

Abstract
Background:
We demonstrated how to comprehensively translate the existing and updated scientific evidence on genomic discovery, tumour phenotype, clinical features, and conventional risk factors in association with breast cancer to facilitate individually tailored screening for breast cancer.
Methods:
We proposed an individual-risk-score-based approach that translates state-of-the-art scientific evidence into the initiators and promoters affecting onset and subsequent progression of breast tumour underpinning a novel multi-variable three-state temporal natural history model. We applied such a quantitative approach to a population-based Taiwanese women periodical screening cohort.
Results:
Risk prediction for pre-clinical detectable and clinical-detected breast cancer was made by the two risk scores to stratify the underlying population to assess the optimal age to begin screening and the inter-screening interval for each category and to ascertain which high-risk group requires an alternative image technique. The risk-score-based approach significantly reduced the interval cancer rate as a percentage of the expected rate in the absence of screening by 30% and also reduced 8.2% false positive cases compared with triennial universal screening.
Conclusion:
We developed a novel quantitative approach following the principle of translational research to provide a roadmap with state-of-the-art genomic discovery and clinical parameters to facilitate individually tailored breast cancer screening.
Keywords: translational research, individually tailored breast cancer screening, multi-state multi-variable model, breast cancer
Translational research to bridge the chasm between basic science, clinical practice, and health decision making has increasingly gained attention (Zerhouni, 2005; Woolf, 2008; Dzau et al, 2010). Personalised medicine on prevention and treatment of breast cancer, the top disease to combat worldwide, is one of such kind of tasks that is urgently needed.
Population-based screening for breast cancer using mammography has been demonstrated by using randomised controlled trials between 1970 and 1990 and has been widely applied in developed and developing countries. However, health-policy makers are concerned that the harm (false negative and false positive cases) and cost of screening should be minimised and the benefits, mainly measured by the reduction of mortality from breast cancer, maximised. This may be relieved by using an individually tailored screening (Esserman et al, 2009) with emphasis on optimal age of screening and inter-screening interval and the expedient use of alternative image technique. These subsidiary issues are related to individual variation on the temporal natural history of breast cancer from free-of-breast-cancer, through the pre-clinical detectable phase (PCDP) and finally to clinical phase (CP), three of which have been well defined by using various detection methods (Shapiro et al, 1974; Prorok, 1976; Walter and Day, 1983; van Oortmarssen et al, 1990). Screen-detected breast cancer represents the PCDP, whereas clinically detected one (such as interval cancer) stands for the CP (Duffy et al, 1995; Chen et al, 1997a, 1997b).
With the advent of genetic and biological markers for breast cancer, individually tailored screening for breast cancer can now be achieved by making use of information on genes, conventional risk factors, clinical attributes, and relevant tumour phenotypes such as HER-2/neu. However, it is very rare to focus on how to link these scientific discoveries with temporal natural history to assess which factors (initiators) affect the onset of breast cancer, that is, the transition from free-of-breast-cancer to the PCDP and which ones (promoters) responsible for the subsequent progression from the PCDP to the CP by comparing the distribution of each factor between screen-detected cases and clinically detected ones with the premise that the distribution of initiators should be identical between both the detection modes.
To this end, we conducted a literature review on state-of-the-art genomic discovery, tumour phenotypes, clinical correlates, and conventional risk factors. These factors were further superimposed into the three-state temporal natural history of breast cancer in the light of the roles of initiators and promoters mentioned above to build a multi-variable and three-state Markov regression model. We then estimated the effects of initiators and promoters to construct composite risk scores by the application of the proposed Markov regression model to the data set generated above. We applied this risk-score-based approach to emulating a scenario of Taiwanese population-based periodic screening programme for breast cancer to demonstrate how such a novel quantitative approach can be used to develop an individually tailored screening strategy that is efficient in terms of inter-screening interval, age at the start of screening and the expedient use of costly but accurate alternative image techniques.
Materials and methods
Study framework on initiators and promoters
As shown in Figure 1, our study design was to quantify the effects of initiators on the occurrence of breast cancer in the PCDP and those of promoters on the transition from the PCDP to CP. The definitions of these three-state outcomes (free of breast cancer (state 1), breast cancer in the PCDP (state 2, sometimes called pre-symptomatic cases), and breast cancer in the CP (state 3, sometimes called symptomatic cases)) are realised by the following three corresponding outcomes, free of breast cancer, screen-detected cases and interval cancer obtained from population-based screening data as defined in the previous studies (Shapiro et al, 1974; Prorok, 1976; Walter and Day, 1983; van Oortmarssen et al, 1990; Duffy et al, 1995; Chen et al, 1997a, 1997b). The initiators in Figure 1 included BRCA1, BRCA2 (Peto et al, 1999; Chen and Parmigiani, 2007), single-nucleotide polymorphisms (Cox et al, 2007; Easton et al, 2007; Hunter et al, 2007; Stacey et al, 2007), breast density (del Carmen et al, 2007; Martin and Boyd, 2008; Cummings et al, 2009), and conventional risk factors (body mass index (BMI) and age at first full-term pregnancy (AP)) (Hsieh et al, 2002; Chen et al, 2004). The promoters were identified by comparing the distributions of relevant tumour phenotypes between screen-detected cases and interval cancer without making any assumption because the initiators affecting the occurrence of breast cancer would not lead to a disparity between screen-detected cases and interval cancer (Dong et al, 2008). According to the literatures of previous studies (Hsieh et al, 2002; Dong et al, 2008), the promoters responsible for the progression from the PCDP to the CP in the current model included BMI, AP, oestrogen receptor (ER), HER-2 immunochemical score, and Ki-67 expression.
