

Abstract
ATP-sensitive K+ (KATP) channels that contain K+ inward rectifier subunits of the 6.2 isotype (Kir6.2) are important regulators of the cardiac response to ischemia-reperfusion (I/R) injury. Opening of these channels is implicated in the cardioprotective mechanism of ischemic preconditioning (IPC), but debate surrounds the contribution of surface KATP (sKATP) versus mitochondrial KATP (mKATP) channels. While responses to I/R injury and IPC have been examined in Kir6.2−/− mice before, breeding methods and other technical obstacles may have confounded interpretations. The aim of this study was to elucidate the role of Kir6.2 in cardioprotection and mKATP activity, using conventionally bred Kir6.2−/− mice with wild-type littermates as controls. We found that perfused hearts from Kir6.2−/− mice exhibited a normal baseline response to I/R injury, were not protected by IPC, and showed a blunted response to the IPC mimetic drug diazoxide. These data suggest that the loss of IPC in Kir6.2−/− hearts is not due to an underlying difference in I/R sensitivity. Furthermore, mKATP channel activity was identical in cardiac mitochondria isolated from wild-type versus Kir6.2−/− mice, suggesting no role for Kir6.2 in the mKATP. Collectively, these data indicate that Kir6.2 is required for the full response to IPC or diazoxide but is not involved in mKATP formation.
Keywords: mitochondria, ischemic preconditioning, KATP channel, diazoxide
the heart and other organs can be protected against ischemia-reperfusion (I/R) injury via ischemic preconditioning (IPC), wherein short periods of I/R can engage protective signaling pathways to reduce the impact of a prolonged ischemic event (23). The protective effects of IPC can be mimicked by openers of ATP-sensitive potassium (KATP) channels. Since changes in cardiomyocyte bioenergetics are known to occur during IPC (11), the metabolic-sensing role of these channels has driven interest in their potential role in cardioprotective signaling (13, 24).
KATP channels comprise octamers of four inward-rectifying potassium channel subunits (Kir6.1 or -6.2) and four sulfonylurea receptors 1, 2A, or 2B (SUR1, -2A, or -2B), with different Kir/SUR combinations giving rise to unique cellular roles, locations, and pharmacological profiles (13). For example, the cardiac surface KATP (sKATP) is Kir6.2/SUR2A. It plays a role in cardiomyocyte volume regulation and stress responses (28) and is activated by cromakalim but not diazoxide (DZX) (11, 33, 42). In contrast, the pancreatic sKATP is Kir6.2/SUR1, which plays a role in insulin secretion and is activated by DZX but not cromakalim (13).
Despite there being no effect of DZX on cardiac sKATP channels (4, 9), DZX is known to mimic IPC and protect the heart against I/R injury. This led to speculation on the existence of another DZX-sensitive KATP channel in the heart, namely, the mitochondrial KATP (mKATP). However, previous studies using Kir6.2−/− mice concluded that there was no role for Kir6.2 in the formation of the mKATP, although channel activity was determined only by the surrogate measurement of mitochondrial flavoprotein oxidation rather than by direct measurements of ion transport (33).
Furthermore, to date cardiac studies on Kir6.2 −/− mice (10, 11, 31–33) have used a pure-line breeding strategy [knockout × knockout, purchasing or breeding a separate line of wild-type (WT) controls]. Thus, while it was previously shown that Kir6.2−/− mice cannot be protected by IPC, it is not clear whether this is represents a true failure of IPC or is simply due to a reported greater sensitivity of the knockout animals to baseline I/R injury (vs. WT controls) (32, 33). Several factors can conspire to alter the sensitivity of mouse hearts to I/R injury (strain, vendor, diet, sex, shipping and handling, and season) (12). Thus we sought to use a conventional breeding strategy (heterozygous × heterozygous, with Mendelian offspring ratios, tail-clip PCR genotyping of all animals) to determine whether Kir6.2 is truly required for IPC. Furthermore, we directly assayed mKATP channel activity in mitochondria isolated from WT and Kir6.2−/− littermate mice to determine the role of this Kir in the mKATP.
The results herein show that Kir6.2−/− mouse hearts exhibited identical sensitivity to baseline injury, compared with WT littermate controls. Furthermore, both IPC and DZX-mediated protection were blunted in Kir6.2−/−, indicating a role for the channel in cardioprotective signaling. Notably, mKATP channel activity was identical between WT and Kir6.2−/−-derived cardiac mitochondria, indicating no role for this Kir in forming the mKATP.
MATERIALS AND METHODS
Animals.
All animals were maintained in an Association for Accreditation of Laboratory Animal Care-accredited pathogen-free barrier facility with food and water available ad libitum. All procedures were in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and were approved by an Institutional Animal Care and Use Committee. Mice harboring deletion of the KCNJ11 gene that encodes Kir6.2 were obtained with permission from their originator Dr. Susumu Seino (Kobe University, Japan) (20). These mice were reported to be backcrossed to the C57BL/6 background for five generations and were backcrossed herein for an additional two generations onto C57BL/6J (JAX, Bar Harbor ME). Mice were conventionally bred (heterozygous Kir6.2+/− × Kir6.2+/−, Mendelian offspring ratios), and resulting male WT and Kir6.2−/− littermates (age, 8–10 wk) were used in all experiments. Mice were genotyped by tail biopsy PCR using a mouse genotyping kit (Kapa Biosystems, Woburn MA) according to manufacturer's protocol. Briefly, 2-mm3 tail fragments were digested in 10 μl of 10× KAPA Express Extract Buffer plus 2 μl of 1 U/μl KAPA Express Enzyme, brought to 100 μl with PCR-grade water and incubated at 75°C for 10 min, followed by 5 min at 95°C. Samples were centrifuged for 1 min at 2,000 g, and 70-μl supernatant were transferred to fresh PCR tubes. The DNA amplification protocol (as per KAPA mouse genotyping kit) was 95°C, 3 min; (95°C, 15 s; 62°C, 15 s; and 72°C, 30 s) × 35 cycles; 72°C, 3 min; and 4°C hold. The following primers were used and yielded products of 390 bp for Kir6.2−/− and 223 bp for WT: forward F1, TCC CTG AGG AAT ATG TGC TGA CC; reverse, AGG AAG GAC ATG GTG AAA ATG AGC; and Neo, TCT GCA CGA GAC TAG TGA GAC G.
