

Abstract
The Saccharomyces cerevisiae Pif1 helicase is the prototypical member of the Pif1 DNA helicase family, which is conserved from bacteria to humans. We show that exceptionally potent G-quadruplex unwinding is conserved amongst Pif1 helicases. Moreover, Pif1 helicases from organisms separated by >3 billion years of evolution suppressed DNA damage at G-quadruplex motifs in yeast. The G-quadruplex-induced damage generated in the absence of Pif1 helicases led to novel genetic and epigenetic changes. Further, when expressed in yeast, human Pif1 suppressed both G-quadruplex-associated DNA damage and telomere lengthening.
G-quadruplex (G4) DNA is a four-stranded DNA structure held together by G-G bps, and most genomes are replete with G4 motifs, i.e., sequences that can form G4 structures in vitro1. Multiple DNA helicases unwind G4 structures in vitro, including several human helicases (WRN, BLM, FANCJ, and hPIF1) whose mutation is associated with genome instability, premature aging, and/or increased cancer risk (Supplementary Table 1).
The S. cerevisiae 5′ to 3′ DNA helicase Pif1 (ScPif1) has multiple nuclear functions, including inhibition of telomerase at both telomeres and double-strand breaks (DSBs)2–5 and preventing replication pausing and DSBs at G4 motifs6, 7. Unlike most eukaryotes, which encode one Pif1 helicase, S. cerevisiae encodes two, ScPif1 and Rrm38. However, ScPif1 and Rrm3 have different functions. Until this paper, the only known nuclear functions of Rrm3 were to promote replication past stable protein complexes9 and to separate converged replication forks8, 10. Although the functions of hPIF1 are not known, mutation of a conserved hPIF1 residue in the Pif1 family signature motif11 is associated with increased cancer risk12.
ScPif1 is a potent G4 binder/unwinder
Thus far, >20 tested helicases, including both ScPif1 and hPIF1, bind and/or unwind G4 structures in vitro (Supplementary Table 1). To determine if ScPif1 is particularly adept at unwinding G4 structures, we analyzed its G4 binding and unwinding activities in a quantitative manner. Filter binding assays were used to quantitate ScPif1 binding to different DNA substrates (Fig. 1 and Supplementary Fig. 1; oligonucleotides in Supplementary Table 2). ScPif1 had a preference for poly-purine tracts (Fig. 1a), which was consistent with its preference for G-rich (Kd = 0.04 nM) over non-G-rich (Kd = 0.2 nM) ssDNA (Fig. 1c). ScPif1 displayed similarly high binding to G4 DNA (average Kd = 0.08 nM for three G4 motifs; Fig. 1e), which was roughly 500-fold better than its binding to Y-structures (Supplementary Fig. 1).
Figure 1.
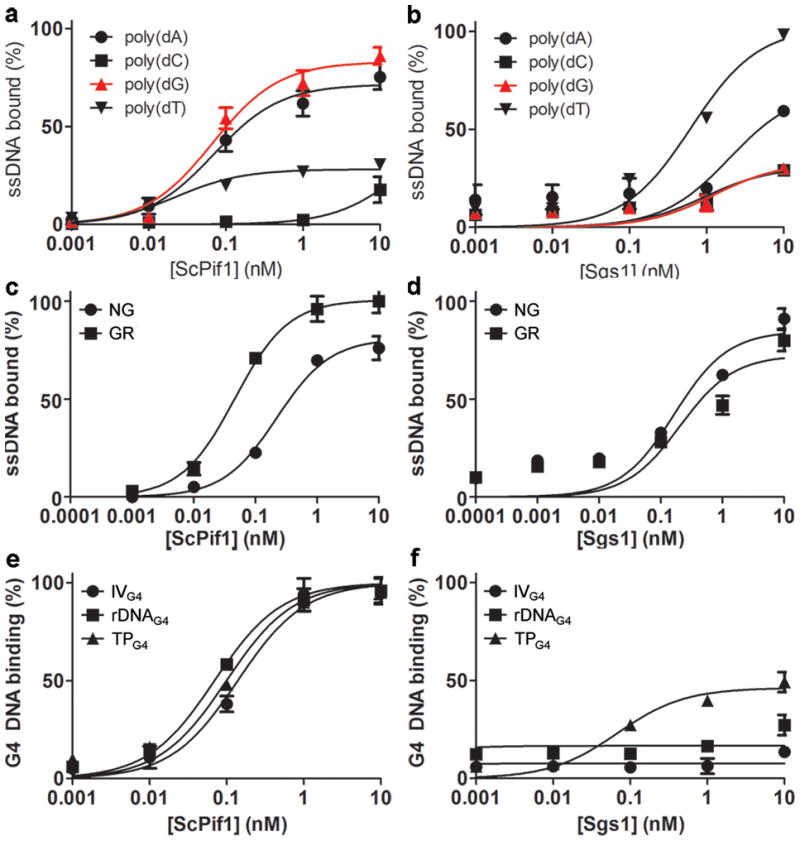
ScPif1 preferentially binds G4 DNA. (a) ScPif1 and (b) Sgs1 binding to homopolymeric oligonucleotides. (c) ScPif1 and (d) Sgs1 binding to 20-mers comprised of 25% each dNTP (non-G-rich, NG) or 75% purines (G-rich, GR; Table S2). (e) ScPif1 and (f) Sgs1 binding to G4 structures. Error bars here and in all figures correspond to one standard deviation of the mean from ≥3 experiments.
ScPif1 efficiently unwound seven of seven G4 substrates, six from S. cerevisiae chromosomal DNA and TPG4, a standard G4 substrate from the mouse immunoglobulin locus (Fig. 2a,d-f and data not shown; sequences in Supplementary Table 2). The apparent KM of unwinding for each G4 structure occurred at equimolar concentrations of ScPif1 and the G4 substrate (0.1 nM) (Fig. 2a,d). In contrast, using the same enzyme preparation, a 5-fold molar excess of ScPif1 was required to unwind Y-structures (Fig. 2a), even though Y-structures are considered preferred ScPif1 substrates13.
Figure 2.
