

Rho GTPases play an important role in cell migration. Here the activity of the Rho GTPase GEF Vav2 is shown to be regulated by a phosphorylation-dependent interaction with PKL (GIT2). PKL is required for Vav2 activation, and, in turn, Vav2 regulates the localization of PKL and β-PIX to focal adhesions and to the leading edge of migrating cells.
Abstract
The Rho family of GTPases plays an important role in coordinating dynamic changes in the cell migration machinery after integrin engagement with the extracellular matrix. Rho GTPases are activated by guanine nucleotide exchange factors (GEFs) and negatively regulated by GTPase-activating proteins (GAPs). However, the mechanisms by which GEFs and GAPs are spatially and temporally regulated are poorly understood. Here the activity of the proto-oncogene Vav2, a GEF for Rac1, RhoA, and Cdc42, is shown to be regulated by a phosphorylation-dependent interaction with the ArfGAP PKL (GIT2). PKL is required for Vav2 activation downstream of integrin engagement and epidermal growth factor (EGF) stimulation. In turn, Vav2 regulates the subsequent redistribution of PKL and the Rac1 GEF β-PIX to focal adhesions after EGF stimulation, suggesting a feedforward signaling loop that coordinates PKL-dependent Vav2 activation and PKL localization. Of interest, Vav2 is required for the efficient localization of PKL and β-PIX to the leading edge of migrating cells, and knockdown of Vav2 results in a decrease in directional persistence and polarization in migrating cells, suggesting a coordination between PKL/Vav2 signaling and PKL/β-PIX signaling during cell migration.
INTRODUCTION
Cell migration plays a critical role in numerous pathological and physiological processes, including embryonic development, wound healing, and tumor cell metastasis (Huttenlocher and Horwitz, 2011). It is well established that the Rho family of small GTPases plays an important role in coordinating the cytoskeletal and cell migration machinery after integrin engagement with the extracellular matrix (ECM). Rac1 and Cdc42 stimulate the formation of nascent adhesion complexes at the leading edge and the development of lamellipodia and filopodia, respectively. Transition to RhoA/C activation subsequently promotes the maturation of adhesions and the formation of associated stress fibers and is also required for focal adhesion disassembly (Webb et al., 2002; Hall, 2005; Broussard et al., 2008).
The activity of Rho GTPases is stimulated by guanine nucleotide exchange factors (GEFs) and negatively regulated by GTPase-activating proteins (GAPs), as well as by guanine nucleotide dissociation inhibitors (GDIs; Bos et al., 2007). Many of the GEFs and GAPs, including β-PIX, Tiam1, and CdGAP, play a role in cell migration (Lee et al., 2005; LaLonde et al., 2006; Pegtel et al., 2007; Feng et al., 2010; Wormer et al., 2012). However, we are only beginning to understand the mechanisms by which the cell coordinates the localization and activity of Rho GTPase regulators. For example, focal adhesion scaffold proteins such as paxillin act to coordinate the spatiotemporal activation of the Rho family of GTPases, including Cdc42, Rac1, and RhoA and their associated effectors to regulate cell adhesion, spreading, and migration (Nobes and Hall, 1999).
Paxillin achieves GTPase regulation by recruiting GEFs and GAPs, along with specific effector proteins that mediate downstream responses to GTP-bound active GTPases, to focal adhesions. For example, paxillin binds the β-PIX-PAK-Nck protein cassette via the ARF GAP PKL (GIT2) and localizes them to adhesions (Turner et al., 1999; Brown et al., 2002). This interaction is required for precise coordination of localized Rac1 activity, presumably via the GEF activity of β-PIX, and likely contributes to PAK regulation of lamellipodia extension and adhesion turnover (Brown et al., 2002).
Signaling via both integrin and growth factor receptors is required for efficient cell migration to occur, and several focal adhesion components, including paxillin, FAK, and Src, are phosphorylated in response to growth factor stimulation (Turner, 2000). PKL is tyrosine phosphorylated downstream of platelet-derived growth factor (PDGF)–receptor activation in fibroblasts, an event that promotes PKL association with paxillin and is required for directional cell migration and polarization, suggesting that PKL is an integral component of growth factor and cell adhesion signaling cross-talk (Yu et al., 2009).
