

Abstract
The medial prefrontal cortex plays a key role in cocaine addiction. However, how chronic cocaine exposure affects cortical networks remains unclear. Most studies have focused on layer 5 pyramidal neurons (the circuit output), while the response of local GABAergic interneurons to cocaine remains poorly understood. Here, we recorded from fast-spiking interneurons (FS-IN) after repeated cocaine exposure and found altered membrane excitability. After cocaine withdrawal, FS-IN showed an increase in the number of spikes evoked by positive current injection, increased input resistance, and decreased hyperpolarization-activated current. We also observed a reduction in miniature excitatory postsynaptic currents, whereas miniature inhibitory postsynaptic current activity was unaffected. We show that, in animals with cocaine history, dopamine receptor D2 activation is less effective in increasing FS-IN intrinsic excitability. Interestingly, these alterations are only observed 1 wk or more after the last cocaine exposure. This suggests that the dampening of D2-receptor-mediated response may be a compensatory mechanism to rein down the excitability of FS-IN.
Keywords: cocaine addiction, intrinsic excitability, interneurons, medial prefrontal cortex
addictive drugs like cocaine act on the mesocorticolimbic dopamine (DA) system. The latter, also termed “reward system,” consists mainly of the ventral tegmental area (VTA) and its targets, the nucleus accumbens and prefrontal cortex (PFC). The PFC is involved in high cognitive processes, such as attention, working memory, and executive function, and also in several neurological disorders, including drug addiction (Goldstein and Volkow 2011; Tzschentke 2001). The PFC is known to be involved in the induction of the primary behavioral drug effects (e.g., increased locomotion, conditioned place preference, self-administration), as well as in drug-craving and relapse (Tzschentke 2001). For instance, direct electrical stimulation of the PFC in animals implanted with a stimulating electrode lead to rewarding effects (Mora 1978; Mora and Myers 1977; Phillips and Fibiger 1978; Robertson et al. 1982; Rolls and Cooper 1973) and enhanced the reward value induced by cocaine (Corbett 1991; McGregor et al. 1992; Moody and Frank 1990). Some studies also report that lesion of the PFC prevents cocaine-induced behavioral sensitization and associated neuroadaptations (Li et al. 1999; Pierce et al. 1998; Tzschentke and Schmidt 1998a, 1998b). In addition, there is evidence showing that a disruption in medial PFC (mPFC) neurotransmission systems, including dopaminergic, but also glutamatergic, GABAergic, acetylcholinergic, noradrenergic, serotoninergic, and peptididergic systems, may affect the development of psychostimulant-induced behavioral sensitization (Steketee 2003).
More specifically, psychostimulant effects of cocaine are mainly attributed to inhibition of the DA transporter that results in an increase of dopaminergic transmission (Sulzer 2011). Chronic drug exposure to psychostimulants like cocaine leads to adaptations in synaptic transmission and cellular intrinsic excitability within the reward circuit that may underlie compulsive drug consumption and relapse even after long periods of abstinence (Hu 2007; Hyman et al. 2006; Luscher and Malenka 2011). In the PFC, these modifications have been mostly characterized in the pyramidal neurons from the deep layer that constitutes the output of the cortical circuit (Ben-Shahar et al. 2009; Ford et al. 2009; Huang et al. 2007; Lu et al. 2009, 2010; McFarland et al. 2003; Nasif et al. 2005a; Steketee 2003). However, mesocortical dopaminergic fibers innervate both pyramidal neurons (Goldman Rakic et al. 1989; Verney et al. 1990) and GABAergic interneurons (IN) (Benes et al. 1993; Sesack et al. 1995). GABAergic IN are very heterogeneous with highly variable intrinsic firing rates (Markram et al. 2004) and degree of recruitment in cerebral rhythms and synaptic kinetics (Gupta et al. 2000). Different subtypes are defined according to immunohistochemical, morphological, and electrophysiological criteria (Ascoli et al. 2008; Cauli et al. 1997; Kawaguchi and Kondo 2002; Klausberger and Somogyi 2008; Markram et al. 2004). Parvalbumin (PV)-positive fast-spiking INs (FS-IN), which constitute one of the major subtypes of IN in cortical layer 5 (Markram et al. 2004; Rudy et al. 2010), synapse close to the soma of pyramidal neurons and other local IN (Kawaguchi and Kubota 1997, 1998).
In drug-naive animals, DA modulates IN activity (Gorelova et al. 2002; Tseng and O'Donnell 2007) and GABAergic synaptic transmission onto pyramidal neurons in the PFC (Gao et al. 2003; Seamans et al. 2001; Trantham-Davidson et al. 2004). Repeated cocaine exposure alters this DA modulation, probably due to a significant decrease in D2 receptors (D2R) expression and/or less effective receptors (Bowers et al. 2004; Briand et al. 2008; Kroener and Lavin 2010). However, little is known about how psychostimulant withdrawal affects GABAergic IN. We report here that both intrinsic excitability and basal synaptic transmission (spontaneous, action-potential independent transmission) are affected in FS-IN after long-term cocaine withdrawal, whereas no change is observed after short-term withdrawal. The increase of intrinsic excitability is associated with a decrease in the hyperpolarization-activated current (Ih) and basal excitatory synaptic transmission.
MATERIALS AND METHODS
Animals and Treatment
All animal procedures were conducted in accordance with US National Institutes of Health guidelines, as approved by the National Institute of Child Health and Human Development Animal Care and Use committee. Male Sprague-Dalwey rats (3 wk old) received repeated administration of cocaine (15 mg·kg−1·day−1 ip) or isovolumetric saline (0.9%) for 5 consecutive days, followed by 3–6 days (short-term withdrawal) or 10–13 days (long-term withdrawal) of abstinence prior to the experiment. Before each injection, animals were habituated for 10–15 min and handled approximately daily until the day of experiment.
Slice Preparation and Electrophysiology
Saline- or cocaine-treated rats were decapitated under chloral hydrate anesthesia (400 mg/kg ip), and the brain was immediately excised and immersed in ice-cold solution containing the following (in mM): 90 sucrose, 80 NaCl, 1.3 KCl, 1 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 10 mM glucose, 3 mM pyruvic acid; pH 7.2–7.3; 310 mOsm/l. Coronal slices (300 μm) containing the mPFC were cut with a vibrotome (Leica VT1200S or DSK Microslicer DTK-1000). After recovery, incubation for ∼15 min at 33°C was followed by ∼45 min at 22°C in artificial cerebrospinal fluid (in mM: 125 NaCl, 2.5 KCl, 1 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 20 glucose, 3 pyruvic acid; pH 7.2–7.3; 310 mOsm/l). Slices were then transferred to the recording chamber and superfused (2–3 ml/min) with artificial cerebrospinal fluid at 32–33°C. All solutions were saturated with 95% O2/5% CO2.
Whole-cell patch recordings were obtained from neurons in layer 5 of the mPFC. FS-IN exhibit clustered and fast-spiking patterns in response to depolarization, high spontaneous activity, and low-input resistance (Fig. 1B). Unless indicated otherwise, the recording electrodes (4–8 MΩ resistance) were filled with an internal solution containing the following (in mM): 125 K-gluconate, 20 KCl, 10 HEPES, 4 NaCl, 0.5 EGTA, 4 Mg ATP, 0.3 GTP, 10 phosphocreatine (pH 7.2, 290 mOsm). To measure the input/output function, depolarizing current steps (1 s, 100–500 pA, 100-pA step) were injected. Additional steps in 10-pA increments were applied to find the rheobase, the minimal current able to induce firing.