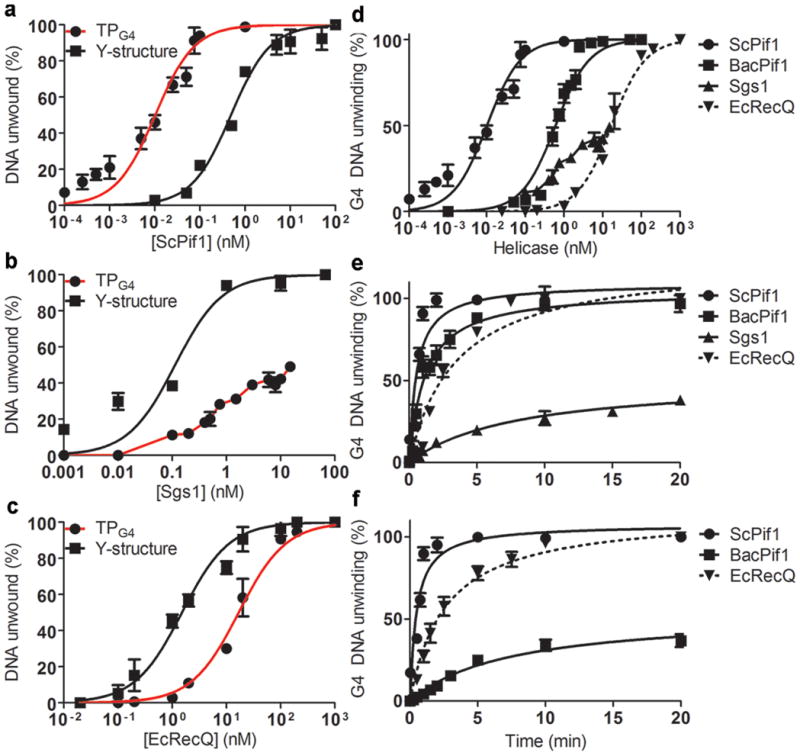
Pif1 helicases preferentially unwind G4 structures. G4 and Y-structure (both 100 pM) unwinding after 20 min (or as indicated) was assessed in standard assays at 37°C (EcRecQ), 30°C (Sgs1, BacPif1), or 25°C (ScPif1 in e and f). A given G4 substrate differed only in the location of the poly(dA) tail (5′ for Pif1 and 3′ for RecQ). For all G4 experiments, graphs show mean unwinding by ScPif1 for three G4 structures (IVG4, rDNAG4, and TPG4) or TPG4 unwinding by BacPif1, Sgs1, and EcRecQ. (a–c) G4 vs. Y-structure unwinding as a function of (a) [ScPif1], (b) [Sgs1], and (c) [EcRecQ]. (d) G4 unwinding as a function of [helicase]. (e) G4 unwinding time course by 100 pM ScPif1, 10 nM BacPif1, 10 nM Sgs1, and 50 nM EcRecQ. Higher Sgs1 concentrations and/or the addition of S. cerevisiae RPA did not increase unwinding (data not shown). (f) G4 unwinding under single-cycle conditions by 100 pM ScPif1, 10 nM BacPif1, and 50 nM EcRecQ.
ScPif1 unwinding rates of G4 structures (Fig. 2a,d) were too fast at 30°C to quantitate. Therefore, time course analyses were performed at a suboptimal temperature (25°C; Fig. 2e). Even at 25°C, ScPif1 unwound 100% of the G4 substrate in 2 min. Although ScPif1 cannot unwind Y-structures under single-cycle conditions14(i.e., in the presence of a 500-fold excess of unlabeled G4 DNA), it unwound G4 structures under single-cycle conditions with no change in kinetics (Fig. 2f). Thus, G4 structures are a preferred ScPif1 substrate.
Bacterial Pif1s are potent G4 unwinders
We are unable to purify full-length Rrm3, Pfh1 (the Schizosaccharomyces pombe Pif1 family helicase), or hPIF111. However, the sequences of many bacterial Pif1 proteins are available15. To determine if vigorous G4 unwinding is conserved amongst Pif1 family helicases, we purified Pif1 proteins from four diverse bacteria and a bacteriophage. All five enzymes robustly unwound the rDNAG4 and TPG4 substrates with apparent KMs in the sub-nanomolar to nanomolar range (Fig. 2d and Supplementary Fig. 2a–f). As with ScPif1, each of these Pif1 family helicases unwound G4 DNA rapidly (Fig. 2e and Supplementary Fig. 2) and under single cycle conditions (though not to completion) (Fig. 2c).
To determine if a helicase is particularly good at G4 DNA unwinding, one can compare its unwinding of G4 DNA to the unwinding of other substrates (e.g., ScPif1 unwinding of G4 DNA vs. Y-structures; Fig. 2a) or compare the unwinding activity of multiple helicases on the same G4 substrate. As several RecQ family helicases unwind G4 structures in vitro (Supplementary Table 1), we tested Sgs1, an S. cerevisiae RecQ helicase, and Escherichia coli RecQ (EcRecQ). Sgs1 bound ssDNA and unwound Y-structures at reported rates16 (Fig. 2b). However, Sgs1 did not bind preferentially to G-rich DNA (Fig. 1b, d), and the apparent Sgs1 binding affinity for four G4 structures was >40-fold lower than that of ScPif1 (Fig. 1f). Likewise, Sgs1 was considerably less efficient than all tested Pif1 family helicases at unwinding G4 structures (e.g., 1000-fold molar excess of Sgs1 was needed to unwind 50% of the G4 structures, Fig. 2e). EcRecQ displayed better unwinding of the TPG4 substrate than Sgs1 (Fig. 2c,e–f), but the apparent KM of this activity was 160-fold greater than that of ScPif1.
Time course experiments revealed slower unwinding of G4 structures by Sgs1 and EcRecQ (Fig. 2e) relative to ScPif1, and Sgs1 was unable to unwind G4 DNA under single cycle conditions. Although EcRecQ did unwind the TPG4 substrate under single-cycle conditions (Fig. 2f), 500-fold more protein relative to ScPif1 was necessary for activity, yielding a t1/2 approximately 10-fold greater than that of ScPif1 in the same assay. The same preparations of Sgs1 and EcRecQ unwound a conventional Y-structure 100- and 10-fold better, respectively, than G4 structures (Fig. 2b,c). With hWRN, a human RecQ helicase, we obtained a similar unwinding rate for TPG4 and rDNAG4 as reported for TPG417; both were similar to unwinding by Sgs1 (Fig. 2e and Supplementary Fig. 3). Thus, three evolutionarily diverse RecQ helicases were much less effective than any tested Pif1 family enzyme at G4 unwinding.
Pif1s suppress G4-induced instability
We developed a quantitative assay to monitor G4-induced genome instability by modifying the gross-chromosomal rearrangement (GCR) assay18. The GCR assay detects complex genome rearrangements by simultaneous selection against URA3 and CAN1 (Fig. 3a). We modified this assay by inserting four strong ScPif1 binding sites6, two G4 motifs (Chr IG4, Chr XG4) and two non-G4 sites (Chr VIING, not G-rich; Chr IGR, G-rich, not G4-forming; Supplementary Table 3), within the PRB1 locus, a non-essential gene that is centromere-proximal to the two counter-selectable genes (Fig. 3a). As reported19, the GCR rate in the “no-insert” WT control was ~1×10−10 events/generation, and none of the inserts affected this rate (Table 1). However, the already high GCR rate in the no-insert pif1-m2 strain was increased ~3-fold in the presence of either of the G4 motifs but was unaffected by either of the other ScPif1 binding sites (Table 1). The G4 inserts did not increase GCR rates in rrm3Δ or sgs1Δ cells compared to no-insert controls. Likewise, the GCR rate in pif1-m2 sgs1Δ cells was not increased by the G4 inserts. However, the GCR rate in pif1-m2 rrm3Δ cells was ~8 times higher in the presence of the G4 motifs compared to the pif1-m2 rrm3Δ strains containing no insert or non-G4 ScPif1 binding sites (1700-fold over the background no-insert WT levels; Table 1). These data suggest that when ScPif1 levels are low, Rrm3 (but not Sgs1) suppresses G4-induced genome instability. Consistent with these findings, Rrm3 bound preferentially to G4 motifs in pif1-m2 but not WT cells20 (Supplemental Fig. 4a).
Figure 3.

Effects of G4 motifs on GCRs. (a) Schematic of Chr V-L in GCR strains. Numbered bars: positions of PCR products in (b). URA3 and CAN1: counter-selectable markers; PCM1: most telomere-proximal essential gene. AlwNI sites are marked. (b) Multiplex PCR analysis before (pre) and after (post) GCR events. +: the strain was WT, had the indicated allele, and/or contained a G4 insert; −: not WT, lacked the allele, and/or did not contain the G4 insert. The numbers indicate the region amplified as shown in Fig. 3a. The loss of band 1 in is consistent with TA.