Figure 1.

Study design ascertaining outcomes from three rounds of screens based on the temporal natural history of breast cancer. BC, breast cancer; Ki-67, Ki-67 proliferation; HER-2/neu, HER-2/neu immunohistochemistry score.
Data sources
We emulated an organised service periodical screening of 1 million Taiwanese women aged 50 years or older assuming the distributions of covariates by various detection methods are identical to the first box in Figure 1. To fit in with this scenario, we tuned baseline pre-clinical incidence rate following age-specific incidence rate, ranging from 0.0017 to 0.021 per women-year during 2001–2005 in Taiwan (Taiwan Cancer Registry Annual Report, 2009). Since it is impossible to have a comprehensive empirical data to directly estimate the parameters underpinning the model, we turned, by using simulation technique, these empirical tabular data obtained from literatures into individual data as if they were collected according to the concept of sufficient statistics (Casella and Berger, 2001). For example, the sufficient statistics of generating data on breast density are the total numbers of each subgroup, which is equivalent to the collection of individual data when a multi-nominal distribution following the percentage of each category. The data on other covariates were spawned in a similar manner. After generating data set with covariates, we further applied a three-state Markov regression model to add information on various detection methods (including screen-detected cases and interval cancer) to capture time to enter the PCDP and time to the CP by using relative risk as specified in Tables 1 and 2. Data on three rounds of screen were generated as illustrated in Figure 1. This completes the creation of data set for parameter estimation (the second box of Figure 2).
Table 1. Percentage and relative risks for initiators affecting the transition rate from free-of-breast cancer to the pre-clinical screen-detectable phase (PCDP).
| Variables | Percentage (%) | Relative risk of being breast cancer in the PCDP (pre-symptomatic cases) | References |
|---|---|---|---|
|
Gene mutation | |||
| BRCA1 gene mutation | 0.11 | 20.26 | Peto et al, 1999; Chen and Parmigiani, 2007 |
|
BRCA2 gene mutation |
0.12 |
15.44 |
|
|
Breast density (BI-RADS) | |||
| Almost entirely fat | 0.92 | 1.00 | del Carmen et al, 2007; Cumming et al, 2009 |
| Scattered fibroglandular densities | 15.86 | 2.03 | |
| Heterogeneously dense | 56.09 | 2.95 | |
| Extremely dense |
27.13 |
4.03 |
|
|
Polygenes (dbSNP no.) per allele | |||
| rs2981582 | 38 | 1.26 | Easton et al, 2007; Hunter et al, 2007 |
| rs3803662 | 25 | 1.20 | Easton et al, 2007 |
| rs889312 | 28 | 1.13 | Easton et al, 2007 |
| rs3817198 | 30 | 1.07 | Easton et al, 2007 |
| rs13281615 | 40 | 1.08 | Stacey et al, 2007 |
| rs13387042 | 50 | 1.20 | Cox et al, 2007 |
| rs1045485 |
86 |
1.13 |
Easton et al, 2007 |
|
BMI (kg m−2) | |||
| ⩽23 | 37.11 | 1.00 | Chen et al, 2004 |
| >23 |
62.89 |
2.59 |
Hsieh et al, 2002 |
|
Age at first full-term pregnancy (years) | |||
| ⩽25 | 66.53 | 1.00 | Chen et al, 2004 |
| >25 | 33.47 | 1.99 | Hsieh et al, 2002 |
Table 2. Percentage and relative risks for promoters affecting the transition rate from the pre-clinical screen-detectable phase (PCDP) to the clinical phase (CP).
| Variables | Percentage (%) of breast cancer in the PCDP (pre-symptomatic cases) | Relative risk of the progression from the PCDP to the CP (symptomatic cases) | References |
|---|---|---|---|
|
BMI (kg m−2) | |||
| ⩽23 | 26.20a | 1.00 | Hsieh et al, 2002 |
| >23 |
73.80a |
2.00 |
Chen et al, 2004 |
|
Age at full-term first pregnancy (years) | |||
| ⩽25 | 57.62a | 1.00 | Hsieh et al, 2002 |
| >25 |
42.38a |
1.56 |
Chen et al, 2004 |
|
ER status | |||
| Positive | 81.00 | 1.00 | Dong et al, 2008 |
| Negative |
19.00 |
1.35 |
|
|
Ki-67 proliferation | |||
| <10% | 30.1 | 1.00 | Dong et al, 2008 |
| 10–30% | 50.7 | 1.40 | |
| >30% |
19.2 |
2.11 |
|
|
HER-2/neu immunohistochemistry score | |||
| 0 or 1+ | 75.6 | 1.00 | Dong et al, 2008 |
| 2+ | 11.9 | 1.28 | |
| 3+ | 12.5 | 1.07 | |
These values are based on the simulated PCDP (pre-symptomatic cases) simulated from free of breast cancer to the PCDP following the distribution of women free of breast cancer in Table 1.
Figure 2.

Data sources, parameter estimation, and validation of three-state Markov regression model.