Fig. 1.

Fast-spiking interneuron (FS-IN) identification. A: confocal image of a FS-IN filled with biocytin and parvalbumin immunostaining (green, biocytin; red, parvalbumin; yellow, overlapping signals). Scale bars: 20 μm. B: representative voltage traces recorded from the FS-IN showed in A. The voltage responses evoked by −200-pA (top left), 0-pA (top right), 400-pA (bottom left), and 500-pA (bottom right) current step (1,000 ms) are shown. Note spontaneous activity (asterisks) on the 0-pA trace. Scale bars: 200 ms/10 mV.
All voltage-clamp current recordings were performed in the presence of 2 mM kynurenate and 100 μM picrotoxin to block glutamatergic and GABAergic transmission, respectively. H-currents were induced with voltage steps (1 s) from −60 mV to −120 mV [holding potential (Vholding) = −50 mV] ± 20 μM ZD7288 to test the activation and with voltage steps from −70 mV to −50 mV (Vholding = −120 mV) to test the deactivation. H-currents were isolated by subtracting traces obtained in ZD7288 from control traces. For analysis of activation curves, the tail current amplitudes (Itail) were normalized to the maximal value (Itail max) evoked by the command step at −120 mV and plotted against the corresponding command step. The resulting data points were fit with a Boltzmann equation. Traces were fitted with a single-exponential function to determine the time constants. K+ inward rectifier (Kir) -mediated and leak K (Kleak) currents were induced with voltage steps (1 s) from −60 mV to −140 mV (Vholding = −50 mV) ± 1 mM Cs+. Kir currents were isolated by subtracting traces obtained in 1 mM Cs+ from control traces (= Kir + Kleak traces). K+ outward rectifier (Kor) currents, which are mostly composed in FS-IN of the fast delayed rectifier [supported by voltage-sensitive K+ (Kv) 3.1 and Kv3.2 subunits] and the slow inactivation outward rectifier (supported by Kv1 subunits) K+ currents (Erisir et al. 1999; Markram et al. 2004), were induced with voltage steps from −40 mV to 0 mV (Vholding = −100 mV). Ca2+ currents were induced by with voltage steps (100 ms) from −80 mV to +20 mV (Vholding = −90 mV) in presence of 1 μM TTX, 10 mM TEA, 10 mM CsCl, and 5 mM BaCl. Pipettes were filled with the following (in mM): 120 Cs-gluconate, 20 CsCl, 10 HEPES, 5 EGTA, 4 Mg ATP, 0.3 GTP, 10 phosphocreatine (pH 7.2, 290 mOsm). For all currents, Kir, Kleak, Kor, Ca2+, and H-currents, the amplitudes of steady-state currents were measured at each hyperpolarizing/depolarizing step and normalized with the whole cell capacitance.
Miniature excitatory postsynaptic currents (mEPSCs) were collected at −60 mV in the presence of 1 μM TTX, 100 μM picrotoxin, and 50 μM dl-2-amino-5-phosphonovaleric acid. For miniature inhibitory postsynaptic currents (mIPSCs), patch electrodes were filled with the following (in mM): 130 KCl, 8.5 NaCl, 5 HEPES, 4 NaCl, 0.5 EGTA, 4 Mg ATP, 0.3 GTP, 10 phosphocreatine (pH 7.2, 290 mOsm; GABA reversal potential = 0 mV). mIPSCs were collected at −70 mV, and 1 μM TTX, 20 μM 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide, and 50 μM dl-2-amino-5-phosphonovaleric acid were included in the perfusion bath. Only events with amplitudes > 5 pA were included in the analysis.
Drugs
All chemicals were purchased from Tocris (Ballwin, MO), Abcam (Cambridge, MA), or Sigma Chemical (St. Louis, MO).
Data Acquisition and Analysis
Electrophysiological recordings were obtained using a multiclamp 700B amplifier and PClamp 10 (Molecular, Devices, Sunnyvale, CA). Data were analyzed using Microsoft Excel, Minianalysis (Synaptosoft, Decatur, GA), and/or IGOR Pro (WaveMetrics, Lake Oswego, OR). Pooled data are presented as either mean ± SE or box plots, and statistical analyses were performed using Mann-Whitney U-test or the Wilcoxon test for paired data. P values are reported in the text or Figs. 2–7 with values < 0.05 considered as significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Fig. 2.
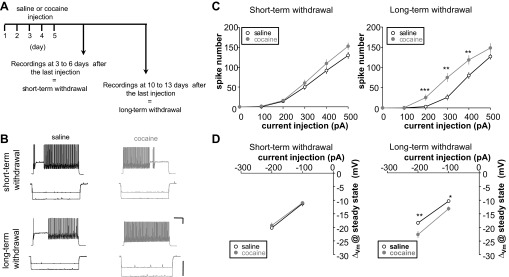
FS-IN intrinsic excitability after short-term and long-term cocaine withdrawal. A: experimental design. B: representative voltage traces recorded in FS-IN from rats repeatedly injected with saline (black) or cocaine (gray) after short-term (top) or long-term withdrawal (bottom). The voltage responses evoked by 300 pA and −200 pA are shown. C: graph showing the number of spikes evoked from saline or cocaine animals for a range of current injections after short-term (left; saline, n = 38, 17 rats; cocaine, n = 35, 16 rats) or long-term withdrawal (right; saline, n = 19, 8 rats; cocaine, n = 18, 7 rats). D: steady-state membrane potentials (Vm) are plotted against current step after short-term (left; saline, n = 38, 17 rats; cocaine, n = 35, 16 rats) or long-term withdrawal (right; saline, n = 19, 8 rats; cocaine, n = 18, 7 rats). Changes in excitability are significant only after long-term withdrawal (C, right, and D, right). Scale bars: 200 ms/20 mV. Δ, Change. Significant difference: *P < 0.05, **P < 0.01, ***P < 0.001.
Fig. 3.

Repeated cocaine injection followed by long-term withdrawal alters H-current. A–D, top: representative traces of membrane currents of FS-IN from repeated saline- (black) or cocaine-treated animals (gray). Current densities, calculated by dividing the amplitude of each steady-state current (dark circle) by membrane capacitance, are plotted against each test potential. A: K+ outward rectifier (Kor) currents were examined by holding cells −100 mV and making depolarizing voltage steps (−40 mV to 0 mV, 10-mV increments) (saline, n = 19, 4 rats; cocaine, n = 11, 3 rats). B: K+ inward rectifier (Kir) + leak K+ (Kleak) mixed currents were examined by holding cells at −50 mV and applying hyperpolarizing voltage steps (−60 to −140 mV, 10-mV increments) (saline, n = 15, 3 rats; cocaine, n = 15, 3 rats). C: Kir currents were isolated by subtracting traces obtained in 1 mM Cs+ from control traces (= Kir + Kleak traces) (saline, n = 15, 3 rats; cocaine, n = 15, 3 rats). D: Ca2+ currents were examined by holding cells at −90 mV and making depolarizing voltage steps (−80 mV to 20 mV, 10-mV increments) (saline, n = 13, 3 rats; cocaine, n = 15, 4 rats).
Fig. 4.

