribution of the hydrogen bonding in the oxyanion hole to the catalytic rate acceleration for KSI has been probed by site directed mutagenesis. The 50,000-fold falloff in kcat for the Y16F mutant of KSI from Pseudomonas putida is reduced to 200-fold for the Y16G, Y16A, Y16S and Y16T mutants.60 It was concluded that the elimination of the hydrogen bond by the Y16F mutation results in a destabilizing interaction between the intermediate oxyanion and the hydrophobic phenyl ring of phenylalanine, and that this destabilization is attenuated for the Y16G, Y16A, Y16S and Y16T mutants.60 The Y16F/D103L double mutation at KSI from P. putida and the Y16F/D103A double mutation at the enzyme from Comamonas testosteroni result in 105- and 108-fold decreases, respectively, in kcat/Km for enzyme-catalyzed isomerization.61,62 These large effects overestimate the magnitude of the stabilizing hydrogen-bonding interactions at KSI compared to those in water, because of the minimal hydrogen-bonding stabilization of the oxyanion at the active site of the mutant enzyme compared with an aqueous solvent. Additional mutations at double mutants of KSI from P. putida and C. testosteroni were made to carve out a ca. 650 Å3 cavity at the enzyme active site that is sufficiently large to hold 15 – 17 water molecules.49 One of these mutant enzymes exhibited only a 1000-fold falloff in kcat/Km compared with wildtype KSI.49 This 1000-fold effect is the best available estimate of the contribution of the oxyanion hole at KSI to the overall enzymatic rate acceleration.
Specificity in Transition State Binding: Interactions with Nonreacting Substrate Fragments
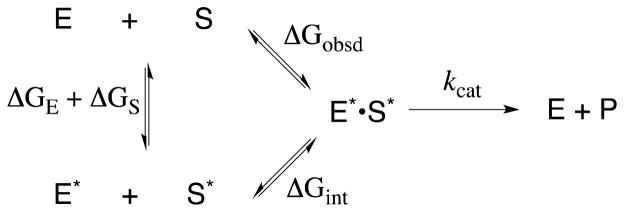
In a classic review published in 1975,16 Bill Jencks considered the possibility that “the intrinsic binding energy that results from the noncovalent interaction of a specific substrate with the active site of the enzyme is considerably larger than is generally believed. An important part of this binding energy may be utilized to provide the driving force for catalysis, so that the observed binding energy represents only what is left over after this utilization.” He went on to propose that “the availability of this energy from binding interactions with nonreacting portions of specific substrates provides a critical difference between enzymic and most nonenzymic catalysis.”16 Specificity in the expression of binding interactions with nonreacting portions of the substrate at an enzymatic transition state results when these interactions drive thermodynamically unfavorable changes in the conformation of the enzyme or substrate that result in an increase in kcat for conversion of the enzyme-substrate complex to product. This is shown in Scheme 5,16 where ΔGE and ΔGS are the unfavorable energetic prices paid to move the enzyme and substrate, respectively, into a conformation with an increased chemical reactivity, ΔGobsd is the observed substrate binding energy, and ΔGint is the total or intrinsic substrate binding energy. The intrinsic substrate binding energy is larger than the observed binding energy by the amount of binding energy that is specifically utilized to drive unfavorable changes in the conformation of the enzyme and/or the substrate, resulting in the conversion of E to E* and S to S* (ΔGE + ΔGS, eq 1). The binding interactions that are utilized in this manner are not expressed as tighter overall binding and a decrease in the experimental Michaelis constant (for simplicity we treat the binding constant KS as being equal to the Michaelis constant Km), but, rather, result in an increase in the chemical reactivity of the E*•S* complex and in the kinetic parameter kcat, compared with that for the unactivated enzyme-catalyzed reaction.
Scheme 5.
| (1) |
Jencks’ review discussed: (1) The utilization of substrate binding energy to induce electrostatic stress, torsional strain or other types of strain into the substrate (S*, Scheme 5), which is then relieved at the transition state for the catalyzed reaction. (2) The utilization of substrate binding energy to induce electrostatic stress, torsional strain or other types of strain into the enzyme (E*, Scheme 5), which is then relieved at the transition state for the catalyzed reaction. He noted that “the induction of a geometric destabilization requires the application of a force and since neither the enzyme nor the substrate have available irresistible forces or substituent groups that are immovable objects, the induction of strain will cause at least a small change in the conformation of both the substrate and the enzyme to give the structures S* and E*, respectively.” (3) The utilization of substrate binding energy to drive thermodynamically unfavorable desolvation of the enzyme and substrate to give E* and S* that are activated at the E*•S* complex for conversion to reaction product.16
Since 2000, our work has focused on defining the mechanisms by which the binding interactions of the nonreacting substrate phosphodianion group are utilized to activate enzymes for catalysis.63 We review here this work, along with that of Jencks and coworkers that was completed after publication of his 1975 review.
Coenzyme A Binding Interactions
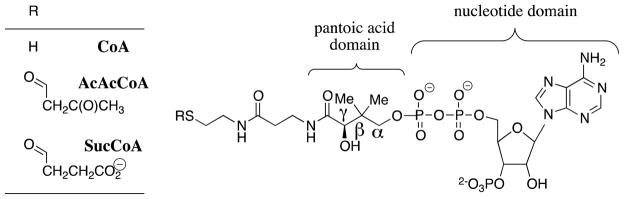
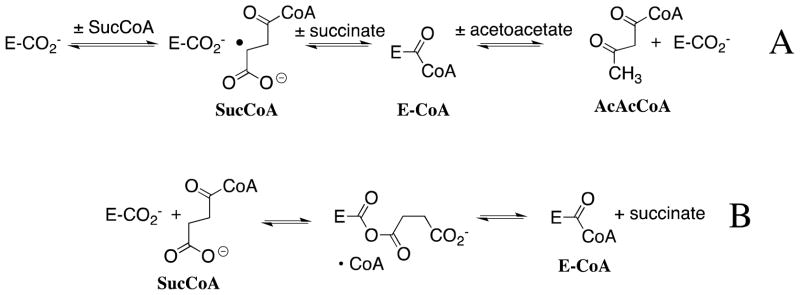
Coenzyme A (CoA, Scheme 6, R = H) functions as an acyl group carrier and is an essential cofactor for all organisms. It serves to activate the carbonyl group for nucleophilic addition and deprotonation at α-carbonyl carbon.64,65 This coenzyme is central to many biochemical transformations and is utilized by an estimated 4% of all known enzymes.66 Succinyl-CoA:3-oxoacid coenzyme A transferase (SCOT) catalyzes the reversible reaction of succinyl-CoA (SucCoA) with acetoacetate to give acetoacetyl-CoA (AcAcCoA) and succinate through a covalent enzyme thiol ester intermediate, E-CoA (Scheme 7A). The CoA transfer reaction catalyzed by class I SCOT from pig heart proceeds by the chemically difficult nucleophilic displacement of a basic thiolate anion by an enzyme carboxylate anion to form an anhydride that partitions between addition of CoA to the substrate-derived carbonyl group to reform substrate and to the enzyme-derived carbonyl group to give the enzyme-based thiol ester intermediate E-CoA (Scheme 7B).67–71
Scheme 6.
Scheme 7.
A comparison of kcat/Km = 9 × 105 M−1 s−1 for transfer of CoA from SucCoA to SCOT to give E-CoA (Scheme 7) and kcat/Km = 3 × 10−7 M−1 s−1 for transfer of the minimal thiol from succinyl-SCH2CH2C(O)OMe shows that interactions between SCOT and the nonreacting nucleotide and pantotheine and nucleotide “domains” of CoA are responsible for a rate acceleration of 3 × 1012-fold,69 which is close to the estimated 5 × 1013-fold total enzymatic rate acceleration. This corresponds to a 17 kcal/mol stabilization of the transition state by binding interactions of SCOT with the nonreacting CoA fragment of SucCoA. By contrast, the large Ki of 1 mM for competitive inhibition of SCOT by CoA, and the small 2-fold difference in the Michaelis constants for the SCOT-catalyzed reactions of acetoacetate and AcAcCoA, show that the observed binding interactions between CoA and the enzyme at the ground state Michaelis complex are weak,72,73 so that these binding interactions are utilized specifically in transition state stabilization.
The strengths of the following specific interactions between SCOT and CoA have been determined:74,75 (1) The interactions between SCOT and the complete CoA fragment that are expressed at the initial Michaelis complex, at the transition state(s) for formation of the E-CoA intermediate, and at the covalent intermediate E-CoA. (2) The interactions between SCOT and the individual nucleotide and pantoic acid domains of CoA that are expressed at the initial Michaelis complex, at the transition state(s) for formation of the E-CoA intermediate, and at the covalent intermediate E-CoA (Scheme 6). The binding interactions with the pantoic acid domain involve only the small region of the CoA molecule that contains the α-β- and γ-carbon atoms of the longer pantotheine moiety (Scheme 6).74
The nucleotide and pantoic acid domains of CoA each provide a small ca. 2 kcal/mol stabilization of the Michaelis complex. However, the nucleotide domain of CoA stabilizes the E-CoA intermediate by 7 kcal/mol, while the pantoic acid domain destabilizes this intermediate by 5 kcal/mol (Figure 2). The strongest stabilizing interactions are observed at the transition states (‡1) and (‡2) for SCOT-catalyzed CoA transfer (Figure 2). Interactions with the nucleotide domain provide a similar 7 – 8 kcal/mol stabilization of both E-CoA and the transition states (‡1) and (‡2) for the half reactions of AcAcCoA and SucCoA. However, the interactions with the pantoic acid domain are stabilizing at these transition states, but destabilizing (!) at the E-CoA covalent intermediate (Figure 2).
