

Abstract
GKAs (glucokinase activators) are promising agents for the therapy of Type 2 diabetes, but little is known about their effects on hepatic intermediary metabolism. We monitored the fate of 13C-labelled glucose in both a liver perfusion system and isolated hepatocytes. MS and NMR spectroscopy were deployed to measure isotopic enrichment. The results demonstrate that the stimulation of glycolysis by GKA led to numerous changes in hepatic metabolism: (i) augmented flux through the TCA (tricarboxylic acid) cycle, as evidenced by greater incorporation of 13C into the cycle (anaplerosis) and increased generation of 13C isotopomers of citrate, glutamate and aspartate (cataplerosis); (ii) lowering of hepatic [Pi] and elevated [ATP], denoting greater phosphorylation potential and energy state; (iii) stimulation of glycogen synthesis from glucose, but inhibition of glycogen synthesis from 3-carbon precursors; (iv) increased synthesis of N-acetylglutamate and consequently augmented ureagenesis; (v) increased synthesis of glutamine, alanine, serine and glycine; and (vi) increased production and outflow of lactate. The present study provides a deeper insight into the hepatic actions of GKAs and uncovers the potential benefits and risks of GKA for treatment of diabetes. GKA improved hepatic bioenergetics, ureagenesis and glycogenesis, but decreased gluconeogenesis with a potential risk of lactic acidosis and fatty liver.
Keywords: gluconeogenesis, glycogenesis, glycolysis, N-acetylglutamate (NAG), tricarboxylic acid (TCA) cycle, ureagenesis
INTRODUCTION
A primary hepatic function is to rapidly clear postprandial glucose that reaches the liver through the portal vein [1]. This function, involving enhanced glycolysis and glycogen synthesis but decreased gluconeogenesis, is essential to overall glucose homoeostasis [1–5]. GK (glucokinase), which mediates the initial step of hepatic glucose metabolism, clearly is pivotal to this role. A network of GK-containing cells is present throughout the body [1,4], most prominently in hepatocytes and pancreatic β-cells [3,4]. This enzyme plays a dual role: in β-cells it is a glucose sensor and in hepatocytes it serves as a pacemaker for glucose storage. The centrality of GK in body glucose homoeostasis has made it a promising drug target for diabetes therapy [3–5]. Enormous strides have been made to develop drugs that activate GK [1–5]. These efforts have been very successful, including limited human clinical trials [6]. However, few of the newly developed GKAs (GK activators) have reached the point of practical clinical application [3,4].
Approximately 99% of GK in the body is located in the liver [4] and hepatic carbohydrate metabolism is profoundly impaired in Type 2 diabetes. Thus a potentially key therapeutic goal is to modify body glucose homoeostasis by utilizing GKAs to modulate GK activity. Little is know about the action of GKAs on hepatic intermediary metabolism, including the effects on PDH (pyruvate dehydrogenase), PC (pyruvate carboxylase), the TCA (tricarboxylic acid) cycle, hepatic energy potential, ureagenesis and lipogenesis. We hypothesized that stimulation of glycolysis by GKA will result in a widespread alteration of hepatic metabolic pathways and intermediary metabolism. Stimulation of GK should increase glycolysis and production of pyruvate, thereby favouring incorporation of glucose carbon into the pathways and metabolic cycles illustrated in Figure 1. In the present study we investigated a new GKA, Piragliatin [3–5], by deploying [U-13C6]glucose as a tracer in a liver perfusion system to evaluate to what extent activation of GK and stimulation of glycolysis would influence: (i) the hepatic redox state; (ii) flux through PC, PDH or the TCA cycle; (iii) glycogen synthesis; (iv) gluconeogenesis; and/or (v) synthesis of amino acids, NAG (N-acetylglutamate) and ureagenesis.
Figure 1. Schematic presentation of the possible action of a GK activator on hepatic metabolic pathways during liver perfusion with [U-13C6 ]glucose, glutamine and ammonia.

This scheme illustrates the potential action of GKAs on the coupling between glycolysis, glycogen synthesis, pentose phosphate shunt (PPS) and nucleotide synthesis, the TCA cycle, the generation of citrate and the resulting lipid synthesis, the generation of amino acids and detoxification of ammonia through the urea cycle. Pyruvate, the end product of glycolysis, is converted into lactate, malate or alanine in the cytosol and/or to acetyl-CoA plus CO2 in the mitochondrion. Acetyl-CoA may be (a) metabolized in the TCA cycle, (b) utilized for NAG synthesis, and/or (c) converted into ketone bodies. In addition, pyruvate may enter the TCA cycle through PC generating oxaloacetate (anaplerosis). Oxaloacetate may be transaminated to aspartate and/or may react with acetyl-CoA to form citrate, thereby facilitating formation of 2-oxoglutarate and glutamate (cataplerosis). Citrate may be transported to the cytosol and metabolized through citrate lyase to malonyl-CoA, leading to enhanced lipid synthesis. The production of glutamate and acetyl-CoA provides substrates for mitochondrial NAG synthesis, and thus activates carbamoyl-phosphate synthetase-1 and up-regulates citrulline and urea synthesis. In addition, the following enzymes participate in the metabolic pathways considered in the present study: (1) PDH; (2) PC; (3) glutamate dehydrogenase; (4) NAG synthetase; (5) ornithine aminotransferase; (6) carbamoyl-phosphate synthetase-1; (7) ornithine transcarbamoylase; (8) aspartate–oxaloacetate aminotransferase; (9) malic enzyme; (10) phosphohexose isomerase; (11) phospho-fructokinase-1; (12) aldolase; (13) glyceraldehyde-3-phosphate dehydrogenase; (14) phosphoglycerate kinase; (15) phosphoglycerate mutase; (16) enolase; (17) pyruvate kinase; and (18) LDH. ARS, argininosuccinate; CP, carbamoyl-phosphate.
Glycolysis articulates with a complex metabolic network (Figure 1). Perfusing the structurally intact liver with a solution similar to portal blood thereby affords a model with which to investigate the action of GKA on hepatic glucose handling. We perfused liver in an antegrade mode with or without Piragliatin at a near-maximal concentration [6,7]. We used livers from fed animals in order to simulate physiological conditions and to avoid hormonal effects, such as an increase of the glucagon/insulin ratio following an overnight fasting state. A separate series of experiments was carried out with isolated hepatocytes in order to determine whether GKA acts specifically on GK or whether it has ‘off-target actions’ directly influencing the metabolism of pyruvate, for example by affecting the PDH and/or PC activity or the cytosolic malic enzyme [8].
The present study provides the first comprehensive insight into the widespread action of GKA on glycolysis and associated hepatic metabolic pathways in the postprandial state. The findings show that GKA has many actions, including improved bioenergetics, up-regulation of hepatic glycogenesis or ammonia detoxification through ureagenesis, increased generation of lactate and decreased gluconeogenesis.
EXPERIMENTAL
Materials and animals
Male Sprague–Dawley rats (Charles River) were fed a standard rat chow diet ad libitum. Chemicals were of analytical grade and obtained from Sigma–Aldrich. Enzymes and cofactors for the analysis of adenine nucleotides, β-OH-butyrate, acetoacetate, NADH, NAD+, urea, lactate, pyruvate and ammonia were obtained from Sigma. 13C-labelled glucose, pyruvate and lactate or 15NH4Cl, 99 MPE (mole percentage excess), were from Isotec and Piragliatin (RO4389620) from Hoffman-La Roche.
Experiments with liver perfusions
Livers from fed male rats were perfused in the non-recirculating mode and antegrade flow of 3–3.5 ml. g−1 · min−1 as described previously [9]. Before initiation of the experiment, the liver was washed with Krebs saline plus 0.1%DMSO and glucose (5 mM) as a metabolic fuel (pH 7.4). The perfusate was continuously gassed with 95% O2/5% CO2 and pO2 (in the influent and effluent media) was monitored throughout. After 15 min of conditioning with basic medium containing 0.1% DMSO and 5 mM glucose in Krebs buffer, pH 7.4, the perfusate was replaced with one containing 0.1% DMSO, 5 mM [U-13C6]glucose, 0.3 mM 15NH4Cl and 1 mM glutamine (Medium-A). After 30 min of perfusion with Medium-A, 3 μmol · l−1 Piragliatin was added (Medium-A + GKA), and perfusion was continued for an additional 30 min. The concentration of Piragliatin was chosen on the basis of previous studies demonstrating that this drug reaches a near-maximal effect within 2–3 min at 2–3 μmol · l−1 [6]. Independent control perfusions without GKA were performed with Medium-A for 50 min after conditioning the liver for 15 min.
In each perfusion, samples were taken from the influent and effluent media for measurement of 13CO2 release, enzymatic assays, GC (gas chromatography)-MS or LC (liquid chromatography)-MS analyses as indicated [9,10]. At the end of perfusion the liver was freeze-clamped with aluminum tongs pre-cooled in liquid N2. The frozen liver was ground into a fine powder and kept at −80 °C. One portion was extracted with perchloric acid, neutralized with KOH, and used for metabolite determination and measurement of 13C enrichment with GC-MS or determination of a positional 13C isotopomer with NMR.
Studies with isolated hepatocytes
Hepatocytes were prepared from liver of fed rats by a two-step procedure as described previously [11,12]. Briefly, after washing the liver with oxygenated Krebs buffer (pH 7.5), isolated liver was perfused for 10–15 min with 0.03% collagenase in Krebs buffer (37 °C, pH 7.5). The liver was then transferred to a flask and the exterior membrane was disrupted. Hepatocytes were filtered through 250 μm mesh. Non-viable cells were removed by differential centrifugations. The final cell viability was between 90–95% as determined by Trypan Blue exclusion. Incubations were carried out with a 3 ml cell suspension containing approximately 8–10 mg · ml−1 hepatocyte protein. Incubations with or without 3 μM GKA were carried out for 60 min at 37 °C in a shaking water bath with Krebs buffer (pH 7.4) containing (mM): [1-13C]lactate (10) plus [1-13C]pyruvate (1.5) or [3-13C]lactate (10) plus [3-13C]pyruvate (1.5). A separate series of experiments was performed with only [1-13C]pyruvate (5), [3-13C]pyruvate (5) or [U-13C6]glucose (5).
At the end of incubation, an aliquot (200–300 μl) was taken for measuring the release of 13CO2. Immediately afterwards, incubation was stopped with HClO4 (70%). Metabolite measurements were performed using neutralized extracts. Three to five independent experiments were carried out per series.
LC-MS, GC-MS or NMR methodology
Measurement of 13C-isotopomer enrichment was performed on either an Agilent Triple Quad 6410 mass spectrometer combined with an Agilent LC 1290 Infinity or Hewlett-Packard 5971 MSD (mass selective detector), coupled with a 5890 HP-GC, GC-MS Agilent System (6890 GC/5973 MSD) or a Hewlett Packard HP-5970 MSD using electron impact ionization with an ionizing voltage of −70eV and an electron multiplier set to 2000V. The following metabolites were analysed.