We sought to determine how PKL tyrosine phosphorylation may contribute to the coordination of Rho-family GTPase signaling downstream of integrin and growth factor receptor activation. Using a high-throughput Src homology 2 (SH2) domain-binding screen (Machida et al., 2007), we identified the Rho-family GEF Vav2 as a new binding partner for phosphorylated PKL. Through the expression of PKL mutants and RNA interference (RNAi) of PKL, we determined that PKL is required for adhesion and growth factor–dependent Vav2 activation. Vav2 activity is subsequently required for epidermal growth factor (EGF)–stimulated enrichment of PKL and β-PIX at focal adhesions during cell spreading and to adhesions at the leading edge of migrating cells, suggesting a feedforward loop that coordinates PKL phosphorylation and activation of Vav2 with the distribution of PKL and β-PIX during cell migration.
RESULTS
PKL interacts with the SH2 domain of Vav2, and coexpression of PKL and Vav2 promotes lamellipodia and stress fiber formation
PKL is tyrosine phosphorylated during cell adhesion, as well as in response to the addition of growth factors. This phosphorylation is necessary for optimal cell migration and spreading (Yu et al., 2009) and for PKL targeting to focal adhesions (Brown et al., 2005). The principal phosphorylation sites Y286, Y392, and Y592 are potential candidates for mediating interactions with SH2 domain–containing proteins (Brown et al., 2005). To identify novel binding partners for these phosphorylation sites, we transfected CHO-K1 cells with green fluorescent protein (GFP) wild-type PKL (PKL) or the nonphosphorylatable GFP-PKL Y286F, Y392F, Y592F mutant (PKL-3YF; Brown et al., 2005) and then spread them on fibronectin to stimulate phosphorylation of the wild-type protein. The GFP-PKL proteins were precipitated and used in a screen of glutathione S-transferase (GST)–SH2 domain fusion proteins (Machida et al., 2007). In addition to confirming previously reported interactions between phospho-PKL and Crk, Nck, and Src (Brown et al., 2005), of particular note was a phosphorylation-dependent interaction between PKL and the SH2 domain of the Rho-family GEF Vav2 (data not shown). Pull-down assays from CHO-K1 lysates using GST-Vav2-SH2 domain confirmed a specific interaction with GFP-PKL but not GFP-PKL-3YF or GFP alone (Figure 1A). Furthermore, cotransfection of CHO-K1 cells with full-length hemagglutinin (HA)-tagged Vav2 along with GFP-tagged PKL constructs followed by spreading on fibronectin and immunoprecipitation of GFP-tagged proteins demonstrated that HA-Vav2 coprecipitated effectively with phosphorylated GFP-PKL, whereas only background levels of HA-Vav2 were observed in the GFP and GFP-PKL-3YF precipitates (Figure 1B).
FIGURE 1:

PKL and Vav2 coexpression induces an “active” Vav phenotype. (A) Representative Western blot showing GFP-PKL binding to GST-Vav2-SH2 domain. In contrast, the nonphosphorylatable GFP-PKL-3YF does not bind. (B) Representative Western blot showing coimmunoprecipitation of HA-Vav2 with GFP-PKL. (C) Fluorescence analysis of cells expressing GFP, GFP-PKL, or GFP-GIT1 together with HA-Vav2 or HA-CA-Vav2 as indicated. Arrows indicate transfected cells exhibiting “active” Vav2 phenotype. Scale bar, 10 μm. (D–F) Cell counts of cells coexpressing the constructs indicated. GFP-positive cells exhibiting an “active” Vav2 phenotype were scored. At least 100 cells per condition were counted, and numbers are mean values ± SEM for three separate experiments. Significance was determined using a Student's t test. **p < 0.005, ***p < 0.0005.