Figure 2.

Diagram illustrating the expression of noncovalent binding energy from interaction of SCOT with domains of the CoA moiety at key points along the reaction coordinate, using the free enzyme and substrate as the ground state at a relative free energy of zero (reproduced with permission from: Role of binding energy with coenzyme A in catalysis by 3-oxoacid coenzyme A transferase, Biochemistry 34, 11678 – 11689 (1995), American Chemical Society). The histograms indicate the binding energies derived from interactions with the pantoic acid domain (black bars) and the terminal nucleotide domain of CoA (white bars). The bold black line illustrates the total noncovalent binding energy derived from interaction of the entire nonreacting CoA moiety.
Figure 3 compares the free energy profile for the SCOT-catalyzed transfer of CoA from SucCoA to SCOT to form E-CoA with that for transfer of a hypothetical thiol from SucSR, for which the binding interactions with only the pantoic acid domain have been eliminated. Figure 3 shows that a portion of the binding energy of the pantoic acid domain is used to destabilize the E-CoA intermediate relative to E-SR by 5 kcal/mol (ΔGstrain = (ΔGE + ΔGS), eq 1), while a portion is used to stabilize the transition state for CoA transfer relative to that for SR transfer by ca. 5 kcal/mol (ΔΔG‡). The result is a 10 kcal/mol smaller barrier for the reaction of E-CoA with succinate than for the reaction of E-SR (ΔG‡SR − ΔG‡CoA = 10 kcal/mol, Figure 3). This corresponds to a 107-fold contribution of the small nonreacting pantoic acid domain of CoA to the rate acceleration for SCOT.74,75
Figure 3.

Free energy profiles for the SCOT-catalyzed transfer of CoA from SucCoA to form the E-CoA covalent intermediate (through transition state (‡)CoA), and for the transfer of a hypothetical thiol from SucSR to give E-SR, for which the binding interactions with only the pantoic acid domain have been eliminated (through transition state (‡)SR). The binding interactions between SCOT and the pantoic acid domain stabilize the transition state for transfer of CoA to SCOT by 5 kcal/mol but destabilize the resulting E-CoA intermediate by 5 kcal/mol. The net effect is a 10 kcal/mol smaller barrier for the reverse transfer to succinate of CoA from E-CoA than for the transfer of SR from E-SR.
These differences in the expression of binding interactions at different enzyme complexes require multiple modes for the binding of CoA to SCOT, in which interactions between the nucleotide and pantoic acid domains are expressed differently. For example, the stabilizing interactions between SCOT and the pantoic acid domain of CoA might be expressed first at the rate-determining transition state for formation of the short-lived and thermodynamically unstable anhydride reaction intermediate (Scheme 7B),67–71 while the destabilizing interactions are expressed only after thermodynamically favorable collapse of this complex to form E-CoA. The destabilizing interactions of the pantoic acid domain at E-CoA then serve to attenuate the large driving force for breakdown of the high energy anhydride, preventing E-CoA from lying in a deep thermodynamic well. X-ray structures of crystals of pig heart SCOT grown in the presence of AcAcCoA show subunits of the dimeric protein as both the free enzyme and as the covalently-linked E-CoA intermediate, in a conformation substantially different from that of the free enzyme.76 This change in enzyme conformation affects interactions between SCOT and both the nucleotide and pantoic acid domains of CoA. Molecular mechanics calculations on X-ray crystal structures of wildtype and mutants of SCOT from Acetobactor aceti provide evidence that the thiol ester functional group of substrate is compressed against the catalytic glutamate.77 The results are consistent with the use of the intrinsic CoA binding energy to induce strain into the reactant complex, that is relieved at the transition for formation of the acyl anhydride intermediate.72
Enzymes that catalyze the deprotonation of the α-carbonyl carbon of CoA thiol esters have solved the baffling catalytic challenge of stabilizing the enolate-like transition state and intermediate.78 The acetyl group is too small to provide the large transition state binding energy required to rationalize the rate acceleration for enzyme-catalyzed deprotonation of the α-carbonyl carbon of these carbon acids.79,80 It is expected that the binding interactions between these enzymes and the CoA moiety of the substrate are utilized to stabilize the transition state for carbon deprotonation. However, X-ray crystal structures of these enzymes provide limited insight into the mechanism for transition state stabilization,48,81,82 and we are not aware of studies to determine the mechanism for enzyme activation by interactions with CoA.
Phosphodianion Binding Interactions
We targeted the small charged phosphodianion group as a stable substrate fragment that should show specificity in transition state binding for the following reasons: (1) This group often provides a substantial fraction of the total transition state binding energy needed to account for the enzymatic rate acceleration.63,83 (2) The closure of phosphodianion “gripper loops” of enzymes over the bound substrate provides a starting point for the development of mechanisms to rationalize the utilization of phosphodianion binding energy in enzyme activation. (3) The wide distribution of phosphate gripper loops,84,85 and of phosphate cups,86 suggests a common function for these structural elements, so that it should be possible to generalize the results of studies of representative enzymes to a broad range of catalysts. We have used the following protocols to quantify the stabilization of the transition states for the reactions catalyzed by TIM,63,87 OMPDC83 and α-glycerol phosphate dehydrogenase (α-GPDH)88 by interactions with the substrate phosphodianion group.
The second-order rate constants for the enzyme-catalyzed reaction of the whole substrates were determined to be ca. 108-fold larger than the corresponding rate constants for the enzyme-catalyzed reactions of truncated substrates that lack a phosphodianion group.63,83,88 This shows that the interactions of the enzyme with the substrate phosphodianion stabilize the respective transition states by 11 – 12 kcal/mol. This phosphodianion binding energy may be utilized to anchor the Michaelis complex to the enzyme so that it is expressed in the binding constant, or to activate the enzyme for catalysis, in which case it is expressed specifically at the transition state for the catalyzed reaction, as an increase in kcat.
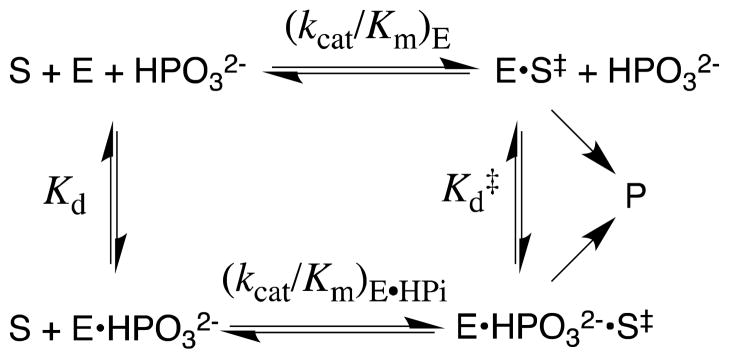
The activation of the enzyme-catalyzed reactions of truncated substrates by added phosphite dianion (HPO32−) was characterized for the reactions catalyzed by TIM, α-GPDH and OMPDC (Scheme 8).83,87,88 Despite the absence of an anchoring covalent connection to the substrate, in each case the binding of HPO32− to the enzyme results in large increases in the observed second-order rate constant (kcat/Km)obsd for the enzyme-catalyzed reaction of the truncated substrate piece. The dependence of (kcat/Km)obsd on [HPO32−] is accounted for by the model in Scheme 9, and the data were used to give values of (kcat/Km)E for the unactivated reaction, Kd for dissociation of phosphite from the E•HPO32− complex, and (kcat/Km)E•HPi for the reaction of the second truncated substrate piece catalyzed by this complex.83,87,88
Scheme 8.

Scheme 9.
| (2) |
Rate constant ratios of (kcat/Km)E•HPi/(kcat/Km)E ≥ 103 were determined for the three enzyme-catalyzed reactions of the “two-part substrates” (Scheme 8A – 8C), which shows that there is a similar strong activation of each enzyme for turnover of the second truncated substrate piece by the binding of phosphite dianion to form the phosphite-liganded enzyme E•HPO32−. This activation reflects the weak binding of HPO32− to the free enzyme (Kd ≥ 0.01 M), but the much higher specificity for binding to the transition state complex (Kd ≫ Kd‡, eq 2).88 The values of Kd‡ were used to calculate total intrinsic phosphite binding energies in the transition state of 6 – 8 kcal/mol, which are 4 – 6 kcal/mol stronger than the binding energies of phosphite dianion at the ground-state Michaelis complex. In other words, the strong interactions between these enzymes and phosphite dianion are utilized specifically to stabilize the respective transition states.83,87–89
Scheme 10 shows the TIM-catalyzed reactions of the whole substrate GAP and the two-part substrate glycolaldehyde (GA) + phosphite. Our results strongly suggest that the reactivities of the binary TIM•GAP complex and the ternary TIM•GA•HPO32− complex are similar [(kcat)GAP ≈ (kcat)GA•HPi], but much higher than that of the binary TIM•GA complex [(kcat)GA]. The 12 kcal/mol total intrinsic phosphodianion binding energy for the whole substrate GAP is larger than the 6 kcal/mol intrinsic phosphite binding energy for the reaction of the substrate pieces.87,88 This difference is due mainly, or entirely, to the entropic advantage provided by the covalent attachment of the pieces at the whole substrate, which undergoes binding to the enzyme with the loss of the translational and rotational entropy of a single molecule, compared with the larger loss in entropy associated with the binding and reaction of two separate pieces.90
Scheme 10.