Statistical analysis
Estimation of parameters
The data created above were used to build up a series of likelihood functions based on our proposed Markov regression model (the third box of Figure 2). The details on the formula of likelihood function (the fourth box of Figure 2) are similar to those used in the previous studies (Hsieh et al, 2002).
Composite risk scores
The maximum likelihood estimation method was used to calculate risk scores (Chen et al, 2000; Hsieh et al, 2002) (the fourth box of Figure 2). The risk scores are the natural logarithms of the estimated transition rates for initiators and promoters. The risk stratification by deciles of risk score was constructed to demonstrate the following individually tailored screening strategies. The median value of 10-year breast cancer risk for the average-risk population at age 50 was used as a criterion to recommend the suitable starting screening age for other deciles, similar to Pharoah et al (2008).
Model validation
Figure 2 also shows model validation including cross-validation and external validation. The former was to divide full data set into the two parts, 2/3 trained data for estimating parameters and 1/3 validated data that were calculated by the application of parameters estimated from 2/3 trained data to the distributions of covariates of the other 1/3 validated data. We then compared the observed with the predicted number of breast cancer by different detection modes (first screen, subsequent screen, and interval cancer).
As far as external validation is concerned, we applied the parameters estimated from full data set we simulated from the empirical tabular data published in literatures to Taiwanese multi-centre screening (TAMCAS). The study design and results have been elaborated in previous studies (Lai et al, 1998; Chen et al, 2000; Wu et al, 2008). 4867 women with family history of breast cancer were invited and attended the first screen. We ascertained 76 breast cancer cases out of 4867 women. The observed breast cancer cases and number of women free of breast cancer identified at subsequent screen and interval cancers are listed in Table 3. Since different populations have different underlying incidence rates of breast cancer, the baseline pre-clinical incidence rate (in the absence of characteristics of relevant covariates) should be tuned to accommodate the underlying incidence rate of breast cancer for the target population of interest when the proposed Markov regression model is attempted. In our TAMCAS study, we let the pre-clinical incidence rate follow the Weibull distribution specified by the shape parameter equal to 1.94 and the scale parameter equal to 0.00015 on the basis of the underlying Taiwanese incidence rate of breast cancer among women with family history of breast cancer (Taiwan Cancer Registry Annual Report, 2009). The distributions of factors such as BMI, and AP were derived from TAMCAS data, whereas those of other attributes were based on Table 1 obtained from literatures, assuming, in addition to adjustment for baseline incidence rate, relative risks of those across studies would not vary with various ethnic groups.
Table 3. Observed and predicted results of cross-validation and external validation.
| Validation by rounds of screens and detection methods | Observed number | Expected number |
|---|---|---|
|
Cross-validation | ||
|
1st round of screen (prevalence screen) | ||
| Free of breast cancer | 333 081 | 333 082.7 |
| Screen-detected cases |
737 |
735.3 |
|
2nd round of screen (subsequent screen) | ||
| Free of breast cancer | 332 349 | 332 360 |
| Screen-detected cases | 463 | 458.67 |
| Interval cancers |
269 |
262.3 |
|
3rd round of screen (subsequent screen) | ||
| Free of breast cancer | 331 624 | 331 587.9 |
| Screen-detected cases | 496 | 484.19 |
| Interval cancers |
229 |
276.93 |
Goodness of fit test: 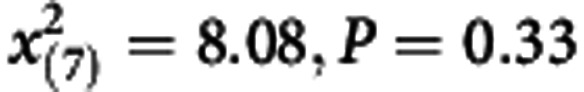 | ||
|
External validation | ||
|
Prevalence screen | ||
| Free of breast cancer | 4,791 | 4,802.66 |
| Screen-detected cases |
76 |
64.34 |
|
Subsequent screen | ||
| Free of breast cancer | 3,700 | 3,676.75 |
| Screen-detected cases | 11 | 18.29 |
| Interval cancers |
7 |
6.19 |
Goodness of fit test: 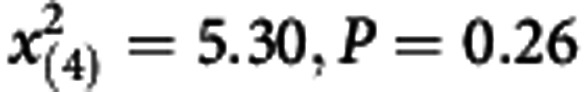
| ||
Applications to individually tailored screening
To determine the optimal inter-screening interval in different high-risk groups, we adopted a triennial screening interval as a reference to identify the number of interval cancer for other risk score percentiles that did not exceed the median-risk score based on a triennial programme for high-risk screening. This is called a risk-score-based approach. We also applied estimates for improving sensitivity by using a combination of ultrasound or magnetic resonance imaging (MRI) with mammography compared with mammography alone borrowing information from previous studies (Berg et al, 2008; Kuhl et al, 2010) to identify the threshold of high-risk categories requiring alternative image techniques. According to Berg et al (2008), sensitivity improved 27.5% when mammography was combined with ultrasound. The sensitivity improved 14.8% when mammography was combined with ultrasound or 66.7% when it was combined with MRI in the light of Kuhl et al (2010). We calculated the incidence rate of interval cancer at which the threshold of risk percentiles combining ultrasound with mammography would be the same as the median population rate using mammography only.