Because both Vav2 and PKL contribute to the regulation of lamellipodia formation during cell migration and spreading (Marignani and Carpenter, 2001; Brown et al., 2005), the importance of the PKL-Vav2 interaction in regulating cell morphology was evaluated. Of interest, the coexpression of Vav2 with PKL produced a dramatic change in cell morphology, with cells displaying a flattened, nonpolarized, pancake-like morphology, as well as a robust array of stress fibers (Figure 1C, arrows). In addition, GFP-PKL localized to focal adhesions in these cells (Figure 1C). This phenotype was remarkably similar to that of cells expressing an oncogenic Vav2 mutant that lacks the amino-terminal calponin homology domain and acidic domain and is constitutively active toward Rac1, Cdc42, and RhoA (Schuebel et al., 1996, 1998; Abe et al., 2000; Liu and Burridge, 2000). Indeed, introduction of a constitutively active Vav2 (CA-Vav2) mutant (Booden et al., 2002) into CHO-K1 cells in the absence of GFP-PKL generated a similar phenotype to cells coexpressing PKL and Vav2 (Figure 1C), suggesting that coexpression of PKL and Vav2 results in the activation of Vav2 exchange activity toward multiple Rho GTPases. Consequently, this phenotype will be described as the “active” Vav2 phenotype. Quantitation of cotransfected cells demonstrated that >30% of cells (33 ± 0.7%) coexpressing PKL and Vav2 displayed the “active” Vav2 phenotype, compared with approximately 10% of cells individually expressing wild-type Vav2 (11.2 ± 0.63%) and <1% of cells expressing GFP-PKL alone (0.67 ± 1.3%). In addition, approximately 50% of GFP-positive cells (52 ± 1.4%) expressing constitutively active Vav2 (Figure 1D) exhibited the “active” Vav2 phenotype. Of interest, coexpression of the closely related protein GIT1, which has also been implicated in the regulation of adhesion-dependent Rac1-activation (Nishiya et al., 2005; Nayal et al., 2006), with Vav2 failed to induce the formation of the “active” Vav2 phenotype (Figure 1, C and D). Furthermore, the “active” Vav2 phenotype was not observed when PKL was coexpressed with Vav1 or Vav3 (Figure 1E), suggesting that the phenotype observed is specific to PKL-dependent activation of Vav2.
To dissect the importance of PKL tyrosine phosphorylation and focal adhesion localization in the activation of Vav2, we coexpressed Vav2 with either the nonphosphorylatable PKL-3YF mutant or the PKL-ΔPBS2 mutant, which is defective in focal adhesion targeting due to the absence of the paxillin interaction domain (West et al., 2001), and counted the number of cells exhibiting the “active” Vav2 phenotype. Coexpression of Vav2 with either of these mutants resulted in a significant decrease in the number of cells exhibiting the “active” Vav2 phenotype (Figure 1, C and F). Because the PKL-ΔPBS2 mutant can still be phosphorylated in response to adhesion (Brown et al., 2005), these results indicate that both PKL tyrosine phosphorylation and its localization to focal adhesions are necessary for the morphological changes observed in the generation of the “active” Vav2 phenotype.
Overexpression of PKL with Vav2 results in Vav2 phosphorylation and activation
To test whether coexpression of PKL and Vav2 did indeed result in the stimulation of Vav2 exchange activity, we assessed the phosphorylation of Vav2 at Y172 by Western blotting. Phosphorylation at this site was reported to be required for Vav2 exchange activity (Schuebel et al., 1998) and thus can be used as a readout for Vav2 activation. Coexpression of PKL with Vav2 resulted in an increase in the PY172 levels compared with Vav2 coexpressed with GFP (Figure 2, A and B). In contrast, coexpression of GIT1 or of PKL 3YF with Vav2 was unable to stimulate Vav2 phosphorylation to the same extent (Figure 2, A and B). To evaluate the influence of PKL on Vav2 exchange activity, we undertook pull-down assays using GST fusion proteins of nucleotide binding–defective mutants of RhoA and Rac1. These mutants bind selectively to the activated form of GEFs (Garcia-Mata et al., 2006; Garrett et al., 2007). The binding of Vav2 to both Rac G15A and Rho G17A mutants was significantly enhanced by coexpression of PKL with Vav2 as compared with GFP (Figure 2, C and D), suggesting increased Vav2 exchange activity toward both Rac1 and RhoA. Conversely, binding of Vav2 was suppressed by expression of the PKL 3YF mutant, in comparison to expression of wild-type PKL (Figure 2, C and D), confirming the role of tyrosine phosphorylation of PKL in PKL-dependent activation of Vav2. The results from the active GEF pull-down assays exhibited a similar trend to those generated using Vav2 phosphorylation as a marker for Vav2 activation, demonstrating that the level of PY172 Vav2 is a viable determinant of Vav2 activity.