Isolated mitochondria, mKATP thallium flux assay.
Mitochondria were isolated from WT and Kir6.2−/− mouse hearts using differential centrifugation in sucrose-based media, as previously described (37). During the procedure, mitochondria were loaded with benzothiazole coumarin-acetyoxymethyl ester (Invitrogen, Carlsbad, CA) for 10 min at 25°C, as previously described (37, 39). Following isolation, benzothiazole coumarin-acetyoxymethyl ester-loaded mitochondria were subject to mKATP thallium flux (Tl+ flux) assay, whereby Tl+ serves as surrogate for K+ and where Tl+ entry into mitochondria results in dye fluorescence (λex, 488 nm, and λem, 525 nm). mKATP activity was monitored as a change in fluorescence following addition of Tl2SO4, as previously described (37, 39).
Ex vivo perfused heart.
Mice were anesthetized with Avertin (100 mg/kg ip), and an EKG reading was obtained. Corrected QT interval was calculated as QT/(RR interval/100)0.5 (17). The aorta was cannulated in situ and rapidly transferred to a perfusion apparatus and perfused without pacing in constant flow mode (4 ml·min−1·100 mg−1) with Krebs-Henseleit buffer (KH, in mM) 118 NaCl, 4.7 KCl, 25 NaHCO3, 10 glucose, 1.2 MgSO4, 1.2 KH2PO4, and 2.5 CaCl2, gassed with 95% O2-5% CO2 at 37°C, as previously described (37). A water-filled balloon connected to a pressure transducer was inserted into the left ventricle via the mitral valve, and left ventricular pressure was monitored using a pressure transducer (Radnoti, Monovia, CA), connected to a digital recording device (DATAQ, Akron OH). Coronary root pressure was also monitored in-line with the perfusion cannula. After a 20-min equilibration period, the following protocols were observed, as shown in Fig. 1: 1) I/R injury: 30 min perfusion, 30 min global ischemia, 60 min reperfusion; 2) IPC + I/R: 3 × 5 min ischemia plus 5 min perfusion, and then I/R as above; 3) 5-hydroxydecanoate (5-HD) + IPC + I/R: 2 min perfusion with 5-HD (300 μM final), 3 × 5 min ischemia plus 5 min perfusion with 5-HD present, 30 s washout, and then I/R as above; 4) 5-HD + I/R: 30 min perfusion with 5-HD (300 μM), 30 s washout, and then I/R as above; 5) DZX 30 + I/R: 10 min perfusion, 20 min perfusion with DZX (30 μM), 30 s washout, and then I/R as above; 6) DZX 100 + I/R: 10 min perfusion, 20 min perfusion with DZX (100 μM), 30 s washout, and then I/R as above; 7) 5-HD + DZX 30 + I/R: 8 min perfusion, 2 min perfusion with 5-HD (300 μM final), 20 min perfusion with DZX (30 μM final) plus 5-HD, 30 s washout, and then I/R as above; and 8) pacing + I/R: I/R as above, but hearts were paced at 420 beats/min (3-V amplitude, 2 ms duration square pulse) during the entire perfusion period except for the initial 2 min of reperfusion. Each protocol was performed on both WT and Kir6.2−/− animals for a total of 16 experimental groups. DZX and 5-HD stock solutions were prepared fresh daily in DMSO and water, respectively, and the final concentration of DMSO in perfusions never exceeded 0.2% (vol/vol). Compounds were delivered into the perfusion cannula immediately above the aorta. Following perfusion protocols, hearts were sliced, stained in 1% (wt/vol) 2,3,5-triphenyltetrazolium chloride (TTC) for 20 min, and then fixed in 10% neutral buffered formalin for 24 h. Digital images of slices were acquired and analyzed using Adobe Photoshop, as previously described (37). Although baseline cardiac function was not different between the entire cohort of WT and Kir6.2−/− hearts (N > 40, see Table 2), individual subgroups with N = 6–9 were somewhat noisier; hence, cardiac functional data in Figs. 2 and 3 were presented as percentages, relative to the initial value for each heart.
Fig. 1.

Heart perfusion protocols. Schematic showing 8 treatment protocols applied to each genotype [wild-type (WT) and K+ inward rectifier subunits of the 6.2 isotype knockout (Kir6.2−/−)]. Gray shading represents ischemia; white bars represent perfusion. Drugs (concentrations in μM) were added for the durations indicated. Pacing was at 7 Hz (420 beats/min) where indicated. I/R, ischemia-reperfusion; IPC, ischemic preconditioning; 5-HD, 5-hydroxydecanoate; DZX, diazoxide; Veh, vehicle.
Table 2.