We further extended the study period until 12 years. We calculated the proportion of interval cancer and the incidence rate of interval cancer as a percentage of the expected incidence rate (0.00185 per year) in the absence of screening (I/E ratio), a reflection of the sensitivity of the screening method, by different inter-screening intervals and risk-score-based strategy. The higher I/E ratio is, the lower the screening programme performance is. This also allowed us to assess how a risk-score-based approach contributes to reducing false positive cases resulting from the low-risk group and false negative cases from the high-risk group compared with universal screening policy. The sensitivity for each risk decile was estimated from the 1−I/E ratio, and each corresponding false-positive fraction from Berg et al (2008) was applied for calculating the false-positive fraction.
Results
Composite score for multi-state risk prediction
To identify the role of initiators and promoters, we therefore compared the frequencies of the risk factors across free of breast cancer, screen-detected cases, and clinically detected cases (such as interval cancers). The percentage (prevalence) and relative risk of having breast cancer in the PCDP (pre-symptomatic cases) for each initiator, which was extracted from the literature, are shown in Table 1. The transition rates from free-of-breast-cancer to the PCDP among carriers were estimated from the results of a meta-analysis (Chen and Parmigiani, 2007). The total law of probability was used to calculate the baseline effect of non-carriers. The estimated results for non-carrier, BRCA1 carrier and BRCA2 carrier were equal to 1.04 × 10−6, 2.10 × 10−5, and 1.60 × 10−5. The corresponding relative risks are equal to 20.26 and 15.44. The percentages listed in Table 2 for three tumour-specific risk factors (ER, Ki-67, and HER-2) are indicative of the distribution of breast cancer in the PCDP (pre-symptomatic cases). The relative risk for the progression from the PCDP to the CP (symptomatic cases) listed accompanied with the percentage of pre-symptomatic cases was used to simulate the corresponding percentage of interval cancers (symptomatic cases). Note that the percentages, of BMI and AP applied to pre-symptomatic (screen-detected) cases were simulated following the distribution from the normal (disease-free) women in Table 1, as these two attributes have been carried for the general women during the lifetime.
The estimated clinical weights of the two-step progression to breast cancer were expressed by the two risk scores
 |
and
 |
Information on risk score 1 provided the basis for risk stratification of the underlying general population. The 10-year risk of having breast cancer in each category is diagrammed in Figure 3. The 90th percentile of risk score of 10-year follow-up was 2.3%. The corresponding figure of lifetime risk was 10.2%. The corresponding relative risk compared with the median value (50th percentile) was 2.24.
Figure 3.

Ten-year risk and lifetime risk of developing breast cancer by risk score percentile.
Model validation
Table 3 shows the results of comparisons between the observed and the predicted number of breast cancer by detection modes for cross-validation and external validation. The proposed regression model may be adequate for risk score 1 and risk score 2, as there was lacking of statistical significance for cross-validation (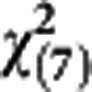 =8.08, P=0.33) and external validation (
=8.08, P=0.33) and external validation (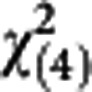 =5.30, P=0.26).
=5.30, P=0.26).
Individually tailored screening policies
Table 4 shows the recommended policies by age at the start of screening and inter-screening intervals by different decile categories. The recommended age for the start of screening was suggested by identifying the equivalent age-specific risk for other percentiles, which is commensurate with the 10-year risk of the 50th percentile of the population and was estimated as 1% or so.
Table 4. The recommend age to start screening and inter-screening interval for two types of screening at different percentiles of risk score 1.
| Percentile | Start screening agea | Inter-screening interval (year)b | Alternative imaging technique (threshold/study)c |
|---|---|---|---|
| 90–100 | 29 | 0.4 (4.8 months) | Mammography+MRI (94th/Kuhl et al (2010)) |
| 80–90 | 34 | 1 | Mammography+Ultrasound (85th/Berg et al, 2008) |
| 70–80 | 39 | 1.5 | Mammography+Ultrasound (76th/Kuhl et al (2010)) |
| 60–70 | 44 | 2 | |
| 50–60 | 50d | 3d | |
| 40–50 | 57 | 4 | |
| 30–40 | 69 | 8 | |
| 20–30 | NA | NA | |
| 10–20 | NA | NA | |
| 0–10 | NA | NA |
Abbreviations: NA=not applicable; MRI=magnetic resonance imaging.
The optimal age was determined by the age at which the 10-year risk equivalent to 1% of the 10-year risk for the 50th percentile group at age 50 years.
The optimal inter-screening interval for each percentile was determined by the rate of interval cases equal to the threshold at the 3-year inter-screening interval for the 50th percentile group.
The optimal percentile cutoff using the combined alternative imaging technique was determined by the improvement of sensitivity derived by reducing the incidence rate of interval cases until the percentile was equal to the threshold of that at the median value of population with triennial mammography only.
Representing average-risk group.
Table 4 also shows the higher the risk of entering the PCDP and further progressing to the CP becomes, the more intensive is the inter-screening interval demanded. In addition to shortening the inter-screening interval to reduce the incidence rate of interval cancer, a higher sensitivity tool could also be considered. When triennial mammography plus ultrasound was applied, the incidence rate for interval cancer of women in the 85th risk percentile was reduced to the same rate as the median-risk women when the triennial mammography screening alone was applied in the light of Berg et al (2008). The thresholds for using ultrasound and MRI based on Kuhl et al (2010) (the sensitivity improved 14.8% when mammography was combined with ultrasound or 66.7% when it was combined with MRI) were at the 76th and 94th percentiles, respectively.