Table 1.
Mean GCR rates (± standard deviation) calculated from ≥3 independent experiments and normalized to the rate (1.5±0.7×10−10 GCR events/generation) in the wild type strain with no insert at the PRB1 locus.
| Sequence inserted at the PRB1 locus | |||||
|---|---|---|---|---|---|
| Genotype | No insert | Chr IG4 | Chr XG4 | G-rich | Non-G-rich |
| Wild type | 1 | 1.2±0.5 | 1.4±0.5 | 1.2±0.2 | 1.3±0.6 |
| pif1-m2 | 76±8 | 200±20 | 210±10 | 70±30 | 60±20 |
| rrm3Δ | 6±5 | 12±6 | 9±4 | 3±1 | 3.2±0.9 |
| sgs1Δ | 16±5 | 12±8 | 19±8 | ND | ND |
| pif1-m2 rrm3Δ | 210±32 | 1500±500 | 1900±200 | 200±10 | 250±40 |
| pif1-m2 sgs1Δ | 20±80 | 190±35 | 200±50 | ND | ND |
ND, not determined.
To determine if ScPif1 suppression of DNA damage at G4 motifs is evolutionarily conserved, we tested diverse Pif1 proteins for their ability to suppress the high GCR rate in pif1-m2 rrm3Δ+G4 cells. Helicases were introduced on a single-copy plasmid and expressed from the RRM3 promoter (see Supplementary Fig. 6 for western analysis of protein expression). A simple spot assay was used to monitor the frequency of GCR events; cells were spotted 150 times at high density on FOA+Can plates and incubated until resistant colonies formed (~20 GCR events/spot for the pif1-m2 rrm3Δ+G4 strain containing no or empty vector; Fig. 4c). As expected, ScPif1 and Rrm3 both suppressed the pif1-m2 rrm3Δ+G4 GCR rate (0.06–0.09 events/spot), while helicase-dead ScPif1 (ScPif1-KA) did not (19 events/spot; Fig. 4c). Remarkably, all seven heterologous Pif1 helicases, including hPIF1 (0.3 events/spot) and six prokaryotic/viral Pif1 helicases (0.07–1.0 events/spot), suppressed the high GCR rate.
Figure 4.
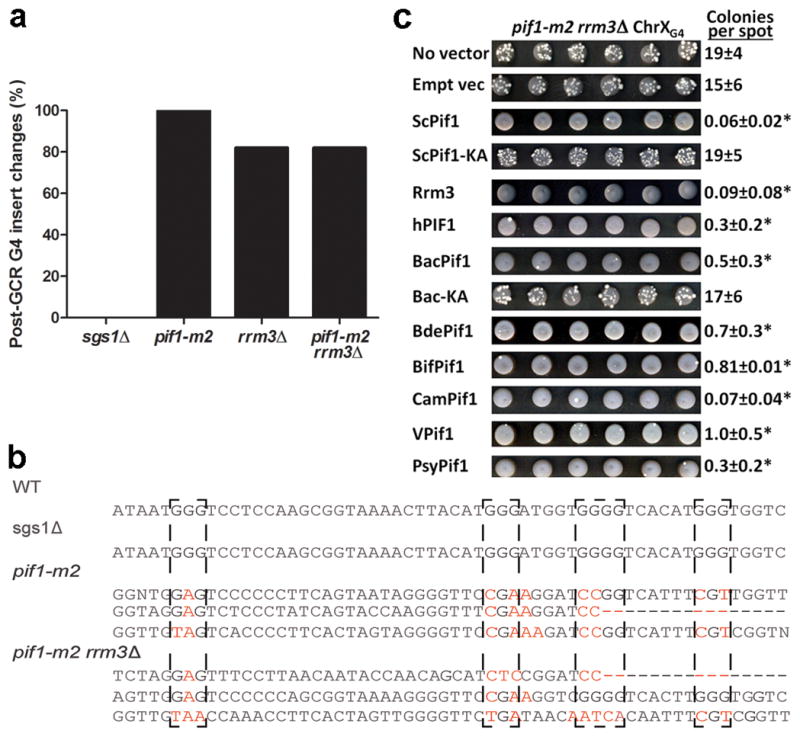
Pif1 family helicases suppress G4-induced GCR events in pif1-m2 rrm3Δ+G4 cells. (a, b) The G4-insert region was PCR-amplified and sequenced from 19 (pif1-m2 rrm3Δ) or 17 (others) GCR clones. (b) Examples of G4 mutations. G-tracts involved in G4 formation are denoted with dashed boxes. Mutated Gs, red; dashes, deletions. (c) GCR events in pif1-m2 rrm3Δ+G4 cells expressing the indicated helicase. Six of 150 spots/strain are shown. GCR events are white colonies on a gray background of non-growing cells. Average±SD colonies/spot is indicated; *, p<0.016 for the colonies/spot vs. the “no vector” control as calculated by Student’s t-test. Empt vec, empty vector; VPif1, V99B1Pif1.
Novel G4-induced events in pif1 cells
We used several methods to determine if G4 motifs affected the structure of the distal portion of chromosome V in the GCR events (Fig. 3, 4a,b, Supplementary Fig. 4c–e, and Supplementary Fig. 5). As predicted21, multiplex PCR (Fig. 3b) and Southern (Supplementary Fig. 4c–e) analyses revealed that almost all GCR events in the no-insert pif1-m2 strain were due to telomere addition (TA) centromere-proximal to CAN1 (52/56 events). However, apparent TA was rare (5/27) or not detected (0/28) in GCR clones from, respectively, pif1-m2+G4 and pif1-m2 rrm3Δ+G4 cells (i.e., CAN1 fragment retained in multiplex PCR, Fig. 3b; new telomere bands were rare in Southerns, Supplementary Fig. 4d,e).
We also sequenced the 1000-bp region around the G4 insert in individual GCR clones (Fig. 4a,b, Supplementary Fig. 5). There were no changes in this region in 17 of 17 GCR clones from sgs1Δ+G4 cells. However, all (19/19) G4 inserts were altered in pif1-m2+G4 GCR clones. These changes included mutations limited to the G4 motif (5% of clones); partial or complete deletion of the G4 motif and/or flanking DNA (10%); and more complex events involving deletions, mutations, and insertions (85%) (Supplementary Fig. 5). A similar pattern was seen in most (82%) of the pif1-m2 rrm3Δ GCR events (Fig. 4a, Supplementary Fig. 5).
As expected, URA3 and CAN1 were lost or moved to new locations in all GCR clones from WT+G4 (8/8) and sgs1Δ+G4 (11/11) cells. However, the positions of URA3 and CAN1 were unchanged in most pif1-m2+G4 (19/27) and pif1-m2 rrm3Δ+G4 (27/28) GCR clones. Based on the high mutation rate of the G4 inserts, we predicted that loss of URA3 and CAN1 expression would be due to mutations in the genes. However, cloning and sequencing of URA3 and CAN1 from six clones each from the pif1-m2+G4 and pif1-m2 rrm3Δ+G4 strains revealed that the URA3 and CAN1 sequences, including ~200 bp up and downstream of the genes, were unchanged, but Chr V-L was unstable in many of these clones. Subsequent analyses of the same clones, e.g., after restreaks or growth in liquid culture, revealed that either URA3 or both URA3 and CAN1 were lost as often as 95% of the time (data not shown). However, some clones maintained WT URA3 and CAN1 genes for ≥200 generations.