Repeated cocaine injection followed by long-term withdrawal alters H-current. A: maximum current density of each studied current. B: representative traces of membrane currents of FS-IN from repeated saline- (black) or cocaine-treated animals (gray). H-currents were isolated by subtracting traces obtained in 20 μM ZD 7288 from control traces [holding potential (Vholding) = −50 mV, step from −60 mV to −120 mV, 10-mV increments] (saline,n = 14, 3 rats; cocaine, n = 17, 4 rats). Current densities, calculated by dividing the amplitude of each steady-state current (dark circle) by membrane capacitance, are plotted against each test potential. Scale bars: 100 ms/200 pA. C: activation curves from repeated saline- (black) or cocaine-treated animals (gray). Itail, tail current; Itail max, maximum tail current. D: H-current kinetics from repeated saline- (black) or cocaine-treated animals (gray). The mean activation (open triangle) and deactivation (filled square) time constants are plotted against the voltage step command. Significant difference: *P < 0.05, **P < 0.01. ns, Not significant.
Fig. 5.

Effects of Ih blockage on FS-IN intrinsic excitability following long-term cocaine withdrawal. A and B: effect of 5 μM ZD7288 on neuronal current-voltage (I-V) relationship. Top traces, representative example. I-V curve is shown in which the steady-state Vm are plotted against each injected current step in control (filled circle) and after ZD7288 application (open circle) in saline (A) and cocaine-pretreated animals (B) (saline, black circle, n = 14, 3 rats; cocaine, gray circle, n = 9, 2 rats). C and D: effect of 5 μM ZD7288 on I-V relationship on input-output function. Top traces, representative example at +300 pA. Input-output curve is shown in control (filled circle) and after 5-μM ZD7288 application (open circle) in saline (C) and cocaine-pretreated animals (D) (saline, black circle, n = 9, 2 rats; cocaine, gray circle, n = 8, 2 rats). ZD7288 only affects saline animals. Scale bars: 200 ms/20 mV. Significant difference: *P < 0.05, ***P < 0.001.
Fig. 6.
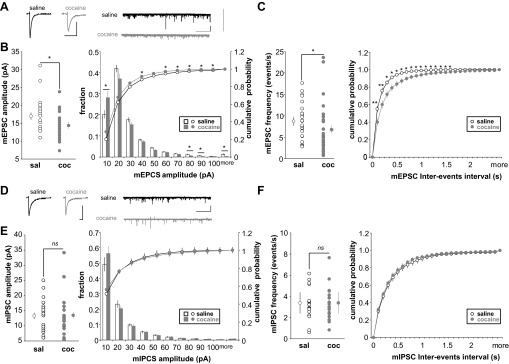
Amplitude and frequency of miniature excitatory postsynaptic currents (mEPSCs) and miniatures inhibitory postsynaptic currents (mIPSCs) in FS-IN following long-term cocaine withdrawal. A: average mEPSC (scale bars: 5 ms/10 pA) and sample traces of mEPSCs (scale bars: 10 s/10 pA) recorded in saline (black) or cocaine-injected (gray) rats. Graph is shown for pooled data values and graph plotting cumulative probability of mEPSC amplitude (B) and frequency (C) (saline, n = 25, 4 rats; cocaine, n = 26, 5 rats). D: average mIPSC (scale bars: 5 ms/10 pA) and sample traces of mIPSCs (scale bars: 10 s/10 pA) recorded in saline (black) or cocaine-injected (gray) rats. Graph is shown for pooled data values and graph plotting cumulative probability of mIPSC amplitude (E) and frequency (F) (saline, n = 20, 3 rats; cocaine, n = 19, 3 rats). Significant difference: *P < 0.05, **P < 0.01.
Fig. 7.

FS-IN spike-firing modulation by D2 receptors (DR2) after short- and long-term cocaine withdrawal. Representative traces show the effects of quinpirole (10 μM for 5 min) on FS-IN spike-firing in saline- and cocaine-treated animals following short- (A) or long-term withdrawal (C). Time course is shown of normalized spike number during quinpirole bath application after short- (B: saline, n = 13, 7 rats; cocaine, n = 12, 5 rats) or long-term withdrawal (D: saline, n = 9, 5 rats; cocaine, n = 8, 6 rats). Note that quinpirole is less effective in cocaine animals after long-term withdrawal. E: representative images for DR2 and parvalbumin immunostaining for saline- and cocaine-injected animals after long-term withdrawal (green, DR2; red, parvalbumin; yellow, overlapping signals). Insets, higher magnification of soma pointed by arrows. Scale bar: 50 μm, 10 μm inset. F: quantification of DR2 expression in parvalbumin+ cells (saline, n = 85, 3 rats; cocaine, n = 84, 3 rats). G: examples of whole-cell traces of Ih and current density graph from saline (black) or cocaine-pretreated (gray) animals after long-term withdrawal in control (filled circle) or after quinpirole (10 μM) application (open circle) (saline, n = 14, 4 rats; cocaine, n = 15, 4 rats). H: quantification of H-current reduction. Significant difference: *P < 0.05, **P < 0.01, ***P < 0.001.
Immunochemistry and Confocal Imaging
From acute slices.
For morphological identification of FS-IN, neurons were filled with biocytin (0.2–0.4%). Additionally, the expression of the Ca2+-binding protein PV, a molecular maker of FS-IN, was also examined (Fig. 1A). After recording, slices were fixed with 4% paraformaldehyde in phosphate-buffered solution (PBS; 0.1 M, pH 7.3) overnight at 4°C. Following wash with PBS, slices were permeabilized and blocked with 0.3% Triton X-100, 5% goat serum, and 1% bovine serum albumin in PBS (blocking solution) for 1 h at 22°C. The blocking solution was also used to dilute primary and secondary antibodies. After incubation overnight at 4°C with a primary monoclonal antibody against PV (P3088, Sigma, St. Louis, MO, 1:1000) and streptavidin-Alexa 488 conjugated (Invitrogen, Carlsbad, CA, 1:1000), slices were washed in PBS and incubated 2 h with secondary antibody (AlexaFluor 546, 1:500; Invitrogen) and streptavidin-Alexa 488 (1:1,000). Slices were coverslipped with Mowiol and imaged. Slices were imaged using a LMS 510 confocal microscope (Carl Zeiss Microimaging, Thornwood, NY) with a ×20 oil objective. All IN analyzed were PV+.
From fixed brain.
Rats were perfused transcardially with 0.9% saline solution followed by 4% paraformaldehyde in PBS (100 ml/100 mg). Brain tissue was then postfixed at 4°C for 24 h, and coronal slices (50 μm) were prepared using a cryostat (Leica CM3050S). Brain sections were treated as previously described. Sections were incubated with mouse monoclonal anti-PV (1:1,000) and rabbit anti-D2R (sc-9113, Santa Cruz Biotechnology, CA, 1:100). Immunoreactivity was imaged with Alexa 488 and Alexa 633 conjugated (1:500). Slices were imaged using a LMS 780 confocal microscope (Carl Zeiss Microimaging, Thornwood, NY) with a ×40 oil objective. PV+ FS-IN were selected and manually traced. D2R analysis was based on measuring the fluorescence along the cells' perimeter. First PV+ FS-IN perimeter was manually traced for maximum accuracy (on the PV immunostaining image), and then intensity of D2R was measured (on the D2R immunostaining image). Intensity was expressed in arbitrary units of fluorescence per micrometer. For the quantification, 8–10 PV+ FS-IN, from 3 different slices, were used (3 saline and 3 cocaine animals).
All image processing was performed using Image-J (National Institutes of Health, Bethesda, MD) and Photoshop 10 (Adobe Systems, Mountain View, CA).
RESULTS
To determine whether chronic exposure to cocaine leads to electrophysiological changes in FS-IN, juvenile male rats were injected during 5 consecutive days with 15 mg/kg of cocaine or saline, and FS-IN were recorded in layer 5 of the prelimbic and infralimbic PFC after short- (3–6 days) or long-term (10–13 days) withdrawal (Fig. 2A).