Hemodynamic parameters for WT and Kir6.2−/− ex vivo perfused hearts exposed to ischemia-reperfusion injury
| WT | Kir6.2−/− | |
|---|---|---|
| Baseline LVDP, mmHg | 124.5 ± 3.5 | 124.4 ± 3.2 |
| Heart rate, beats/min | 335.5 ± 8.4 | 327.6 ± 8.6 |
| Time to contracture, min | 4.6 ± 1.2 | 2.1 ± 0.5* |
| Degree of contracture, mmHg | 45.2 ± 3.7 | 59.0 ± 8.4* |
Values are means ± SE. LVDP, left ventricular developed pressure (systolic − diastolic). Time to contracture was calculated as the time from onset of ischemia to the start of ischemic hypercontracture. Degree of contracture was the peak ventricular pressure during ischemia minus the pressure immediately before the onset of ischemic hypercontracture. LVDP and heart rate were collected from the initial parameters of all WT or knockout animals before treatment protocols (N > 40), whereas other parameters relating to ischemic hypercontracture were from ischemia-reperfusion-alone groups (N >6).
P < 0.05 between WT and Kir6.2−/− hearts (ANOVA).
Fig. 2.
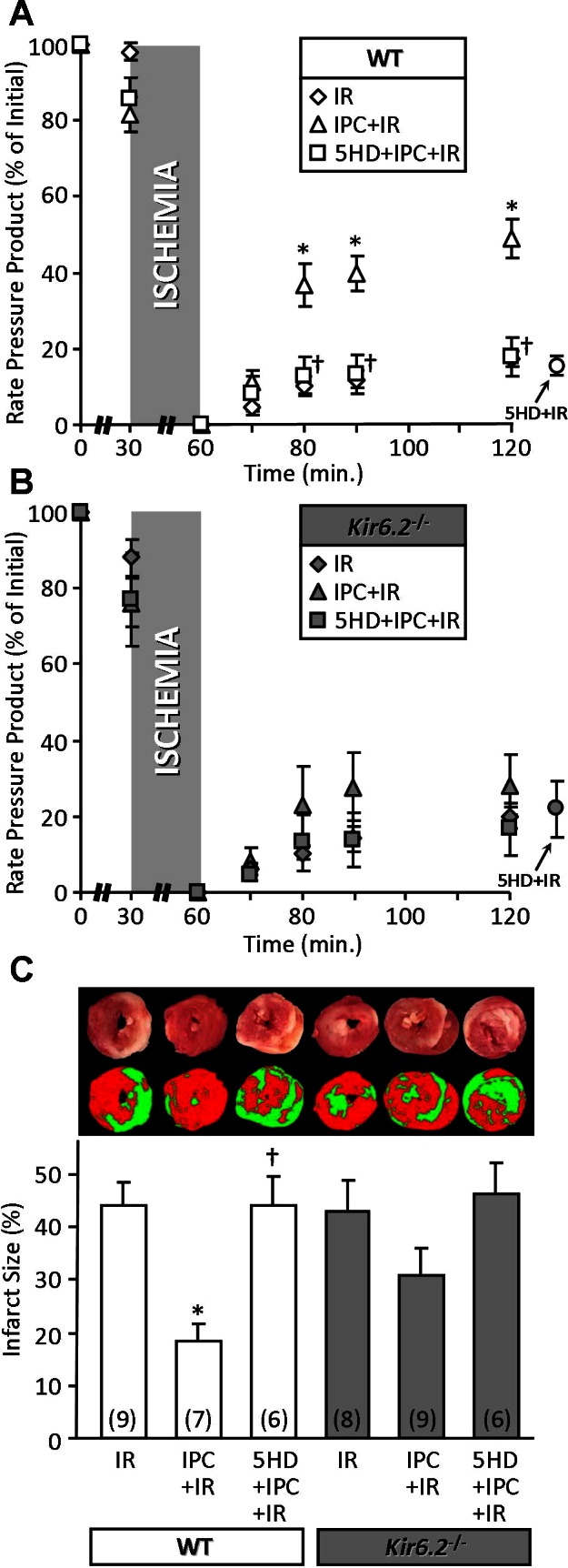
Blunted protective effect of IPC in Kir6.2−/− mouse hearts. Mouse hearts were subjected to I/R injury, I/R with IPC, or I/R with both 5-HD and IPC, as described in materials and methods. A and B, left: ventricular function [heart rate × pressure product (RPP)] was monitored throughout perfusion and expressed as percentage of the initial value (see Table 2). WT (A) and Kir6.2−/− (B) littermates are shown on separate axes for clarity. Separate groups of hearts were subjected to I/R with 5-HD alone (no IPC), and the final recovery data (120 min) are shown to the right of each graph. C: following perfusion, hearts were sliced, stained with TTC, and formalin fixed. The resulting live (red) and infarcted (white) tissue was quantified by planimetry to determine infarct size, expressed as a percentage of area at risk (100% in this global ischemia model). Images above the graph show representative heart slices in actual color (top) and pseudocolor obtained by thresholding and used for planimetry (bottom). All data are means ± SE, and N for each group is shown in parentheses (the same N values apply to A and B also). *P < 0.05 vs. I/R and †P < 0.05 vs. IPC + I/R (ANOVA).
Fig. 3.