Mechanism of G4-induced silencing
Given that the sequences and positions of URA3 and CAN1 were unchanged in the pif1-m2+G4, pif1-m2 rrm3Δ+G4, and pif1-m2 rrm3Δ GCR clones that retained these genes, their FOAR CANR phenotypes must be due to epigenetic silencing. To determine if the silencing occurred at the transcriptional level, we used real-time quantitative PCR to assess the amounts of URA3 and CAN1 mRNA in four independent pif1-m2 rrm3Δ+G4 GCR clones and the parental pre-GCR strain. Depending on the clone, URA3 mRNA levels ranged from 9–24% of the levels in the pre-GCR strain; CAN1 mRNA ranged from 20–53% of the control in the same clones (Fig. 5a,b). Thus, silencing was not due to translational regulation.
Figure 5.
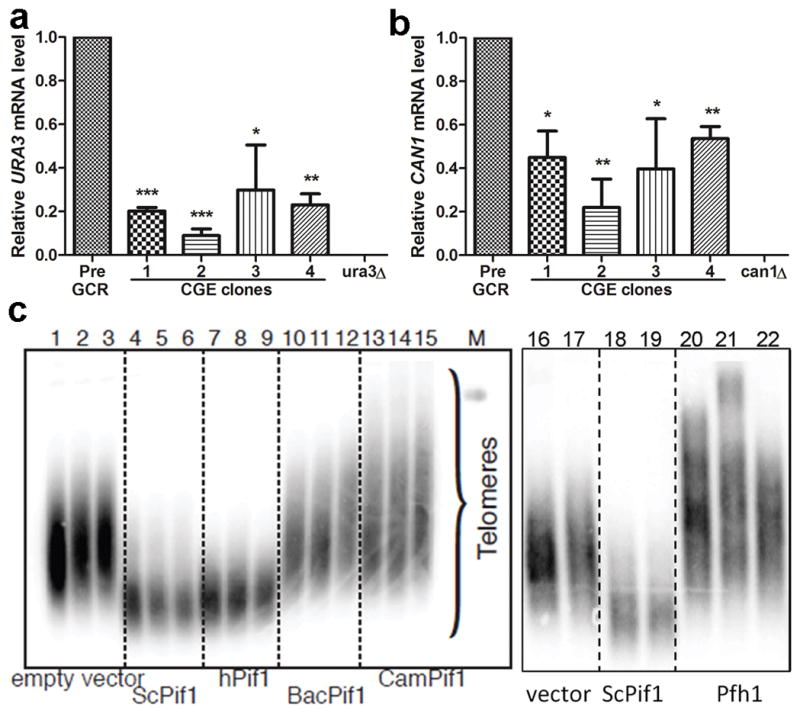
Mechanism of CGE silencing and effect of Pif1 helicases on telomere length. (a) URA3 and (b) CAN1 mRNA levels in pif1-m2 rrm3Δ+G4 CGE clones and controls (pre-GCR parental strains and ura3Δ and can1Δ cells). qRT-PCR was used to determine the ACT1, URA3, or CAN1 mRNA levels in the indicated strains. URA3 and CAN1 values were normalized to ACT1 levels in each strain; the 2−ΔΔCt method38 was used to determine URA3 and CAN1 levels relative to parental pre-GCR cells. *, **, and ***: p<0.05, 0.01, and 0.001, respectively. (c) Telomere blot of DNA from pif1-m2 spore clones expressing vector only (lanes 1–3,16–17), ScPif1 (lanes 4–6,18–19), hPIF1 (7–9), two different bacterial Pif1s (10–12,13–15), or Pfh1 (20–22). M, markers. DNA was prepared ~50, 75, and 100 generations after sporulation (first, second, and third lanes in each set) or 100 generations after sporulation from two or three spore clones (lanes 16–22). See Supplementary Fig. 7 for full gel images.
In many organisms, including S. cerevisiae and humans22, 23, genes that are near telomeres are transcriptionally repressed (telomere position effect, TPE; reviewed in24). To determine if the silencing observed in the pif1-m2+G4 GCR clones was due to TPE, we deleted SIR2, which encodes a protein that is required for TPE, in two independent pif1-m2+G4 GCR clones that retained URA3 and CAN1 in their original positions. Both clones lost their FOAR CanR phenotypes, suggesting that silencing was due to a TPE-like mechanism.
hPIF1 inhibits telomere lengthening
Telomeres are longer in Pif1-deficient cells owing to its ability to remove telomerase from DNA ends2, 3. To determine if other Pif1 helicases inhibit yeast telomerase, we determined telomere length in pif1-m2 cells expressing heterologous Pif1 helicases (Fig. 5c). Empty vector or the expression of bacterial Pif1s (BacPif1 and CamPif1) or S. pombe Pfh1 did not suppress the long telomere phenotype of pif1-m2 cells. Indeed, telomeres were even longer in pif1-m2 cells expressing bacterial Pif1s or Pfh1 than in pif1-m2 cells alone. However, hPIF1 was nearly as effective as ScPif1 in restoring telomere length to pif1-m2 cells (Fig. 5c), even though it was expressed at much lower levels (Supplementary Fig. 6).
Discussion
ScPif1 and five prokaryotic Pif1 helicases were extremely proficient at unwinding G4 structures (Fig. 2 and Supplementary Fig. 2), while three RecQ helicases had ~1000-fold lower G4 unwinding activity than the Pif1 helicases (Fig. 2; Supplementary Fig. 2–3). Moreover, while ScPif1 unwound G4 structures much better than Y-structures (Fig. 2a), which are themselves a preferred ScPif1 substrate13, Sgs1 and EcRecQ were more active on Y-structure than G4 substrates (Fig. 2b,c). Thus, vigorous G4 unwinding is a conserved feature of Pif1 helicases.
Suppression of G4-induced DNA instability was also conserved (Table 1). In pif1-m2 cells, GCR rates were increased when the substrate contained a G4 motif but not when it contained other strong Pif1 binding sites; this effect is likely underestimated as pif1-m2 is not a null allele3. Similarly, the human minisatellite CEB1, a tandem array of ~40 GC-rich repeats, increases the GCR rate in pif1Δ cells25. In contrast to no-insert pif1-m2 cells, G4-mediated GCR events were rarely due to TA (52 TA/56 GCR events, pif1-m2 vs. 5 TA/27 GCR events, pif1-m2+G4 cells). In both pif1-m2 and pif1-m2 rrm3Δ cells, the G4-induced events were usually associated with mutation of the G4 insert so that it could no longer form a G4 structure (Fig. 4a), suggesting that the process enabling cells to replicate and/or repair a G4 motif in the absence of Pif1 helicases is error-prone. Remarkably, the double drug-resistant phenotype of the G4-induced clones recovered from pif1-m2+G4 (75/104 clones) and pif1-m2 rrm3Δ+G4 (50/64 clones) cells was usually due to epigenetic silencing, though the genes could be lost during further outgrowth. Although silencing of URA3 and CAN1 in CGE clones was Sir2-dependent, as is TPE26, this silencing was considerably more effective than classical TPE. When URA3 is immediately adjacent to the Chr VII-L telomere, mRNA levels are ~20% of control levels, but when URA3 is ~20 kb from the same telomere, FOAR colonies are not detected (<6×10−7). In contrast, in CGE clones, the average URA3 mRNA level was 19% of the control, even though URA3 was 21 kb from the telomere. The extension of silencing to more internal sites may be associated with impaired replication through a G4 structure, as changes in silencing occur in translesion polymerase-defective avian DT40 cells27. The unusual URA3 and CAN1 silencing seen here also required or was enhanced by lack of Pif1 and/or Rrm3, as it was not detected in sgs1Δ+G4 GCR clones (0/17 clones). Additionally, it was enhanced by a nearby G4 motif as it was not seen in GCR clones from the no-insert pif1-m2 cells (0/56 clones). The novel events at both G4 motifs and structural genes in the absence of Pif1 family helicases are distinct from previously described GCR events. Thus, we term them CGE (complex genetic-epigenetic) events. The epigenetic silencing of URA3 and CAN1 is reminiscent of the gene silencing that occurs in some human tumors that can lead to loss of heterozygosity.