Repeated Administration of Cocaine Increase FS-IN Excitability
The passive and active membrane properties of mPFC FS-IN were studied after short- (3–6 days) or long-term withdrawal (10–13 days) (Fig. 2 and Table. 1). A series of depolarizing current steps was injected into the cell to evoke spikes and assess membrane excitability. We found that the number of spikes elicited upon current injection into FS-IN cells is similar between saline- and cocaine-injected animals after short-term withdrawal (Fig. 2, B and C). Neither did we observe a change difference in FS-IN input resistance (Fig. 2D). However, after long-term withdrawal, there was a significant leftward-shift in the input-output curve, indicating an increase in intrinsic excitability (Fig. 2C). Spike firing was increased in cocaine-injected animals, and the rheobase, defined as the minimal current to evoke a single spike, was significantly smaller (saline 258.60 ± 13.41 pA vs. cocaine 169.63 ± 13.46 pA, P < 0.001) (Table 1). First spike parameters such as onset time, threshold, rise time, afterhyperpolarization, and width were measured both at rheobase and 15-Hz firing. No significant changes in any of these parameters were observed after short- (data not shown) or long-term withdrawal (Table 1). Passive membrane properties measured by the input resistance at −100 pA were also affected by cocaine. Input resistance was significantly enhanced in cocaine animals after long-term withdrawal (Fig. 2D, Table 1). Resting membrane potential remained unaffected, although the cells were slightly more depolarized after long-term withdrawal (Table 1).
Table 1.
Passive and active membrane properties of FS-IN in layer 5 mPFC neurons from saline- and cocaine-treated rats after long-term withdrawal
| Pretreatment | Saline | Cocaine | P Value |
|---|---|---|---|
| n | 19 | 18 | |
| Passive membrane properties | |||
| RMP, mV | −78.03 ± 0.98 | −75.76 ± 0.91 | ns |
| Input resistance, MΩ | 103.58 ± 4.65 | 131.25 ± 8.29 | <0.05* |
| Active membrane properties | |||
| Rheobase, pA | 258.60 ± 13.41 | 169.63 ± 13.46 | <0.001* |
| First spike parameters @ rheobase | |||
| Onset, ms | 206.72 ± 51.79 | 258.78 ± 49.98 | ns |
| Threshold, mV | −40.73 ± 1.07 | −41.28 ± 3.36 | ns |
| Rise time 10–90%, ms | 0.29 ± 0.01 | 0.33 ± 0.04 | ns |
| Maximum, mV | 9.86 ± 1.38 | 13.01 ± 1.56 | ns |
| AHP amplitude, mV | −21.72 ± 1.03 | −19.79 ± 0.88 | ns |
| Width, ms | 0.56 ± 0.02 | 0.54 ± 0.01 | ns |
| First spike parameters @ 15-Hz firing | |||
| Onset, ms | 42.19 ± 16.87 | 70.02 ± 29.59 | ns |
| Threshold, mV | −44.64 ± 1.35 | −46.84 ± 1.44 | ns |
| Rise time 10–90%, ms | 0.34 ± 0.04 | 0.34 ± 0.03 | ns |
| Maximum, mV | 9.05 ± 1.75 | 11.16 ± 1.65 | ns |
| AHP amplitude, mV | −15.78 ± 1.24 | −16.32 ± 1.61 | ns |
| Width, ms | 0.57 ± 0.02 | 0.55 ± 0.0 | ns |
| Accommodation index | 13.08 ± 4.01 | 5.36 ± 1.34 | ns |
Values are means ± SE; n, no. of neurons (saline, n = 19, 8 rats; cocaine, n = 18, 7 rats). RMP, resting membrane potential; AHP, afterhyperpolarization. Parameters of the first spike were measured during step current injections corresponding at the rheobase or when neurons fired at approximately 15 Hz (i.e., 12–18 action potentials during the 1-s current step). Accommodation is defined by (tspike 2 − tspike 1)/(tspike n − tspike n−1), where t is time.
Significant difference. ns, Not significant.
Cocaine Alters Ih After Long-term Withdrawal
Change in neuronal intrinsic excitability results from modification in properties and/or number of voltage-gated ion channels (Spitzer 1999). Previous studies showed that, in layer 5 pyramidal neurons from cocaine-withdrawn animals, Na+ and Ca2+ spikes are increased (Dong et al. 2005; Ford et al. 2009; Nasif et al. 2005a) due to decreased voltage-gated K+ and Kir currents (Dong et al. 2005; Nasif et al. 2005b). In addition, it was previously reported that DA increases FS-IN excitability by reducing different K+ currents, such as Kleak, Kir, and Kor (Gorelova et al. 2002). We isolated and measured the major ionic currents expressed in FS-IN using voltage-clamp protocols and pharmacological manipulations. We found no difference in K+ current properties between groups (Fig. 3, A–C). Neither did we find changes in Ca2+ currents (Fig. 3D). The maximum current density recorded from all of these currents exhibited no difference between the saline and cocaine groups (Kor: saline 97.09 ± 18.53 pA/pF vs. cocaine 93.02 ± 22.86 pA/pF, P > 0.05; Kleak + Kir: saline −47.7 ± 3.61 pA/pF vs. cocaine −48.8 ± 2.77 pA/pF, P > 0.05; Kir: saline −21.84 ± 2.34 pA/pF vs. cocaine −23.23 ± 1.79 pA/pF, P > 0.05; Fig. 4A). We did, however, observe a significant decrease in the Ih after cocaine (saline −11.64 ± 0.86 pA/pF vs. cocaine −8.74 ± 1.13 pA/pF, P < 0.05; Fig. 4, A and B). No shift in the activation curve was noted (Fig. 4C and Table 2), and Ih kinetics were not affected in cocaine animals (Fig. 4D). These results suggest that Ih reduction is due to a decrease in the maximal conductance.
Table 2.
Comparaison of Ih properties in FS-IN in saline and cocaine pretreated animals after long-term withdrawal
| Pretreatment | Saline | Cocaine | P Value |
|---|---|---|---|
| V1/2, mV | −77.19 ± 1.03 | −78.80 ± 1.32 | ns |
| Slope factor | 14.04 ± 1.02 | 15.26 ± 1.35 | ns |
| τ Activation @ −120 mV, ms | 36.62 ± 6.41 | 47.23 ± 4.84 | ns |
| τ inactivation @ −5 mV, ms | 26.21 ± 4.05 | 29.02 ± 4.30 | ns |
| Ih density at −110 mV, pA/pF | −8.35 ± 0.58 | −6.08 ± 0.75 | <0.05* |
Values are means ± SE. V1/2, half-activation voltage; τ, time constant; Ih, hyperpolarization-activated current.
Significant difference.
To confirm 1) the role of Ih on FS-IN intrinsic excitability and 2) its implication in increased excitability after long-term cocaine withdrawal, we measured the effect of Ih blockade, using its specific blocker ZD7288, on FS-IN membrane properties and input-output function in saline and cocaine animals after long-term withdrawal. We found that current/voltage and input/output curves are significantly affected by Ih blockade in saline animals. Suppression of Ih enhanced the voltage response to a given current step (Fig. 5A) and increased the number of spikes evoked by supralinear depolarizing current step (Fig. 5C). In contrast, there is no significant effect of Ih blockage in cocaine animals (Fig. 5, B and D). Our results therefore indicate that Ih plays an important role in FS-IN intrinsic excitability and is altered in cocaine animals after long-term withdrawal.