Measurement of 13C-labelled aldose or aldose-P
We used the LC-MS system to determine the 13C isotopomers as well as the concentration by an isotope-dilution approach. 13C-labelled glucose, G6P (glucose 6-phosphate) and GA3P (glyceraldehyde 3-phosphate) were prepared as described previously [13]. Briefly, 50–200 μl of tissue extract were dried, and thereafter 50 μl of NaOH (0.3 M) and 50 μl of PMP (1-phenyl-3-methyl-5-pyrazolone) (0.5 M) were added. The mixture was incubated for 30 min at 70 °C. After cooling to room temperature (25°C), 50 μl of 0.3 M HCl and 0.5 ml of H2O were added. The excess of PMP was removed by two extractions with ethyl acetate. The water fraction was dried down and dissolved in 200 μl of LC solvent A (water with 0.1% formic acid). A 1 μl volume of derivatized sample was injected into the LC-MS instrument for analysis. The LC gradient was as follows: Solution A, water with 0.1% of formic acid; Solution B, acetonitrile with 0.1% formic acid and 0.005% TFA (trifluoroacetic acid). Running program: 0–1 min, 25% B; 1–4 min, 28% B; 5–7 min, 100% B; 7–9 min, 0% B; and 9–10 min, 25% B. The flow rate was 0.5 ml · min−1 throughout. The retention time for G6P was 2.2 min, for glucose 2.95 min and for GA3P 3.10 min. The column used was Poroshell 120 EC-C18, 4.6 mm×50 mm with a particle size of 2.7 μm. We followed the precursor–product links for the aldoses studied with a collision energy of 30 V and fragmentor voltage of 150 V. For glucose in MRM (multiple reaction monitoring) mode, we measured ion-pairs 511-175, 512-175, 513-175, 514-175, 515-175, 516-175 and 517-175 for determination of M0, M1, M2, M3, M4, M5 and M6 respectively (containing one to six 13C atoms above M0, the natural abundance). For G6P, we measured ion-pairs 591-175, 592-175, 593-175, 594-175, 595-175, 596-175 and 597-175 for determination of M0, M1, M2, M3, M4, M5 and M6 respectively (containing one to six 13C atoms above M0, the natural abundance). For GA3P we measured ion-pairs at 501-175, 502-175, 503-175 and 504-175 for determination of M0, M1, M2, M3 respectively (containing one to three 13C atoms above M0, the natural abundance). After determining the 13C enrichment in G6P and GA3P, the samples were spiked with known concentrations of unlabelled G6P and GA3P, then samples were analysed by LC-MS to determine the dilution of 13C enrichment for calculation of the metabolite levels.
Measurement of 13CO2 release
The production of 13CO2 during liver perfusion or following incubation of isolated hepatocytes was monitored as follows: 100–200 μl samples were placed into autosampler tubes. The capped tubes with sample were filled with helium to remove any atmospheric CO2. Then, 100 μl of 20% phosphoric acid was injected through the stopper to liberate the CO2. Isotopic enrichment in 13CO2 was determined by an isotope-ratio mass spectrometer (Thermoquest Finnigan Delta Plus) using an m/z ratio of 45/44.
Measurement of 13C-labelled glycogen synthesis
A separate portion of the frozen liver after perfusion was ground into powder and samples were incubated in the presence or absence of amyloglucosidase as described previously [14]. The liberated glucose from glycogen was determined by glucose oxidase assay [15]. The background glucose content was subtracted to obtain the tissue glycogen content, and 13C enrichment in glucose was determined by LC-MS as described above. Results obtained were expressed as μmol of 13C-labelled glucose released from glycogen per g of wet mass of liver.
Measurement of 13C-labelled isotopomers in amino or organic acids
For measurement of the 13C enrichment in amino acids or organic acids, samples were prepared as described previously [9]. Briefly, an aliquot (500 μl) of effluent, liver or hepatocyte extracts was purified by passage through an AG-1 column (Cl−: 100–200 mesh; 0.5 cm×2.5 cm) for separation of organic acids and glutamate and aspartate residues or by passage through an AG-50 column (H+: 100–200 mesh) for separation of alanine, serine, glycine and glutamine residues and urea. Then, samples were converted into the t-butyldimethylsilyl derivatives [9,10]. Isotopic enrichment in [13C]alanine isotopomers was monitored using ions at m/z 260, 261, 262 and 263 for M0, M1, M2 or M3 (containing one to three 13C atoms above M0, the natural abundance) respectively. Isotopic enrichment for [13C]serine isotopomers was monitored at m/z 390, 391, 392 and 393 for M0, M1, M2 or M3 (containing one to three 13C atoms above M0, the natural abundance) respectively. Isotopic enrichment in [13C]glycine isotopomers was monitored using ions at m/z 218, 219 and 220 for M0, M1 or M2 (containing one or two 13C atoms above M0, the natural abundance) respectively. Isotopic enrichment in glutamine isotopomers was monitored using ions at m/z 431, 432, 433, 434, 435, 436 and 437 for M0, M1, M2, M3, M4, M5 and M6 (containing one to five 13C atoms plus 15N in the amido-N) respectively. Isotopic enrichment in [13C]glutamate isotopomers was monitored using ions at m/z 432, 433, 434, 435, 436 and 437 for M0, M1, M2, M3, M4 or M5 (containing one to five 13C atoms above M0, the natural abundance) respectively. Isotopic enrichment in [13C]aspartate isotopomers was monitored using ions at m/z 418, 419, 420, 421 and 422 for M0, M1, M2, M3 and M4 (containing one to four 13C atoms above M0, the natural abundance) respectively. Isotopic enrichment in [13C]lactate was monitored using ions at m/z 261, 262, 263 and 264 for M0, M1, M2 and M3 (containing one to three 13C atoms above natural abundance) respectively. Since lactate and pyruvate are in equilibrium, we used the enrichment of 13C isotopomers of lactate as surrogate for 13C enrichment in pyruvate. Isotopic enrichment in 13C malate isotopomers was monitored using ions at m/z 419, 420, 421, 422 and 423 for M0, M1, M2, M3 and M4 (containing one to four 13C atoms above the natural abundance) respectively, and 13C enrichment in [13C]citrate isotopomers was monitored using ions at m/z 459, 460, 461, 462, 463, 464 and 465 for M0, M1, M2, M3, M4, M5 and M6 (containing one to six 13C atoms above the natural abundance) respectively.
Measurement of 13C-labelled NAG or 15N-labelled urea synthesis
The concentration and 13C enrichment in NAG isotopomers in liver extracts were determined using LC-MS. A 200 μl volume of sample was dried down, mixed with 200 μl of 3 M butanolic HCl and heated at 60 °C for 15 min. Then, samples were cooled to room temperature, dried and mixed with 0.5 ml of water. The derivatized NAG was extracted with 2 ml of ethyl acetate. The organic fraction was dried under a stream of N2, and then reconstituted with 200 μl of 0.1% formic acid in H2O. Finally, samples were filtered and 1 μl was injected into the LC instrument. The chromatogram gradient was as follows: Solution A, 0.1% formic acid in H2O; Solution B, acetonitrile with 0.1% formic acid and 0.005% TFA. Times: 0–1 min, 25% B; 1–2 min, 35% B; 2–3 min, 45% B; 3–4 min, 55% B; 4–5 min, 65% B; 5–6 min, 75% B; 6–7 min, 85% B; and 7–9 min, 100% B. The flow rate was 0.5 ml · min−1 throughout. The retention time of the NAG derivative was 3.92 min. The column used was Poroshell 120 EC-C18, 4.6 mm×50 mm with a particle size of 2.7 μm. The ion-pairs monitored were 302-228, 303-229, 304-230, 305-231, 306-232, 307-233, 308-234 and 309-235 for determination of M0, M1, M2, M3, M4, M5, M6 and M7 respectively (containing one to seven 13C atoms above M0, i.e. five carbons from glutamate and two carbons from acetyl-CoA). This link reflects the loss of a fragment weighing 73 (i.e. CH3-CH2-CH2-CH2-OH). After the initial measurement of 13C enrichment in NAG isotopomers, the samples were spiked with a known amount of NAG for determination of the NAG concentration by isotope dilution [10]. The production of 15N-labelled urea isotopomers (containing one or two 15N atoms) from 15NH4Cl and its output in the perfusate were determined as described previously [16].
NMR analysis
To further evaluate the action of GKA on positional isotopomers in the intermediary metabolites, a portion (approximately 1 g of wet mass) of the neutralized liver extracts was analysed by 1H NMR, 13C NMR and 31P NMR methodology using a Bruker Avance III 400 wide bore equipped with a Hewlett Packard workstation running the Bruker Topspin™ NMR software. The chemical shifts of 13C signals were measured relative to the resonance of trimethylsilylpropionic acid at −2.7 p.p.m. in the 13C-NMR spectra. Data acquisition and calculation of percentage 13C enrichment in various glutamate carbons were performed as described in [10,17]. Pi or compounds containing phosphate were identified and their levels were measured relative to the resonance of methylene diphosphonate (16.7 p.p.m.) contained in an external capillary [10,17].
Measurement of metabolite levels
The concentration of amino acids was determined with an Agilent 1260 Infinity LC system, utilizing pre-column derivatization with o-phthalaldehyde [18]. The levels of ammonia and urea were measured as described in [9,10,16], those of ATP as in [19], and those of of ADP and AMP as in [20]. We also measured the level of NAD and NADH as described in [21] using a BioVision quantification kit, levels of lactate [22], pyruvate [23], acetoacetate and β-OH-butyrate [24], malate [25], glucose [15], glycogen [14] and fructose 1,6-bisphosphate [26], and levels of triglycerides in liver extract using an Infinity™ Triglycerides Reagent kit.
Calculations and statistical analyses
Production and output of 13C-labelled mass isotopomers or [15N]urea
During liver perfusions, the rate of uptake or the output of metabolites was determined by the measurement of metabolite concentration in the influent and effluent (nmol · ml−1), normalized to the flow rate (ml · min−1) and liver wet mass (g) as described previously [9,16].
13C enrichment in 13C-labelled mass isotopomers is expressed as MPE, which is the molar fraction (%) of analyte containing 13C atoms above natural abundance. The MPE of an individual 13C-labelled mass isotopomer (containing i 13C atoms, i.e. in the case of glutamate i = 1–5, for citrate i = 1–6 etc.) was calculated as in [27,28]. Briefly, MPE (M + i) = percentage AM + i/(AM + ΣAM + i), where AM and AM + i represents the peak area from LC-MS or GC-MS ions corrected for natural abundance as described in [27,29], and corresponding to the unlabelled (M0) and 13C-labelled (M + i) mass isotopomer respectively. The output of 13C-labelled mass isotopomer was calculated by the product of (MPE/100) multiplied by the concentration (nmol · min−1 per g of wet mass) and is expressed as nmol of 13C-labelled metabolite · min−1 per g of wet mass. Similarly, the concentration of 13C-labelled mass isotopomer in the liver was calculated by the product of (MPE/100) multiplied by the concentration (nmol · g−1 of wet mass) and is expressed as nmol of 13C-labelled metabolite per g of wet mass. The output of 13CO2 (nmol · min−1 per g) was calculated by the product of [13CO2 enrichment (atom percentage excess)/100] multiplied by flow rate (ml · min−1 per g) multiplied by 25 mM (i.e. the concentration of NaHCO3 in the perfusate). The output of 15N-labelled urea isotopomers was calculated as indicated in [9,16].