Though ScPif1 and Rrm3 have largely non-overlapping functions11, they both suppressed damage at G4 motifs, as did seven of seven heterologous Pif1 helicases (Fig. 4c). This suppression was efficient. For example, hPIF1 suppressed CGE events ~20% as effectively as ScPif1, even though it was expressed at considerably lower levels (Supplementary Fig. 6). Thus, activity at G4 DNA by both in vitro and in vivo assays is a conserved feature of Pif1 family helicases.
ScPif1 (but not Rrm3 or Pfh1) inhibits telomerase4, 28, 29. hPIF1 (but not prokaryotic Pif1 helicases or Pfh1) restored telomere length in pif1-m2 cells (Fig. 5c), suggesting that hPIF1 is a regulator of both telomerase and G4 structures in its endogenous context. One or both of these activities might explain why mutation of hPIF1 is associated with cancer12.
Methods
Yeast strains
All experiments were performed in the YPH500 background30. Yeast strains are listed in Supplementary Table 4, except for those used in the gross-chromosomal rearrangement (GCR) assays (Supplementary Table 5). Gene disruptions and epitope tagging of proteins were confirmed by colony PCR, sequencing, Southern blotting, and/or phenotypic analysis. The pif1-m2 allele was introduced as previously described3 (see Supplementary Table 6 for oligonucleotide sequences used for cloning). The C-terminus of Rrm3 was tagged at its endogenous locus with 13 Myc epitopes using PCR39. Tagged Rrm3 was expressed from its own promoter as the only version of the protein in the strain. Plasmids are listed in Supplementary Table 8. All GCR strains (Supplementary Table 5) are derivatives of YPH500 in which HXT13 was deleted with the Kluveromyces lactis URA3 gene using pUG7240 and oligonucleotides MB262 and MB277 (see Supplementary Table 6 for the sequences of oligonucleotides used in GCR strain construction). The partial loss of nuclear function pif1-m2 allele was used instead of pif1Δ because pif1-m2 cells are mitochondrial proficient3. RRM3 was deleted with HIS3MX6 using pFA6a-His3MX641 and oligonucleotides MB30 and MB31. SGS1 was deleted with the Schizosaccharomyces pombe his5+gene using pUG2740 and oligonucleotides MB32 and MB33 (Supplementary Table 6). Strains containing “inserts” (Supplementary Table 5) were made by deleting PRB1 with LEU2 marked cassettes using oligonucleotides KP321f and KP321r (Supplementary Table 8). The LEU2 marked cassettes were derived from pRS415-based plasmids30 containing the designated inserts cloned into the XbaI and BamHI sites (Supplementary Table 8).
Biochemical methods
Full-length S. cerevisiae Pif1 and Sgs1 and Escherichia coli RecQ were purified as previously described14, 16, 31. In vitro analyses of independent protein preparations revealed little to no prep-to-prep variability and that these preparations had similar biochemical activities (i.e., ssDNA binding and Y-structure DNA unwinding, see below) to previously published values14, 16, 31.
Bacterial Pif1 helicases were cloned as follows. Emma Allen-Vercoe (University of Guelph), Craig Parker (U.S. Department of Agriculture), Roger Johnson (Public Health Agency of Canada), and Hector L. Ayala-del-Rio (University of Puerto Rico at Humacao) kindly provided genomic DNA from Bacteroides sp. 2_1_16, Campylobacter jejuni subsp. jejuni NCTC 11168, Escherichia coli phage rv5, and Psychrobacter sp. PRwf-1, respectively. A pUC19-based plasmid containing the gene encoding the Bdellovibrio bacteriovorus HD100 Pif1 helicase (Supplementary Table 8) was a gift from Elizabeth Sockett (University of Nottingham). PCR primers were designed to amplify the Pif1-like helicase genes from the above mentioned organisms (see Supplementary Table 6) with iProof HF Master Mix (BioRad). PCR products were then digested and ligated into a modified pET21d vector (pMB116; Supplementary Table 8) such that they were in-frame with an N-terminal 4x Strep-tag II sequence and a C-terminal 6x His tag. Additional cloning details and nucleotide sequences are available upon request.
Expression plasmids were transformed into Rosetta™ 2(DE3) pLysS cells and selected for at 37°C on LB medium supplemented with 100 μg/mL ampicillin and 34 μg/mL chloramphenicol. Fresh transformants were used to inoculate one or more 5-mL LB cultures supplemented with antibiotics and incubated at 30°C for ~6 h with agitation. These starter cultures were then diluted 1:100 in ZYP-5052 autoinduction medium containing 1x trace metals mix42, 100 μg/mL ampicillin, and 34 μg/mL chloramphenicol and incubated at 30°C with agitation to OD600 >3 (~18 h). Cells were harvested by centrifugation for 10 min in a GS-3 rotor at 5,000 rpm and 4°C. Cell pellets were weighed and frozen at −80°C prior to lysis or for long-term storage.
The cells were thawed at room temperature and resuspended in 2 mL/g cell pellet buffer A (50 mM Na-HEPES (pH 8), 10% (v/v) glycerol, 300 mM NaCl, and 5 mM MgCl2) supplemented with 1x protease inhibitor cocktail (Sigma), 20 μg/mL DNase I, and 2.5 μg/mL RNase A. Cells were lysed for 10 min at room temperature by adding methyl 6-O-(N-heptylcarbamoyl)-α-D-glucopyranoside (HECAMEG; Sigma) to a final concentration of 0.05% (w/v) and 1x BugBuster (Novagen) or FastBreak (Promega) with gentle stirring. Subsequent steps were performed at 4°C.
The soluble fraction was clarified by centrifugation for 30 min in an SA-600 rotor at 13,000 rpm followed by filtering the supernatant through a 0.22-μm membrane. This mixture was then loaded onto a Strep-Tactin Sepharose gravity column (IBA) pre-equilibrated in buffer A. The column was washed with four column volumes each of buffer W1 (50 mM Na-HEPES, pH 8, 10% (v/v) glycerol, 500 mM NaCl, 5 mM MgCl2, and 0.05% (v/v) HECAMEG), W2 (50 mM Na-HEPES, pH 8, 10% (v/v) glycerol, 300 mM NaCl, 5 mM MgCl2, 0.05% (v/v) HECAMEG, and 5 mM ATP), and W3 (50 mM Na-HEPES, pH 8, 10% (v/v) glycerol, 300 mM NaCl, 5 mM MgCl2, and 0.01% (v/v) HECAMEG). Protein was eluted with three column volumes of buffer W3 supplemented with 1 mM desthiobiotin. Column fractions were examined on 10% SDS-PAGE gels run at 20 V/cm and stained with Coomassie Brilliant Blue R-250 (BioRad).