Cocaine Effects on Basal Synaptic Transmission After Long-term Withdrawal
In the mPFC, cocaine is known to induce many changes in neurotransmission (Steketee 2003), and synaptic background activity has been shown to influence the neuronal input/output function (Destexhe and Pare 1999; Wolfart et al. 2005). Therefore, we examined potential effects of cocaine on basal synaptic transmission by recording mEPSCs from FS-IN from saline- and cocaine-injected rats (Fig. 6). The amplitude of mEPSCs was decreased in cocaine animals compared with saline-treated animals, indicating that α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptor properties and/or expression were affected by cocaine exposure (saline 16.99 ± 0.92 pA vs. cocaine 14.36 ± 0.70 pA, P < 0.05) (Fig. 6, A and B). Interestingly, we found more small mEPSCs (<10 pA, P < 0.05) and fewer large mEPSCs (>80 pA, P < 0.05) in cocaine-injected animals (Fig. 6B, right). The frequency of mEPSCs observed was also affected by cocaine exposure with the number of events per second being significantly decreased in cocaine animals (saline 8.74 ± 0.85 Hz vs. cocaine 6.81 ± 1.17 Hz, P < 0.05) (Fig. 6C). Since FS-IN receive also local inhibitory inputs, we also measured inhibitory activity. We found no change in either mIPSC amplitude or frequency (amplitude: saline 13.33 ± 1.16 pA vs. cocaine 13.49 ± 1.57, P > 0.05; frequency: saline 3.39 ± 0.35 vs. cocaine 3.39 ± 0.36, P > 0.05) (Fig. 6, D and F).
Altered D2R Intrinsic Excitability Modulation After Long-term Cocaine Withdrawal
Consistent with previous studies, bath application of the D2R agonist quinpirole increased spike firing evoked by a positive step of current in FS-IN after long-term withdrawal (Kroener and Lavin 2010; Tseng and O'Donnell 2007). During short-term withdrawal, no difference was observed in the enhancement of spike numbers after 5 min quinpirole between saline and cocaine groups (saline 179 ± 4% vs. cocaine 184 ± 4%, P > 0.05) (Fig. 7, A and B). However, after long-term withdrawal, cocaine-injected animals were found to be less sensitive to quinpirole application (saline 261 ± 6% vs. cocaine 197 ± 5%, P < 0.05) (Fig. 7, C and D). These data suggest that, as for our observed changes in intrinsic excitability (Fig. 2, Table 1), changes in D2R activity occurred only after a long withdrawal period.
It was previously shown that D2R expression and/or activity is decreased in mPFC after cocaine exposure (Briand et al. 2008). However, in this study, mPFC was analyzed in its entirety. Therefore, we examined D2R expression after long-term withdrawal specifically in layer 5 FS-IN with double immunochemistry labeling for D2R and PV, a specific cell maker for FS-IN. As expected D2R labeling was mostly found at the cell periphery (Vincent et al. 1995) (Fig. 7E). After long-term withdrawal, D2R expression in FS-IN+ and PV+ neurons from cocaine-injected animals was decreased compared with saline-injected animals (saline 131 ± 7 vs. cocaine 105 ± 5, P < 0.01) (Fig. 7F). This observation is consistent with our previous results showing that intrinsic excitability modulation induced by D2R agonist application is less effective in cocaine animals compared with saline animals (Fig. 7, C and D).
We next tested the effect of D2R activation on Ih, which we showed here to be crucial in FS-IN intrinsic excitability. We measured Ih in control conditions and after quinpirole application after long-term cocaine withdrawal. In both saline and cocaine-treated animals, quinpirole induced a significant reduction in H-current density (Fig. 7G). However, this reduction is significantly larger in saline compared with cocaine animals (Fig. 7H). Taken together, our data suggest that, after long-term cocaine withdrawal, the ability of D2R to control FS-IN activity and H-current are reduced due to a decrease in D2R expression.
DISCUSSION
In this study, we have examined the excitability of FS-IN in the mPFC after a period of short-term or long-term cocaine withdrawal. We found that modifications in FS-IN occurred only after long-term withdrawal. Indeed, no significant changes are observed during short-term withdrawal. We showed that 5 days of repeated cocaine exposure followed by 10–13 days of withdrawal facilitated spike firing, reduced Ih current, decreased mEPSC activity, and reduced D2R expression in FS-IN.
FS-IN Intrinsic Plasticity Induced By Cocaine Exposure
Withdrawal from repeated cocaine exposure is associated with altered membrane properties of FS-IN in the mPFC. It has been previously shown that chronic cocaine increased mPFC intrinsic excitability of L5 pyramidal neurons by modulating K+ and Ca2+ channel properties (Dong et al. 2005; Ford et al. 2009; Nasif et al. 2005a, 2005b). This increased intrinsic excitability has also been attributed to a decrease in GABAergic inhibition by reducing the surface expression of GABAA receptors in juvenile cocaine-treated animals or after in utero cocaine injection (Huang et al. 2007; Lu et al. 2009, 2010). Unlike pyramidal neurons that exhibit these cocaine-induced electrophysiological adaptations after only 3 days of withdrawal, we observed changes in FS-IN appeared after several days of cocaine withdrawal (requiring more than 6 days). This increase in FS-IN excitability is consistent with the reported increase in extracellular concentration of GABA and GABAergic transmission in the mPFC during cocaine withdrawal, as previously described (Jayaram and Steketee 2005).
Mechanisms of FS-IN Increased Intrinsic Excitability After Cocaine Exposure
We observed cocaine-induced changes in FS-IN firing, along with a decrease in Ih. Pharmacological Ih blockade significantly affected current/voltage and input/output curves in saline animals, while there was no significant effect of Ih blockage in cocaine animals (Fig. 5, B and D). Our results, therefore, indicate that Ih plays an important role in FS-IN intrinsic excitability and is altered in cocaine animals after long-term withdrawal. Although we did not observe significant changes in any other ionic currents measured (Figs. 3 and 4A), the discrepancy between the magnitude of firing frequency change after long-term cocaine withdrawal (Fig. 2C) and that found after Ih block (Fig. 3C) may indicate that other unresolved factors are involved.
In FS-IN, Ih controls the resting membrane potential and the input resistance, and therefore shapes the dendritic integration and the input-output function (Aponte et al. 2006). Ih also plays an important role in rhythmogenesis (Aponte et al. 2006). Recent studies showed that regulation of Ih might contribute to drug sensitization neuroadaptations. For instance, an increase in VTA DA neuron excitability is observed in association with a downregulation of Ih after withdrawal from repeated ethanol (but see also Bandyopadhyay and Hablitz 2007; Hopf et al. 2007; Okamoto et al. 2006) or cocaine exposure (Arencibia-Albite et al. 2012). Our observed reduction in Ih amplitude is caused by a reduced number of H-channels at the cell membrane, as we found no change in its activation curve or kinetic properties.
It has been previously shown that stimulation of D2R in VTA neurons produces a cAMP-independent decrease in the maximal conductance of HCN currents without changing the activation voltage dependence (Jiang et al. 1993). This mechanism is relatively unlikely here, since Ih downregulation was associated with decreased D2R expression. One possible explanation of our results would be via the activation of protein kinase C. It has been previously shown that inhibition of Ih by neuromodulators involves protein kinase C (Cathala and Paupardin-Tritsch 1997). Regulation by other kinases, like protein kinase A or calcium/calmodulin kinase II is also possible, since H-channels have a putative site for phosphorylation (Santoro et al. 1997; Zong et al. 2005) and D1/D2R pathways involve many different kinases (Beaulieu and Gainetdinov 2011). It has been previously reported that DA through D1R and/or D2R affects K+ currents (Dong et al. 2004, 2005; Gorelova et al. 2002). For instance, activation of D1R suppresses a Cs-sensitive Kir and a resting Kleak in FS-IN (Gorelova et al. 2002). However, we did not find any change in K+ currents after cocaine exposure in this study.