Glycogen synthesis
Hepatic glycogen synthesis may be mediated through a direct pathway from glucose and an indirect pathway from gluconeogenic precursors (lactate, pyruvate etc.) [30,31]. In the present study, the synthesis of glycogen from the perfusate [U-13C]glucose, i.e. the ‘direct pathway’, was calculated as follows: G-direct = [Glycogen]×[U-13C6]glucose (MPE/100), where [glycogen] is the total glycogen concentration in freeze-clamped liver, MPE is the 13C enrichment in [U-13C6]glucose derived from glycogen hydrolysis and C6 refers to the number of carbon atoms labelled. The synthesis from 13C-labelled lactate/pyruvate, i.e. the ‘indirect pathway’, was calculated as follows: G-indirect = [Glycogen]×[13C3]glucose (MPE/100), where MPE is the 13C enrichment in C3-glucose (glucose labelled at 3 carbons) derived from glycogen hydrolysis.
Phosphorylation potential and redox state
Phosphorylation potential was calculated from the measured concentrations of ATP, ADP and Pi as: ([ATP]/[ADP])× [Pi]. The adenylate energy charge was calculated as: ([ATP] + [ADP]/2)/([ATP] + [ADP] + [AMP]).
Because the direct measurement of NAD+, NADH, NADP+ and NADPH is not informative in terms of the control of metabolism, the free cytosolic NAD+/NADH and NADP+/NADPH ratios were calculated from metabolite concentrations and the equilibrium constants of LDH (lactate dehydrogenase) and malic enzyme as described in [32,33], as follows: cytoplasm NAD+/NADH = [pyruvate]/[lactate]×1/kld, where kld is the equilibrium constant of LDH (kld = 1.11 × 10−4 M) [32]. Cytoplasmic NADP+/NADPH = ([pyruvate]×[CO2]/[malate])×1/kme, where kme, is the equilibrium constant of malic enzyme (kme = 3.44×10−2 M). The CO2 concentration was taken as 1.16 mM [32]. The mitochondrial NAD+/NADH ratio was calculated as follows: NAD+/NADH = [β-OH-butyrate]/[acetoacetate]×1/kBHB, where kBHB is the equilibrium constant of β-OH-butyrate dehydrogenase (kBHB = 4.93×10−2 M) [32].
Each series of experiments was repeated three times with an individual liver perfusion system as outlined above. Statistical analysis was carried out using Prism 5 software. A Student’s t test or ANOVA test was employed to compare two groups or differences among groups as needed.
RESULTS
O2 consumption following near-maximal GK activation with Piragliatin
Figure 2 represents O2 consumption during perfusions with or without GKA. In control studies without GKA, O2 consumption reached a plateau of approximately 2.1 ± 0.08 μmol · min−1 · g−1 (mean ± S.D.; n = 3) between 30 and 65 min of perfusion with [U-13C6]glucose. In the presence of Piragliatin (3 μmol · l −1), O2 consumption increased within the first 5 min and reached a plateau of 2.5 ± 0.1 μmol · min−1 · g−1 (mean ± S.D., n = 3) between 10 and 35 min (55–75 min of perfusion). The plateau levels of O2 consumption in both study groups indicate that equilibrium is reached during the course of liver perfusion. The increased O2 consumption within 3–5 min following supplementation of Piragliatin also demonstrates an immediate and direct response to the drug, probably subsequent to increased glycolysis and oxidative phosphorylation.
Figure 2. O2 consumption during liver perfusion with or without a GKA.

Liver perfusions were carried out as detailed in the Experimental section. Briefly, after a 15 min conditioning of the liver with a basic medium containing 0.1 % DMSO and 5 mM glucose in Krebs buffer, pH 7.4 (WASH), the perfusate was replaced with one containing 0.1 % DMSO, 5 mM [U-13C]glucose, 0.3 mM 15NH4 Cl and 1 mM glutamine in Krebs buffer (Medium-A). At 45 min, perfusion was continued with Medium-A plus 3 mmol · l −1 Piragliatin, [(+) GKA]. Independent control perfusions without GKA (Control) were performed with Medium-A only throughout the course of the perfusion. O2 consumption was monitored polarographically using a Clark Electrode connected to an Instech Oxygen Monitoring System coupled with WinDAq/Lite Waveform software. Results are means ± S.D. for three independent liver perfusions per study group. *P < 0.05, compared with control.
[U-13C6]glucose uptake and metabolite output following GK activation with Piragliatin
Table 1 summarizes rates of [U-13C6]glucose uptake and 13C-labelled metabolite output during perfusion with or without GKA. In the perfusion with GKA, there was a near-4-fold increased uptake of [U-13C6]glucose compared with controls (Table 1). The hepatic uptake of glucose is in line with the notion that the liver switches from an organ of net glucose output to one of net glucose uptake in response to a ‘threshold’ concentration of portal venous glucose (approximately 5.5 mM) [30]. The increased uptake of [U-13C6]glucose by GKA was accompanied by a 3-fold increased output of 13CO2 (Table 1 and Figure 3A) and an even larger increase of 13C-labelled pyruvate and lactate output (Table 1, Figures 3B and 3C, and Supplementary Figure S1 at http://www.BiochemJ.org/bj/444/bj4440537add.htm). Similarly, with the addition of GKA there was a 6–7-fold increased outflow of 13C-labelled alanine (Table 1, Figure 4A, and Supplementary Figure S2 at http://www.BiochemJ.org/bj/444/bj4440537add.htm) accompanied by approximately 30% lower output of 13C-labelled glutamate (Figure 4B). These results demonstrate that the action of GKA on hepatic metabolism occurred within 3–5 min following exposure to the drug.
Table 1. [U-13C]Glucose uptake and the output of the primary 13C-labelled glycolytic products during the course of perfusion with or without GKA.
The rate of glucose uptake was determined by the differences between glucose concentrations in the influent and effluent (nmol · ml−1), normalized to the flow rate (ml · ml−1) and liver wet mass (g), as described previously [9,16]. The rates of 13C-labelled metabolite output are the steady-state values obtained between 30 and 65 min of perfusion without GKA (Control) or 55–75 min of perfusion with GKA [(+) GKA], as presented in Figures 3 and 4. These rates were determined by measuring of metabolite concentrations in the effluent (nmol/ml), normalized to the flow rate (ml · ml−1) and liver wet mass (g), as in [9,16]. Experimental details are as described in the legends to Figure 2. Results are means ± S.D. of three livers per group. P values were determined by Student’s t test using Prism 5 software.
| Metabolite | Control (nmol · min−1 · g−1) | (+) GKA (nmol · min−1 · g−1) | P value |
|---|---|---|---|
| [U-13C6 ]Glucose uptake | 568 ± 230 | 2233 ± 739 | <0.01 |
| 13CO2 output | 131 ± 12 | 448 ± 36 | <0.01 |
| [13C]Lactate output | 22 ± 4 | 452 ± 46 | <0.01 |
| [13C]Pyruvate output | 3.1 ± 1.3 | 67 ± 10.5 | <0.01 |
| [13C]Alanine output | 2.5 ± 0.9 | 15 ± 1.5 | <0.01 |
Figure 3. Action of a GKA on output of 13CO2 (A) and 13C-labelled pyruvate (B) or 13C-labelled lactate (C) during liver perfusion.
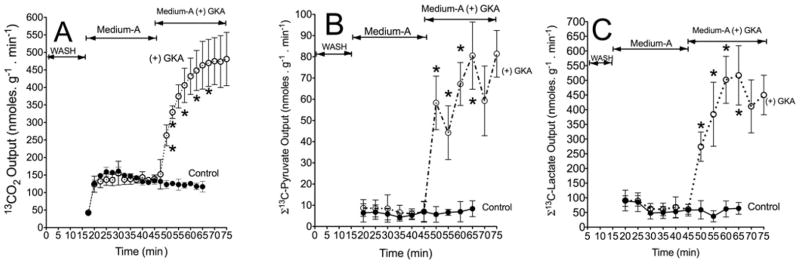
Liver perfusions were carried out as detailed in the legend to Figure 2. The output of 13C-labelled pyruvate or lactate is the sum of the 13C enrichment (MPE/100) in M2 and M3 isotopomers of these metabolites multiplied by total output (nmol · min−1 per g of wet mass). Results are means ± S.D. for three independent liver perfusions per study group. *P < 0.05, compared with control.
Figure 4. Action of a GKA on the output of 13C-labelled alanine (A) or 13C-labelled glutamate (B) during liver perfusion.
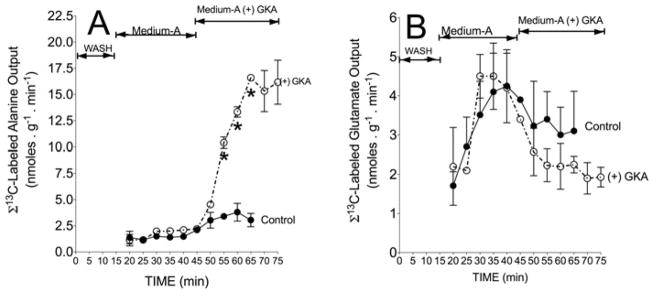
Liver perfusions were carried out as detailed in the legend to Figure 2. The output of 13C-labelled alanine or glutamate is the sum of the 13C enrichment (MPE/100) in M2 and M3 (13C isotopomers of alanine) and M1, M2, M3, M4 and M5 (13C isotopomers of glutamate) multiplied by total output (nmol · min−1 per g of wet mass). Results are means ± S.D. for three independent liver perfusions per study group. *P < 0.05, compared with control.
Metabolite profiles, energy potential and redox state of liver tissue following near-maximal GK activation with Piragliatin
The results in Table 2 represent the energy state of freeze-clamped livers perfused with 5 mM glucose in the presence and absence of a GKA. The increased O2 consumption (Figure 2) during perfusions with GKA was coupled with increased [ATP] and a lowering of [Pi], thereby resulting in a higher phosphorylation potential and adenylate energy charge (Table 2). [ATP] was increased (P = 0.03) compared with the basal condition, and [AMP] and [Pi] were decreased, with only little concomitant change in [ADP]. Calculation of the phosphorylation potential ([ATP]/[ADP]×[Pi]) indicates an increase (P = 0.03) upon stimulation of glucose metabolism, a finding in accordance with the modest increase of adenylate energy charge (P = 0.06). These results imply that GKA improved the energy metabolism in the liver, since both the ATP/ADP ratio and adenylate energy charge are indicative of the availability of high-energy phosphates for metabolic and functional needs.
Table 2. Levels of adenine nucleotides, Pi, phosphorylation potential and adenylate energy charge in freeze-clamped liver following perfusion with or without GKA.