FIGURE 2:

PKL induces Vav2 phosphorylation and increased GEF activity. (A, B) Representative Western blots demonstrating that coexpression of GFP-PKL with HA-Vav2 induces elevated phosphorylation of Vav2 at Y172. In contrast, expression of GFP-GIT1 (A) or GFP-PKL-3YF (B) does not induce Vav2 phosphorylation to the same extent. (C, D) Coexpression of GFP-PKL with HA-Vav2 induces Vav2 GEF activity toward Rac1 and RhoA as determined by binding to either GST-Rac G15A (C) or GST-Rho G17A (D).
PKL is required for activation of endogenous Vav2 in response to adhesion and EGF stimulation
Because expression of PKL and Vav2 in CHO-K1 cells resulted in the activation of Vav2, we investigated a potential role for PKL in the activation of endogenous Vav2. When HT1080 human fibrosarcoma cells were spread on fibronectin (FN) in the absence of serum, no significant change in phosphorylation of Vav2 was observed in comparison to cells spread in poly-l-lysine (PL; Figure 3A). This suggests that Vav2 activation requires additional stimulation in this cell type. Vav2 activity, as measured by its tyrosine phosphorylation at Y172, is stimulated in response to EGF, PDGF, and vascular endothelial growth factor (VEGF; Liu and Burridge, 2000; Marcoux and Vuori, 2003; Garrett et al., 2007), and therefore HT1080 cells were spread on either PL or FN in the absence or presence of 30 ng/ml EGF. Robust phosphorylation of Vav2 was observed in cells spread on FN in the presence of EGF, with maximal phosphorylation being observed after 30 min (Figure 3B). Phosphorylation of Vav2 was not stimulated in cells spread on PL, even in the presence of EGF, demonstrating that Vav2 activation requires both adhesion- and growth factor–dependent signaling. Furthermore, coimmunoprecipitation of endogenous Vav2 with PKL was observed in HT1080 cells spread on fibronectin in the presence of EGF, whereas spreading on fibronectin alone did not promote an interaction between Vav2 and PKL (Figure 3C). Of interest, maximal PKL tyrosine phosphorylation in fibroblasts stimulated with PDGF also requires adhesion to FN and growth factor stimulation (Yu et al., 2009).
FIGURE 3:
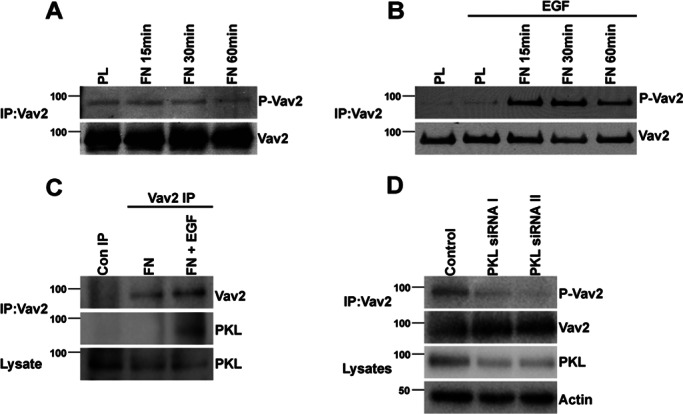
PKL is required for fibronectin- and EGF-dependent phosphorylation and activation of endogenous Vav2. HT1080 cells were spread on PL or Fibronectin FN in the absence (A) or presence (B) of EGF for the time periods indicated. FN alone was unable to induce phosphorylation of immunoprecipitated Vav2 (A), whereas elevated phosphorylation of Vav2 was observed when cells were spread on FN in the presence of 30 ng/ml EGF. (C) PKL coimmunoprecipitates with Vav2 when cells are spread on FN in the presence of EGF but not in the absence of EGF. (D) PKL knockdown by RNAi inhibits the stimulation of Vav2 phosphorylation in response to FN and EGF.