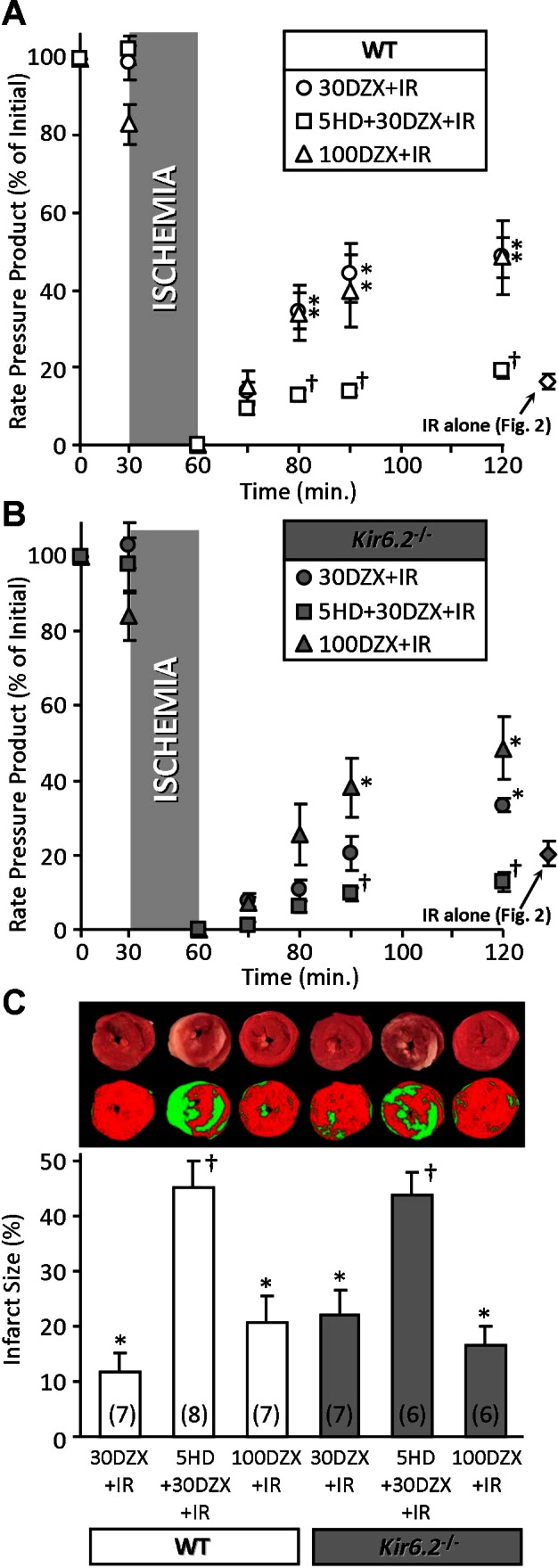
Blunted protective effect of diazoxide in Kir6.2−/− mouse hearts. Mouse hearts were subjected to I/R injury in the presence of 30 or 100 μM DZX, or 30 μM DZX plus 300 μM 5-HD, as described in materials and methods (Fig. 1). A and B: left ventricular function (RPP) was monitored throughout perfusion and expressed as percentage of the initial value (see Table 2). WT (A) and Kir6.2−/− (B) littermates are shown on separate axes for clarity. For comparative purposes, functional recovery data for I/R injury alone (from Fig. 2) are shown to the right of each graph. C: following perfusion, hearts were sliced, stained with TTC, and formalin fixed. The resulting live (red) and infarcted (white) tissue was quantified by planimetry to determine infarct size, expressed as a percentage of area at risk (100% in this global ischemia model). Images above the graph show representative heart slices in actual color (top) and pseudocolor obtained by thresholding and used for planimetry (bottom). All data are means ± SE; N for each group is shown in parentheses. *P < 0.05 vs. I/R (Fig. 1) and †P < 0.05 vs. 30 DZX + I/R (ANOVA).
Statistics.
Statistical significance (P < 0.05) between multiple groups was determined using analysis of variance (ANOVA) with Bonferroni correction.
RESULTS
Blunted protective effect of IPC in Kir6.2−/− mouse hearts.
Following anesthesia (Avertin) and before heart isolation, EKG was recorded on anesthetized mice. No significant differences in measured parameters were observed between WT and Kir6.2−/− (Table 1). Upon heart perfusion, all hearts conformed to recently described criteria for perfused heart studies (2), and no differences were observed in baseline cardiac functional parameters (Table 2), consistent with previous reports (33). Upon exposure to global ischemia, Kir6.2−/− hearts entered ischemic hypercontracture significantly faster than WT and exhibited a greater degree of contracture (Table 2), consistent with previous reports (10).
Table 1.
Electrocardiogram measurements from avertin-anesthetized wild-type and Kir6.2−/− mice
| WT | Kir6.2−/− | |
|---|---|---|
| Heart rate, beats/min | 488 ± 14 | 488 ± 9 |
| PR interval, ms | 46.3 ± 1.5 | 45.0 ± 1.1 |
| QRS duration, ms | 16.9 ± 0.5 | 16.6 ± 0.5 |
| Corrected QT, ms | 52.0 ± 0.9 | 52.7 ± 1.1 |
Values are means ± SE (N > 11) and were acquired using a rodent 3-lead EKG. No significant differences were observed in any parameter between wild type (WT) and K+ inward rectifier subunits of the 6.2 isotype knockout (Kir6.2−/−) mice (ANOVA).
As Fig. 2 shows, WT and Kir6.2−/− hearts exhibited similar sensitivity to baseline I/R injury, yielding the same recovery of function (rate × pressure product) and the same infarct size. However, while IPC afforded a significant improvement in functional recovery and infarct size in WT hearts, this effect was blunted in Kir6.2−/− hearts, such that it did not reach significance. These data are consistent with previous findings in vivo (33). Furthermore, the protective effect of IPC was blocked by the mKATP antagonist 5-HD in both WT and Kir6.2−/− hearts, indicating that the remaining mild cardioprotection afforded in the absence of Kir6.2 was likely due to a mKATP channel. 5-HD alone was without effect on baseline I/R injury (functional recovery: WT, 15 ± 3%, and Kir6.2−/−, 22 ± 7%; infarct size: WT, 41 ± 5%, and Kir6.2−/−, 37 ± 5%; data shown to the right of graphs in Fig. 2).