Peak fractions were pooled and loaded onto a His60 Ni Gravity column (Clontech) pre-equilibrated in buffer W3. The column was washed with five column volumes of buffer W3 supplemented with 25 mM imidazole, and protein was eluted with five column volumes of W3 supplemented with 250 mM imidazole. Fractions were analyzed by SDS-PAGE as above, and peak fractions were pooled and extensively dialyzed against storage buffer (50 mM Na-HEPES (pH 8), 30% (v/v) glycerol, 50 mM NaCl, 150 mM NaOAc (pH 7.6), 25 mM (NH4)2SO4, 5 mM MgOAc, 1 mM DTT, and 0.01% (v/v) HECAMEG) using 30- (Slide-A-Lyzer; Pierce) or 50-kDa (Tube-O-DIALYZER; G-Biosciences) MWCO membranes. Protein concentration and purity in the final dialysates were determined on SYPRO orange (Sigma)-stained SDS-PAGE gels using known amounts of a standard protein for comparison. In all cases, protein purity was ≥95%.
For some protein preparations, the N-terminal 4x Strep-II tag was removed by PreScission Protease (GE Healthcare) digestion (2 U protease/mL protein at 4°C overnight) prior to His60 column chromatography. In all cases, removal of the tag had little effect on subsequent protein purity and no effect on the in vitro activities examined. However, tag cleavage occasionally resulted in precipitation of a considerable portion of the protein. Thus, recombinant proteins containing both N- and C-terminal tags were used for all experiments shown.
For preparation of substrates, various S. cerevisiae G4 motifs were chosen from the >500 identified G4 motifs in the budding yeast genome32 (see Supplementary Tables 2 and 3 for sequences). Oligonucleotides of G4 motifs were synthesized by IDT (USA). The concentrations of all oligonucleotides were estimated using extinction coefficients provided by the manufacturer. G4 DNA structures were formed in vitro as described33. Formation of G4 structures was confirmed by non-denaturing PAGE. After G4 structure formation, the substrates were 5′-labeled with T4 polynucleotide kinase (NEB) and γ32P-ATP, purified via 7% non-denaturing PAGE, and visualized using phosphoimaging.
In all biochemical assays, 100 pM radioactively labeled DNA was used, unless noted otherwise, and the reaction buffers used were previously described for ScPif114 Sgs116, and EcRecQ31. Briefly, protein-DNA binding was analyzed using a BioDot SF apparatus (Bio-Rad) by the double-filter binding method34. Reactions were set up as for helicase assays, but ATP was omitted. The reactions were incubated on ice for 30 min, filtered through the membranes, and then the membranes were washed with additional reaction buffer. The membranes were dried and analyzed by phosphoimaging. ScPif1, Sgs1, and EcRecQ helicase activity assays were performed essentially as described in14, 16, 31. The helicase activity of non-yeast Pif1 enzymes was also measured as described for ScPif1 in14. hWRN helicase assays were performed as described in35. For protein titrations, reactions were incubated for 30 min at helicase at 25°C (ScPif1), 30°C (Sgs1), or 37°C (EcRecQ, hWRN, and non-yeast Pif1s). In time course experiments, 100 pM ScPif1 10 nM BacPif1, 10 nM Sgs1, or 50 nM EcRecQ was added to the reaction; 100 pM Sgs1 or EcRecQ displayed only basal levels of unwinding in our G4 unwinding assays. For single cycle conditions, we used a 500x excess of either G4 DNA or ssDNA as a protein trap. The excess trap DNA was added together with ATP to start the reactions.
The data were fit with rectangular hyperbolic curves using GraphPad Prism 5 and equation 1:
where X is the helicase concentration or time (as indicated), Y is either DNA binding or unwinding (as indicated), Bmax is the maximum level of binding or unwinding (as indicated), and K is the midpoint of the curve. When a log10-scale x-axis is used, the hyperbolic curve assumes a sigmoidal shape.
GCR assays
GCR assays were cloned and performed essentially as described36 (primer sequences for cloning are listed in Supplementary Table 6). Briefly, sets of five or more 5-mL cultures of each S. cerevisiae GCR strain (Supplementary Table 5) were grown to saturation in YPD medium at 30°C for 36–48 h. A final dilution of 1×10−7 of each culture was plated on YPD and incubated at room temperature for 4 days to determine the viable cell count. Cells (1.5 or 2 mL) from each culture were pelleted, resuspended in sterile water, plated on drop-out medium lacking uracil and arginine (US Biologicals) supplemented with 1g/L 5-FOA and 60 mg/L canavanine sulfate (FOA+Can), and incubated at 30°C for ~4 days. GCR rates were calculated using the FALCOR web server and MMS Maximum Likelihood Method37 and normalized to WT rate of 10−10 GCR events/cell division. The rates presented in Table 1 are the means ± standard deviations of ≥3 experiments per strain. We define GCR clones as colonies that grew on the FOA+Can plates. Such FOARCanR clones were selected for post-GCR analyses (below).
G4 inserts were sequenced from samples of genomic DNA from FOARCanR clones prepared using a MasterPure Yeast DNA Purification kit (Epicentre) following the manufacturer’s instructions. The oligonucleotides used for sequencing the Watson and Crick strands are MB540 (CAATAGGCCGAAATCGGCAAAATCCC) and MB537 (CTCCTATGTTGTGTGGAATTGTGAGCGG), respectively, which amplified a ~1-kb region containing the inserts. The results were analyzed using the Biology Workbench tools (http://workbench.sdsc.edu/) and classified into five different categories, as indicated in Fig. S5: no change, the G4 inserts were identical to the starting strain in the GCR clones; Recombination, the G4 insert was replaced with a partially homologous sequence from Chr VII; Mutation and Deletion, several of the guanines responsible for forming the G4 structures were either mutated to other residues or deleted; and Mut/Del/Ins, the G4 inserts experienced a variety of events, including substitution mutations and short deletions and/or insertions.
Suppression analyses of the pif1-m2 rrm3Δ+G4 GCR phenotype were performed by transforming strain KP326 (Supplementary Table 5) with TRP1-marked plasmids containing C-terminally 3xFLAG-tagged helicase genes expressed under control of the RRM3 promoter (Supplementary Table 8). Three independent colonies were used to inoculate 5 mL synthetic complete medium lacking tryptophan (SC-Trp) and grown on a roller drum for ~48 h at 30°C. The OD660 for each culture was determined with a spectrophotometer, and the cells were pelleted by centrifugation and resuspended to OD660= 10 in sterile H2O. Then, a repeat pipettor was used to spot 10-μL samples of each strain 50 times on a FOA+ Can plate, and the plates were incubated at 30°C for 4 days. This process was repeated ≥3 times for each strain. When colonies appeared on the FOA+ Can plates, the number of colonies per 10-μL spot was counted, and the average number of colonies in the 50 spots per plate was calculated. The mean of these values (± the standard deviation) from the ≥3 plates per strain was determined and reported in the right column of Fig. 4c.