To determine whether PKL is required for phosphorylation and activation of endogenous Vav2, an RNAi-based approach was used to suppress the levels of PKL in HT1080 cells. Phosphorylation of Y172 Vav2 was decreased in cells after PKL knockdown and spreading on FN for 30 min in the presence of EGF (Figure 3D). These data therefore demonstrate the requirement for PKL in stimulating activation of Vav2 in response to integrin ligation and EGF stimulation.
Vav2 activity promotes PKL adhesion localization
Recruitment of PKL to adhesions is promoted by expression of constitutively active Rac1 and Cdc42 during cell spreading (Brown et al., 2002). Because Vav2 can demonstrate exchange activity toward both Rac1 and Cdc42, we hypothesized that Vav2 might play a role in stimulating PKL localization to adhesions in response to EGF. We spread HT1080 cells expressing GFP-PKL on fibronectin for 30 min in the absence or presence of EGF, the conditions in which PKL-dependent activation of Vav2 is observed, and assessed the amount of GFP-PKL colocalization with paxillin at focal adhesions by confocal microscopy. In cells expressing similar levels of GFP-PKL as determined by cell mean fluorescence intensity (not shown), the mean Pearson's r per cell between GFP-PKL and paxillin was significantly increased in the presence of EGF (Figure 4, A and B), suggesting that EGF stimulation is able to promote the localization of GFP-PKL to focal adhesions. We previously demonstrated that PKL association with paxillin and recruitment to adhesions is specifically regulated by growth factor stimulation in NIH 3T3 cells in comparison to GIT1, which remains constitutively associated (Yu et al., 2009). In HT1080 cells, GFP-GIT1 localized to paxillin-positive focal adhesions in spread cells, but this localization was unaltered in the presence of EGF (Figure 4, A and B), suggesting that EGF is able to specifically regulate recruitment of GFP-PKL to focal adhesions in this cell type. To assess the effect of EGF on endogenous PKL localization, we undertook confocal microscopy using an antibody reported to cross-react with both PKL and GIT1. More robust adhesion staining of PKL/GIT1 was observed in the presence of EGF (Figure 4C), and line profiles through individual adhesions demonstrate that although paxillin intensity is similar in the absence or presence of EGF, the intensity of PKL/GIT1 staining in adhesions increases (Figure 4C). To quantify changes in PKL/GIT1 at focal adhesions in a nonsubjective manner, we determined the relative intensity of PKL/GIT1 to paxillin for all paxillin-positive adhesions in the cell and generated a mean value per cell. With this approach it was apparent that after stimulation with EGF, the mean intensity of endogenous PKL/GIT1 staining in relation to paxillin at focal adhesions per cell was significantly increased (Figure 4, C and E), with no change in average focal adhesion size per cell being observed (Figure 4D). Taken together with the specific effect of EGF on GFP-PKL localization to focal adhesions (Figure 4A), these data suggest that the changes in endogenous staining observed using the PKL/GIT1 antibody in HT1080 cells occur as a result of EGF promoting the enrichment of PKL at paxillin-positive focal adhesions.