Following the spirit of translational research, we showed two cases to demonstrate how to implement individually tailored screening as suggested in Table 4. For a woman having average risk, she would be classified into the 40th∼50th percentile by calculating her risk score 1. By using Table 4, the recommend age to start screening is around 57 years and the recommended inter-screening interval is 4 years. For a woman in the 90th∼100th percentile, age at start of screening should not be older than 30 years, and the recommended inter-screening interval should not be over 6 months and the screening tool may consider using alternative image technique in addition to mammography.
The number of screenings, incidence of screen-detected cases, interval cancer, the proportion of interval cancer among breast cancer cases, and the I/E ratio for 12 years are simulated and listed in Table 5, which compares the risk-score-based policies with traditional universal screening policies. In terms of an indicator of I/E ratio, the risk-score-based approach (the optimal inter-screening interval was determined from Table 4) was equivalent to a universal biennial screen. The risk-score-based approach significantly reduced the interval caner rate as a percentage of expected rate in the absence of screening by 30% compared with triennial screenings, and it also reduced the false positive cases by 8.2%.
Table 5. A comparison of different screening policies for 1 million women during 12 years.
| Screening policies | Number of Screening | Incident screen-detected cases (A) | Interval cancers (B) |
Interval cancers proportion
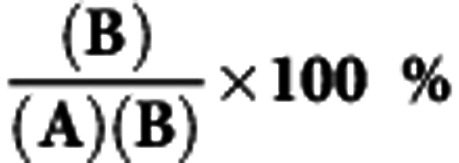
|
I/E ratio |
|---|---|---|---|---|---|
| Universal | |||||
| Annual | 12 895 635 | 11 444 | 2989 | 20.71 | 0.14 |
| Biennial | 6 947 795 | 9344 | 5089 | 35.26 | 0.23 |
| Triennial |
4 964 306 |
7739 |
6694 |
46.38 |
0.30 |
| Risk-score-based approach | 7 297 357 | 9017 | 4670 | 34.12 | 0.21 |
Discussion
An individually tailored screening strategy can be developed by applying empirical evidence on genomic discovery, tumour phenotype, clinical correlates, and epidemiological profiles to a periodical population-based screening for breast cancer with a multi-variable and three-state Markov regression model. Developing this model is potentially helpful to ascertain the roles of different risk factors responsible for different phases in breast cancer progression, and also for targeting individually based preventive or surveillance interventions.
Individual susceptibility to breast cancer is determined by identifying initiators. Because high-risk women tend to develop breast cancer at an earlier age, screening age should be lower in the higher-risk group to detect early onset cases, especially in Asian countries, where younger breast cancer cases are predominant (Anderson et al, 2006). Our study demonstrated that the determination of an inter-screening interval using the risk-score-based approach was more effective for reducing interval cancer and false positive cases than the universal triennial screening programme. A risk-stratification approach truly reflects the priority for targeting individuals invited to be screened. However, denser breast tissues in younger women may lead to rapidly growing malignancies due to the low sensitivity of mammography. Although more intensive screening can reduce interval cancer, an alternative image technique may be a better solution to reduce interval cancer for some high-risk women. Our risk assessment model can be also applied to ascertaining women with the risk higher than 75% of the underlying population that may require mammography in combination with ultrasound and 90% that may need mammography combined with MRI, which were consistent with the previous findings (Kriege et al, 2004; Boyd et al, 2007).
In terms of translational research, our study is the first to demonstrate how to translate genetic markers, tumour phenotypes, clinical attributes, and conventional risk factors to facilitate individually tailored screening for breast cancer. Our approach can help women who have the BRCA1 or BRCA2 mutations in conjunction with other risk factors to reduce the breast cancer risk by intensive surveillance, chemotherapy (tamoxifen), oral contraceptives, prophylactic mastectomy, and prophylactic oophorectomy (Anderson et al, 2006). In addition, the lower risk with less-intensive screening or the lower likelihood of using alternative image techniques can also reduce false positive cases as demonstrated in our study. A cost-effectiveness study on testing breast cancer susceptibility genes showed that test and no-test strategies are very similar in cost and effectiveness, even with a small mutation probability (Holland et al, 2009).
In our current model, there are five promoters responsible for the progression from the PCDP (pre-symptomatic disease) to the CP (symptomatic disease) (including BMI, age at first full-term pregnancy (AP), ER, Ki-67, and HER-2). Although BMI and AP have been reported to have the role of promoters (Hsieh et al, 2002), the other three tumour-specific risk factors (ER, Ki-67, and HER-2) associated with the role of promoter have not been validated yet. It is therefore very interesting to assess whether and how the three tumour-specific factors (ER, Ki-67, HER-2) makes contribution to the progression from the PCDP to the CP. By using the likelihood ratio test, we compared the full model with the reduced model (excluding three biomarkers) and found the contribution of three tumour-specific factors was statistically significant (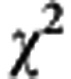 =137.06, P<0.0001). This confirms that these three biomarkers have an important role of promoter.
=137.06, P<0.0001). This confirms that these three biomarkers have an important role of promoter.