Blunted protective effect of DZX in Kir6.2−/− mouse hearts.
The antidiabetic drug DZX is a KATP channel opener and has been extensively used to study the role of KATP channels in cardioprotection. At appropriate concentrations (10–30 μM), it is somewhat selective for the mKATP over the cardiac sKATP (Kir6.2/SUR2A) (8, 18), although at higher concentrations it has off-target effects such as inhibition of mitochondrial complex II (18, 26). As shown in Fig. 3, we found that 30 μM DZX was protective in WT hearts (improved functional recovery and reduced infarct size), but this protection was blunted in Kir6.2−/− hearts. Overall, results with 30 μM DZX (loss of cardioprotection in Kir6.2−/−) were not as impressive as those seen with IPC (Fig. 2) but trended in the same direction. Importantly, the protective effects of 30 μM DZX were blocked by the mKATP inhibitor 5-HD in both WT and Kir6.2−/− hearts (Fig. 3), suggesting that the mitochondrial channel is the bona fide target of low concentrations of DZX. As we previously reported (35), 5-HD alone was without effect on basal I/R injury.
When a higher concentration of DZX (100 μM) was used, WT hearts exhibited no further improvement in post-I/R functional recovery and even a slightly worse infarct size, compared with hearts with 30 μM DZX. However, in Kir6.2−/− hearts, 100 μM DZX elicited a significant improvement in post-I/R functional recovery and a slight reduction in infarct size, compared with 30 μM DZX (Fig. 3). Thus, in the Kir6.2−/− heart, a higher concentration of DZX (associated with off-target effects) is required to elicit cardioprotection.
mKATP channel activity is independent of Kir6.2.
Despite recent advances in this area (5), the molecular identity of the mKATP remains unclear, and pharmacological evidence has suggested it may be derived from a canonical KATP involving Kir6.2 (3, 7, 26). However, mKATP channel activity has never been directly measured in Kir6.2−/− mice. Herein we used a mitochondrial Tl+ flux assay, which follows K+ channel-mediated electrophoretic uptake of Tl+ as a surrogate for K+ (37, 39) to investigate mKATP activity in mitochondria isolated from WT and Kir6.2−/− hearts. This assay has been extensively validated in mammalian and nematode mitochondria, with genetic deletion of candidate proteins leading to ablation of specific pharmacophore-stimulated Tl+ fluxes (36, 37, 39).
The data in Fig. 4 show that baseline Tl+ flux was inhibited by ATP and that previously validated concentrations of the mKATP channel openers DZX and atpenin A5 (39) were able to overcome this inhibition and open the channel in both WT and Kir6.2−/− mitochondria. Channel activity was blocked by 5-HD in both genotypes, and similar blockage was obtained with the KATP blocker glyburide (data not shown). Overall, these data indicate no role for Kir6.2 in mKATP activity.
Fig. 4.

Mitochondrial ATP-sensitive K+ (mKATP) channel activity is independent of Kir6.2. mKATP activity was determined using thallium (Tl+) flux assay in mitochondria isolated from WT or Kir6.2−/− littermate hearts. mKATP activators [DZX and atpenin A5 (AA5)] or inhibitor (5-HD) were present as listed below the x-axis, before Tl+ addition. Addition of Tl+ resulted in a Δfluorescence. The baseline Δfluorescence [control (Ctrl), 100%] was 37.6 ± 4.7 and 34.7 ± 5.9 arbitrary units in WT (white bars) and Kir6.2−/− (gray bars), respectively. Data are means ± SE; N for each group is shown in parentheses . *P < 0.05 vs. Ctrl, †P < 0.05 vs. ATP, and ‡P < 0.05 vs. ATP + DZX or ATP + AA5 (ANOVA). Like symbols are not significantly different.
DISCUSSION
The key findings of this study are that the cardioprotective effects of IPC and DZX are blunted in Kir6.2−/− hearts, relative to WT hearts from littermate controls. Furthermore, mKATP channel activity is identical in mitochondria from WT and Kir6.2−/− hearts. While these results are largely confirmatory with regard to previous findings (33), there are a number of important methodological distinctions that render the current study more conclusive.
First, all cardiac studies to date on Kir6.2−/− mice have used pure-bred knockout lines, comparing these animals with separately bred lines of WT mice (10, 11, 31–33). Thus it was concluded that part of the loss of IPC observed in Kir6.2−/− mice could have been due to a greater baseline I/R sensitivity in the knockouts (33). Our data using littermate control animals (Fig. 1) show this is not the case, and the loss of IPC in Kir6.2−/− hearts is due to loss of a component in IPC signaling, not a change in baseline I/R sensitivity. Such a pattern is common in the cardioprotection field; for example, ablation of STAT3 abolishes protection by IPC while having no impact on baseline I/R injury (29, 40). It is also notable that genetic ablation of SUR2 increases resistance to I/R injury (30), but this may be due to expression of short-form SUR2 splice variants in the SUR2−/− knockout mouse, which are thought to independently mediate cardioprotection (25, 41).
A second key difference between this and other previous Kir6.2−/− heart studies was baseline cardiac function. Previously, a low perfusion rate of 3 ml/min was used, resulting in starting left ventricular developed pressure (LVDP) values in the 50–60-mmHg range (33). This is less than half of our starting LVDP (Table 2), and a recent review on the perfused heart technique recommended that hearts exhibiting LVDPs < 60mmHg should be excluded from study (2). It is known that Kir6.2−/− mice have an impaired response to stress (e.g., swimming) (14), and the lower ex vivo perfusion rate and resulting LVDP may also indicate the hearts were stressed, such that baseline I/R sensitivity would be greater in Kir6.2−/− (33).