ATP, ADP, AMP and Pi were determined in extracts of freeze-clamped liver after perfusion without (Control) or with GKA [(+) GKA] as indicated in the Experimental section. The phosphorylation potential was calculated from the measured total concentrations of ATP, ADP and Pi as: [ATP]/[ADP]×[Pi ]. The adenylate energy charge was calculated as: ([ATP] + [ADP]/2)/([ATP] + [ADP] + [AMP]). Results are means ± S.D. of three livers per group. P values were determined by Student’s t test using Prism 5 software.
| Control (μmol per g of wet mass) | (+) GKA (μmol per g of wet mass) | P value | |
|---|---|---|---|
| ATP | 3.4 ± 0.1 | 4.1 ± 0.4 | 0.03 |
| ADP | 1.4 ± 0.2 | 1.1 ± 0.4 | N.S. |
| AMP | 0.7 ± 0.1 | 0.5 ± 0.1 | 0.08 |
| Pi | 4.8 ± 0.5 | 3.9 ± 0.6 | 0.07 |
| Phosphorylation potential | 0.52 ± 0.14 | 1.05 ± 0.32 | 0.03 |
| Adenylate energy charge | 0.74 ± 0.01 | 0.82 ± 0.03 | 0.06 |
N.S., not significant.
Table 3 presents levels of intermediary metabolites in freeze-clamped livers at the end of perfusion. We found a significant accumulation of lactate (P = 0.02) and a slight increase in pyruvate (P = 0.06) in the presence of GKA, but strikingly constant levels of citrate, malate, acetoacetate, β-OH-butyrate, NAD+ or NADH (Table 3). However, the level of triglycerides was increased marginally (P = 0.07) (Table 3). In addition, the levels of G6P and GA3P rose significantly with little change in the levels of fructose 1,6-bisphosphate. The 31P-NMR spectra of freeze-clamped liver corroborate the LC-MS measurement and demonstrate a higher peak of G6P following GKA (Supplementary Figure S3 at http://www.BiochemJ.org/bj/444/bj4440537add.htm). Similarly, 31P-NMR spectra show an approximately 2-fold higher peak of glycerol 3-phosphate in the liver when GKA was present.
Table 3. Metabolite profiling and redox state in freeze-clamped liver following perfusion with or without GKA.
Metabolites were determined in extracts of freeze-clamped liver after perfusion without (Control) or with GKA [(+) GKA] as indicated in the Experimental section. The redox state is represented by the free cytosolic NAD+/NADH and NADP+/NADPH ratios calculated from concentrations of pyruvate, lactate or malate. The mitochondrial NAD+/NADH ratio was calculated from concentrations of β-OH-butyrate and acetoacetate as indicated [32]. Results are means ± S.D. of three livers per group. P values were determined by Student’s t test using Prism 5 software.
| (a) Metabolite level
| |||
|---|---|---|---|
| Control (μmol per g of wet mass) | (+) GKA (μmol per g of wet mass) | P value | |
| Lactate | 1.51 ± 0.06 | 1.81 ± 0.15 | 0.02 |
| Pyruvate | 0.28 ± 0.03 | 0.42 ± 0.12 | 0.06 |
| Malate | 0.18 ± 0.05 | 0.26 ± 0.03 | N.S. |
| Citrate | 0.25 ± 0.04 | 0.29 ± 0.04 | N.S. |
| Acetoacetate | 0.35 ± 0.03 | 0.37 ± 0.13 | N.S |
| β-OH-butyrate | 0.23 ± 0.04 | 0.25 ± 0.04 | N.S. |
| NAD | 0.18 ± 0.06 | 0.26 ± 0.03 | N.S. |
| NADH | 0.083 ± 0.05 | 0.081 ± 0.02 | N.S. |
| Control (nmol per g of wet mass) | (+) GKA (nmol per g of wet mass) | P value | |
| G6P | 33.7 ± 6.2 | 103.2 ± 15.9 | 0.007 |
| GA3P | 3.2 ± 1.3 | 7.2 ± 0.9 | 0.014 |
| Fructose 1,6-bisphosphate | 65.7 ± 21 | 75.4 ± 5.8 | N.S. |
| Control (mg per g of wet mass) | (+) GKA (mg per g of wet mass) | P value | |
| Triglycerides | 0.26 ± 0.03 | 0.38 ± 0.16 | N.S. |
| (b) Redox state
| |||
|---|---|---|---|
| Control | (+) GKA | P value | |
| Cytoplasmic NAD+/NADH | 1684 ± 126 | 2170 ± 819 | N.S. |
| Cytoplasmic NADP+/NADPH | 0.055 ± 0.01 | 0.066 ± 0.02 | N.S. |
| Mitochondrial NAD+/NADH | 32 ± 7.2 | 31 ± 12.5 | N.S. |
N.S., not significant.
The ratios of NAD+/NADH as calculated from the levels of these cofactors show little change following GKA (Table 3). In addition, the free cytosolic NAD+/NADH and NADP+/NADPH ratios were calculated from concentrations of pyruvate, lactate or malate, and mitochondrial NAD+/NADH from concentrations of β-OH-butyrate and acetoacetate [32,33]. These results demonstrate that GKA has little effect on cytoplasmic NADP+/NADPH or cytoplasmic and mitochondrial NAD+/NADH (Table 3), suggesting a minor effect on the redox state.
13C-isotopomer profiling in liver tissue following GK activation with Piragliatin
Figure 5 represents the GC-MS analysis of 13C-isotopomer profiling of lactate (panel A), citrate (panel B) and malate (panel C) in liver extracts. These results demonstrate that GKA remarkably increased 13C enrichment (MPE) in all mass isotopomers of these intermediates compared with control perfusions. The results in Figures 5(D)–5(F) demonstrate that the incorporation of [U-13C6]glucose carbon into lactate, citrate and malate was approximately 5-fold higher following perfusion with GKA compared with controls, and that the generation of 13C-labelled lactate is the primary ‘sink’ into which glucose carbon flows following stimulation of glycolysis by GKA.
Figure 5. Action of a GKA on the generation and profiling of 13C mass isotopomers of lactate, citrate or malate in the liver.

Perfusions were carried out as indicated in the legend to Figure 2. At the end of perfusion, the liver was freeze-clamped, extracted with perchloric acid, neutralized and analysed with either GC-MS or LC-MS as detailed in the Experimental section. (A–C) M2, M3 etc. are the mass isotopomers of each metabolite in perfusion with GKA [(+) GKA] or without (CON). The levels of 13C-labelled metabolites (D–F) are the sum of 13C enrichment (MPE/100) of mass isotopomers of the indicated metabolite multiplied by concentration (nmol · g−1 of wet mass). Results are means ± S.D. for three individual livers per study group. P values were determined by Student’s t test using Prism 5 software.
The 13C3-lactate (M3) isotopomer was the major product of glycolysis, and was probably derived from [U-13C3]pyruvate mediated through the LDH reaction. Furthermore, a readily detectable amount of doubly 13C-labelled lactate (M2) was observed with or without GKA (Figure 5A). M2 lactate can not be produced from [U-13C6]glucose through glycolysis alone and it may be generated through the sequence glycolysis → TCA cycle → cytosolic malic enzyme → gluconeogenesis, followed by glycolysis of newly formed 13C-labelled glucose. An additional possibility is that the pyruvate cycling through pyruvate kinase may be responsible for production of M2 lactate.
Figures 5(B) and 5(C) depict the 13C-isotopomer profile of citrate and malate, the chief metabolites produced through incorporation of 13C2-labelled acetyl-CoA into the TCA cycle and/or by anaplerosis mediated through PC. An additional possibility for generation of M2 and M4 isotopomers of [13C]citrate and [13C]malate is through incorporation into the TCA cycle of [13C2]acetyl-CoA followed by one turn of the cycle. The M3 isotopomer of citrate is probably produced through PC from [U-13C3]pyruvate and CO2. After another turn of the TCA cycle the M5 isotopomer will be generated. The M6 isotopomer of citrate was approximately 1 MPE (results not shown). Figures 5(E) and 5(F) demonstrate a significant generation of 13C-labelled citrate and malate in livers perfused with GKA, but to a much lesser extent than that of lactate.
Figure 6 represents the 13C isotopomer profile of alanine, serine or glycine in the liver. The M3 labelling pattern for the [13C]alanine isotopomer (Figure 6A) was generally similar to that of lactate (Figure 5A), the alanine having formed from transamination of [U-13C3]pyruvate. Activation of GK significantly increased the generation and accumulation of 13C-labelled alanine and resulted in a marginal elevation of serine and glycine from [U-13C6]glucose (Figures 6D–6F respectively).
Figure 6. Action of a GKA on the generation and profiling of 13C mass isotopomers of alanine, serine and glycine in the liver.

The same liver extracts described in the legend to Figure 5 were analysed for 13C mass isotopomers of the indicated amino acids. (A–C) M2, M3 etc are the mass isotopomers of each metabolite in perfusions with GKA [(+) GKA] or without (CON). The concentrations of 13C-labelled metabolites (D–F) are the sum of 13C enrichment (MPE/100) of mass isotopomers of the indicated metabolite multiplied by concentration (nmol · g−1 of wet mass). Results are means ± S.D. for three individual livers per study group. P values were determined by Student’s t test using Prism 5 software.
Figure 7 represents the 13C mass isotopomer profiles of glutamate, glutamine and aspartate. 13C mass isotopomers of glutamate and aspartate (Figures 7A and 7B) are cataplerotic metabolites deriving from the TCA cycle [27,28]. The M2, M3, M4 and M5 13C mass isotopomers of glutamine resemble that of glutamate (Figure 7C). Because 15NH4Cl was added to the perfusate, the M1 isotopomer of glutamine must have been derived from 15NH4Cl and unlabelled glutamate in hepatocytes. The sum of 13C mass isotopomer production of glutamate, aspartate and glutamine from [U-13C6]glucose is presented in Figures 7(D)–7(F) respectively and demonstrates a significantly increased generation of these amino acids following GKA-induced stimulation of hepatic glycolysis.
Figure 7. Action of a GKA on the generation and profiling of 13C mass isotopomers of glutamate, aspartate and glutamine in the liver.

The same liver extracts described in the legend to Figure 5 were analysed for 13C mass isotopomers of the indicated amino acids. (A–C) M2, M3 etc are the mass isotopomers of the indicated metabolite in experiments with [(+) GKA)] or without (CON) the GKA. For glutamine, the M1 isotopomer represents the synthesis of glutamine from 15NH4 Cl and unlabelled glutamate. (D–F) The concentrations of 13C-labelled glutamate, aspartate and glutamine are the sum of 13C enrichment (MPE/100) of mass isotopomers multiplied by concentration (nmol · g−1 of wet mass). Bars are means ± S.D. for three individual livers per study group. P values were determined by Student’s t test using Prism 5 software.