Promoters identified from screen-detected cases and interval cancer can be used as prognostic or predictive factors for personalised early treatment or prophylactic intervention. Other than the interval cancer resulting from false negatives, rapid tumour progression also accounts for the failure of detection by screening. The identification of promoters affecting the transition rate between the PCDP and the CP leading to interval cancer with rapid progression has an important role in personalised treatment and therapy (Dowsett and Dunbier, 2008). Perou et al (2000) proposed four main breast cancer molecular classes categorised by histological grade and gene-expression profile, including ER, PR, and HER-2/neu statuses, all of which may be defined as promoters and provide a better prediction of prognosis for optimal treatment (Dowsett and Dunbier, 2008). For example, trastuzumab is a first-line treatment for HER-2/neu-positive metastatic breast cancer (Viani et al, 2007). The other new high-throughput genomic technologies, such as Oncotype DX and MammaPrint, are already commercialised as prognostic tests (Dowsett and Dunbier, 2008). By identifying different risk groups at different stages, our risk assessment model provides guidance for implementing all these new therapies to achieve efficacy and efficiency.
The concept of individually tailored breast cancer screening created in the current study is also helpful for a woman to prevent breast cancer using knowledge of her genes as well as how she lives her life by the application of our method after gene testing and health consultation. Our proposed method and application can also provide a platform to facilitate the personal genetic-testing industry that has been highly debated for the time being but seems promising for the industry of direct-to-consumer genetics firms on health prevention of cancer. All these efforts have driven the use of new cancer markers adopted by clinical practice and health-policy makers to make translational research feasible and create medical enterprise as proposed in the previous study (Dalton and Friend, 2006).
In spite of strength in using our proposed approach to link the state-of-the-art research findings to clinical practice, several concerns and limitations should be clarified. Although the methodology developed in the current study is based on a multi-variable three-state Markov regression model, the definition of three-state outcome is in accordance with population-based screening data and the clinical weight (relative risk) of each factor are given in the light of current evidence from literatures. We therefore think the simulated results given such evidence-based information do not rely on mathematical model assumptions. Notably, factors responsible for the distinction between initiators and promoters were identified by the comparison of distributions between screen-detected cases and interval cancer without making any assumption. Nonetheless, the application of molecular findings to clinical practice through our novel quantitative approach may be still in a preliminary stage as initiators and promoters are ethnic-specific and may change from time to time. The current results may not be directly applied to other populations. Hence, the proposed model should be updated by modulating clinical weights with new empirical evidence to adapt the change. In addition, the determination of age of the start of screening and inter-screening interval and the threshold of screening modality with alternative image technique for high-risk group may vary with different populations. The direct application of our results on Table 5 based on our multi-variable Markov regression may be limited and should be modified. Moreover, to be comprehensive in making allowance for all factors affecting the two-step transitions of the three-state Markov regression model, those proposed in literatures were included as either initiators or promoters in our Markov regression model. The concern arises as to whether the results on the associations between certain factors, in particular genetic variants, and onset or the subsequent progression of breast cancer may be still too preliminary or costly to be applied to population-based screening. To this end, it is therefore necessary to corroborate the validity of each factor by adding more empirical evidence and extending the application of our risk-score-based algorithm to population-based screening by reducing costs involved in determining these genetic variants in economic scale.
In conclusion, this is the first to demonstrate how to translate state-of-the-art biological information on genetic, tumour, and clinical attributes as well as conventional risk factors into the realisation of the concept of an individually tailored screening strategy. The proposed risk-score-based approach can be readily applied to other screening programs in different countries and areas by tuning their own genetic susceptibility factors, tumour phenotypes, clinical attributes, and risk factors.
Acknowledgments
This study was funded by National Science Council, Taiwan (NSC 96-2314-B-002-002 and NSC 97-2314-B-002-019-MY3).
The authors declare no conflict of interest.
Footnotes
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