Another important distinction between this and previous studies is the pacing of hearts. Previous studies on ex vivo Kir6.2−/− hearts (10, 33, 42) employed pacing at frequencies of 420–600 beats/min, which may represent an additional stress. Our studies (Figs. 2 and 3; and Tables 1 and 2) used nonpaced hearts. When we examined the impact of pacing at 420 beats/min on the response of WT and Kir6.2−/− hearts to I/R injury, a trend toward more damage in the knockouts was observed, although this effect did not reach statistical significance (Fig. 5). Overall, it is possible that the combination of pacing, underperfusion, and nonlittermate WT controls may have led to the erroneous conclusion that Kir6.2−/− hearts are more sensitive to I/R injury.
Fig. 5.

Effect of pacing at 7 Hz (420 beats/min) on I/R injury in WT vs. Kir6.2−/− hearts. Hearts were subjected to I/R injury as in Figs. 2 and 3 (see materials and methods). A: recovery of RPP at 120 min time point, as percentage of initial. B: infarct size (% of area at risk). All data are means ± SE. Data for nonpaced hearts (white bars) are reproduced from Fig. 2 (N = 8 to 9). Data for paced hearts (gray-shaded bars) are N = 6. Although a trend to greater injury was seen in paced Kir6.2−/− hearts, no significant differences were observed between paced/nonpaced groups in either parameter (ANOVA).
At the level of isolated mitochondrial studies, further distinctions can be made between previous and current efforts. Previously, a role for Kir6.2−/− in the activity of mKATP had been precluded largely based on measurements of flavoprotein fluorescence (33), which is an indirect measure of mitochondrial complex II redox state, related to mKATP by an unknown mechanism (19). This indirect measure of K+ channel activity was further complicated by the use of a high concentration of DZX (100 μM), which may have mKATP independent effects [e.g., uncoupling or complex II inhibition (18, 26)]. We measured mitochondrial Tl+ flux, which is a more direct measure of actual mKATP activity, and used a low dose of DZX (10 μM) or the potent mKATP agonist atpenin A5 (1 nM). This enables a greater degree of confidence in the conclusion that Kir6.2−/− is not part of the mKATP channel.
The molecular origin of the small amount of cardioprotection still present in Kir6.2−/− hearts exposed to IPC or 30 μM DZX is unclear. It is known that Kir6.2−/− mice exhibit large changes in the metabolic proteome (1) and IPC is known to modulate energy metabolism (11). In addition loss of Kir6.2 has been reported to elicit a shortened action potential duration in cardiac myocytes (33), which may alter responses to I/R injury. Thus, long term effects of Kir6.2 loss may upregulate other protective signaling pathways that can be recruited by IPC or DZX in the absence of the channel. Furthermore, as Fig. 3 shows, higher concentrations of DZX were able to override the lack of protection, possibly via recruitment of nonspecific pathways. Notably, the fact that we saw a blunted cardioprotective responses in Kir6.2−/− using a blood-free perfused heart (Fig. 2) suggests the segment of protection that is attributable to Kir6.2 is a cardiac phenomenon and does not involve leukocytes or other factors absent from the perfused heart.
Examining the data in our study altogether, an important paradox arises: Kir6.2−/− is not part of the mKATP channel and is not a target of DZX in isolated mitochondria, but DZX-mediated protection (at appropriate concentrations) is still blunted in Kir6.2−/− hearts. The off-target effects of DZX such as inhibition of mitochondrial complex II (18, 26) are generally thought to occur at high concentrations (100 μM), whereas lower concentrations (e.g., 30 μM) are thought to be specific for mKATP (4, 9). Notably, however, the sKATP blocker HRM1098 can prevent DZX-induced cardioprotection in the mouse (32) and can partially block DZX-induced cardioprotection in the rabbit (34). While this might suggest sKATP (which undoubtedly contains Kir6.2) is a target of DZX, the fact that 100 μM DZX was protective in Kir6.2−/− hearts suggests this is not the case. It is also possible that the different effects of low versus high doses of DZX in WT versus Kir6.2−/− hearts may represent a simple difference in the DZX dose response.
The present conclusions that Kir6.2 is not involved in mKATP and only minimally involved in IPC- or low-dose DZX-mediated protection are somewhat consistent with a recent report that the mKATP channel is derived from Kir 1.1 (the renal outer medullary K+ channel, ROMK) (5). However, the assignment of ROMK as the mKATP conflicts with pharmacological data on the sensitivity of either channel to ATP and fluoxetine (15, 16, 22, 38, 39).
Regarding the possibility that the mKATP channel may contain the 6.1 isotype of Kir, although this channel is DZX sensitive, several pieces of evidence rule down this protein as a mKATP channel candidate, including studies on the pharmacological properties of the mKATP (39), dominant-negative gene transfer models (27) and indirect measures of channel activity (21). Moreover, commonly used antibodies that originally used to implicate Kir 6.1 as the mKATP were recently shown to recognize off-target mitochondrial proteins (6). Furthermore, we found that mKATP activity and IPC are independent of all isotypes of Kir channel in Caenorhabditis elegans (36), which suggests the mKATP might not contain a Kir at all. Clearly, cardiac-specific knockouts of ROMK and other K+ channels may aid in elucidating the identity of mKATP and its role in cardioprotection.