Enzyme Activation by Phosphite Dianion
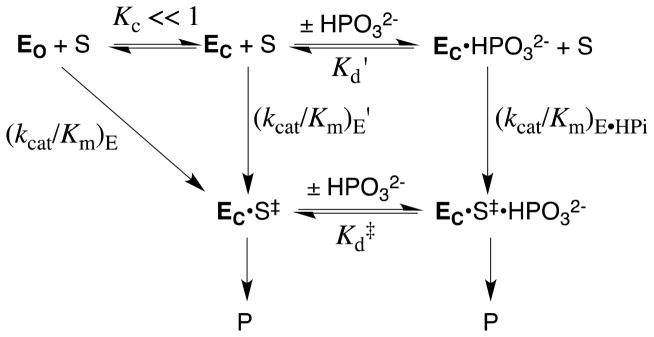
In developing a physical model to rationalize phosphite activation of three radically different enzyme-catalyzed reactions, we decided to emphasize the common catalytic features of OMPDC,23,26,91 TIM,92,93 and α-GPDH.88,94,95 Each enzyme utilizes a flexible loop that interacts with the phosphodianion group of bound substrate, and in each case loop closure over the substrate sequesters the ligand from interaction with bulk solvent. These observations guided our proposal of a minimal general model in which the binding of phosphite dianion drives this loop closure and converts an inactive open enzyme EO to an active closed form EC (Scheme 11).
Scheme 11.
| (3) |
| (4) |
The observed binding of HPO32− to EO to give EC•HPO32− (Scheme 11) is weak (Kd = Kd′/Kc), because much of the ligand binding energy is used to drive the unfavorable enzyme conformational change that converts EO to EC (Kc ≪ 1). Once the cost for the conformational change has been paid, the full intrinsic binding energy of the second ligand is observed in the formation of EC•S‡•HPO32− from EC•HPO32− [barrier for (kcat/Km)E•HPi, Scheme 11], or from EC•S‡ (Kd‡, Scheme 11). This model may be supplanted by enzyme-specific models that consider the energetic cost for moving both the enzyme and the substrate into a reactive conformation (Scheme 5). Such models have been considered in detail for the catalysis of decarboxylation of OMP catalyzed by OMPDC.23,96,97
Enzyme Activation: TIM
Figure 4 shows a free energy diagram that was constructed for Scheme 11 using the kinetic parameters for the unactivated and phosphite-activated TIM-catalyzed reactions of the truncated substrate glycolaldehyde (GA).87 The intrinsic binding energy for association of HPO32− with the transition state complex EC•S‡ to give EC•S‡• HPO32−, ΔGint = −5.8 kcal/mol, is obtained as the difference in the barriers to the activated reaction (defined by (kcat/Km)E•HPi/Kd) and the unactivated reaction (defined by (kcat/Km)E), according to eq 3. The difference between this intrinsic phosphite binding energy and the observed binding energy of phosphite dianion in the ground state of −1.9 kcal/mol (Kd ≈ 40 mM), is then the binding energy that is utilized to drive the conversion of EO to EC, ΔGC = 3.9 kcal/mol, as shown by eq 4.87 Phosphite is proposed to act as a “spectator” in holding the enzyme in the active EC conformation and it does not affect the second-order rate constant for the enzyme-catalyzed reaction of GA catalyzed by the closed form of the enzyme, so that (kcat/Km)E′ = (kcat/Km)E•HPi (Scheme 11). Essentially, the barrier for conversion of EO to EC (ΔGC) is included in the observed barrier for the reaction of GA catalyzed by the free enzyme EO [(kcat/Km)E = Kc(kcat/Km)E′], but not in the barrier for the reaction catalyzed by EC•HPO32− [(kcat/Km)E•HPi, Figure 4].87
Figure 4.

Free energy diagram for the turnover of the truncated substrate glycolaldehyde (S) by free TIM (EO) and by TIM that is saturated with phosphite dianion (EC•HPO32−, Scheme 11) constructed using data from Ref. 87. The observed tighter binding of phosphite dianion to the transition state complex EC•S‡ to give EC•HPO32−•S‡ (ΔGint = −5.8 kcal/mol, eq 3) than to the free enzyme EO to give EC•HPO32− (ΔGobsd = −1.9 kcal/mol) represents the “interaction energy” of 3.9 kcal/mol.90 This can be attributed to ΔGC = +3.9 kcal/mol for the unfavorable conformational change that converts the inactive open ground state enzyme EO to the active closed enzyme EC (eq 4). The observed value of ΔG‡ = +18.3 kcal/mol for turnover of glycolaldehyde with second order rate constant (kcat/Km)E can be partitioned into ΔG‡ = +14.4 kcal/mol for proton transfer from glycolaldehyde to EC and ΔGC = +3.9 kcal/mol for the unfavorable conformational change that converts EO to EC.
The conversion of EO to EC in Scheme 11 and Figure 4 is formally equivalent to the conversion of E to E* shown in Scheme 5.16 We emphasize, for the sake of simplicity, a minimal model where the dianion binding energy is utilized entirely to drive an unfavorable change in the enzyme conformation, but the intrinsic substrate binding energy might also be utilized to drive the unfavorable change from S to S* (Scheme 5). The thermodynamic barrier to the EO to EC conformational change might represent strain induced in EC by loop closure, or an entropic price for loop closure arising from the requirement to obtain optimal alignment of the catalytic side chains relative to the bound substrate.98,99 However, the report that the thermodynamic barrier to loop closure at unliganded TIM is only ca. 1.4 kcal/mol, suggests that the protein conformational strain induced by loop closure is small.100 Enzyme active sites must be open to the bulk solvent, in order for the substrate to bind. However, catalysis of polar reactions is favored at desolvated active sites of low effective dielectric constant (D), because the magnitude of stabilizing polar/electrostatic interactions between the enzyme and the reaction transition state increases with 1/D.34,101,102 Therefore, we propose that a large fraction of the free energy barrier for the conversion of EO to EC is the barrier to desolvation of the enzyme active site that accompanies ligand binding and loop closure.34
The displacement of water from the active site of TIM that is driven by the binding of phosphite dianion will result in a decrease in the effective local dielectric constant of the enzyme active site that will enhance all stabilizing polar interactions between the enzyme and bound ligand. We have proposed that desolvation of the active site of TIM results in a large 8 kcal/mol stabilizing interaction between the cationic alkyl ammonium side chain of Lys-12 and the transition state for formation of the trianionic enediolate-phosphate reaction intermediate of the isomerization of GAP.42,43 The specific desolvation of the carboxylate side chain of the catalytic base Glu-167 at TIM should lead to an increase in the basicity of this side chain and enhance its reactivity in the deprotonation of bound carbon acids. Our studies of the effects of the I172A and L232A mutations at TIM from Trypanosoma brucei brucei on the kinetic parameters for the enzyme-catalyzed reactions of the whole substrates GAP and DHAP and the substrate pieces GA + phosphite provided direct evidence for the activation of the side chain of Glu-167 by loop closure at TIM.103,104 These results were summarized in a recent review.34
Enzyme Activation: Amino Acid Racemases
When the protein catalyst is relatively rigid, enzyme activation may be due entirely to ligand driven desolvation of the substrate and the enzyme active site that occurs on formation of the Michaelis complex. In this case, Esol and Edesol could be substituted for EO and EC in Scheme 11, in order to emphasize the importance of desolvation. Deprotonation of the α-amino carbon of an N-protonated amino acid to form a zwitterionic amino acid carbanion is the first step in the reactions catalyzed by proline racemase (Scheme 12),105 glutamate racemase,106,107 and diaminopimelate epimerase.108
Scheme 12.

Proline racemase provides a ca. 19 kcal/mol stabilization of the transition state for deprotonation of bound proline to form the zwitterionic carbanion reaction intermediate (Scheme 12).109 However, the complete desolvation of N-protonated proline by transfer from aqueous solution to the gas phase is expected to result in a much larger 100 kcal/mol increase in the thermodynamic driving force for this proton transfer.110 Proline racemase,105 glutamate racemase,106,107 and diaminopimelate epimerase108 each bind their substrate at the bottom of a deep hydrophobic cavity, so that partial desolvation of the amino acid is expected to accompany substrate binding from water.111–113 A 19 kcal/mol reduction in the barrier for deprotonation of partly desolvated proline at such a water-free enzyme active site is much smaller than the limiting 100 kcal/mol difference in the driving force for proton transfer in the gas phase compared to water, and it could account for most or all of the rate acceleration for enzyme-catalyzed deprotonation.102,109,110,114 We propose that binding interactions between amino acid racemases and their amino acid substrates are utilized to drive the desolvation of the enzyme active site and the substrate, so that proton transfer occurs at an active site of low effective dielectric constant.109,110 This proposal is supported by the results of a QM/MM study about the “importance of being zwitterionic” for the amino acid racemization catalyzed by glutamate racemase.114
Enzyme Activation: OMPDC
We have proposed: (1) That the closed conformation of OMPDC (EC) exercises the enzyme’s full catalytic power for catalysis of decarboxylation and C-6 deuterium exchange reactions (Scheme 8C and 8D).115 (2) That the turnover of the truncated substrates 1-(β-D-erythrofuranosyl)orotic acid (EO, Scheme 8C) and 1-(β-D-erythrofuranosyl)-5-fluorouracil (FEU, Scheme 8D) is slow in the absence of HPO32, because the fraction of the enzyme in the active form EC is low (Kc ≪ 1, Scheme 11).115 (3) That the function of the three amino acid side chains of OMPDC that interact with the phosphodianion group of OMP, Gln-215, Tyr-217, and Arg-235 (Figure 5) is to drive the conversion of OMPDC from EO to the active form EC•OMP or EC•HPO32−.116
Figure 5.

X-ray crystal structure of yeast OMPDC liganded with 6-hydroxyuridine 5′-monophosphate (Protein Data Bank entry 1DQX),26 showing interactions of side chains of the protein with the bound phosphodianion group of the ligand.