The positional 13C isotopomers in liver tissue: measurement with 13C-NMR
We found that the LC-MS and GC-MS data (Figures 5–7) were corroborated by 13C-NMR analysis (Supplementary Figure S4 at http://www.BiochemJ.org/bj/444/bj4440537add.htm). 13C-NMR spectra demonstrate higher peaks of 13C isotopomers of lactate, alanine, serine, glycine, malate, glutamate and aspartate with GKA (Supplementary Figure S4B) compared with controls (Supplementary Figure S4A). The 13C-coupling pattern of C-3 and C-2 of lactate and alanine were doublet and quartet, indicating that lactate and alanine were largely labelled at all three carbon positions. This is consistent with the abundance of the M3 mass isotopomers of lactate and alanine as determined by GC-MS (Figures 5A and 6A). Similarly, the doublet or multiplet pattern of C-2, C-3 and C-4 of glutamate, C-2, C-3 of aspartate or C-3 of malate are in agreement with the GC-MS measurement of 13C mass isotopomer profile (Figures 5–7). Of special interest is the observation that the peaks corresponding to C-3 and C-2 glutamate isotopomers are higher than the C-4 isotopomer. Glutamate is labelled at the C-4 and C-5 positions through the forward cycle reactions following incorporation into the TCA cycle of [13C2]acetyl-CoA, the product of PDH. However, glutamate is labelled at C-2 and C-3 mainly through anaplerosis by pyruvate carboxylation. In perfusions with GKA, the percentage 13C enrichment in C-2, C-3 and C-4 was approximately 9, 16 and 9 respectively. In controls without GKA, the percentage 13C enrichment in C-2, C-3 and C-4 was approximately 2, 3 and 1 respectively. Thus the percentage enrichment of 13C isotopomers indicate that: (i) the incorporation of 13C-labelled pyruvate into the TCA cycle through the PC was remarkably higher compared with the incorporation of [13C2]acetyl-CoA, the product of PDH activity; and (ii) the incorporation of 13C-labelled pyruvate into the TCA cycle and the flux of carbons through the cycle were elevated in livers perfused with GKA compared with control.
The 13C-NMR spectra also revealed important information regarding the relationship between hepatic glycolysis and glutamine metabolism. C-3 of glutamine is mainly singlet in control studies (Supplementary Figure S4), suggesting that the glutamine peak was largely related to the natural abundance of 13C in unlabelled glutamine added to the perfusate. However, following perfusion with GKA, C-2, C-3 or C-4 of glutamine resembled the 13C pattern of glutamate. It is likely that an increase in glycolysis induced by GKA together with increased availability of 13C-labelled glutamate and ATP led to increased synthesis of 13C[glutamine] from 13C[glutamate] in the perivenous hepatocytes [34,35].
Hepatic glycogenesis following GK activation with Piragliatin
Figure 8(A) indicates that the total glycogen in freeze-clamped liver at the end of perfusion was slightly elevated following perfusion with GKA, being approximately 161 ± 38 and 180 ± 18 μmol · g−1 of wet mass (means ± S.D., n = 3) in liver perfused with or without GKA respectively. Figure 8(B) demonstrates that GKA significantly (P = 0.045) stimulated the synthesis of glycogen from perfusate [U-13C6]glucose as determined by the appearance of [U-13C6]glucose in glycogen. However, the synthesis of glycogen with M3 (indirect pathway) was significantly (P = 0.02) decreased by GKA as determined by the appearance of [13C3]glucose (M3) in hepatic glycogen following perfusion with GKA (Figure 8B). The amount of [U-13C6]glucose derived through the amyloglucosidase reaction was 3.97 ± 1 and 2.3 ± 1.1 μmol · g−1 of wet mass liver in perfusions with or without GKA respectively (means ± S.D., n = 3, P = 0.04). The amount of [13C3]glucose was 0.45 ± 0.1 and 0.71 ± 0.11 μmol · g−1 of wet mass liver in perfusions with or without GKA respectively (means ± S.D., n = 3, P = 0.02) (Figure 8B). Thus, in liver perfused with GKA, approximately 87% and 13% of glycogen synthesis was derived from glucose and a 3-carbon precursor respectively. In the perfusion without GKA, the fraction of direct and indirect pathways was approximately 77% and 23% respectively. In addition, the 13C-NMR spectra demonstrate a remarkably higher [13C]glycogen peak (C1 glucosyl at approximately 100 p.p.m.) in liver following perfusion with GKA (Supplementary Figure S4), indicating that GKA augmented the synthesis of glycogen from 13C6-labelled glucose. The finding that GKA increased the direct and decreased the indirect pathway of glycogen synthesis is further supported by the observation that when liver was perfused only with 13C-labelled lactate plus pyruvate the rate of gluconeogenesis from these 3-carbon precursors was decreased by GKA (Supplementary Figure S5 at http://www.BiochemJ.org/bj/444/bj4440537add.htm).
Figure 8. Action of a GKA on the direct and indirect pathways of glycogen synthesis.

(A) Total amount of glucose derived from glycogen at the end of perfusions. (B) Direct and indirect synthesis of 13C-labelled glycogen from [U-13C]glucose following perfusion with or without GKA. The direct pathway was calculated as [glycogen]×[U-13C6 ]glucose (MPE/100), where [glycogen] is the total glucose derived from glycogen hydrolysis (μmol · g−1 of wet mass) in freeze-clamped liver extract and MPE is the 13C enrichment in [U-13C6 ]glucose derived from glycogen hydrolysis. The indirect pathway was calculated as [glycogen] (μmol · g−1 of wet mass)×[13C3 ]glucose (MPE/100), where MPE is the 13C enrichment in C3 -glucose (glucose labelled at three carbons) derived from glycogen hydrolysis. Results are means ± S.D. for three individual livers per control without GKA (CON) or with (+) GKA. P values were determined by Student’s t test using Prism 5 software.
GKA increases hepatic NAG and stimulates urea synthesis
The results in Figure 9(A) indicate that the total [NAG] in the liver was increased in perfusions with GKA. This increase is mainly due to a significantly augmented (P = 0.028) synthesis of 13C-labelled NAG. Because NAG is synthesized from acetyl-CoA and glutamate [12], the 13C mass isotopomers profile for NAG indicates that [13C]NAG was generated from [13C2]acetyl-CoA and the various 13C mass isotopomers of glutamate, as depicted in Figure 7(A). Thus, consistent with the augmented production of 13C-labelled metabolites by GKA (Figures 5–7), the increased production of 13C-labelled NAG was probably mediated by increased availability of [13C]pyruvate and its metabolism to acetyl-CoA.
Figure 9. Action of a GKA on the level and synthesis of 13C-NAG and the resulting synthesis and output of total and 15N-labelled urea.
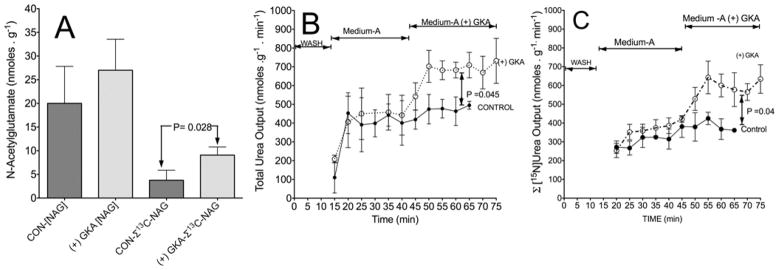
Liver perfusions with [(+) GKA] or without (CON) were performed as indicated in the legend to Figure 2. (A) Total levels of NAG in the liver at the end of perfusion and sum (Σ) of 13C enrichment (MPE/100) of mass isotopomers (M2–M7) of NAG multiplied by total concentration (nmol · g−1 of wet mass). (B) Rate of total urea output (nmol · g−1 · min−1) during the perfusion. (C) Output of the sum of [15N]urea containing one or two 15N calculated by the 15N enrichment (MPE/100) multiplied by the rate of total urea output (nmol · g−1 · min−1) as indicated previously [9]. Results are means ± S.D. for three individual livers per control without GKA (CON) or with [(+) GKA]. P values were determined by Student’s t test using Prism 5 software.
The increased NAG synthesis in perfusions with GKA was accompanied by a significant increased output of total and [15N]urea synthesized from 15NH4Cl added to the perfusate (Figures 9B and 9C). In the control perfusion between 35 and 65 min, the rate of [15N]urea output was approximately 368 ± 33 nmol · g−1 · min−1, (mean ± S.D.). In the perfusion with GKA between 55 and 75 min, the rate of [15N]urea output was approximately 599 ± 43 nmol · g−1 · min−1 (mean ± S.D.) (Figure 9C). The data suggest that the stimulation of glycolysis by GKA furnished both acetyl-CoA and glutamate for synthesis of NAG, and consequently up-regulation of hepatic ureagenesis.
Experiments with isolated hepatocytes
To determine whether GKAs act on the multiple pathways of pyruvate metabolism (Figure 1), experiments were carried out using isolated hepatocytes incubated with [1-13C]pyruvate or [3-13C]pyruvate. The release of 13CO2 from [1-13C]pyruvate was approximately 2.2 μmol · mg−1 hepatocyte protein, with or without GKA, suggesting that GKA has no direct effect on PDH activity. In addition, there was approximately 35 MPE M1 aspartate isotopomer, 27 MPE M1 citrate isotopomer and 24 MPE M1 malate isotopomer with or without GKA. The M1 mass isotopomer is formed following incorporation of [1-13C]pyruvate into the TCA cycle through PC, resulting in the generation of M1 oxaloacetate and then M1 aspartate. M1 citrate is formed through the first turn of the TCA cycle, whereas M1 malate is formed through malic enzyme [8,27,28]. In experiments with [3-13C]pyruvate, M1 13C-acetyl-CoA is formed through PDH and 13CO2 is released only through the TCA cycle. In this series of experiments, the release of 13CO2 was approximately 4.8 or 5.1 μmol · mg−1 hepatocyte protein without or with GKA respectively. Incubation with GKA had little effect on either the concentration or enrichment (MPE) of 13C mass isotopomers of citrate, glutamate, succinate, malate and aspartate (results not shown). Thus when an equimolar amount of pyruvate (i.e. 5mM) was used, there was little difference in the release of 13CO2 or in the generation of M1 mass isotopomers of aspartate, citrate and malate, suggesting that GKA has no direct effect on the activity of PDH, PC or malic enzyme.
To explore whether Piragliatin has ‘off-target’ effects, experiments were carried out with 5 mM [U-13C6]glucose as the sole substrate. We first determined the time course of GKA action. The results indicated that within the first 2 min of incubation, there was a near-2-fold increase in 13CO2 release from 5 mM [U-13C6]glucose (results not shown), in agreement with previous studies demonstrating an immediate activation of GK when β-cells were exposed to Piragliatin [2–4]. A separate series of experiments demonstrated a 2-fold increased release of 13CO2 from [U-13C6]glucose, i.e. 11.8 ± 2.4 and 21.8 ± 3.1 nmol · mg−1 hepatocyte protein (means ± S.D., n = 4) without or with GKA respectively (Figure 10A). Cumulatively, the results indicate that the addition of 3 μmol · l−1 Piragliatin activated GK and stimulated glycolysis in experiments with isolated hepatocytes in a manner similar to that observed in a liver perfusion system.
Figure 10. Action of a GKA on glycolysis and gluconeogenesis by isolated hepatocytes.