FIGURE 4:

EGF enhances PKL localization to adhesions. (A) HT1080 cells transfected with either GFP-PKL or GFP-GIT1 were spread on FN in the absence or presence of EGF for 30 min and then stained for paxillin. Images are contrast enhanced to equal degrees for presentation. Scale bar, 10 μm. (B) Mean Pearson's r between GFP-PKL or GFP-GIT1 and paxillin per cell. (C) HT1080 cells were spread on FN in the absence or presence of EGF for 30 min and then stained for paxillin and PKL/GIT1. Images are contrast enhanced to equal degrees for presentation. Scale bar, 10 μm. Line profiles through individual adhesions demonstrate increased intensity of PKL/GIT1 in paxillin-positive adhesions in the presence of EGF, whereas paxillin intensity remains unchanged. The average focal adhesion size per cell (D) and the average ratio of PKL/GIT1 intensity to paxillin intensity in adhesions per cell (E) were quantified in background-subtracted raw images using ImageJ. Values are means ± SEM for three experiments and at least 10 cells per experiment. Significance was determined using Student's t test.
To determine whether Vav2 was required for this recruitment to occur, we spread HT1080 cells expressing GFP-PKL alone or GFP-PKL along with CA-Vav2 on FN for 30 min in the absence of EGF. In the presence of CA-Vav2, we observed an increase in Pearson's r between paxillin and GFP-PKL (Figure 5, A and B), comparable to cells stimulated with EGF. In addition, we transfected HT1080 cells with GFP alone or GFP together with CA-Vav2 and determined the relative intensity of endogenous PKL to paxillin staining at adhesions. Compared to cells expressing GFP alone, CA-Vav2–expressing cells demonstrated a significant increase in PKL/GIT1 staining at focal adhesions (Figure 5, C and E), with no associated change in mean adhesion size per cell (Figure 5D). Conversely, expression of dominant-negative L342R/L343S Vav2 (RS-Vav2), which lacks nucleotide exchange activity (Marignani and Carpenter, 2001), or small interfering RNA (siRNA)–mediated knockdown of Vav2 (Figure 6C) suppressed EGF-stimulated recruitment of PKL to focal adhesions during cell spreading, as shown by a reduction in PKL/GIT1 staining intensity at adhesions (Figure 6, A, B, and E). These treatments had no effect on the mean focal adhesion size per cell (Figure 6D).
FIGURE 5:

Expression of constitutively active CA-Vav2 promotes PKL localization to adhesions. (A) HT1080 cells transfected with GFP-PKL or GFP-PKL along with HA-CA-Vav2 were spread on FN in the absence of EGF for 30 min and then stained for paxillin. Images are contrast enhanced to equal degrees for presentation. Scale bar, 10 μm. (B) Mean Pearson's r between GFP-PKL and paxillin per cell was calculated using ImageJ. (C) HT1080 cells transfected with either GFP alone or GFP along with HA-CA-Vav2 were spread on FN in the absence of EGF for 30 min and then stained for paxillin and PKL/GIT1. Scale bar, 10 μm. Line profiles through adhesions demonstrate increased intensity of PKL/GIT1 in paxillin-positive adhesions. The average focal adhesion size per cell (D) and the average ratio of PKL/GIT1 intensity to paxillin intensity in adhesions per cell (E) were quantified in background-subtracted raw images using Image J. Values are means ± SEM for three experiments and at least 10 cells per experiment. Significance was determined using Student's t test. *p < 0.05, *** p < 0.005.
FIGURE 6:

Vav2 activity is required for EGF-stimulated localization of PKL to adhesions. (A) HT1080 cells transfected with GFP or GFP plus dominant-negative L342R/L343S Vav2 (RS-Vav2), which lacks nucleotide exchange activity, were spread on FN for 30 min in the presence of EGF and then stained for PKL/GIT1 and paxillin. Images are contrast enhanced to equal degrees for presentation. Scale bars, 10 μm. (B) HT1080 cells were transfected with either control siRNA or siRNA targeting Vav2. After 72 h cells were spread on FN for 30 min in the presence of EGF and then stained for PKL/GIT1 and paxillin. Images are contrast enhanced to equal degrees for presentation. Scale bars, 10 μm. Line profiles through adhesions indicated in A and B demonstrate decreased intensity of PKL in paxillin-positive adhesions. (C) Representative blot showing efficient knockdown of Vav2 in HT1080 cells. The average focal adhesion size (D) and the average ratio of PKL/GIT1 intensity to paxillin intensity in adhesions per cell (E) were quantified in background-subtracted raw images using ImageJ. Values are means ± SEM for three experiments and at least 10 cells per experiment. Significance was determined using Student's t test. *P < 0.05.