These proposals were tested by preparing all possible of yeast OMPDCs containing the single (Q215A, Y217F, R235A), double (Q215A/Y217F, Q215A/R235A, Y217F/R235A) and triple (Q215A/Y217F/R235A) substitutions for alanine of the side chains of Gln-215, Tyr-217 and Arg-235.116 The single mutations result in up to a 20,000-fold decrease in kcat/Km for decarboxylation of the whole substrate OMP, but only very small < 3-fold decreases in (kcat/Km)E (Scheme 9) for decarboxylation of the truncated substrate EO that lacks a phosphodianion group. Essentially the entire effect of these mutations is expressed as up to a 3 × 105-fold decrease in the third-order rate constant (kcat/Km)E•HPi/Kd for the phosphite-activated reaction of EO (Scheme 9). The small effect of these mutations on (kcat/Km)E for the unactivated decarboxylation of EO shows that there is no significant direct stabilization of the transition state for decarboxylation by long-range interactions of the side chains of Gln-215, Tyr-217 and Arg-235 with the remote pyrimidine ring of EO.116
We concluded that the activation of OMPDC by phosphite dianion is the result of the development of loop-dianion interactions that drive loop closure over OMP or EO. This provides direct evidence that the conversion of unliganded enzyme EO to EC•HPO32− activates OMPDC for catalysis of decarboxylation (Scheme 11). We find a clean separation of the functions of the pyrimidine and phosphodianion binding sites of OMPDC. The pyrimidine binding site has the potential to provide effective catalysis of the decarboxylation of EO (Scheme 8C) and of C-6 carbon deprotonation of FEU (Scheme 8D), but the observed catalysis is weak because the fraction of the enzyme in the active form EC is low. The phosphodianion binding site functions to enable the pyrimidine site to reach its full catalytic potential, by using stabilizing interactions between the protein and a bound phosphodianion to drive the conversion of EO to EC•HPO32− or EC•OMP, resulting in an increase in the fraction of the enzyme in the active form EC.116
Part of the intrinsic binding energy of the phosphodianion group of the natural substrate OMP might be used to induce destabilizing “electrostatic stress” between the substrate carboxylate group and the carboxylate group of the side chain of Asp-91 at the E•OMP Michaelis complex, and which is then relieved at the transition state for decarboxylation.23 However, we have shown that the phosphodianion groups of OMP and 5-fluorouridine 5′-monophosphate (FUMP) both provide a similar stabilization of the transition state for the OMPDC-catalyzed decarboxylation of OMP and the C-6 deuterium exchange reaction of FUMP of ca. 11 and 9 kcal/mol, respectively.115 There are recoveries of 8 and 6 kcal/mol of this intrinsic phosphodianion binding energy as phosphite dianion activation of the OMPDC-catalyzed decarboxylation and deuterium exchange reactions of the corresponding truncated substrates EO (Scheme 8C) and FEU (Scheme 8D), respectively.115 The binding energy of HPO32− is utilized mainly to stabilize features of the transition state that are common to the OMPDC-catalyzed decarboxylation and deuterium exchange reactions, rather than to introduce electrostatic stress into complexes between OMPDC and EO or OMP.23 Part of the intrinsic binding energy of the phosphodianion group of OMP may also be utilized to induce destabilizing strain into the protein catalyst that is relieved at the transition state for the enzyme-catalyzed decarboxylation and deuterium exchange reactions.96,97
Enzyme Activation: Reaction of the Whole Substrate
The reactions of truncated substrates catalyzed by TIM, OMPDC and α-GPDH are activated by phosphite dianion.83,87,88 These results uncover dianion activation for the reactions of the whole phosphorylated substrates, which was not detected in direct kinetic studies using the natural substrates, or in analyses of X-ray crystal structures. In fact, earlier intense mechanistic studies on these enzymes uniformly failed to suggest the possibility of such enzyme activation in the reaction of the whole phosphodianion substrate. However, it is necessary to extend the model shown in Scheme 11, developed to rationalize the results of our studies on catalysis of the reaction of substrates in pieces, to enzymatic catalysis of the reaction of the whole substrate.
Figure 6 shows the free energy profile for the enzymatic reaction of a whole phosphorylated substrate S-PO32−. For simplicity, the Figure shows only the binding energy of the phosphodianion portion of the substrate, ΔGPi. Once again, part of this available binding energy is utilized to drive the conversion of the inactive open enzyme EO to the active loop-closed form EC that occurs on the binding of S-PO32− to give the reactive EC•S-PO32− ground state Michaelis complex, so that the observed substrate binding energy, ΔGobsd, differs from ΔGPi by an amount ΔGC. Therefore, this portion of the available phosphodianion binding energy is not expressed in the Michaelis constant Km, but is manifested at the transition state for catalysis by the “activated” enzyme EC, as a lowered barrier to the reaction of the bound substrate, ΔG‡chem, and hence an increase in kcat for conversion of the Michaelis complex to product (Figure 6). Now, if this phosphodianion binding energy were not used to drive the unfavorable enzyme conformational change, but rather was fully expressed as tight binding of the substrate in the Michaelis complex with the open enzyme, EO•S-PO32−, then the barrier for reaction of the bound substrate through the transition state complex containing the active closed enzyme would be increased by an amount ΔGC, resulting in a smaller value of kcat (Figure 6). In other words, coupling of the thermodynamically unfavorable loop closure and conformational change to the development of interactions between the catalyst and substrate phosphodianion group provides a mechanism to circumvent tight and irreversible substrate binding, by providing a high specificity for binding of the reaction transition state.
Figure 6.

Free energy profile for the reaction of a whole phosphorylated substrate S-PO32− catalyzed by an enzyme that exists predominantly in a stable inactive open form EO. For simplicity, the Figure shows only the binding energy of the phosphodianion portion of the substrate, ΔGPi. The relative small observed binding energy of the phosphodianion group (ΔGobsd) is the sum of the much larger total available binding energy of this group (ΔGPi) and the binding energy that is utilized to drive the unfavorable enzyme conformational change (ΔGC). Catalysis occurs at a higher energy active closed form EC, which is stabilized by interactions with the phosphodianion group of substrate at the EC•S-PO32− Michaelis complex (upper profile). In the case that the phosphodianion binding energy is not used to drive the unfavorable conformational change at the ground state Michaelis complex (lower profile), the activation barrier ΔG‡chem for turnover of the enzyme-bound substrate will increase by an amount ΔGC (see text).
At first glance, it might appear inefficient for a catalyst to exist mainly in an inactive form ([EO] ≫ [EC], Figure 6). However, the utilization of substrate binding energy to drive an uphill enzyme conformational change will not result in a large decrease in kcat/Km, provided the reaction is close to the diffusion-controlled limit, as is the case for OMPDC and TIM.20,21,117 The binding of substrate to EO to form EC•S-PO32− (Scheme 13) will occur at the diffusion limit, so long as the initial encounter complex EO•S-PO32− undergoes loop closure to form the reactive Michaelis complex EC•S-PO32− faster than release of substrate back to solution. It is not clear that natural selection would favor the evolution of a predominantly inactive enzyme (Kc ≪ 1) over an enzyme for which Kc ≈ 1 for the activating conformational change (Scheme 11). However, we note the requirement that enzyme active sites be solvent-exposed and presumably well solvated, in order to allow access for the substrate, and that there are clear catalytic advantages (see above) to desolvation of the substrate and active site. This serves as an imperative for the utilization of substrate binding energy to drive the necessary desolvation of enzymes and substrates. In the case of TIM, we have proposed that desolvation is driven by an enzyme conformational change that results in solvent being swept out of the enzyme active site, and that the barrier for this active site desolvation makes a large contribution to ΔGC (Figure 6).34,101,103,104
Scheme 13.
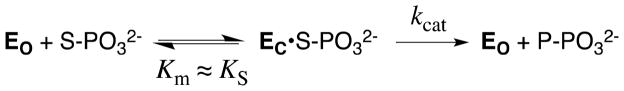
Enolase and Other Superfamilies
The many members of the enolase superfamily catalyze a common half-reaction in which an active site base abstracts a proton from an α-carboxylate carbon to generate an enolate dianion intermediate stabilized by coordination to one or two essential Mg2+.118,119 The reaction may be completed by reprotonation of the carbanion intermediate to give the product of a racemization or epimerization reaction, or by the loss of an oxygen- or nitrogen-based leaving group from the β-carbon to give an alkene (Figure 7). This catalytic diversity is provided by a two-domain structure. A modified TIM barrel domain [(β/α)7β barrel], for which the active site lies at the C-terminal end of the β-strands that provide the functionalities that carry out the reaction chemistry. These include a Mg2+ [or two Mg2+ for enolase]120 which stabilizes the developing negative charge at the enolate intermediate, and one or more amino acid side chains that function as Brønsted acid/base catalysts. Variations in the acid/base functional groups located at the ends of the second, third, fifth, sixth, and seventh β-strands of the catalytic domain contribute to the enormous range of reactions catalyzed by the enolase superfamily members. A second variable capping domain, composed of segments at the N- and C-termini of the polypeptide, plays a role similar to that of phosphodianion gripper loops. This capping domain interacts with nonreacting portions of the substrate and acts to sequester the substrate from interaction with bulk solvent.119,121 Specificity for the nonreacting portions of the substrate is provided by variations in the capping domain. An important question is whether or not interactions with the capping domain of the enolase superfamily provide specificity for formation of the Michaelis complex, or if a part of the available substrate binding energy is expressed specifically at the transition state. This would result if ligand-driven closure of the capping domain activates these enzymes for catalysis of proton transfer, in a manner similar to that observed for the activation of TIM for deprotonation of GAP by phosphodianion-driven closure of loop 6 over the substrate.34
Figure 7.