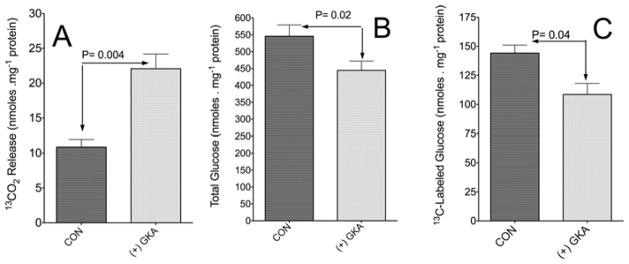
(A) Release of 13CO2 following incubation with [U-13C]glucose. Incubations of isolated hepatocytes were carried out for 60 min with 5 mM [U-13C]glucose and 3 μmol · l−1 Piragliatin [(+) GKA] or without Piragliatin (CON). Experimental details are as described in the Experimental section. Results are means ± S.D. of three or four individual experiments. (B and C) Action of a GKA on gluconeogenesis by isolated hepatocytes. Incubations of isolated hepatocytes were carried out for 60 min with 5 mM [3-13C]pyruvate and 3 μmol · l−1 Piragliatin [(+) GKA] or without Piragliatin (CON). (B) Total generation of glucose (nmol · mg−1 hepatocyte protein). (C) Sum of the production of M1 and M2 isotopomers of 13C-labelled glucose (glucose with one or two 13C atoms repectively), calculated by 13C enrichment (MPE/100) multiplied by concentration (nmol · mg−1 protein). Results are means ± S.D. of three to five independent experiments per control without Piragliatin (CON) or with (+) GKA. P values were determined by Student’s t test using Prism 5 software.
It remained unresolved whether the GKA effect on gluconeogenesis represents an indirect effect, perhaps secondary to the induction of changes in levels of metabolites, or a direct effect that corresponds to GKA-induced modification of critical enzymatic steps in the gluconeogenic pathway. This question was addressed in experiments with isolated hepatocytes incubated with 5 mM [3-13C]pyruvate as a precursor. LC-MS analysis indicated the production of 13C-labelled glucose mainly at one (M1) or two (M2) carbons. The sum of M1 and M2 13C-labelled glucose produced was 108 ± 9 and 144 ± 7 (means ± S.D.) nmol · mg−1 hepatocyte protein with or without GKA respectively (Figures 10B and 10C), indicating that GKA decreased gluconeogenesis from pyruvate. These findings and those noted with liver perfusion indicate that GKA reduced gluconeogenesis (Figure 10 and Supplementary Figure S5), even in the absence of added glucose.
DISCUSSION
The contribution of the present study to understanding the action of GKA on hepatic metabolism
GK, a unique isoform of the hexokinase enzymes, phosphorylates D-glucose and other hexoses [1,4]. GK activators were recently identified as new promising drugs targeting Type 2 diabetes [1–4]. The basic enzymology, structure and mechanism of action of GK have been described [3,4,7]. The effectiveness of GKA is determined by the level of free cytosolic GK, by the ability of the drug to dissociate the enzyme from a nuclear regulatory protein [GKRP (GK regulatory protein)–GK complex] [7] and, perhaps most importantly, by the level of free intracellular glucose. A network of GK-containing cells contributes to glucose homoeostasis, the most important regulators being the pancreatic β-cell and the liver, which contains approximately 99% of total body GK [3,4]. Despite the key role of GK, knowledge about the effects of GKAs on hepatic intermediary metabolism and metabolic pathways remains limited [1,3,4,7]. There has been little research on the impact of GKA treatment on flux through PDH, PC or the TCA cycle, the hepatic redox or energy states, ureagenesis or lipogenesis. We have investigated how a new GKA, Piragliatin [2,3,4], influences glycolysis and related pathways of hepatic glucose metabolism. We chose Piragliatin because of its known efficacy in Type 2 diabetics [6]. In accordance with our hypothesis, the present findings demonstrate that GKA stimulation of glycolysis had wide effects on the hepatic metabolic network, including: (i) increased hepatic glucose uptake (Table 1) and subsequent augmentation of the output of 13CO2, 13C-labelled lactate and alanine, the products of glycolysis (Table 1, and Figures 3 and 4A); (ii) augmented anaplerosis and increased flux through the TCA cycle and cataplerosis of glutamate and aspartate (Figures 5–7); (iii) increased glycogen synthesis through the direct pathway and evidence for decreased gluconeogenesis and synthesis of glycogen through the indirect pathway (Figures 8 and 10, and Supplementary Figure S5); (iv) increased synthesis of NAG and augmented ureagenesis (Figure 9); (v) a tendency towards elevation of hepatic triglycerides (Table 3); and (vi) improved hepatic O2 consumption, energetic state and phosphorylation potential (Figure 2 and Table 2). Taken together, the results of the present study provide the deepest insight to date into the action of GKA on hepatic intermediary metabolism, in particular the recognition of close coupling between hepatic glycolysis and nitrogen metabolism, as illustrated in Figure 11.
Figure 11. Schematic illustration of the potential benefits and risks of a GKA stimulation of hepatic glycolysis.

On the left-hand side are listed the potential benefits (i.e. glycogen synthesis, ammonia detoxification and improved energy balance), and on the right-hand side are given the potential risks of GKA (lactic acidosis, lipogenesis and the possibility of triglyceride accumulation in the liver). +, stimulation and/or increased generation; −, inhibition and/or decreased generation. The present findings demonstrate that the stimulation of glycolysis by GKA resulted in numerous changes in normal hepatic metabolism, including: (i) enhanced direct pathway of glycogen synthesis, but decreased gluconeogenesis; (ii) augmented anaplerosis and, associated with this, increased flux through the TCA cycle and generation of glutamate, aspartate or citrate (cataplerosis); (iii) the increased flux through the TCA may enhance the generation of NADH and thus ATP synthesis, resulting in an improved hepatic phosphorylation potential and energetic state; (iv) stimulated synthesis of NAG, and enhanced detoxification of NH3 through ureagenesis; (v) increased generation and output of lactate, which could lead to lactic acidosis; and (vi) increased levels of citrate may augment hepatic lipogenesis and thus accumulation of triglycerides. OXA, oxaloacetate.
The GKA-induced alteration in hepatic intermediary metabolism is accompanied by a near-3-fold increase in glucose uptake (Table 1). These results indicate that approximately 50% and 30% of glucose uptake was accounted for by the output of 13C-labelled pyruvate, CO2, lactate and alanine in perfusions with or without GKA respectively (Table 1). The remainder of glucose uptake was probably used for synthesis of glycogen, other amino acids, organic acids and, possibly, triglycerides, glycerol 3-phosphate, G3AP and G6P, as demonstrated in Figures 5–8, Supplementary Figures S3 and S4, and Table 3.
The increased release of 13CO2 during liver perfusion with [U-13C6]glucose probably represents the sum of 13CO2 generated through the cytosolic pentose phosphate shunt and 13CO2 generated through mitochondrial PDH activity and the TCA cycle (Figure 1). Similarly, quantitatively significant amounts of 13C-labelled lactate were generated from pyruvate and an increased flow of glucose carbon into alanine through transamination and into glutamate, possibly through reductive amination of 2-oxoglutarate (Figure 4). The perfused liver released these products of glycolysis within 3–5 min of adding GKA. It is important to note that the apparent decreased output of 13C-labelled glutamate (nmol · min−1 per g of wet mass) (Figure 4B) was not a result of decreased 13C enrichment in glutamate isotopomers (Figure 7A). Rather, this decrease may result from increased transfer of the amino group from glutamate to form alanine. This explanation is supported by the increased output of 13C-labelled alanine with GKA (Figure 4A), which is inversely correlated with decreased [13C]glutamate output. This pattern resembles the glucose–alanine cycle [36]. Additionally, the augmented availability of pyruvate probably increased acetyl-CoA production and incorporation of 13C derived from glucose into the TCA cycle. This notion is evident from elevated cataplerosis of citrate, glutamate and aspartate (Figures 5–7), secondary to increased incorporation of 13C-labelled pyruvate into the TCA cycle (anaplerosis).
An important question is how GKA increases flux through the TCA cycle. The present results with isolated hepatocytes demonstrate that GKA has no direct effect on the activity of PC or PDH, as is evident by unchanged release of 13CO2 or in the generation of 13C mass isotopomer of aspartate, citrate or malate from 13C-labelled pyruvate as precursor. Cumulatively, the data suggest that the significantly increased production of 13CO2 or 13C-labelled mass isotopomers of citrate, malate, glutamate or aspartate from [U-13C6]glucose during liver perfusion with GKA (Figures 3–7) is probably mediated entirely through elevated generation of 13C-labelled pyruvate as an end product of glycolysis and its incorporation into the TCA cycle through an effective feed-forward mechanism.
In addition to increased generation of 13C-labelled glutamate and aspartate (Figures 7A and 7B), the results indicate that GKA also elevated the synthesis of serine and glycine secondary to stimulation of glycolysis. In addition, the results of the present study indicate increased synthesis of 13C-labelled glutamine from 13C-labelled glutamate (Figure 7F). In the liver, glutamine synthesis occurs mainly in the perivenous zone [34]. Previous studies show a significant GK activity in this region [34,35]. It is possible that increased glycolysis by GKA and the resultant increased availability of glutamate and ATP (Figure 7D and Table 1) led to activation of glutamine synthesis in perivenous hepatocytes.
The present study demonstrates for the first time that GKA stimulates the synthesis of NAG and ureagenesis (Figure 9). This may be explained by the fact that GKA stimulated the generation of pyruvate and its metabolism to acetyl-CoA, thus furnishing more acetyl-CoA for NAG synthesis. In addition, the results demonstrate that the production of 13C-labelled glutamate through the TCA cycle was significantly increased (Figure 7). Together, increased mitochondrial levels of glutamate and acetyl-CoA, the precursors of NAG [12], led to increased NAG synthesis, activation of carbamoylphosphate synthetase-I and greater mitochondrial citrulline synthesis [12,37,38]. The results of the present study are consistent with the concept that the synthesis of citrulline and urea is determined in large measure by NAG synthesis, with which the rate of ureagenesis is linearly correlated.
As discussed above, the results of the present study provide strong evidence supporting the conclusion that increased glycolysis is directly responsible for the widespread metabolic alterations that occurred in liver exposed to GKA. We base this conclusion on the following observations.
In the present study, 5 mM [U-13C6]glucose was infused at a rate of 3.5 ml · g−1 · min−1, or 17 μmol · g− · min−1. Assuming a glycogenolysis rate of 0.5% · min−1 [39], glucose would be derived from glycogenolysis at approximately 1.5 μmol · g−1 · min−1, or 8% of perfusate [U-13C6]glucose (i.e. 1.5 compared with 17 μmol · g−1 · min−1). Indeed, the 13C enrichment in the outflow [U-13C6]glucose during 30–70 min of perfusion was approximately 90–94 MPE, with or without GKA, suggesting a minor dilution of perfusate [U-13C6]glucose with unlabelled glucose coming from glycogenolysis.