To determine which GTPases were required for Vav2-mediated PKL localization to focal adhesions, we spread HT1080 cells coexpressing CA-Vav2 and either vector control or dominant-negative Cdc42, Rac1, or RhoA on fibronectin in the absence of EGF for 30 min and quantified the relative intensity of PKL/GIT1 staining to paxillin at focal adhesions per cell. Expression of either dominant-negative Cdc42 or Rac1 was able to significantly suppress CA-Vav2–stimulated PKL/GIT1 staining at focal adhesions, whereas dominant-negative RhoA was ineffective in this regard (Figure 7, A and C). No change in mean focal adhesion size per cell was observed in cells coexpressing dominant-negative Cdc42, Rac1, or RhoA with CA-Vav2 (Figure 7B), suggesting that any change in PKL distribution is not a result of global changes in focal adhesion size. These data therefore demonstrate that stimulation of Vav2 exchange activity is required to promote recruitment of PKL to adhesions downstream of adhesion to fibronectin and stimulation of cells with EGF, likely through activation of Cdc42 and Rac1 small GTPases.
FIGURE 7:

Activation of Cdc42 and Rac1, but not RhoA, is required for Vav2-stimulated recruitment of PKL/GIT1 to paxillin-positive adhesions. (A) HT1080 cells were transfected with GFP or GFP plus HA-CA-Vav2 in the absence or presence of dominant-negative Cdc42, Rac1, or RhoA and then spread on FN in the absence of EGF for 30 min. After fixation, cells were stained for paxillin and PKL/GIT1. Images are contrast enhanced to equal degrees for presentation. Scale bar, 10 μm. Line profiles through adhesions indicated in A demonstrate increased intensity of PKL/GIT1 in paxillin-positive adhesions when CA-Vav2 is expressed and that this is suppressed by coexpression of dominant-negative Cdc42 and Rac1. The average focal adhesion size per cell (B) and the average ratio of PKL/GIT1 intensity to paxillin intensity in adhesions per cell (C) were quantified in background-subtracted raw images using ImageJ. Values are means ± SEM for three experiments and at least 10 cells per experiment. Significance was determined using Student's t test. *P < 0.05, **P < 0.005.
PKL is able to directly bind the Rac-GEF β-PIX (Turner et al., 1999) and facilitate recruitment of β-PIX and the Rac effector PAK to focal adhesions (Turner et al., 1999; Brown et al., 2002). Furthermore, PKL tyrosine phosphorylation is required for the polarized distribution of β-PIX to the leading edge of migrating NIH 3T3 fibroblasts (Yu et al., 2009). We therefore hypothesized that EGF-stimulated Vav2 activity may also promote β-PIX recruitment to focal adhesions. Indeed, as observed with PKL, treatment with EGF or expression of CA-Vav2 resulted in an increase in β-PIX at focal adhesions (Supplemental Figure S1). In addition, expression of dominant-negative RS-Vav2 or depletion of Vav2 by RNAi suppressed the recruitment of β-PIX to paxillin-positive adhesions in cells spread on FN for 30 min in the presence of EGF (Supplemental Figure S2 and Figure 8).