Reactions catalyzed by members of the enolase superfamily, for which an active site base abstracts an α-carboxylate proton to generate an enolate dianion stabilized by coordination with one or two essential Mg2+ ions. (A) Deprotonation of mandelate and trifluorolactate, the first step in racemization catalyzed by mandelate racemase. (B) Deprotonation of 2-phosphoglycerate, the first step in the elimination reaction catalyzed by enolase. (C) Deprotonation of aspartate, the first step in the elimination reaction catalyzed by aspartate ammonia-lyase.
The capping domain of mandelate racemase (MR) interacts with the phenyl group of mandelate. The specificity of this interaction for stabilization of the Michaelis complex is low, as shown by the small effect of mutations at the capping domain on substrate binding affinity,122 and by the similar values of Km ≈ KS for the MR-catalyzed racemization of mandelate and trifluorolactate (Figure 7A).123 By contrast, the same α-CF3 for α-phenyl substitution results in a 400-fold decrease in kcat for racemization of (R)-mandelate. A detailed comparison of the free energy profiles for the nonenzymatic and MR-catalyzed racemizations of mandelate and trifluorolactate shows that the enzyme provides a 6 kcal/mol greater stabilization of the transition state for racemization of mandelate, the physiological substrate.123 These results show that there is a large specific stabilization of the transition state for the reaction catalyzed by MR, from interactions of the capping domain with the phenyl group of mandelate. They are consistent with the conclusion that closure of this domain, which is driven by stabilizing interactions with the phenyl group, activates the enzyme for catalysis of racemization of the physiological substrate. It is likely that closure of the capping domain provides a similar activation of other members of the enolase superfamily.
Members of the haloalkanoic acid dehalogenase superfamily (>19,000 unique sequences) function mainly to catalyze the hydrolysis of adenosine triphosphate and phosphate monesters.124 These enzymes bind phosphate monoesters with the phosphoryl group oriented toward the Rossmann-fold core, and with the leaving group projected towards a “capping” domain that sequesters the bound substrate from bulk solvent and also provides substrate specificity. It would be informative to carry out experiments to test the proposal that interactions between this capping domain and the substrate leaving group activate members of this superfamily for catalysis.
Concluding Remarks
We show in this review that a significant fraction of the enzymatic rate accelerations for polar reactions of substrates which contain a phosphodianion moiety may be rationalized by Pauling’s simple model,1,2 and consider the mechanism for the utilization of binding interactions of the nonreacting substrate phosphodianion group in enzyme activation. The possibility of enzyme activation by the substrate phosphodianion was not suggested by X-ray crystallographic analysis, computational studies, or by the other tools of mechanistic enzymology. Rather, this possibility arose from an intuitive sense of the power of the Pauling model to rationalize the rate acceleration for enzymatic reactions. We expect that the catalytic rate acceleration for many other enzymatic reactions will eventually be rationalized by experiments that characterize enzyme activation by interactions with nonreacting substrate fragments. Such activation is one important function of enzyme loops, capping domains, and other structural elements that function to sequester the substrate from interaction with bulk solvent at the interior of the protein catalyst.101
Debates about the origins of the catalytic rate acceleration for enzymes should focus on distinguishing the effects that make essential contributions to the rate acceleration, from those that are incidental. X-ray crystal structures of enzymes that catalyze polar reactions show extensive networks of interactions between the protein and ligand; and, the free energy profiles for these reactions show that the enzymes provide a much larger stabilization of the transition state than of the ground state Michaelis complex. Understanding of the role of protein-ligand interactions in transition state stabilization, and of the mechanism for their differential expression in the binding of ground and transition state is essential to understanding the enzymatic rate acceleration. These relatively simple issues have not yet been resolved for most enzymes that catalyze polar reactions. Their resolution may not provide us with a complete understanding of enzymatic catalysis, but it would represent a significant step forward.
Abbreviations
- TPP
thiamine pyrophosphate
- NAD
nicotinamide adenine dinucleotide
- PLP
pyridoxal 5′-phosphate
- OMPDC
orotidine 5′-monophosphate decarboxylase
- OMP
orotidine 5′-monophosphate
- UMP
uridine 5′-monophosphate
- TIM
triosephosphate isomerase
- GAP
glyceraldehyde 3-phosphate
- DHAP
dihydroxyacetone phosphate
- KSI
3-Oxo-Δ5-steroid isomerase (ketosteroid isomerase)
- CoA
coenzyme A
- SucCoA
succinyl-CoA
- AcAcCoA
acetoacetyl-CoA
- SCOT
succinyl-CoA:3-oxoacid coenzyme A transferase
- α-GPDH
α-glycerol phosphate dehydrogenase
- GA
glycolaldehyde
- EO
1-(β-D-erythrofuranosyl)orotic acid
- FEU
1-(β-D-erythrofuranosyl)-5-fluorouracil
- FUMP
5-fluorouridine 5′-monophosphate
- MR
mandelate racemase
References
- 1.Pauling L. Nature of forces between large molecules of biological interest. Nature. 1948;161:707–709. doi: 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- 2.Pauling L. Molecular Architecture and Biological Reactions. Chem Eng News. 1946;24:1375–1377. [Google Scholar]
- 3.Wolfenden RV. Binding of substrate and transition state analogs to triosephosphate isomerase. Biochemistry. 1970;9:3404–3407. doi: 10.1021/bi00819a018. [DOI] [PubMed] [Google Scholar]
- 4.Hartman FC, LaMuraglia GM, Tomozawa Y, Wolfenden R. Influence of pH on the interaction of inhibitors with triosephosphate isomerase and determination of the pKa of the active-site carboxyl group. Biochemistry. 1975;14:5274–5279. doi: 10.1021/bi00695a007. [DOI] [PubMed] [Google Scholar]
- 5.Collins KD. An Activated Intermediate Analogue. J Biol Chem. 1974;249:136–142. [PubMed] [Google Scholar]
- 6.Wolfenden R. Analog approaches to the structure of the transition state in enzyme reactions. Acc Chem Res. 1972;5:10–18. [Google Scholar]
- 7.Cardinale GJ, Abeles RH. Purification and mechanism of action of proline racemase. Biochemistry. 1968;7:3970–3978. doi: 10.1021/bi00851a026. [DOI] [PubMed] [Google Scholar]
- 8.Evans B, Wolfenden R. Potential transition state analog for adenosine deaminase. J Am Chem Soc. 1970;92:4751–4752. doi: 10.1021/ja00718a056. [DOI] [PubMed] [Google Scholar]
- 9.Lienhard GE. Enzymatic Catalysis and Transition-State Theory. Science. 1973;180:149–154. doi: 10.1126/science.180.4082.149. [DOI] [PubMed] [Google Scholar]
- 10.Ho MC, Shi W, Rinaldo-Matthis A, Tyler PC, Evans GB, Clinch K, Almo SC, Schramm VL. Four generations of transition-state analogues for human purine nucleoside phosphorylase. Proc Natl Acad Sci U S A. 2010;107:4805–4812. doi: 10.1073/pnas.0913439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schramm VL. Enzymatic transition state theory and transition state analog design. J Biol Chem. 2007;282:28297–28300. doi: 10.1074/jbc.R700018200. [DOI] [PubMed] [Google Scholar]
- 12.Peters KS, Kim G. Characterization of Solvent and Deuterium Isotope Effects on Nonadiabatic Proton Transfer in the Benzophenone/N,N-Dimethylaniline Contact Radical Ion Pair. J Phys Chem A. 2004;108:2598–2606. [Google Scholar]