In conclusion, Kir6.2 plays a role in cardioprotection by IPC, but not by forming the mKATP channel. Furthermore, Kir6.2 appears to play only a partial role in cardioprotection by low doses of DZX, with the remainder of DZX-mediated cardioprotection being accounted for by other Kir6.2-independent mechanisms, which are as yet unknown. Recent advances in the genetic identification of mKATP components (5, 36) suggest that future approaches should explore both Kir and non-Kir genes as diverse candidates for both mKATP activity and cardioprotection.
GRANTS
This work was supported in part by an American Heart Association, Founder's Affiliate Postdoctoral Fellowship award 11POST7290028 (to A. P. Wojtovich) and by National Institute of General Medical Sciences Grant GM-087483 (to P. S. Brookes and K. Nehrke).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.P.W. and W.R.U. performed experiments; A.P.W. interpreted results of experiments; A.P.W. prepared figures; A.P.W. drafted manuscript; S.C., A.B.F., K.N., and P.S.B. conception and design of research; S.C., A.B.F., K.N., and P.S.B. edited and revised manuscript; K.N. and P.S.B. analyzed data; P.S.B. approved final version of manuscript.
REFERENCES
- 1. Arrell DK, Zlatkovic LJ, Yamada S, Terzic A. K(ATP) channel-dependent metaboproteome decoded: systems approaches to heart failure prediction, diagnosis, and therapy. Cardiovasc Res 90: 258–266, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bell RM, Mocanu MM, Yellon DM. Retrograde heart perfusion: the Langendorff technique of isolated heart perfusion. J Mol Cell Cardiol 50: 940–950, 2011 [DOI] [PubMed] [Google Scholar]
- 3. Facundo HT, Fornazari M, Kowaltowski AJ. Tissue protection mediated by mitochondrial K+ channels. Biochim Biophys Acta 1762: 202–212, 2006 [DOI] [PubMed] [Google Scholar]
- 4. Faivre JF, Findlay I. Effects of tolbutamide, glibenclamide and diazoxide upon action potentials recorded from rat ventricular muscle. Biochim Biophys Acta 984: 1–5, 1989 [DOI] [PubMed] [Google Scholar]
- 5. Foster DB, Ho AS, Rucker J, Garlid AO, Chen L, Sidor A, Garlid KD, O′Rourke B. Mitochondrial ROMK channel is a molecular component of mitoKATP. Circ Res 111: 446–454, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Foster DB, Rucker JJ, Marban E. Is Kir6.1 a subunit of mitoK(ATP)? Biochem Biophys Res Commun 366: 649–656, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garlid KD, Halestrap AP. The mitochondrial K(ATP) channel—fact or fiction? J Mol Cell Cardiol 52: 578–583, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D'Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res 81: 1072–1082, 1997 [DOI] [PubMed] [Google Scholar]
- 9. Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial KATP channel as a receptor for potassium channel openers. J Biol Chem 271: 8796–8799, 1996 [DOI] [PubMed] [Google Scholar]
- 10. Gumina RJ, O'Cochlain DF, Kurtz CE, Bast P, Pucar D, Mishra P, Miki T, Seino S, Macura S, Terzic A. KATP channel knockout worsens myocardial calcium stress load in vivo and impairs recovery in stunned heart. Am J Physiol Heart Circ Physiol 292: H1706–H1713, 2007 [DOI] [PubMed] [Google Scholar]
- 11. Gumina RJ, Pucar D, Bast P, Hodgson DM, Kurtz CE, Dzeja PP, Miki T, Seino S, Terzic A. Knockout of Kir6.2 negates ischemic preconditioning-induced protection of myocardial energetics. Am J Physiol Heart Circ Physiol 284: H2106–H2113, 2003 [DOI] [PubMed] [Google Scholar]
- 12. Guo Y, Flaherty MP, Wu WJ, Tan W, Zhu X, Li Q, Bolli R. Genetic background, gender, age, body temperature, and arterial blood pH have a major impact on myocardial infarct size in the mouse and need to be carefully measured and/or taken into account: results of a comprehensive analysis of determinants of infarct size in 1,074 mice. Basic Res Cardiol 107: 288, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366, 2010 [DOI] [PubMed] [Google Scholar]
- 14. Kane GC, Behfar A, Yamada S, Perez-Terzic C, O'Cochlain F, Reyes S, Dzeja PP, Miki T, Seino S, Terzic A. ATP-sensitive K+ channel knockout compromises the metabolic benefit of exercise training, resulting in cardiac deficits. Diabetes 53, Suppl 3: S169–S175, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by fluoxetine (Prozac). Br J Pharmacol 138: 1119–1128, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kobayashi T, Washiyama K, Ikeda K. Modulators of G protein-activated inwardly rectifying K+ channels: potentially therapeutic agents for addictive drug users. Ann NY Acad Sci 1025: 590–594, 2004 [DOI] [PubMed] [Google Scholar]
- 17. Korte T, Fuchs M, Guener Z, Bonin J, de Sousa M, Niehaus M, Tebbenjohanns J, Drexler H. In-vivo electrophysiological study in mice with chronic anterior myocardial infarction. J Interv Card Electrophysiol 6: 121–132, 2002 [DOI] [PubMed] [Google Scholar]
- 18. Kowaltowski AJ, Seetharaman S, Paucek P, Garlid KD. Bioenergetic consequences of opening the ATP-sensitive K+ channel of heart mitochondria. Am J Physiol Heart Circ Physiol 280: H649–H657, 2001 [DOI] [PubMed] [Google Scholar]
- 19. Liu Y, Sato T, O'Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation 97: 2463–2469, 1998 [DOI] [PubMed] [Google Scholar]