Between 15 and 45 min of perfusion with or without GKA, there was little difference in O2 consumption and outflow of 13C-labelled metabolites (Figures 2–4), or total and 15N-urea output (Figure 9). However, within 3–5 min of adding GKA (the latter was added to the perfusate at 45 min), there was a significant (P < 0.01) stimulation in the output of 13CO2 and 13C-labelled lactate and alanine (Figures 3 and 4, Supplementary Figures S1 and S2, and Table 1). The increased outflow of 13C-labelled metabolites was associated with augmented O2 consumption in the perfusion with GKA (Figure 2). In addition, experiments with isolated hepatocytes indicate that the addition of 3 μmol · l−1 Piragliatin activated GK and stimulated glycolysis (Figure 10) in a manner similar to that observed in a liver perfusion system.
In both controls and perfusion with GKA, the 13C enrichment (MPE) in 13CO2, lactate and alanine in the outflow was increased within 5–10 min after the initiation of perfusion with [U-13C]glucose. Thereafter, the 13C enrichment reached a steady-state (Supplementary Figures S1 and S2), suggesting that the intermediary metabolites were mainly derived from perfusate [U-13C]glucose, and that the metabolic differences between control and perfusion with GKA are a result of GK activation.
G6P is a key precursor for the 13C-labelled metabolites presented in Figures 3–8. At the end of perfusion, the average 13C enrichment in M6 G6P was approximately 41 and 58 MPE and the sum of 13C isotopomers of G6P was approximately 56 and 69 MPE, indicating that approximately 56% and 69% of the hepatic G6P pool was derived from perfusate [U-13C6] glucose in control and perfusion with GKA respectively (Supplementary Table S1 at http://www.BiochemJ.org/bj/444/bj4440537add.htm). Furthermore, there is an apparent correlation between the levels of G6P (Table 3) and the synthesis and output of 13C-labelled metabolites (Figures 5–7 and Table 1) as well as glycogen synthesis (Figure 8). Taken together, the results suggest that Piragliatin is responsible for increased generation of 13C-labelled G6P and the resulting production of 13C-labelled metabolites through various metabolic pathways (Figures 3–8). This conclusion is in accordance with a previous suggestion that G6P arising from the catalytic action of GK is responsible for the activation of liver glycogen synthesis in addition to providing a substrate for several other pathways [40].
Implications of the results of the present study for the potential of GKAs in drug therapy of Type 2 diabetes
The results of the present study provide novel and deepened insights into the effects of Piragliatin on hepatic metabolism. Such information is essential in order to assess the therapeutic potential of this class of drugs. We emphasize that we did not study diabetic animals, but healthy rats in a postabsorptive state We therefore caution against facile extrapolation of our data to a liver that is either deprived of insulin or resistant to this hormone. Bearing this caveat in mind, the results of the present study support the following conclusions.
Systemic glucose homoeostasis depends on the balance between hepatic glycolysis versus gluconeogenesis and glycogenesis versus glycogenolysis. The results of the present study demonstrate that the GKA decreased glycogen synthesis from 3-carbon precursors (i.e. pyruvate and lactate) and increased glycogen formed directly from glucose (Figure 8), suggesting reduced recycling of pyruvate back to glucose through gluconeogenesis. The notion that GKAs inhibit gluconeogenesis is supported by the results in Figure 10 and Supplementary Figure S5. The conclusion that GKAs inhibited gluconeogenesis and augmented synthesis of glycogen is in agreement with a previous study demonstrating increased glycogenesis and lowered hepatic glucose production in Zucker diabetic rats following treatment with an adenovirus that directs the expression of hepatic GK [41]. Taken together, the results of the present study suggest that GKA modifies glucose homoeostasis by diminishing hepatic gluconeogenesis and stimulating the direct pathway of glycogenesis.
The present study provides the first evidence that GKAs enhance hepatic ureagenesis and thus ammonia detoxification. Hepatic urea synthesis is the only metabolic pathway capable of removing waste nitrogen. Liver metabolism in diabetes is profoundly impaired [1–5], and hyperammonaemia may occur in Type 2 diabetes, a phenomenon noted in Zucker rats [42]. GKA may improve the detoxification of excess systemic ammonia through enhanced NAG and urea synthesis, and thus minimize metabolic complications and deleterious effects on the central nervous system secondary to diabetes.
GKA might cause lactic acidosis by causing overproduction of pyruvate. In contrast with β-cells, which have a relatively low activity of LDH [3], hepatic parenchymal cells are rich in this enzyme. Production of lactate might then afford a ‘sink’ for excess generation of pyruvate. The marked stimulation of 13C-labelled lactate outflow by the GKA (Figure 3C) suggests that these drugs may cause lactic acidosis. In addition, the inhibition of gluconeogenesis by GKA (Figure 10 and Supplementary Figure S5) may diminish hepatic metabolism of lactate and subsequently lead to elevation of systemic lactate. However, further in vivo study in normal and diabetic animals is required to examine this possibility.
The striking GKA stimulation of citrate production (Figures 5B and 5E) as well as glycerol 3-phosphate (Supplementary Figure S3) and triglycerides (Table 3) raises the possibility of increased hepatic lipogenesis as another liability of long-term treatment with this drug, as illustrated in Figure 11. Citrate is a crucial intermediate and metabolic signal in the synthesis of cholesterol and complex lipids. In addition, previous studies showed that lipogenic genes are induced by G6P [43], which is significantly elevated in liver perfused with GKA (Table 2 and Supplementary Figure S3), again supporting the formulation that GKA augments hepatic lipogenesis. A relationship between activation of hepatic GK and increased synthesis of triglycerides is consistent with the finding that a higher GK can stimulate lipogenic genes [44], a conclusion similar to that of other studies [43–46]. However, we must emphasize that the present short-term experiments with GKA indicate an insignificant elevation of triglycerides (Table 3). Thus further long-term in vivo study in normal and diabetic animals is required to examine this possibility.
Conclusions and long-term perspectives
We found that the GKA Piragliatin causes widespread changes in hepatic metabolism in normal post-absorptive rats (Figure 11). The present study confirmed favourable therapeutic effects of GKA, such as stimulation of hepatic glycogenesis, inhibition of gluconeogenesis, improvement of the energy potential of liver tissue and facilitation of ammonia detoxification through ureagenesis. However, the results of the present study also identify lactic acidosis and/or fatty liver as potential risks of GKA treatment. Details of the dose-dependencies of these positive and negative actions of the GKA remain to be elucidated. Additional studies also may reveal whether it is feasible to dissociate these positive and negative effects. Studies also are needed in animals with diabetes of diverse aetiology in order to learn whether GKA effects on liver metabolism are similar to those we found in unaffected rats. In this regard, it is significant that long-term studies with haploinsufficient, mildly diabetic, GK-knockout mice fed a high-fat diet [47,48] demonstrate that GKA can normalize hyperglycaemia without causing the side effects seen in the present study.
Supplementary Material
Acknowledgments
We thank Dr Joseph Grimsby (Department of Metabolic Diseases, Roche, Nutley, NJ, U.S.A.) and Dr Ramakanth Sarabu (Department of Discovery Chemistry, Roche, Nutley, NJ, U.S.A.) for essential contributions to this study, including the provision of the GKA Piragliatin and many insightful discussions of the biochemical pharmacology of this new class of antidiabetic agents.
FUNDING
This work was supported by the National Institutes of Health [grant numbers DK-053761 (to I.N.), PO1HD26979 (to M.Y.) and DK22122 (to F.M.M.)], and in part by the Elke Matschinsky Glucokinase Fund (to I.N.).
Abbreviations used
- G6P
glucose 6-phosphate
- GA3P
glyceraldehyde 3-phosphate
- GC
gas chromatography
- GK
glucokinase
- GKA
GK activator
- LC
liquid chromatography
- LDH
lactate dehydrogenase
- MPE
mole percentage excess
- MSD
mass selective detector
- NAG
N-acetylglutamate
- PC
pyruvate carboxylase
- PDH
pyruvate dehydrogenase
- PMP
1-phenyl-3-methyl-5-pyrazolone
- TCA
tricarboxylic acid
- TFA
trifluoroacetic acid
Footnotes
AUTHOR CONTRIBUTION
Itzhak Nissim and Franz Matschinsky initiated and conceived the study design; Oksana Horyn conducted liver perfusions, prepared samples for analysis, and performed enzyme assays and data summary; Ilana Nissim performed all HPLC analyses, and assisted with enzyme assays, sample preparation and data summary; Yevgeny Daikhin performed GC-MS and LC-MS/MS analysis; Suzanne Wehrli performed NMR analysis; Marc Yudkoff assisted with writing the paper and in discussions; Itzhak Nissim and Franz Matschinsky wrote the paper.