- 13.Oyola Y, Singleton DA. Dynamics and the Failure of Transition State Theory in Alkene Hydroboration. J Am Chem Soc. 2009;131:3130–3131. doi: 10.1021/ja807666d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagel Z, Klinman J. A 21st century revisionist’s view at a turning point in enzymology. Nat Chem Biol. 2009;5:543–550. doi: 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- 15.Antoniou D, Schwartz SD. Protein dynamics and enzymatic chemical barrier passage. J Phys Chem B. 2011;115:15147–15158. doi: 10.1021/jp207876k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jencks WP. Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 17.Tsang WY, Wood BM, Wong FM, Wu W, Gerlt JA, Amyes TL, Richard JP. Proton Transfer from C-6 of Uridine 5′-Monophosphate Catalyzed by Orotidine 5′-Monophosphate Decarboxylase: Formation and Stability of a Vinyl Carbanion Intermediate and the Effect of a 5-Fluoro Substituent. J Am Chem Soc. 2012;134:14580–14594. doi: 10.1021/ja3058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. Formation and Stability of a Vinyl Carbanion at the Active Site of Orotidine 5′-Monophosphate Decarboxylase: pKa of the C-6 Proton of Enzyme-Bound UMP. J Am Chem Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radzicka A, Wolfenden R. A proficient enzyme. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 20.Wood BM, Chan KK, Amyes TL, Richard JP, Gerlt JA. Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Effect of Solvent Viscosity on Kinetic Constants. Biochemistry. 2009;48:5510–5517. doi: 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porter DJT, Short SA. Yeast Orotidine-5′-Phosphate Decarboxylase: Steady-State and Pre-Steady-State Analysis of the Kinetic Mechanism of Substrate Decarboxylation. Biochemistry. 2000;39:11788–11800. doi: 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- 22.Wu N, Gillon W, Pai EF. Mapping the Active Site-Ligand Interactions of Orotidine 5′-Monophosphate Decarboxylase by Crystallography. Biochemistry. 2002;41:4002–4011. doi: 10.1021/bi015758p. [DOI] [PubMed] [Google Scholar]
- 23.Wu N, Mo Y, Gao J, Pai EF. Electrostatic stress in catalysis: structure and mechanism of the enzyme orotidine monophosphate decarboxylase. Proc Natl Acad Sci U S A. 2000;97:2017–2022. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu N, Pai EF. Crystal structures of inhibitor complexes reveal an alternate binding mode in orotidine-5′-monophosphate decarboxylase. J Biol Chem. 2002;277:28080–28087. doi: 10.1074/jbc.M202362200. [DOI] [PubMed] [Google Scholar]
- 25.Appleby TC, Kinsland C, Begley TP, Ealick SE. The crystal structure and mechanism of orotidine 5′-monophosphate decarboxylase. Proc Natl Acad Sci U S A. 2000;97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc Natl Acad Sci U S A. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris P, Poulsen J-CN, Jensen KF, Larsen S. Structural basis for the catalytic mechanism of a proficient enzyme: orotidine 5′-monophosphate decarboxylase. Biochemistry. 2000;39:4217–4224. doi: 10.1021/bi992952r. [DOI] [PubMed] [Google Scholar]
- 28.Wittmann JG, Heinrich D, Gasow K, Frey A, Diederichsen U, Rudolph MG. Structures of the Human Orotidine-5′-Monophosphate Decarboxylase Support a Covalent Mechanism and Provide a Framework for Drug Design. Structure. 2008;16:82–92. doi: 10.1016/j.str.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 29.Fujihashi M, Wei LH, Kotra LP, Pai EF. Structural Characterization of the Molecular Events during a Slow Substrate-Product Transition in Orotidine 5′-Monophosphate Decarboxylase. J Mol Biol. 2009;387:1199–1210. doi: 10.1016/j.jmb.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinrich D, Diederichsen U, Rudolph M. Lys314 is a Nucleophile in Non-Classical Reactions of Orotidine-5′-Monophosphate Decarboxylase. Chem Eur J. 2009;15:6619–6625. doi: 10.1002/chem.200900397. [DOI] [PubMed] [Google Scholar]
- 31.Stanton CL, Kuo IF, Mundy CJ, Laino T, Houk KN. QM/MM metadynamics study of the direct decarboxylation mechanism for orotidine-5′-monophosphate decarboxylase using two different QM regions: acceleration too small to explain rate of enzyme catalysis. J Phys Chem B. 2007;111:12573–12581. doi: 10.1021/jp074858n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Donoghue AC, Amyes TL, Richard JP. Hydron Transfer Catalyzed by Triosephosphate Isomerase. Products of Isomerization of Dihydroxyacetone Phosphate in D2O. Biochemistry. 2005;44:2622–2631. doi: 10.1021/bi047953k. [DOI] [PubMed] [Google Scholar]
- 33.O’Donoghue AC, Amyes TL, Richard JP. Hydron Transfer Catalyzed by Triosephosphate Isomerase. Products of Isomerization of (R)-Glyceraldehyde 3-Phosphate in D2O. Biochemistry. 2005;44:2610–2621. doi: 10.1021/bi047954c. [DOI] [PubMed] [Google Scholar]
- 34.Richard JP. A Paradigm for Enzyme-Catalyzed Proton Transfer at Carbon: Triosephosphate Isomerase. Biochemistry. 2012;51:2652–2661. doi: 10.1021/bi300195b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knowles JR. To build an enzyme. Philos Trans R Soc, B. 1991;332:115–121. doi: 10.1098/rstb.1991.0039. [DOI] [PubMed] [Google Scholar]
- 36.Knowles JR. Enzyme catalysis: not different, just better. Nature. 1991;350:121–124. doi: 10.1038/350121a0. [DOI] [PubMed] [Google Scholar]
- 37.Morrow JR, Amyes TL, Richard JP. Phosphate Binding Energy and Catalysis by Small and Large Molecules. Acc Chem Res. 2008;41:539–548. doi: 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crugeiras J, Richard JP. A Comparison of the Electrophilic Reactivities of Zn2+ and Acetic Acid as Catalysts of Enolization: Imperatives for Enzymatic Catalysis of Proton Transfer at Carbon. J Am Chem Soc. 2004;126:5164–5173. doi: 10.1021/ja049489c. [DOI] [PubMed] [Google Scholar]
- 39.Mathews RA, Rossiter CS, Morrow JR, Richard JP. A minimalist approach to understanding the efficiency of mononuclear Zn(II) complexes as catalysts of cleavage of an RNA analog. Dalton Trans. 2007:3804–3811. doi: 10.1039/b707409c. [DOI] [PubMed] [Google Scholar]
- 40.O’Donoghue AM, Pyun SY, Yang MY, Morrow JR, Richard JP. Substrate Specificity of an Active Dinuclear Zn(II) Catalyst for Cleavage of RNA Analogues and a Dinucleoside. J Am Chem Soc. 2006;128:1615–1621. doi: 10.1021/ja056167f. [DOI] [PubMed] [Google Scholar]
- 41.Iranzo O, Kovalevsky AY, Morrow JR, Richard JP. Physical and Kinetic Analysis of the Cooperative Role of Metal Ions in Catalysis of Phosphodiester Cleavage by a Dinuclear Zn(II) Complex. J Am Chem Soc. 2003;125:1988–1993. doi: 10.1021/ja027728v. [DOI] [PubMed] [Google Scholar]
- 42.Go MK, Koudelka A, Amyes TL, Richard JP. Role of Lys-12 in Catalysis by Triosephosphate Isomerase: A Two-Part Substrate Approach. Biochemistry. 2010;49:5377–5389. doi: 10.1021/bi100538b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Go MK, Amyes TL, Richard JP. Rescue of K12G Triosephosphate Isomerase by Ammonium Cations: The Reaction of an Enzyme in Pieces. J Am Chem Soc. 2010;132:13525–13532. doi: 10.1021/ja106104h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henderson R. Structure of crystalline α-chymotrypsin: IV. The structure of indoleacryloyl-α-chymotrypsin and its relevance to the hydrolytic mechanism of the enzyme. J Mol Biol. 1970;54:341–354. doi: 10.1016/0022-2836(70)90434-1. [DOI] [PubMed] [Google Scholar]
- 45.Kraut J. Serine Proteases: Structure and Mechanism of Catalysis. Annu Rev Biochem. 1977;46:331–358. doi: 10.1146/annurev.bi.46.070177.001555. [DOI] [PubMed] [Google Scholar]
- 46.Whiting AK, Peticolas WL. Details of the acyl-enzyme intermediate and the oxyanion hole in serine protease catalysis. Biochemistry. 1994;33:552–561. doi: 10.1021/bi00168a021. [DOI] [PubMed] [Google Scholar]
- 47.Lo YC, Lin SC, Shaw JF, Liaw YC. Crystal structure of Escherichia coli thioesterase I/protease I/lysophospholipase L1: consensus sequence blocks constitute the catalytic center of SGNH-hydrolases through a conserved hydrogen bond network. J Mol Biol. 2003;330:539–551. doi: 10.1016/s0022-2836(03)00637-5. [DOI] [PubMed] [Google Scholar]
- 48.Merilainen G, Poikela V, Kursula P, Wierenga RK. The Thiolase Reaction Mechanism: The Importance of Asn316 and His348 for Stabilizing the Enolate Intermediate of the Claisen Condensation. Biochemistry. 2009;48:11011–11025. doi: 10.1021/bi901069h. [DOI] [PubMed] [Google Scholar]
- 49.Schwans JP, Sunden F, Gonzalez A, Tsai Y, Herschlag D. Evaluating the catalytic contribution from the oxyanion hole in ketosteroid isomerase. J Am Chem Soc. 2011;133:20052–20055. doi: 10.1021/ja208050t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma S, Bhaumik P, Schmitz W, Venkatesan R, Hiltunen JK, Conzelmann E, Juffer AH, Wierenga RK. The Enolization Chemistry of a Thioester-Dependent Racemase: The 1.4 Å Crystal Structure of a Reaction Intermediate Complex Characterized by Detailed QM/MM Calculations. J Phys Chem B. 2012;116:3619–3629. doi: 10.1021/jp210185m. [DOI] [PubMed] [Google Scholar]
- 51.Robertus JD, Kraut J, Alden RA, Birktoft JJ. Subtilisin. Stereochemical mechanism involving transition-state stabilization. Biochemistry. 1972;11:4293–4303. doi: 10.1021/bi00773a016. [DOI] [PubMed] [Google Scholar]