- 20. Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci USA 95: 10402–10406, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miki T, Suzuki M, Shibasaki T, Uemura H, Sato T, Yamaguchi K, Koseki H, Iwanaga T, Nakaya H, Seino S. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med 8: 466–472, 2002 [DOI] [PubMed] [Google Scholar]
- 22. Mironova GD, Negoda AE, Marinov BS, Paucek P, Costa AD, Grigoriev SM, Skarga YY, Garlid KD. Functional distinctions between the mitochondrial ATP-dependent K+ channel (mitoKATP) and its inward rectifier subunit (mitoKIR). J Biol Chem 279: 32562–32568, 2004 [DOI] [PubMed] [Google Scholar]
- 23. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74: 1124–1136, 1986 [DOI] [PubMed] [Google Scholar]
- 24. Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature 440: 470–476, 2006 [DOI] [PubMed] [Google Scholar]
- 25. Pu JL, Ye B, Kroboth SL, McNally EM, Makielski JC, Shi NQ. Cardiac sulfonylurea receptor short form-based channels confer a glibenclamide-insensitive KATP activity. J Mol Cell Cardiol 44: 188–200, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schafer G, Wegener C, Portenhauser R, Bojanovski D. Diazoxide, an inhibitor of succinate oxidation. Biochem Pharmacol 18: 2678–2681, 1969 [PubMed] [Google Scholar]
- 27. Seharaseyon J, Ohler A, Sasaki N, Fraser H, Sato T, Johns DC, O'Rourke B, Marban E. Molecular composition of mitochondrial ATP-sensitive potassium channels probed by viral Kir gene transfer. J Mol Cell Cardiol 32: 1923–1930, 2000 [DOI] [PubMed] [Google Scholar]
- 28. Sellitto AD, Al-Dadah AS, Schuessler RB, Nichols CG, Lawton JS. An open sarcolemmal adenosine triphosphate-sensitive potassium channel is necessary for detrimental myocyte swelling secondary to stress. Circulation 124: S70–S74, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith RM, Suleman N, Lacerda L, Opie LH, Akira S, Chien KR, Sack MN. Genetic depletion of cardiac myocyte STAT-3 abolishes classical preconditioning. Cardiovasc Res 63: 611–616, 2004 [DOI] [PubMed] [Google Scholar]
- 30. Stoller D, Kakkar R, Smelley M, Chalupsky K, Earley JU, Shi NQ, Makielski JC, McNally EM. Mice lacking sulfonylurea receptor 2 (SUR2) ATP-sensitive potassium channels are resistant to acute cardiovascular stress. J Mol Cell Cardiol 43: 445–454, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suzuki M, Li RA, Miki T, Uemura H, Sakamoto N, Ohmoto-Sekine Y, Tamagawa M, Ogura T, Seino S, Marban E, Nakaya H. Functional roles of cardiac and vascular ATP-sensitive potassium channels clarified by Kir6.2-knockout mice. Circ Res 88: 570–577, 2001 [DOI] [PubMed] [Google Scholar]
- 32. Suzuki M, Saito T, Sato T, Tamagawa M, Miki T, Seino S, Nakaya H. Cardioprotective effect of diazoxide is mediated by activation of sarcolemmal but not mitochondrial ATP-sensitive potassium channels in mice. Circulation 107: 682–685, 2003 [DOI] [PubMed] [Google Scholar]
- 33. Suzuki M, Sasaki N, Miki T, Sakamoto N, Ohmoto-Sekine Y, Tamagawa M, Seino S, Marban E, Nakaya H. Role of sarcolemmal K(ATP) channels in cardioprotection against ischemia/reperfusion injury in mice. J Clin Invest 109: 509–516, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanno M, Miura T, Tsuchida A, Miki T, Nishino Y, Ohnuma Y, Shimamoto K. Contribution of both the sarcolemmal K(ATP) and mitochondrial K(ATP) channels to infarct size limitation by K(ATP) channel openers: differences from preconditioning in the role of sarcolemmal K(ATP) channels. Naunyn Schmiedebergs Arch Pharmacol 364: 226–232, 2001 [DOI] [PubMed] [Google Scholar]
- 35. Wojtovich AP, Brookes PS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol 104: 121–129, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wojtovich AP, DiStefano P, Sherman T, Brookes PS, Nehrke K. Mitochondrial ATP-sensitive potassium channel activity and hypoxic preconditioning are independent of an inwardly rectifying potassium channel subunit in Caenorhabditis elegans. FEBS Lett 586: 428–434, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wojtovich AP, Sherman TA, Nadtochiy SM, Urciuoli WR, Brookes PS, Nehrke K. SLO-2 is cytoprotective and contributes to mitochondrial potassium transport. PLoS One 6: e28287, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wojtovich AP, Smith CO, Haynes CM, Nehrke KW, Brookes PS. Physiological consequences of complex II inhibition for aging, disease, and the mKATP channel. Biochim Biophys Acta 1827: 598–611, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wojtovich AP, Williams DM, Karcz MK, Lopes CM, Gray DA, Nehrke KW, Brookes PS. A novel mitochondrial KATP channel assay. Circ Res 106: 1190–1196, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci USA 98: 9050–9055, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ye B, Kroboth SL, Pu JL, Sims JJ, Aggarwal NT, McNally EM, Makielski JC, Shi NQ. Molecular identification and functional characterization of a mitochondrial sulfonylurea receptor 2 splice variant generated by intraexonic splicing. Circ Res 105: 1083–1093, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, Pucar D, Bienengraeber M, Dzeja PP, Miki T, Seino S, Alekseev AE, Terzic A. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA 99: 13278–13283, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]