References
- 1.Agius LA. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J. 2008;414:1–18. doi: 10.1042/BJ20080595. [DOI] [PubMed] [Google Scholar]
- 2.Pal M. Recent advances in glucokinase activators for the treatment of type 2 diabetes. Drug Discovery Today. 2009;14:784–792. doi: 10.1016/j.drudis.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 3.Leighton B, Atkinson A, Coghlan MP. Assessing the potential of glucokinase activators in diabetes therapy. Biochem Soc Trans. 2005;33:371–374. doi: 10.1042/BST0330371. [DOI] [PubMed] [Google Scholar]
- 4.Matschinsky FM. Allosteric activators of glucokinase: potential role in diabetes therapy. Nat Rev Drug Discov. 2009;8:399–416. doi: 10.1038/nrd2850. [DOI] [PubMed] [Google Scholar]
- 5.Grimsby J, Sarabu R, Corbett WL, Haynes NE, Bizzarro FT, Coffey JW, Guertin KR, Hilliard DW, Kester RF, Mahaney PE, et al. Allosteric activators of glucokinase: potential role in diabetes therapy. Sci Signaling. 2003;301:370–373. doi: 10.1126/science.1084073. [DOI] [PubMed] [Google Scholar]
- 6.Bonadonna RC, Heise T, Arbet-Engels C, Kapitza C, Avogaro A, Grimsby J, Zhi J, Grippo JF, Balena RJ. Piragliatin [RO4389620], a noval glucokinase activator, lowers plasma glucose both in the postabsorptive state and after glucose challenge in patients with type 2 diabetes mellitus: a mechanistic study. J Clin Endocrinol Metab. 2010;95:5028–5036. doi: 10.1210/jc.2010-1041. [DOI] [PubMed] [Google Scholar]
- 7.Brocklehurst KJ, Payne VA, Davis RA, Carrol D, Vertigan HL, Wightman HJ, Aiston S, Waddell ID, Leighton B, Coghlan MP, Agius L. Stimulation of hepatocyte glucose metabolism by novel small molecule glucokinase activators. Diabetes. 2004;53:535–541. doi: 10.2337/diabetes.53.3.535. [DOI] [PubMed] [Google Scholar]
- 8.Li C, Nissim I, Chen P, Buettger C, Najafi H, Daikhin Y, Collins HW, Yudkoff M, Stanley CA, Matschinsky FM. Elimination of KATP channels in mouse islets results in elevated [U-13C]glucose metabolism, glutaminolysis and pyruvate cycling but a decreased γ-aminobutyric acid shunt. J Biol Chem. 2008;283:17238–17249. doi: 10.1074/jbc.M709235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nissim I, Luhovyy B, Horyn O, Daikhin Y, Nissim I, Yudkoff M. The role of mitochondrially bound arginase in the regulation of urea synthesis: studies with [U-15N4 ]arginine, isolated mitochondria and perfused rat liver. J Biol Chem. 2005;280:17715–17724. doi: 10.1074/jbc.M500607200. [DOI] [PubMed] [Google Scholar]
- 10.Nissim I, Horyn O, Nissim I, Daikhin Y, Wehrli SL, Yudkoff M. 3-Isobutylmethylxanthine inhibits hepatic urea synthesis: protection by agmatine. J Biol Chem. 2008;283:15063–15071. doi: 10.1074/jbc.M800163200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berry MN, Edwards AM, Barritt GJ. Isolated hepatocytes: preparation, properties and applications. Lab Tech Biochem Mol Biol. 1991;21:15–58. [Google Scholar]
- 12.Nissim I, Horyn O, Nissim I, Daikhin Y, Caldovic L, Barcelona B, Cervera J, Tuchman M, Yudkoff M. Down-regulation of hepatic urea synthesis by oxypurines: xanthine and uric acid inhibit N-acetylglutamate synthase. J Biol Chem. 2011;282:22055–22068. doi: 10.1074/jbc.M110.209023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Perreault H. Characterization of carbohydrates using a combination of derivatization, high-performance liquid chromatography and mass spectrometry. J Chromatogr, A. 1998;81:47–59. doi: 10.1016/s0021-9673(98)00238-6. [DOI] [PubMed] [Google Scholar]
- 14.Fernández-Fígares I, Shannon AE, Wray-Cahen D, Caperna TJ. The role of insulin, glucagon, dexamethasone, and leptin in the regulation of ketogenesis and glycogen storage in primary cultures of porcine hepatocytes prepared from 60 kg pigs. Domest Anim Endocrinol. 2004;27:125–140. doi: 10.1016/j.domaniend.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Bergmeyer HU, Bernt E, Schmidt F, Stork H. D-glucose: determination with hexokinase and glucose-6-phosphate dehydrogenase. Methods Enzym Anal. 1974;3:1196–1201. [Google Scholar]
- 16.Nissim I, Brosnan ME, Yudkoff M, Nissim I, Brosnan JT. Insulin and glucagon regulation of hepatic glutamine kinetics and metabolism: studies in perfused liver with 15N. J. Biol. Chem. 1999;274:28958–28965. doi: 10.1074/jbc.274.41.28958. [DOI] [PubMed] [Google Scholar]
- 17.Wehrli S, Reynolds R, Chen J, Yager C, Segal S. Galactose metabolism in normal human lymphoblasts studied by 1H, 13C and 31P NMR spectroscopy of extracts. NMR Biomed. 2001;14:192–198. doi: 10.1002/nbm.694. [DOI] [PubMed] [Google Scholar]
- 18.Jones BN, Gilligan JP. Ortho-phthaldialdehyde precolumn derivatization and reversed-phase high-performance liquid-chromatography of polypeptide hydrolysates and physiological fluids. J Chromatogr. 1983;266:471–482. doi: 10.1016/s0021-9673(01)90918-5. [DOI] [PubMed] [Google Scholar]
- 19.Jaworek D, Welsch J. Adenosine 5′-triphosphate. UV-method with phosphoglycerate kinase. Methods Enzym Anal. 1985;7:340–346. [Google Scholar]
- 20.Jaworek D, Gruber W, Bergmeyer HU. Adenosine-5′-diphosphate and adenosine-5′-monophosphate. Methods Enzym Anal. 1974;2:2127–2131. [Google Scholar]
- 21.Gibon Y, Larher F. Cycling assay for nicotinamide adenine dinucleotides: NaCl precipitation and ethanol solubilization of the reduced tetrazolium. Anal Biochem. 1997;251:153–157. doi: 10.1006/abio.1997.2283. [DOI] [PubMed] [Google Scholar]
- 22.Gutmann I, Wahlefeld AW. L-(+)-Lactate. Methods Enzym Anal. 1974;3:1464–1472. [Google Scholar]
- 23.Kientsch-Engel RI, Siess EA. D-(−)-3-Hydroxybutyrate and acetoacetate. Methods Enzym Anal. 1985;8:60–69. [Google Scholar]
- 24.Czok R, Lamprecht W. Pyruvate, phosphoenolpyruvate and D-glycerate-2-phosphate. Methods Enzym Anal. 1974;3:1446–1451. [Google Scholar]
- 25.Gutmann I, Wahlefeld AW. L-(−)-Malate. Determination with malate dehydrogenase and NAD. Methods Enzym Anal. 1974;3:1585–1589. [Google Scholar]
- 26.Michal G, Beutler HO. D-Fructose-1,6-bisphosphate, dihydroxyacetone phosphate and D-glyceraldehyde-3-phosphate. Methods Enzym Anal. 1974;3:1314–1319. [Google Scholar]
- 27.Des Rosiers C, Di Donato L, Comte B, Laplante A, Marcoux C, David F, Fernandez CA, Brunengraber H. Isotopomer analysis of citric acid cycle and gluconeogenesis in rat liver. Reversibility of isocitrate dehydrogenase and involvement of ATP-citrate lyase in gluconeogenesis. J Biol Chem. 1995;270:10027–10036. doi: 10.1074/jbc.270.17.10027. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Kasumov T, Kombu RS, Shu-Han Z, Cendrowski AV, David F, Anderson VE, Kelleher JK, Brunengraber H. Metabolomic and mass isotopomer analysis of liver gluconeogenesis and citric acid cycle. II. Heterogeneity of metabolite labeling pattern. J Biol Chem. 2008;233:21988–21996. doi: 10.1074/jbc.M803455200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolfe RR. Biomedicine. Wiley-Liss; New York: 1992. Radioactive and Stable Isotope Tracers. [Google Scholar]
- 30.Cline GW, Shulman GI. Mass and positional isotopomer analysis of glucose metabolism inperiportal and pericentral hepatocytes. J Biol Chem. 1995;270:28062–28067. doi: 10.1074/jbc.270.47.28062. [DOI] [PubMed] [Google Scholar]
- 31.Landau BR. Methods for measuring glycogen cycling. Am J Physiol Endocrinol Metab. 2001;281:E413–E419. doi: 10.1152/ajpendo.2001.281.3.E413. [DOI] [PubMed] [Google Scholar]
- 32.Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J. 1967;103:514–527. doi: 10.1042/bj1030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krebs HA, Veech RL. Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Adv Enzyme Regul. 1969;7:397–413. doi: 10.1016/0065-2571(69)90030-2. [DOI] [PubMed] [Google Scholar]
- 34.Jungermann K, Keitzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev Nutr. 1996;16:179–203. doi: 10.1146/annurev.nu.16.070196.001143. [DOI] [PubMed] [Google Scholar]
- 35.Trus M, Zawalich K, Gaynor D, Matschinsky F. Hexokinase and glucokinase distribution in the liver lobule. J Histochem Cytochem. 1980;28:579–581. doi: 10.1177/28.6.7391551. [DOI] [PubMed] [Google Scholar]
- 36.Lehninger AL. Lehninger Principles of Biochemistry. 4. W. H Freeman; New York: 2005. [Google Scholar]
- 37.Gropman AL, Summar M, Leonard JV. Neurological implications of urea cycle disorders. J Inherit Metab Dis. 2007;30:865–869. doi: 10.1007/s10545-007-0709-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahuja V, Powers-Lee SG. Human carbamoyl-phosphate synthetase: insight into N-acetylglutamate interaction and the functional effects of a common single nucleotide polymorphism. J Inherit Metab Dis. 2008;31:481–490. doi: 10.1007/s10545-008-0913-y. [DOI] [PubMed] [Google Scholar]
- 39.Baillet-Blanco L, Beauvieux MC, Gin H, Rigalleau V, Gallis JL. Insulin induces a positive relationship between the rates of ATP and glycogen changes in isolated rat liver in presence of glucose; a 31P and 13C NMR study. Nutr Metab. 2005;32:1–9. doi: 10.1186/1743-7075-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomis RR, Faver C, Garcia-Roch M, Fernandez-Novell JM, Ferrer JC, Guinovart JJ. Glucose 6-phosphate produced by gluconeogenesis and by glucokinase is equally effective in activating hepatic glycogen synthase. J Biol Chem. 2003;278:9740–9746. doi: 10.1074/jbc.M212151200. [DOI] [PubMed] [Google Scholar]
- 41.Torres TP, Catlin RL, Chan R, Fujimoto Y, Sasaki N, Printz RL, Newgard CB, Shiota M. Restoration of hepatic glucokinase expression corrects hepatic glucose flux and normalizes plasma glucose in Zucker diabetic fatty rats. Diabetes. 2009;58:78–86. doi: 10.2337/db08-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maswoswe SM, Tremblay GC. Biosynthesis of hippurate, urea and pyrimidines in the fatty liver: studies with rats fed orotic acid or a diet deficient in choline and inositol, and with genetically obese (Zucker) rats. J Nutr. 1989;119:273–279. doi: 10.1093/jn/119.2.273. [DOI] [PubMed] [Google Scholar]
- 43.Postic C, Dentin R, Girard J. Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab. 2004;30:398–408. doi: 10.1016/s1262-3636(07)70133-7. [DOI] [PubMed] [Google Scholar]
- 44.Pollin TI, Jablonski JB, McAteer JB, Saxena R, Kathiresan S, Kahn SE, Goldberg RB, Altshuler D, Florez JC, et al. Triglyceride response to an intensive lifestyle intervention is enhanced in carriers of the GCKR Pro446Leu polymorphism. J Clin Endocrinol Metab. 2001;96:E1142–E1147. doi: 10.1210/jc.2010-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agius L. Targeting hepatic glucokinase in type 2 diabetes: weighing the benefits and risks. Diabetes. 2009;58:18–20. doi: 10.2337/db08-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kozlan DH, Barthel A, Cousin E, Brunnhofer R, Anderka O, Marz W, Bohm B, Winkelmann B, Bornstein SR, Schmoll D. Glucokinase-activating GCKR polymorphisms increase plasma levels of triglycerides and free fatty acids, but do not elevate cardiovascular risk in the Ludwigshafen Risk and Cardiovascular Health Study. Horm Metab Res. 2010;42:502–506. doi: 10.1055/s-0030-1249637. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura A, Terauchi Y, Ohyama S, Kubota J, Shimazaki H, Nambu T, Takamoto I, Kubota N, Eiki J, Yoshioka N, et al. Impact of small-molecule glucokinase activator on glucose metabolism and β-cell mass. Endocrinology. 2009;150:1147–1154. doi: 10.1210/en.2008-1183. [DOI] [PubMed] [Google Scholar]
- 48.Nakamura A, Shimazaki H, Ohyama S, Eiki J, Terauchi YJ. Effect of long-term treatment with a small-molecule glucokinase activator on glucose metabolism, lipid profiles and hepatic function. Diabetes Invest. 2011;2:276–279. doi: 10.1111/j.2040-1124.2011.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.