- 52.Bryan P, Pantoliano MW, Quill SG, Hsiao HY, Poulos T. Site-directed mutagenesis and the role of the oxyanion hole in subtilisin. Proc Natl Acad Sci U S A. 1986;83:3743–3745. doi: 10.1073/pnas.83.11.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kamerlin SC, Chu ZT, Warshel A. On catalytic preorganization in oxyanion holes: highlighting the problems with the gas-phase modeling of oxyanion holes and illustrating the need for complete enzyme models. J Org Chem. 2010;75:6391–6401. doi: 10.1021/jo100651s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamerlin S, Sharma P, Chu Z, Warshel A. Ketosteroid isomerase provides further support for the idea that enzymes work by electrostatic preorganization. Proc Natl Acad Sci U S A. 2010;107:4075–4080. doi: 10.1073/pnas.0914579107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pollack RM. Enzymatic mechanisms for catalysis of enolization: ketosteroid isomerase. Bioorg Chem. 2004;32:341–353. doi: 10.1016/j.bioorg.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 56.Hawkinson DC, Eames TCM, Pollack RM. Energetics of 3-oxo-Δ5-steroid isomerase: source of the catalytic power of the enzyme. Biochemistry. 1991;30:10849–10858. doi: 10.1021/bi00109a007. [DOI] [PubMed] [Google Scholar]
- 57.Zeng B, Pollack RM. Microscopic rate constants for the acetate ion catalyzed isomerization of 5-androstene-3,17-dione to 4-androstene-3,17-dione: a model for steroid isomerase. J Am Chem Soc. 1991;113:3838–3842. doi: 10.1021/ja035957r. [DOI] [PubMed] [Google Scholar]
- 58.Schwans JP, Kraut D, Herschlag D. Determining the catalytic role of remote substrate binding interactions in ketosteroid isomerase. Proc Natl Acad Sci U S A. 2009;106:14271–14275. doi: 10.1073/pnas.0901032106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Richard JP. The Enhancement of Enzymic Rate Accelerations by Bronsted Acid-Base Catalysis. Biochemistry. 1998;37:4305–4309. doi: 10.1021/bi972655r. [DOI] [PubMed] [Google Scholar]
- 60.Kraut D, Sigala P, Fenn T, Herschlag D. Dissecting the paradoxical effects of hydrogen bond mutations in the ketosteroid isomerase oxyanion hole. Proc Natl Acad Sci U S A. 2010;107:1960–1965. doi: 10.1073/pnas.0911168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi G, Ha N-C, Kim SW, Kim D-H, Park S, Oh B-H, Choi KY. Asp-99 Donates a Hydrogen Bond Not to Tyr-14 but to the Steroid Directly in the Catalytic Mechanism of Δ5–3-Ketosteroid Isomerase from Pseudomonas putida Biotype B. Biochemistry. 2000;39:903–909. doi: 10.1021/bi991579k. [DOI] [PubMed] [Google Scholar]
- 62.Thornburg LD, Goldfeder YR, Wilde TC, Pollack RM. Selective Catalysis of Elementary Steps by Asp-99 and Tyr-14 of 3-Oxo-Δ5-Steroid Isomerase. J Am Chem Soc. 2001;123:9912–9913. doi: 10.1021/ja016683f. [DOI] [PubMed] [Google Scholar]
- 63.Amyes TL, O’Donoghue AC, Richard JP. Contribution of phosphate intrinsic binding energy to the enzymatic rate acceleration for triosephosphate isomerase. J Am Chem Soc. 2001;123:11325–11326. doi: 10.1021/ja016754a. [DOI] [PubMed] [Google Scholar]
- 64.Amyes TL, Richard JP. Determination of the pKa of Ethyl Acetate: Brønsted Correlation for Deprotonation of a Simple Oxygen Ester in Aqueous Solution. J Am Chem Soc. 1996;118:3129–3141. [Google Scholar]
- 65.Amyes TL, Richard JP. Generation and stability of a simple thiol ester enolate in aqueous solution. J Am Chem Soc. 1992;114:10297–10302. [Google Scholar]
- 66.Begley TP, Kinsland C, Strauss E. The biosynthesis of coenzyme A in bacteria. In: Begley T, Litwack G, editors. Vitamins & Hormones. Elsevier; 2001. pp. 157–171. [DOI] [PubMed] [Google Scholar]
- 67.Moore SA, Jencks WP. Model reactions for CoA transferase involving thiol transfer. Anhydride formation from thiol esters and carboxylic acids. J Biol Chem. 1982;257:10882–10892. [PubMed] [Google Scholar]
- 68.Moore SA, Jencks WP. Reactions of acyl phosphates with carboxylate and thiol anions. Model reactions for CoA transferase involving anhydride formation. J Biol Chem. 1982;257:10874–10881. [PubMed] [Google Scholar]
- 69.Moore SA, Jencks WP. Formation of active site thiol esters of CoA transferase and the dependence of catalysis on specific binding interactions. J Biol Chem. 1982;257:10893–10897. [PubMed] [Google Scholar]
- 70.Falcone AB, Boyer PD. Studies concerning the mechanism of action of acetoacetyl succinic thiophorase by use of O18. Arch Biochem Biophys. 1959;83:337–344. doi: 10.1016/0003-9861(59)90039-6. [DOI] [PubMed] [Google Scholar]
- 71.Pickart CM, Jencks WP. Formation of Stable Anhydrides from CoA Transferase and Hydroxamic Acids. J Biol Chem. 1979;254:9120–9129. [PubMed] [Google Scholar]
- 72.White H, Jencks WP. Mechanism and Specificity of Succinyl-CoA:3-Ketoacid Coenzyme A Transferase. J Biol Chem. 1976;251:1688–1699. [PubMed] [Google Scholar]
- 73.Blair JB. Skeletal Muscle Coenzyme A Transferase: Purification and Properties. J Biol Chem. 1969;244:951–954. [PubMed] [Google Scholar]
- 74.Whitty A, Fierke CA, Jencks WP. Role of binding energy with coenzyme A in catalysis by 3-oxoacid coenzyme A transferase. Biochemistry. 1995;34:11678–11689. doi: 10.1021/bi00037a005. [DOI] [PubMed] [Google Scholar]
- 75.Fierke CA, Jencks WP. Two functional domains of coenzyme A activate catalysis by coenzyme A transferase. Pantetheine and adenosine 3′-phosphate 5′-diphosphate. J Biol Chem. 1986;261:7603–7606. [PubMed] [Google Scholar]
- 76.Fraser ME, Hayakawa K, Brown WD. Catalytic role of the conformational change in succinyl-CoA:3-oxoacid CoA transferase on binding CoA. Biochemistry. 2010;49:10319–10328. doi: 10.1021/bi100659s. [DOI] [PubMed] [Google Scholar]
- 77.Mullins EA, Kappock TJ. Crystal Structures of Acetobacter aceti Succinyl-Coenzyme A (CoA):Acetate CoA-Transferase Reveal Specificity Determinants and Illustrate the Mechanism Used by Class I CoA-Transferases. Biochemistry. 2012;51:8422–8434. doi: 10.1021/bi300957f. [DOI] [PubMed] [Google Scholar]
- 78.Gerlt JA, Kozarich JW, Kenyon GL, Gassman PG. Electrophilic Catalysis Can Explain the Unexpected Acidity of Carbon Acids in Enzyme-Catalyzed Reactions. J Am Chem Soc. 1991;113:9667–9669. [Google Scholar]
- 79.Amyes TL, Richard JP. Proton transfer to and from carbon in model reactions. In: Hynes JT, Klinman JP, Limbach H-H, Schowen RL, editors. Hydrogen-Transfer Reactions. WILEY-VCH; Weinheim: 2007. pp. 949–973. [Google Scholar]
- 80.Richard JP, Amyes TL. Proton transfer at carbon. Curr Opin Chem Biol. 2001;5:626–633. doi: 10.1016/s1367-5931(01)00258-7. [DOI] [PubMed] [Google Scholar]
- 81.Steussy CN, Robison AD, Tetrick AM, Knight JT, Rodwell VW, Stauffacher CV, Sutherlin AL. A Structural Limitation on Enzyme Activity: The Case of HMG-CoA Synthase. Biochemistry. 2006;45:14407–14414. doi: 10.1021/bi061505q. [DOI] [PubMed] [Google Scholar]
- 82.Skaff DA, Ramyar KX, McWhorter WJ, Barta ML, Geisbrecht BV, Miziorko HM. Biochemical and Structural Basis for Inhibition of Enterococcus faecalis Hydroxymethylglutaryl-CoA Synthase, mvaS, by Hymeglusin. Biochemistry. 2012;51:4713–4722. doi: 10.1021/bi300037k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amyes TL, Richard JP, Tait JJ. Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J Am Chem Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 84.Chan K, Fedorov AA, Fedorov EV, Almo SC, Gerlt JA. Structural Basis for Substrate Specificity in Phosphate Binding (β/α)8-Barrels: D-Allulose 6-Phosphate 3-Epimerase from Escherichia coli K-12. Biochemistry. 2008;47:9608–9617. doi: 10.1021/bi800821v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Arsenieva D, Jeffery CJ. Conformational changes in phosphoglucose isomerase induced by ligand binding. J Mol Biol. 2002;323:77–84. doi: 10.1016/s0022-2836(02)00892-6. [DOI] [PubMed] [Google Scholar]
- 86.Denesyuk AI, Denessiouk KA, Korpela T, Johnson MS. Phosphate group binding “cup” of PLP-dependent and non-PLP-dependent enzymes: leitmotif and variations. Biochim Biophys Acta. 2003;1647:234–238. doi: 10.1016/s1570-9639(03)00057-8. [DOI] [PubMed] [Google Scholar]
- 87.Amyes TL, Richard JP. Enzymatic catalysis of proton transfer at carbon: activation of triosephosphate isomerase by phosphite dianion. Biochemistry. 2007;46:5841–5854. doi: 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsang W-Y, Amyes TL, Richard JP. A Substrate in Pieces: Allosteric Activation of Glycerol 3-Phosphate Dehydrogenase (NAD+) by Phosphite Dianion. Biochemistry. 2008;47:4575–4582. doi: 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Malabanan MM, Go MK, Amyes TL, Richard JP. Wildtype and engineered monomeric triosephosphate isomerase from Trypanosoma brucei: Partitioning of reaction intermediates in D2O and activation by phosphite dianion. Biochemistry. 2011;50:5767–5779. doi: 10.1021/bi2005416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jencks WP. On the attribution and additivity of binding energies. Proc Natl Acad Sci U S A. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Begley TP, Appleby TC, Ealick SE. The structural basis for the remarkable catalytic proficiency of orotidine 5′-monophosphate decarboxylase. Curr Opin Struct Biol. 2000;10:711–718. doi: 10.1016/s0959-440x(00)00148-2. [DOI] [PubMed] [Google Scholar]