Chronic cocaine is well known to induce changes in glutamatergic neurotransmission within the mesocorticolimbic system (Bowers et al. 2010). Here, we reported a decrease in mEPSC amplitude in cocaine-treated animals that might be the consequence of a downregulation of AMPA receptors (AMPARs) at the FS-IN cell surface. A decrease in the number of AMPARs is consistent with the increased input resistance that we observed. It has also been shown that DA regulates AMPAR surface and synaptic expression in pyramidal neurons of the PFC (Sun et al. 2005). The reduction in mEPSC frequency could also reflect a presynaptic effect from the L2–3 or L5 pyramidal neurons, the two main glutamatergic input into FS-IN. Bidirectional changes such as increased excitability vs. reduced synaptic input might result from homeostatic compensation mechanisms.
Altered DA Modulation After Cocaine Exposure
DA controls cell excitability and synaptic transmission in the PFC (Seamans and Yang 2004), and, as a result, DA also regulates local network activity (Bandyopadhyay and Hablitz 2007). DA receptors thus represent an important subject of interest in drug addiction research (Di Chiara and Bassareo 2007; Le Foll et al. 2009; Nicola et al. 2000). For instance, D2R activation plays an important role in cocaine sensitization. Intracortical D2R agonist injection in the mPFC blocked the initiation and blunted the expression of cocaine-induced behavioral and neurochemical sensitization (Seamans and Yang 2004). As with L5 pyramidal neurons, FS-IN express both D1 and D2 classes of DA receptors (Gaspar et al. 1995; Vincent et al. 1995, 1993). D1-class DA receptors (D1R and D5R) activate the Gαs/olf family of G proteins to stimulate cAMP production by adenylate cyclase, whereas D2-class DA receptors (D2R, D3R, and D4R) couple to Gαi/o family of G proteins and thus induce inhibition of adenylate cyclase. In neurons, through its Gβγ subunits, D2-class DA receptors are known to directly regulate L- and N-type calcium channels, as well as some K+ channels (Beaulieu and Gainetdinov 2011). It is difficult to predict the effect of DA on neurons, as the final effect will depend on the dynamic interaction between DA receptors. For instance, a previous study showed that DA concentration is a critical determinant for cortical inhibition. Low DA concentration activated D1R, whereas a high concentration activated both D1 and D2R (Trantham-Davidson et al. 2004).
An interesting finding in our study is the altered DA modulation in cocaine-treated rats. Our data confirmed that, after long-term withdrawal, FS-IN D2R-mediated increase of current-evoked firing is reduced (Kroener and Lavin 2010). Like changes in spike firing and input resistance, changes in D2R modulation are only observed after long-term withdrawal, suggesting that D2Rs are responsible for these changes in excitability. Accordingly, while we found the D2R agonist quinpirole to reduce Ih in both saline- and cocaine-treated animals after long-term withdrawal, the effect was significantly greater in saline conditions (Fig. 5, G and H).
Physiological and Functional Relevance of Altered Inhibition
Drug addiction is defined as a chronic disorder that has been characterized by compulsion to seek and take the drug, loss of control in limiting intake, and emergence of a negative emotional state (e.g., anxiety, irritability, anhedonia), reflecting a withdrawal syndrome (Koob and Volkow 2010). More specifically, in cocaine users, executive cognitive functions like short-term memory, attention, or decision making, known to require the PFC, are impaired (Bolla et al. 2004; Briand et al. 2008; Garavan et al. 2008; Hoff et al. 1996; Jovanovski et al. 2005; Kantak et al. 2005; Miller 1985). Pharmacological studies showed that inactivation of dorsal mPFC blocks cue-induced (McLaughlin and See 2003), cocaine-primed (McFarland and Kalivas 2001), and stress-induced (Capriles et al. 2003) drug reinstatement in animal models of addiction. Structural and functional imaging studies also confirmed that prefrontal function is altered in addicts (Goldstein and Volkow 2011). For instance, during withdrawal, activity in the orbifrontal cortex (including the mPFC) is remarkably decreased (Volkow 2004; Volkow et al. 2004). The increase in FS-IN excitability that we described may contribute to this prefrontal hypoactivity. Indeed, inhibition plays a key role in the global network activity by controlling the level of excitation (Pouille and Scanziani 2001) and fine-tuning action potential precision in pyramidal neurons (Caillard 2011). Activity levels in orbifrontal cortex have also been correlated with the availability of D2R (Volkow et al. 2004). More generally, it has been suggested that PFC activity states depend on the balance between D1R and D2R activation (Durstewitz and Seamans 2008). After cocaine, D1R signaling is enhanced, whereas D2R is reduced, resulting in attenuated PCF output (Hu 2007). Behaviorally, this D1/D2 imbalance in the PFC may affect the motivational process of assignment of saliency value to a stimulus as a function of it context and as a result reduced motivation to respond to nondrug-related stimuli (Durstewitz and Seamans 2008). Therefore, change in FS-IN intrinsic excitability and altered DA modulation observed after chronic cocaine exposure may participate in behavioral sensitization and withdrawal effects.
GRANTS
This work was supported by the National Institute of Child Health and Human Development Intramural Research Program.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: E.C. and D.A.H. conception and design of research; E.C. performed experiments; E.C. analyzed data; E.C. interpreted results of experiments; E.C. prepared figures; E.C. drafted manuscript; E.C. and D.A.H. edited and revised manuscript; E.C. and D.A.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Veronica Alverez and David Lovinger for critical review of the manuscript. We thank the Dr. Craig Blackstone Laboratory for the use of Zeiss LMS 780 confocal microscope.