References
- Anderson K, Jacobson JS, Heitjan DF, Zivin JG, Hershman D, Neugut AI, Grann VR. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med. 2006;144:397–406. doi: 10.7326/0003-4819-144-6-200603210-00006. [DOI] [PubMed] [Google Scholar]
- Berg WA, Blume JD, Cormack JB, Mendelson EB, Lehrer D, Bohm-Velez M, Pisano ED, Jong RA, Evans WP, Morton MJ, Mahoney MC, Larsen LH, Barr RG, Farria DM, Marques HS, Boparai K. Combined screening with ultrasound and mammography vs mammography alone in women at elevated risk of breast cancer. JAMA. 2008;299:2151–2163. doi: 10.1001/jama.299.18.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E, Jong RA, Hislop G, Chiarelli A, Minkin S, Yaffe MJ. Mammographic density and the risk and detection of breast cancer. N Engl J Med. 2007;356:227–236. doi: 10.1056/NEJMoa062790. [DOI] [PubMed] [Google Scholar]
- Casella G, Berger RL.2001Statistical Inference2nd edn,271–300.Duxbury Press: California [Google Scholar]
- Chen TH, Chiu YH, Luh DL, Yen MF, Wu HM, Chen LS, Tung TH, Huang CC, Chan CC, Shiu MN, Yeh YP, Liou HH, Liao CS, Lai HC, Chiang CP, Peng HL, Tseng CD, Yen MS, Hsu WC, Chen CH. Community-based multiple screening model: design, implementation, and analysis of 42,387 participants. Cancer. 2004;100:1734–1743. doi: 10.1002/cncr.20171. [DOI] [PubMed] [Google Scholar]
- Chen THH, Duffy SW, Day NE. Markov chain models for progression of breast cancer. Part I: tumor attributes and the preclinical screen-detectable phase. J Epidemiol Biostat. 1997a;2:9–23. [Google Scholar]
- Chen THH, Duffy SW, Tabar L, Day NE. Markov chain models for progression of breast cancer. Part II: prediction of outcomes for different screening regimes. J Epidemiol Biostat. 1997b;2:25–35. [Google Scholar]
- Chen THH, Kuo HS, Yen MF, Lai MS, Tabar L, Duffy SW. Estimation of sojourn time in chronic disease screening without data on interval cases. Biometrics. 2000;56:167–172. doi: 10.1111/j.0006-341x.2000.00167.x. [DOI] [PubMed] [Google Scholar]
- Chen SN, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329–1333. doi: 10.1200/JCO.2006.09.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox A, Dunning AM, Garcia-Closas M, Balasubramanian S, Reed MW, Pooley KA, Scollen S, Baynes C, Ponder BA, Chanock S, Lissowska J, Brinton L, Peplonska B, Southey MC, Hopper JL, McCredie MR, Giles GG, Fletcher O, Johnson N, dos Santos Silva I, Gibson L, Bojesen SE, Nordestgaard BG, Axelsson CK, Torres D, Hamann U, Justenhoven C, Brauch H, Chang-Claude J, Kropp S, Risch A, Wang-Gohrke S, Schurmann P, Bogdanova N, Dork T, Fagerholm R, Aaltonen K, Blomqvist C, Nevanlinna H, Seal S, Renwick A, Stratton MR, Rahman N, Sangrajrang S, Hughes D, Odefrey F, Brennan P, Spurdle AB, Chenevix-Trench G, Beesley J, Mannermaa A, Hartikainen J, Kataja V, Kosma VM, Couch FJ, Olson JE, Goode EL, Broeks A, Schmidt MK, Hogervorst FB, Van't Veer LJ, Kang D, Yoo KY, Noh DY, Ahn SH, Wedren S, Hall P, Low YL, Liu J, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Sigurdson AJ, Stredrick DL, Alexander BH, Struewing JP, Pharoah PD, Easton DF. A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet. 2007;39:352–358. doi: 10.1038/ng1981. [DOI] [PubMed] [Google Scholar]
- Cummings SR, Tice JA, Bauer S, Browner WS, Cuzick J, Ziv E, Vogel V, Shepherd J, Vachon C, Smith-Bindman R, Kerlikowske K. Prevention of breast cancer in postmenopausal women: approaches to estimating and reducing risk. J Natl Cancer Inst. 2009;101:384–398. doi: 10.1093/jnci/djp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton WS, Friend SH. Cancer biomarkers--an invitation to the table. Science. 2006;312:1165–1168. doi: 10.1126/science.1125948. [DOI] [PubMed] [Google Scholar]
- del Carmen MG, Halpern EF, Kopans DB, Moy B, Moore RH, Goss PE, Hughes KS. Mammographic breast density and race. Am J Roentgenol. 2007;188:1147–1150. doi: 10.2214/AJR.06.0619. [DOI] [PubMed] [Google Scholar]
- Dong W, Berry DA, Bevers TB, Kau SW, Hsu L, Theriault RL, Shen Y. Prognostic role of detection method and its relationship with tumor biomarkers in breast cancer: the university of Texas M.D. Anderson Cancer Center experience. Cancer Epidemiol Biomarkers Prev. 2008;17:1096–1103. doi: 10.1158/1055-9965.EPI-08-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowsett M, Dunbier AK. Emerging biomarkers and new understanding of traditional markers in personalized therapy for breast cancer. Clin Cancer Res. 2008;14:8019–8026. doi: 10.1158/1078-0432.CCR-08-0974. [DOI] [PubMed] [Google Scholar]
- Duffy SW, Chen HH, Tabar L, Day NE. Estimation of mean sojourn time in breast cancer screening using a Markov chain model of both entry to and exit from the preclinical detectable phase. Stat Med. 1995;14:1531–1543. doi: 10.1002/sim.4780141404. [DOI] [PubMed] [Google Scholar]
- Dzau VJ, Ackerly DC, Sutton-Wallace P, Merson MH, Williams RS, Krishnan KR, Taber RC, Califf RM. The role of academic health science systems in the transformation of medicine. Lancet. 2010;375:949–953. doi: 10.1016/S0140-6736(09)61082-5. [DOI] [PubMed] [Google Scholar]
- Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N, Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG, Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den Ouweland A, Uitterlinden A, Rivadeneira F, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X, Mannermaa A, Kosma VM, Kataja V, Hartikainen J, Day NE, Cox DR, Ponder BA. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esserman L, Shieh Y, Thompson I. Rethinking screening for breast cancer and prostate cancer. JAMA. 2009;302:1685–1692. doi: 10.1001/jama.2009.1498. [DOI] [PubMed] [Google Scholar]
- Hsieh HJ, Chen THH, Chang SH. Assessing chronic disease progression using non-homogeneous exponential regression Markov models: an illustration using a selective breast cancer screening in Taiwan. Stat Med. 2002;21:3369–3382. doi: 10.1002/sim.1277. [DOI] [PubMed] [Google Scholar]
- Holland ML, Huston A, Noyes K. Cost-effectiveness of testing for breast cancer susceptibility genes. Value Health. 2009;12:207–216. doi: 10.1111/j.1524-4733.2008.00418.x. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr., Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl C, Weigel S, Schrading S, Arand B, Bieling H, Konig R, Tombach B, Leutner C, Rieber-Brambs A, Nordhoff D, Heindel W, Reiser M, Schild HH. Prospective multicenter cohort study to refine management recommendations for women at elevated familial risk of breast cancer: the EVA trial. J Clin Oncol. 2010;28:1450–1457. doi: 10.1200/JCO.2009.23.0839. [DOI] [PubMed] [Google Scholar]
- Kriege M, Brekelmans CT, Boetes C, Besnard PE, Zonderland HM, Obdeijn IM, Manoliu RA, Kok T, Peterse H, Tilanus-Linthorst MM, Muller SH, Meijer S, Oosterwijk JC, Beex LV, Tollenaar RA, de Koning HJ, Rutgers EJ, Klijn JG. Efficacy of MRI and mammography for breast-cancer screening in women with a familial or genetic predisposition. N Engl J Med. 2004;351:427–437. doi: 10.1056/NEJMoa031759. [DOI] [PubMed] [Google Scholar]
- Lai MS, Yen MF, Kuo HS, Koong SL, Chen TH, Duffy SW. Efficacy of breast-cancer screening for female relatives of breast-cancer-index cases: Taiwan multicentre cancer screening (TAMCAS) Int J Cancer. 1998;78:21–26. doi: 10.1002/(sici)1097-0215(19980925)78:1<21::aid-ijc5>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Boyd NF. Mammographic density. Potential mechanisms of breast cancer risk associated with mammographic density: hypotheses based on epidemiological evidence. Breast Cancer Res. 2008;10:201. doi: 10.1186/bcr1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro S, Goldberg JD, Hutchison GB. Lead time in breast cancer detection and implications for periodicity of screening. Am J Epidemiol. 1974;100:357–366. doi: 10.1093/oxfordjournals.aje.a112046. [DOI] [PubMed] [Google Scholar]
- Stacey SN, Manolescu A, Sulem P, Rafnar T, Gudmundsson J, Gudjonsson SA, Masson G, Jakobsdottir M, Thorlacius S, Helgason A, Aben KK, Strobbe LJ, Albers-Akkers MT, Swinkels DW, Henderson BE, Kolonel LN, Le Marchand L, Millastre E, Andres R, Godino J, Garcia-Prats MD, Polo E, Tres A, Mouy M, Saemundsdottir J, Backman VM, Gudmundsson L, Kristjansson K, Bergthorsson JT, Kostic J, Frigge ML, Geller F, Gudbjartsson D, Sigurdsson H, Jonsdottir T, Hrafnkelsson J, Johannsson J, Sveinsson T, Myrdal G, Grimsson HN, Jonsson T, von Holst S, Werelius B, Margolin S, Lindblom A, Mayordomo JI, Haiman CA, Kiemeney LA, Johannsson OT, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007;39:865–869. doi: 10.1038/ng2064. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, Easton DF, Evans C, Deacon J, Stratton MR. Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst. 1999;91:943–949. doi: 10.1093/jnci/91.11.943. [DOI] [PubMed] [Google Scholar]
- Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Polygenes, risk prediction, and targeted prevention of breast cancer. N Engl J Med. 2008;358:2796–2803. doi: 10.1056/NEJMsa0708739. [DOI] [PubMed] [Google Scholar]
- Prorok P. The theory of periodic screening II: doubly bounded recurrence times and mean lead time and detection probability estimation. Adv Appl Probab. 1976;8:460–476. [Google Scholar]
- Taiwan Cancer Registry Annual Report 2009. Taiwan: Department of Health, Executive Yuan. http://crs.cph.ntu.edu.tw
- Walter SD, Day NE. Estimation of the duration of a pre-clinical disease state using screening data. Am J Epidemiol. 1983;118:865–886. doi: 10.1093/oxfordjournals.aje.a113705. [DOI] [PubMed] [Google Scholar]
- Woolf SH. The meaning of translational research and why it matters. JAMA. 2008;299:211–213. doi: 10.1001/jama.2007.26. [DOI] [PubMed] [Google Scholar]
- Wu GH, Chang SH, Hsiu-Hsi Chen T. A bayesian random-effects markov model for tumor progression in women with a family history of breast cancer. Biometrics. 2008;64:1231–1237. doi: 10.1111/j.1541-0420.2007.00979.x. [DOI] [PubMed] [Google Scholar]
- van Oortmarssen GJ, Habbema JD, Lubbe JT, van der Maas PJ. A model-based analysis of the HIP project for breast cancer screening. Int J Cancer. 1990;46:207–213. doi: 10.1002/ijc.2910460211. [DOI] [PubMed] [Google Scholar]
- Viani GA, Afonso SL, Stefano EJ, De Fendi LI, Soares FV. Adjuvant trastuzumab in the treatment of her-2-positive early breast cancer: a meta-analysis of published randomized trials. BMC Cancer. 2007;7:153. doi: 10.1186/1471-2407-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerhouni EA. Translational and clinical science--time for a new vision. N Engl J Med. 2005;353:1621–1623. doi: 10.1056/NEJMsb053723. [DOI] [PubMed] [Google Scholar]