- 92.Davenport RC, Bash PA, Seaton BA, Karplus M, Petsko GA, Ringe D. Structure of the triosephosphate isomerase-phosphoglycolohydroxamate complex: an analog of the intermediate on the reaction pathway. Biochemistry. 1991;30:5821–5826. doi: 10.1021/bi00238a002. [DOI] [PubMed] [Google Scholar]
- 93.Lolis E, Petsko GA. Crystallographic analysis of the complex between triosephosphate isomerase and 2-phosphoglycolate at 2.5-Å resolution: implications for catalysis. Biochemistry. 1990;29:6619–6625. doi: 10.1021/bi00480a010. [DOI] [PubMed] [Google Scholar]
- 94.Suresh S, Turley S, Opperdoes FR, Michels PAM, Hol WGH. A potential target enzyme for trypanocidal drugs revealed by the crystal structure of NAD-dependent glycerol-3-phosphate dehydrogenase from Leishmania mexicana. Structure. 2000;8:541–552. doi: 10.1016/s0969-2126(00)00135-0. [DOI] [PubMed] [Google Scholar]
- 95.Ou X, Ji C, Han X, Zhao X, Li X, Mao Y, Wong LL, Bartlam M, Rao Z. Crystal structures of human glycerol 3-phosphate dehydrogenase 1 (GPD1) J Mol Biol. 2006;357:858–869. doi: 10.1016/j.jmb.2005.12.074. [DOI] [PubMed] [Google Scholar]
- 96.Gao J, Byun KL, Kluger R. Catalysis by enzyme conformational change. Top Curr Chem. 2004;238:113–136. [Google Scholar]
- 97.Gao J. Catalysis by enzyme conformational change as illustrated by orotidine 5′-monophosphate decarboxylase. Curr Opin Struct Biol. 2003;13:184–192. doi: 10.1016/s0959-440x(03)00041-1. [DOI] [PubMed] [Google Scholar]
- 98.Warshel A. Electrostatic Origin of the Catalytic Power of Enzymes and the Role of Preorganized Active Sites. J Biol Chem. 1998;273:27035–27038. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 99.Jogl G, Rozovsky S, McDermott AE, Tong L. Optimal alignment for enzymatic proton transfer: structure of the Michaelis complex of triosephosphate isomerase at 1.2-Å resolution. Proc Natl Acad Sci U S A. 2003;100:50–55. doi: 10.1073/pnas.0233793100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Williams JC, McDermott AE. Dynamics of the Flexible Loop of Triose-Phosphate Isomerase: The Loop Motion Is Not Ligand Gated. Biochemistry. 1995;34:8309–8319. doi: 10.1021/bi00026a012. [DOI] [PubMed] [Google Scholar]
- 101.Malabanan MM, Amyes TL, Richard JP. A role for flexible loops in enzyme catalysis. Curr Opin Struct Biol. 2010;20:702–710. doi: 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Richard JP, Amyes TL. On the importance of being zwitterionic: enzymic catalysis of decarboxylation and deprotonation of cationic carbon. Bioorg Chem. 2004;32:354–366. doi: 10.1016/j.bioorg.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 103.Malabanan MM, Koudelka AP, Amyes TL, Richard JP. Mechanism for Activation of Triosephosphate Isomerase by Phosphite Dianion: The Role of a Hydrophobic Clamp. J Am Chem Soc. 2012;134:10286–10298. doi: 10.1021/ja303695u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Malabanan MM, Amyes TL, Richard JP. Mechanism for Activation of Triosephosphate Isomerase by Phosphite Dianion: The Role of a Ligand-Driven Conformational Change. J Am Chem Soc. 2011;133:16428–16431. doi: 10.1021/ja208019p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rudnick G, Abeles RH. Reaction mechanism and structure of the active site of proline racemase. Biochemistry. 1975;14:4515–4522. doi: 10.1021/bi00691a028. [DOI] [PubMed] [Google Scholar]
- 106.Glavas S, Tanner ME. Catalytic acid/base residues of glutamate racemase. Biochemistry. 1999;38:4106–4113. doi: 10.1021/bi982663n. [DOI] [PubMed] [Google Scholar]
- 107.Tanner ME, Gallo KA, Knowles JR. Isotope effects and the identification of catalytic residues in the reaction catalyzed by glutamate racemase. Biochemistry. 1993;32:3998–4006. doi: 10.1021/bi00066a021. [DOI] [PubMed] [Google Scholar]
- 108.Koo CW, Blanchard JS. Chemical mechanism of Haemophilus influenzae diaminopimelate epimerase. Biochemistry. 1999;38:4416–4422. doi: 10.1021/bi982911f. [DOI] [PubMed] [Google Scholar]
- 109.Williams G, Maziarz EP, Amyes TL, Wood TD, Richard JP. Formation and stability of the enolates of N-protonated proline methyl ester and proline zwitterion in aqueous solution: A nonenzymatic model for the first step in the racemization of proline catalyzed by proline racemase. Biochemistry. 2003;42:8354–8361. doi: 10.1021/bi0345992. [DOI] [PubMed] [Google Scholar]
- 110.Rios A, Amyes TL, Richard JP. Formation and stability of organic zwitterions in aqueous solution: enolates of the amino acid glycine and its derivatives. J Am Chem Soc. 2000;122:9373–9385. [Google Scholar]
- 111.Buschiazzo A, Goytia M, Schaeffer F, Degrave W, Shepard W, Gregoire C, Chamond N, Cosson A, Berneman A, Coatnoan N, Alzari PM, Minoprio P. Crystal structure, catalytic mechanism, and mitogenic properties of Trypanosoma cruzi proline racemase. Proc Natl Acad Sci U S A. 2006;103:1705–1710. doi: 10.1073/pnas.0509010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hwang KY, Cho CS, Kim SS, Sung HC, Yu YG, Cho Y. Structure and mechanism of glutamate racemase from Aquifex pyrophilus. Nat Struct Biol. 1999;6:422–426. doi: 10.1038/8223. [DOI] [PubMed] [Google Scholar]
- 113.Pillai B, Cherney MM, Diaper CM, Sutherland A, Blanchard JS, Vederas JC, James MNG. Structural insights into stereochemical inversion by diaminopimelate epimerase: an antibacterial drug target. Proc Natl Acad Sci U S A. 2006;103:8668–8673. doi: 10.1073/pnas.0602537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Puig E, Garcia-Viloca M, González-Lafont A, Lluch JM. On the ionization state of the substrate in the active site of glutamate racemase. A QM/MM study about the importance of being zwitterionic. J Phys Chem A. 2006;110:717–725. doi: 10.1021/jp054555y. [DOI] [PubMed] [Google Scholar]
- 115.Goryanova B, Amyes TL, Gerlt JA, Richard JP. OMP Decarboxylase: Phosphodianion Binding Energy Is Used To Stabilize a Vinyl Carbanion Intermediate. J Am Chem Soc. 2011;133:6545–6548. doi: 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Amyes TL, Ming SA, Goldman LM, Wood BM, Desai BJ, Gerlt JA, Richard JP. Orotidine 5′-monophosphate decarboxylase: Transition state stabilization from remote protein-phosphodianion interactions. Biochemistry. 2012;51:4630–4632. doi: 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lim W, Raines R, Knowles J. Triosephosphate isomerase catalysis is diffusion controlled. Appendix: Analysis of triose phosphate equilibria in aqueous solution by phosphorus-31 NMR. Biochemistry. 1988;27:1165–1167. doi: 10.1021/bi00404a013. [DOI] [PubMed] [Google Scholar]
- 118.Gerlt JA, Babbitt PC, Rayment I. Divergent evolution in the enolase superfamily: the interplay of mechanism and specificity. Arch Biochem Biophys. 2005;433:59–70. doi: 10.1016/j.abb.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 119.Babbitt PC, Hasson MS, Wedekind JE, Palmer DRJ, Barrett WC, Reed GH, Rayment I, Ringe D, Kenyon GL, Gerlt JA. The Enolase Superfamily: A General Strategy for Enzyme-Catalyzed Abstraction of the α-Protons of Carboxylic Acids. Biochemistry. 1996;35:16489–16501. doi: 10.1021/bi9616413. [DOI] [PubMed] [Google Scholar]
- 120.Larsen TM, Wedekind JE, Rayment I, Reed GH. A carboxylate oxygen of the substrate bridges the magnesium ions at the active site of enolase: structure of the yeast enzyme complexed with the equilibrium mixture of 2-phosphoglycerate and phosphoenolpyruvate at 1.8 A resolution. Biochemistry. 1996;35:4349–4358. doi: 10.1021/bi952859c. [DOI] [PubMed] [Google Scholar]
- 121.Gerlt JA, Babbitt PC. Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu Rev Biochem. 2001;70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- 122.Bourque JR, Bearne SL. Mutational Analysis of the Active Site Flap (20s Loop) of Mandelate Racemase. Biochemistry. 2007;47:566–578. doi: 10.1021/bi7015525. [DOI] [PubMed] [Google Scholar]
- 123.Nagar M, Narmandakh A, Khalak Y, Bearne SL. Redefining the minimal substrate tolerance of mandelate racemase. Racemization of trifluorolactate. Biochemistry. 2011;50:8846–8852. doi: 10.1021/bi201188j. [DOI] [PubMed] [Google Scholar]
- 124.Allen KN, Dunaway-Mariano D. Markers of fitness in a successful enzyme superfamily. Curr Opin Struct Biol. 2009;19:658–665. doi: 10.1016/j.sbi.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]