REFERENCES
- Aponte Y, Lien CC, Reisinger E, Jonas P. Hyperpolarization-activated cation channels in fast-spiking interneurons of rat hippocampus. J Physiol 574: 229–243, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arencibia-Albite F, Vazquez R, Velasquez-Martinez MC, Jimenez-Rivera CA. Cocaine sensitization inhibits the hyperpolarization-activated cation current Ih and reduces cell size in dopamine neurons of the Ventral Tegmental Area. J Neurophysiol 107: 2271–2282, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascoli GA, Alonso-Nanclares L, Anderson SA, Barrionuevo G, Benavides-Piccione R, Burkhalter A, Buzsaki G, Cauli B, Defelipe J, Fairen A, Feldmeyer D, Fishell G, Fregnac Y, Freund TF, Gardner D, Gardner EP, Goldberg JH, Helmstaedter M, Hestrin S, Karube F, Kisvarday ZF, Lambolez B, Lewis DA, Marin O, Markram H, Munoz A, Packer A, Petersen CC, Rockland KS, Rossier J, Rudy B, Somogyi P, Staiger JF, Tamas G, Thomson AM, Toledo-Rodriguez M, Wang Y, West DC, Yuste R. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci 9: 557–568, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Hablitz JJ. Dopaminergic modulation of local network activity in rat prefrontal cortex. J Neurophysiol 97: 4120–4128, 2007 [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63: 182–217, 2011 [DOI] [PubMed] [Google Scholar]
- Ben-Shahar O, Obara I, Ary AW, Ma N, Mangiardi MA, Medina RL, Szumlinski KK. Extended daily access to cocaine results in distinct alterations in Homer 1b/c and NMDA receptor subunit expression within the medial prefrontal cortex. Synapse 63: 598–609, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Vincent SL, Molloy R. Dopamine-immunoreactive axon varicosities form nonrandom contacts with GABA-immunoreactive neurons of rat medial prefrontal cortex. Synapse 15: 285–295, 1993 [DOI] [PubMed] [Google Scholar]
- Bolla K, Ernst M, Kiehl K, Mouratidis M, Eldreth D, Contoreggi C, Matochik J, Kurian V, Cadet J, Kimes A, Funderburk F, London E. Prefrontal cortical dysfunction in abstinent cocaine abusers. J Neuropsychiatry Clin Neurosci 16: 456–464, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MS, Chen BT, Bonci A. AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron 67: 11–24, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MS, McFarland K, Lake RW, Peterson YK, Lapish CC, Gregory ML, Lanier SM, Kalivas PW. Activator of G protein signaling 3: a gatekeeper of cocaine sensitization and drug seeking. Neuron 42: 269–281, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand LA, Flagel SB, Garcia-Fuster MJ, Watson SJ, Akil H, Sarter M, Robinson TE. Persistent alterations in cognitive function and prefrontal dopamine D2 receptors following extended, but not limited, access to self-administered cocaine. Neuropsychopharmacology 33: 2969–2980, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillard O. Pre and postsynaptic tuning of action potential timing by spontaneous gabaergic activity. PLos One 6: e22322, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriles N, Rodaros D, Sorge RE, Stewart J. A role for the prefrontal cortex in stress- and cocaine-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 168: 66–74, 2003 [DOI] [PubMed] [Google Scholar]
- Cathala L, Paupardin-Tritsch D. Neurotensin inhibition of the hyperpolarization-activated cation current (Ih) in the rat substantia nigra pars compacta implicates the protein kinase C pathway. J Physiol 503: 87–97, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Audinat E, Lambolez B, Angulo MC, Ropert N, Tsuzuki K, Hestrin S, Rossier J. Molecular and physiological diversity of cortical nonpyramidal cells. J Neurosci 17: 3894–3906, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett D. Cocaine enhances the reward value of medial prefrontal cortex self-stimulation. Neuroreport 2: 805–808, 1991 [DOI] [PubMed] [Google Scholar]
- Destexhe A, Pare D. Impact of network activity on the integrative properties of neocortical pyramidal neurons in vivo. J Neurophysiol 81: 1531–1547, 1999 [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Bassareo V. Reward system and addiction: what dopamine does and doesn't do. Curr Opin Pharmacol 7: 69–76, 2007 [DOI] [PubMed] [Google Scholar]
- Dong Y, Cooper D, Nasif F, Hu XT, White FJ. Dopamine modulates inwardly rectifying potassium currents in medial prefrontal cortex pyramidal neurons. J Neurosci 24: 3077–3085, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT, Malenka RC, White FJ. Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci 25: 936–940, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durstewitz D, Seamans JK. The dual-state theory of prefrontal cortex dopamine function with relevance to catechol-o-methyltransferase genotypes and schizophrenia. Biol Psychiatry 64: 739–749, 2008 [DOI] [PubMed] [Google Scholar]
- Erisir A, Lau D, Rudy B, Leonard CS. Function of specific K(+) channels in sustained high-frequency firing of fast-spiking neocortical interneurons. J Neurophysiol 82: 2476–2489, 1999 [DOI] [PubMed] [Google Scholar]
- Ford KA, Wolf ME, Hu XT. Plasticity of L-type Ca2+ channels after cocaine withdrawal. Synapse 63: 690–697, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WJ, Wang Y, Goldman-Rakic PS. Dopamine modulation of perisomatic and peridendritic inhibition in prefrontal cortex. J Neurosci 23: 1622–1630, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavan H, Kaufman JN, Hester R. Acute effects of cocaine on the neurobiology of cognitive control. Philos Trans R Soc Lond B Biol Sci 363: 3267–3276, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P, Bloch B, Le Moine C. D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur J Neurosci 7: 1050–1063, 1995 [DOI] [PubMed] [Google Scholar]
- Goldman Rakic PS, Leranth C, Williams SM, Mons N, Geffard M. Dopamine synaptic complex with pyramidal neurons in primate cerebral-cortex. Proc Natl Acad Sci U S A 86: 9015–9019, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci 12: 652–669, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelova N, Seamans JK, Yang CR. Mechanisms of dopamine activation of fast-spiking interneurons that exert inhibition in rat prefrontal cortex. J Neurophysiol 88: 3150–3166, 2002 [DOI] [PubMed] [Google Scholar]
- Gupta A, Wang Y, Markram H. Organizing principles for a diversity of GABAergic interneurons and synapses in the neocortex. Science 287: 273–278, 2000 [DOI] [PubMed] [Google Scholar]
- Hoff AL, Riordan H, Morris L, Cestaro V, Wieneke M, Alpert R, Wang GJ, Volkow N. Effects of crack cocaine on neurocognitive function. Psychiatry Res 60: 167–176, 1996 [DOI] [PubMed] [Google Scholar]
- Hopf FW, Martin M, Chen BT, Bowers MS, Mohamedi MM, Bonci A. Withdrawal from intermittent ethanol exposure increases probability of burst firing in VTA neurons in vitro. J Neurophysiol 98: 2297–2310, 2007 [DOI] [PubMed] [Google Scholar]
- Hu XT. Cocaine withdrawal and neuro-adaptations in ion channel function. Mol Neurobiol 35: 95–112, 2007 [DOI] [PubMed] [Google Scholar]
- Huang CC, Lin HJ, Hsu KS. Repeated cocaine administration promotes long-term potentiation induction in rat medial prefrontal cortex. Cereb Cortex 17: 1877–1888, 2007 [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598, 2006 [DOI] [PubMed] [Google Scholar]
- Jayaram P, Steketee JD. Effects of cocaine-induced behavioural sensitization on GABA transmission within rat medial prefrontal cortex. Eur J Neurosci 21: 2035–2039, 2005 [DOI] [PubMed] [Google Scholar]
- Jiang ZG, Pessia M, North RA. Dopamine and baclofen inhibit the hyperpolarization-activated cation current in rat ventral tegmental neurones. J Physiol 462: 753–764, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovski D, Erb S, Zakzanis KK. Neurocognitive deficits in cocaine users: a quantitative review of the evidence. J Clin Exp Neuropsychol 27: 189–204, 2005 [DOI] [PubMed] [Google Scholar]