Western and Southern blotting
Cell extracts for western blotting were prepared as described in43. Briefly, cells were grown overnight in Sc-Trp liquid medium at 30°C with aeration. Then, 1 mL of OD600 = 2.5 cells was harvested, resuspended in 200 μL 0.1 N NaOH, incubated at room temperature for 5 min, pelleted, resuspended in 50 μL SDS-PAGE sample buffer, boiled for 3 min, and pelleted again. Subsequently, 6 μL of the supernatants was loaded onto an 8% (37.5:1 polyacrylamide:bis-acrylamide) SDS-PAGE gel and run at 20 V/cm. The proteins were transferred to a nitrocellulose membrane at 4°C and blocked with 5% non-fat milk in TBST at room temperature using standard protocols. The blot was probed with a monoclonal anti-FLAG antibody (F1804, Sigma) and visualized with an HRP-conjugated secondary antibody and ECL detection reagents (GE Healthcare). The blot was then stripped and reprobed with an anti-tubulin antibody (G094, ABM) to verify equivalent protein loading.
For telomere blots, cells containing a plasmid with either a helicase gene (pMB267, 270, 282, and 292) or no insert (empty vector control; pMB13) (Supplementary Table 8) were transformed into the PIF1/pif1-m2 diploid (KP448). The diploids were sporulated, and the pif1-m2 spore clones carrying the plasmids were recovered. Genomic DNA was isolated from cells from restreaks 1–6 after sporulation (corresponding to approximately 25 generations/restreak) using a MasterPure Yeast DNA Isolation kit (Epicentre Biotechnologies), digested overnight with PstI and XhoI (NEB), and telomere length was determined by Southern analysis as described previously44. Results from DNA from restreaks 1–3 are shown in Figure 5d, but the same results were also observed after 6 restreaks.
When colonies arose after GCR events, single colonies were restreaked onto FOA+Can plates, and genomic DNA was isolated from the survivors using a MasterPure Yeast DNA Isolation kit. The DNA was digested overnight at 37°C with AlwN1 and run on a 0.7% agarose gel. The DNA was transferred to Hybond membranes (GE Life Sciences) by capillary action and blotted using the 400 bp CIN8 PCR product as a probe. The CIN8 PCR probe hybridizes to both sides of the AlwNI cut site in CIN8 (see Fig. 3a), generating bands of 3.2 kb and 6.9 kb in the original strain (PRE; Fig. 4d,e,f). In most of the FOARCanR strains (Fig. 3), the centromere-proximal 3.2 kb band is retained, indicating that any sequence loss does not extend to this region. Retention of the 6.9 kb band in FOARCanR strains indicates that CAN1 function has been lost in the absence of an overt DNA rearrangement. In contrast, bands of <6.9 kb are indicative of a GCR event, with fuzzy bands showing sites of telomere addition. In theory, the two bands in Fig. 4d that are between the 6.9 and 3.2 kb bands (in the fifth and seventh lanes from the left) could be either telomere additions of rearrangements. However, based on sequencing results and multiplex PCR, such bands are likely not telomere additions.
Chromatin immunoprecipitation (ChIP)
ChIP of asynchronous yeast cells growing in rich medium was performed as described6, 20 and analyzed using an iCycleriQ Real-Time PCR detection system (Bio-Rad Laboratories). Rrm3 was C-terminally tagged with 13 Myc epitopes39. An anti-Myc monoclonal antibody (Clontech #631206) was used as the anti-serum in ChIP. The amount of DNA in the immunoprecipitate was normalized to the amount in input samples. The ChIP experiment was analyzed by quantitative PCR (qPCR) in duplicate or triplicate to obtain an average value for each sample. The ChIP experiment was repeated ≥3 times at each locus. For each qPCR experiment, the amount of signal in the Rrm3 immunoprecipitate was normalized to input and to the immunoprecipitated signal from ARO1, a sequence that contains no candidate G4 DNA motif and that has low Rrm3 association20.
Multiplex PCR
Briefly, genomic DNA isolated from S. cerevisiae strains before and after GCR events was analyzed by multiplex PCR using the primer pairs in Supplementary Table 7 and the following cycling parameters: initial denaturation for 5 min at 95°C, 35 cycles of 95°C for 30 s, 56°C for 30 s, and 72°C for 45 s, and a final extension at 72°C for 10 min. PCR products (10 μL/reaction) were run at 90 V on 2.5% agarose gels containing ethidium bromide and visualized by UV transillumination (for primer sequences, see Supplementary Table 7).
Real-time quantitative PCR
The indicated strains were grown in FOA+Can liquid media for 12 h and then transferred to YEPD for 12 h until reaching a concentration of OD600 = 0.5, and total RNA was isolated using a Quick-RNA MiniPrep kit, including the DNase I treatment, as described by the manufacturer (Zymo Research). cDNA was synthesized from 200 ng DNase I-treated RNA using an iScript One-Step RT-PCR kit with SYBR Green (Bio-Rad) and analyzed by real-time quantitative PCR using a Bio-Rad CFX96 Real-time system. URA3 cDNA was synthesized using the primers 5′-GTTCGACTGATGAGCTATTGAAACT-3′ and 5′-CGACAGTACCCTCATAACTGAAATC-3′. CAN1 cDNA was synthesized using the primers 5′-AATATACATCGGGCGTTTAC-3′ and 5′-TCAGCAAGCATCAATAATCC-3′. For the actin control, ACT1 cDNA was synthesized using the primers 5′-GTAACATCGTTATGTCCGGTGGTAC-3′ and primer 5′-CCAAGATAGAACCACCAATCCAGAC-3′. The cycling parameters were: 50°C for 10 min, 95°C for 5 min, and 40 cycles of 95°C for 10 s followed by 57°C (ACT1), 50°C (CAN1), or 55°C (URA3) for 30 s. The data were analyzed by the 2−ΔΔCt method 45.
Supplementary Material
Acknowledgments
This article is dedicated to Dr. J.G. Gall in honor of his 85th birthday. We thank J.B. Boule for early work on ScPif1 biochemistry, P. Opresko for a generous gift of purified hWRN, E. Allen-Vercoe, K. Bidle, C. Parker, R. Johnson, H. L. Ayala-del-Rio, and E. Sockett for materials for cloning non-yeast Pif1 helicases, M. Platts for multiplex PCR and Southern analysis methods to characterize GCR clones, and financial support from National Institutes of Health (VZ, SK), National Science Foundation (KF), DFG and NJCCR (KP) and American Cancer Society (MB).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author contributions This paper is dedicated to J. G. Gall for his 85th birthday. K.P. and M.L.B. purified Pif1 helicases and performed biochemical and GCR experiments; P.D.G. and M.L.B. did the silencing experiments; P.C. purified Sgs1; S.C.K. aided in the analysis and interpretation of the biochemistry and provided purified EcRecQ; K.L.F. aided in the analysis and interpretation of GCR events; K.P., M.L.B., and V.A.Z. designed the study, analyzed data, and wrote the manuscript. K.P. and M.L.B. contributed equally. All authors discussed the results and commented on the manuscript.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Bochman ML, Paeschke K, Zakian VA. DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet. 2012 doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boule J, Vega L, Zakian V. The Yeast Pif1p helicase removes telomerase from DNA. Nature. 2005;438:57–61. doi: 10.1038/nature04091. [DOI] [PubMed] [Google Scholar]
- 3.Schulz VP, Zakian VA. The Saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell. 1994;76:145–155. doi: 10.1016/0092-8674(94)90179-1. [DOI] [PubMed] [Google Scholar]
- 4.Zhou JQ, Monson EM, Teng SC, Schulz VP, Zakian VA. The Pif1p helicase, a catalytic inhibitor of telomerase lengthening of yeast telomeres. Science. 2000;289:771–774. doi: 10.1126/science.289.5480.771. [DOI] [PubMed] [Google Scholar]
- 5.Myung K, Chen C, Kolodner RD. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature. 2001;411:1073–1076. doi: 10.1038/35082608. [DOI] [PubMed] [Google Scholar]
- 6.Paeschke K, Capra JA, Zakian VA. DNA Replication through G-Quadruplex Motifs Is Promoted by the Saccharomyces cerevisiae Pif1 DNA Helicase. Cell. 2011;145:678–91. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopes J, et al. G-quadruplex-induced instability during leading-strand replication. The EMBO journal. 2011;30:4033–46. doi: 10.1038/emboj.2011.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivessa AS, Zhou JQ, Zakian VA. The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell. 2000;100:479–489. doi: 10.1016/s0092-8674(00)80683-2. [DOI] [PubMed] [Google Scholar]
- 9.Ivessa AS, et al. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell. 2003;12:1525–1536. doi: 10.1016/s1097-2765(03)00456-8. [DOI] [PubMed] [Google Scholar]
- 10.Fachinetti D, et al. Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol Cell. 2010;39:595–605. doi: 10.1016/j.molcel.2010.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bochman ML, Sabouri N, Zakian VA. Unwinding the functions of the Pif1 family helicases. DNA Repair (Amst) 2010;9:237–249. doi: 10.1016/j.dnarep.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chisholm KM, et al. A Genomewide Screen for Suppressors of Alu-Mediated Rearrangements Reveals a Role for PIF1. PloS one. 2012;7:e30748. doi: 10.1371/journal.pone.0030748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lahaye A, Leterme S, Foury F. PIF1 DNA helicase from Saccharomyces cerevisiae. Biochemical characterization of the enzyme. J Biol Chem. 1993;268:26155–26161. [PubMed] [Google Scholar]
- 14.Boule JB, Zakian VA. The yeast Pif1p DNA helicase preferentially unwinds RNA DNA substrates. Nucleic Acids Res. 2007;35:5809–5818. doi: 10.1093/nar/gkm613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bochman ML, Judge CP, Zakian VA. The Pif1 family in prokaryotes: what are our helicases doing in your bacteria? Molecular biology of the cell. 2011;22:1955–9. doi: 10.1091/mbc.E11-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cejka P, Kowalczykowski SC. The full-length Saccharomyces cerevisiae Sgs1 protein is a vigorous DNA helicase that preferentially unwinds holliday junctions. The Journal of biological chemistry. 2010;285:8290–301. doi: 10.1074/jbc.M109.083196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohaghegh P, Karow JK, Brosh RM, Jr, Bohr VA, Hickson ID. The Bloom’s Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt KH, Pennaneach V, Putnam CD, Kolodner RD. Analysis of gross-chromosomal rearrangements in Saccharomyces cerevisiae. Methods Enzymol. 2006;409:462–476. doi: 10.1016/S0076-6879(05)09027-0. [DOI] [PubMed] [Google Scholar]
- 19.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 20.Azvolinsky A, Giresi P, Lieb J, Zakian V. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell. 2009;34:722–734. doi: 10.1016/j.molcel.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith S, et al. Mutator genes for suppression of gross chromosomal rearrangements identified by a genome-wide screening in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9039–44. doi: 10.1073/pnas.0403093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell. 1990;63:751–762. doi: 10.1016/0092-8674(90)90141-z. [DOI] [PubMed] [Google Scholar]
- 23.Baur JA, Zou Y, Shay JW, Wright WE. Telomere position effect in human cells. Science. 2001;292:2075–2077. doi: 10.1126/science.1062329. [DOI] [PubMed] [Google Scholar]
- 24.Mondoux M, Zakian V. In: Telomeres. 2. de Lange T, Lundblad V, Blackburn EH, editors. CSHL Press, Cold Spring Harbor; New York: 2005. pp. 261–316. [Google Scholar]
- 25.Piazza A, et al. Stimulation of gross chromosomal rearrangements by the human CEB1 and CEB25 minisatellites in Saccharomyces cerevisiae depends on G-quadruplexes or Cdc13. PLoS Genet. 2012;8:e1003033. doi: 10.1371/journal.pgen.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aparicio OM, Billington BL, Gottschling DE. Modifiers of position effect are shared between telomeric and silent mating-type loci in S. cerevisiae. Cell. 1991;66:1279–1287. doi: 10.1016/0092-8674(91)90049-5. [DOI] [PubMed] [Google Scholar]
- 27.Sarkies P, Reams C, Simpson LJ, Sale JE. Epigenetic instability due to defective replication of structured DNA. Molecular cell. 2010;40:703–13. doi: 10.1016/j.molcel.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivessa AS, Zhou JQ, Schulz VP, Monson EM, Zakian VA. Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and sub-telomeric DNA. Genes Dev. 2002;16:1383–1396. doi: 10.1101/gad.982902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinter SF, Aubert SD, Zakian VA. The Schizosaccharomyces pombe Pfh1p DNA helicase is essential for the maintenance of nuclear and mitochondrial DNA. Mol Cell Biol. 2008;28:6594–6608. doi: 10.1128/MCB.00191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harmon FG, Kowalczykowski SC. RecQ helicase, in concert with RecA and SSB proteins, initiates and disrupts DNA recombination. Genes Dev. 1998;12:1134–1144. doi: 10.1101/gad.12.8.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capra JA, Paeschke K, Singh M, Zakian VA. G-quadruplex DNA sequences are evolutionarily conserved and associated with distinct genomic features in Saccharomyces cerevisiae. PLoS Comput Biol. 2010;6:e1000861. doi: 10.1371/journal.pcbi.1000861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bachrati CZ, Hickson ID. Analysis of the DNA unwinding activity of RecQ family helicases. Methods Enzymol. 2006;409:86–100. doi: 10.1016/S0076-6879(05)09005-1. [DOI] [PubMed] [Google Scholar]
- 34.Wong I, Lohman TM. A double-filter method for nitrocellulose-filter binding: application to protein-nucleic acid interactions. Proc Natl Acad Sci U S A. 1993;90:5428–32. doi: 10.1073/pnas.90.12.5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brosh RM, Jr, Opresko PL, Bohr VA. Enzymatic mechanism of the WRN helicase/nuclease. Methods in enzymology. 2006;409:52–85. doi: 10.1016/S0076-6879(05)09004-X. [DOI] [PubMed] [Google Scholar]
- 36.Putnam CD, Kolodner RD. Determination of gross chromosomal rearrangement rates. Cold Spring Harbor protocols. 2010 doi: 10.1101/pdb.prot5492. pdb prot5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall BM, Ma CX, Liang P, Singh KK. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics. 2009;25:1564–5. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 39.Azvolinsky A, Dunaway S, Torres J, Bessler J, Zakian VA. The S. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev. 2006;20:3104–3116. doi: 10.1101/gad.1478906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002;30:e23. doi: 10.1093/nar/30.6.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Longtine MS, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 42.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein expression and purification. 2005;41:207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 43.Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16:857–60. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 44.Runge KW, Zakian VA. Introduction of extra telomeric DNA sequences into Saccharomyces cerevisiae results in telomere elongation. Mol Cell Biol. 1989;9:1488–1497. doi: 10.1128/mcb.9.4.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.