- Kantak KM, Udo T, Ugalde F, Luzzo C, Di Pietro N, Eichenbaum HB. Influence of cocaine self-administration on learning related to prefrontal cortex or hippocampus functioning in rats. Psychopharmacology (Berl) 181: 227–236, 2005 [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kondo S. Parvalbumin, somatostatin and cholecystokinin as chemical markers for specific GABAergic interneuron types in the rat frontal cortex. J Neurocytol 31: 277–287, 2002 [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex 7: 476–486, 1997 [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. Neurochemical features and synaptic connections of large physiologically-identified GABAergic cells in the rat frontal cortex. Neuroscience 85: 677–701, 1998 [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321: 53–57, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology 35: 217–238, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroener S, Lavin A. Altered dopamine modulation of inhibition in the prefrontal cortex of cocaine-sensitized rats. Neuropsychopharmacology 35: 2292–2304, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Foll B, Gallo A, Le Strat Y, Lu L, Gorwood P. Genetics of dopamine receptors and drug addiction: a comprehensive review. Behav Pharmacol 20: 1–17, 2009 [DOI] [PubMed] [Google Scholar]
- Li Y, Hu XT, Berney TG, Vartanian AJ, Stine CD, Wolf ME, White FJ. Both glutamate receptor antagonists and prefrontal cortex lesions prevent induction of cocaine sensitization and associated neuroadaptations. Synapse 34: 169–180, 1999 [DOI] [PubMed] [Google Scholar]
- Lu H, Cheng PL, Lim BK, Khoshnevisrad N, Poo MM. Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron 67: 821–833, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Lim B, Poo MM. Cocaine exposure in utero alters synaptic plasticity in the medial prefrontal cortex of postnatal rats. J Neurosci 29: 12664–12674, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69: 650–663, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu CZ. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci 5: 793–807, 2004 [DOI] [PubMed] [Google Scholar]
- McFarland K, Kalivas PW. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 21: 8655–8663, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23: 3531–3537, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor IS, Atrens DM, Jackson DM. Cocaine facilitation of prefrontal cortex self-stimulation: a microstructural and pharmacological analysis. Psychopharmacology (Berl) 106: 239–247, 1992 [DOI] [PubMed] [Google Scholar]
- McLaughlin J, See RE. Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology (Berl) 168: 57–65, 2003 [DOI] [PubMed] [Google Scholar]
- Miller L. Neuropsychological assessment of substance abusers: review and recommendations. J Subst Abuse Treat 2: 5–17, 1985 [DOI] [PubMed] [Google Scholar]
- Moody CA, Frank RA. Cocaine facilitates prefrontal cortex self-stimulation. Pharmacol Biochem Behav 35: 743–746, 1990 [DOI] [PubMed] [Google Scholar]
- Mora F. The neurochemical substrates of prefrontal cortex self-stimulation: a review and an interpretation of some recent data. Life Sci 22: 919–929, 1978 [DOI] [PubMed] [Google Scholar]
- Mora F, Myers RD. Brain self-stimulation: direct evidence for the involvement of dopamine in the prefrontal cortex. Science 197: 1387–1389, 1977 [DOI] [PubMed] [Google Scholar]
- Nasif FJ, Hu XT, White FJ. Repeated cocaine administration increases voltage-sensitive calcium currents in response to membrane depolarization in medial prefrontal cortex pyramidal neurons. J Neurosci 25: 3674–3679, 2005a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasif FJ, Sidiropoulou K, Hu XT, White FJ. Repeated cocaine administration increases membrane excitability of pyramidal neurons in the rat medial prefrontal cortex. J Pharmacol Exp Ther 312: 1305–1313, 2005b [DOI] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci 23: 185–215, 2000 [DOI] [PubMed] [Google Scholar]
- Okamoto T, Harnett MT, Morikawa H. Hyperpolarization-activated cation current (Ih) is an ethanol target in midbrain dopamine neurons of mice. J Neurophysiol 95: 619–626, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AG, Fibiger HC. The role of dopamine in maintaining intracranial self-stimulation in the ventral tegmentum, nucleus accumbens, and medial prefrontal cortex. Can J Psychol 32: 58–66, 1978 [DOI] [PubMed] [Google Scholar]
- Pierce RC, Reeder DC, Hicks J, Morgan ZR, Kalivas PW. Ibotenic acid lesions of the dorsal prefrontal cortex disrupt the expression of behavioral sensitization to cocaine. Neuroscience 82: 1103–1114, 1998 [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293: 1159–1163, 2001 [DOI] [PubMed] [Google Scholar]
- Robertson A, Laferriere A, Milner PM. Development of brain stimulation reward in the medial prefrontal cortex: facilitation by prior electrical stimulation of the sulcal prefrontal cortex. Physiol Behav 28: 869–872, 1982 [DOI] [PubMed] [Google Scholar]
- Rolls ET, Cooper SJ. Activation of neurones in the prefrontal cortex by brain-stimulation reward in the rat. Brain Res 60: 351–368, 1973 [DOI] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 71: 45–61, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Grant SG, Bartsch D, Kandel ER. Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to Eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci U S A 94: 14815–14820, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Gorelova N, Durstewitz D, Yang CR. Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J Neurosci 21: 3628–3638, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol 74: 1–58, 2004 [DOI] [PubMed] [Google Scholar]
- Sesack SR, Snyder CL, Lewis DA. Axon terminals immunolabeled for dopamine or tyrosine-hydroxylase synapse on gaba-immunoreactive dendrites in rat and monkey cortex. J Comp Neurol 363: 264–280, 1995 [DOI] [PubMed] [Google Scholar]
- Spitzer NC. New dimensions of neuronal plasticity. Nat Neurosci 2: 489–491, 1999 [DOI] [PubMed] [Google Scholar]
- Steketee JD. Neurotransmitter systems of the medial prefrontal cortex: potential role in sensitization to psychostimulants. Brain Res Brain Res Rev 41: 203–228, 2003 [DOI] [PubMed] [Google Scholar]
- Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron 69: 628–649, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci 25: 7342–7351, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trantham-Davidson H, Neely LC, Lavin A, Seamans JK. Mechanisms underlying differential D1 versus D2 dopamine receptor regulation of inhibition in prefrontal cortex. J Neurosci 24: 10652–10659, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb Cortex 17: 1235–1240, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschentke TM. Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol 63: 241–320, 2001 [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Schmidt WJ. The development of cocaine-induced behavioral sensitization is affected by discrete quinolinic acid lesions of the prelimbic medial prefrontal cortex. Brain Res 795: 71–76, 1998a [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Schmidt WJ. Discrete quinolinic acid lesions of the rat prelimbic medial prefrontal cortex affect cocaine- and MK-801-, but not morphine- and amphetamine-induced reward and psychomotor activation as measured with the place preference conditioning paradigm. Behav Brain Res 97: 115–127, 1998b [DOI] [PubMed] [Google Scholar]
- Verney C, Alvarez C, Geffard M, Berger B. Ultrastructural double-labeling study of dopamine terminals and GABA-containing neurons in rat anteromedial cerebral-cortex. Eur J Neurosci 2: 960–972, 1990 [DOI] [PubMed] [Google Scholar]
- Vincent SL, Khan Y, Benes FM. Cellular colocalization of dopamine D1 and D2 receptors in rat medial prefrontal cortex. Synapse 19: 112–120, 1995 [DOI] [PubMed] [Google Scholar]
- Vincent SL, Khan Y, Benes FM. Cellular distribution of dopamine D1 and D2 receptors in rat medial prefrontal cortex. J Neurosci 13: 2551–2564, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND. Imaging the addicted brain: from molecules to behavior. J Nucl Med 45: 13N–24N, 2004 [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ. The addicted human brain viewed in the light of imaging studies: brain circuits and treatment strategies. Neuropharmacology 47, Suppl 1: 3–13, 2004 [DOI] [PubMed] [Google Scholar]
- Wolfart J, Debay D, Le Masson G, Destexhe A, Bal T. Synaptic background activity controls spike transfer from thalamus to cortex. Nat Neurosci 8: 1760–1767, 2005 [DOI] [PubMed] [Google Scholar]
- Zong X, Eckert C, Yuan H, Wahl-Schott C, Abicht H, Fang L, Li R, Mistrik P, Gerstner A, Much B, Baumann L, Michalakis S, Zeng R, Chen Z, Biel M. A novel mechanism of modulation of hyperpolarization-activated cyclic nucleotide-gated channels by Src kinase. J Biol Chem 280: 34224–34232, 2005 [DOI] [PubMed] [Google Scholar]