FIGURE 8:

Vav2 is required for EGF-stimulated localization of β-PIX to adhesions. (A) HT1080 cells were transfected with either control siRNA or siRNA targeting Vav2. After 72 h cells were spread on FN for 30 min in the presence of EGF and then stained for β-PIX and paxillin. Images are contrast enhanced to equal degrees for presentation. Scale bar, 10 μm. Line profiles through adhesions demonstrate decreased intensity of β-PIX in paxillin-positive adhesions in the absence of Vav2. The average focal adhesion size (B) and the average ratio of β-PIX intensity to paxillin intensity per adhesion (C) were quantified in background-subtracted raw images using ImageJ. Values are means ± SEM for three experiments and at least 10 cells per experiment. Significance was determined using Student's t test. **p < 0.005.
Vav2 is required for polarized distribution of PKL and β-PIX during directional cell migration
To assess a role for PKL-dependent activation of Vav2 in directed cell migration, we evaluated the polarization and motility of PKL- and Vav2-RNAi–depleted HT1080 cells by scrape wound assays. Cells migrating on a two-dimensional (2D) substrate typically develop a morphological polarization associated with reorientation of the Golgi complex and microtubule-organizing center toward the leading edge (Nabi, 1999). Knockdown of PKL or expression of nonphosphorylatable PKL in NIH 3T3 cells results in loss of Golgi reorientation in migrating cells (Yu et al., 2009), demonstrating that PKL facilitates the establishment of a polarized morphology during migration. Similarly, knockdown of Vav2 suppressed the ability of migrating cells to reorient the Golgi (Figure 9A), suggesting that the regulation of Vav2 is also required to establish cell polarity in migrating cells.
FIGURE 9:

Vav2 is required for localization of PKL/GIT1 and β-PIX to the leading edge of migrating cells. (A) HT1080 cells were transfected with siRNAs targeting Vav2. After 72 h cells were replated to form a confluent monolayer and wounded. Cells were allowed to migrate into a scrape wound for 2 h before fixation and staining for the Golgi marker GM130, paxillin, DAPI, and rhodamine–phalloidin. Quantitation of polarized Golgi at the wound edge demonstrates a significant decrease after Vav2 knockdown. *p < 0.05. Scale bar, 50 μm. (B, D) HT1080 cells depleted of Vav2 show perturbed localization of PKL/GIT1 and β-PIX to the leading edge of migrating cells. Scale bars, 10 μm. Line profiles through adhesions indicated in B and D demonstrate decreased intensity of PKL/GIT1 and β-PIX in paxillin-positive adhesions at the leading edge. (C, E) Quantitation of the number of cells at the wound edge, demonstrating polarized distribution of PKL/GIT1 and β-PIX. Values are means ± SEM for three experiments and at least 30 cells per experiment. Significance was determined using Student's t test. **p < 0.05, ***p < 0.005. (F, G) HT1080 or (H, I) NIH 3T3 cells were transfected with siRNA targeting PKL or Vav2 as indicated. After 72 h cells were replated to form a confluent monolayer and wounded. Cell migration was allowed to proceed for 16 h and individual cells subsequently tracked using ImageJ. Values for directionality (F, H) and velocity (G, I) were determined for at least 40 cells per experiment and three separate experiments. ***p < 0.0005.
Having described a role for Vav2 in regulating recruitment of PKL and β-PIX to adhesions in spreading cells, we tested whether Vav2 activity might also be required for the polarized distribution of PKL and β-PIX in migrating cells. HT1080 cells migrating into a scrape wound in serum-containing media generally exhibit a single broad lamellipodium, and the distribution of PKL/GIT1 (Figure 9B) and β-PIX (Figure 9D) is polarized to adhesions at the leading edge. In contrast, after Vav2 RNAi, the polarized distribution of both PKL/GIT1 and β-PIX to the leading edge was greatly reduced (Figure 9, B–E). These data therefore suggest that Vav2 is required for the establishment of cell polarity and thus for the polarized distribution of PKL/GIT1 and β-PIX to adhesions at the leading edge of migrating cells.
In addition to a role in establishing cell polarity, PKL RNAi has been reported to perturb cell migration in a scrape wound, with a reduction in cell speed and a loss of directionality being observed in NIH 3T3 cells (Yu et al., 2009). Similarly, knockdown of PKL in HT1080 cells also resulted in a loss of directionality, but in contrast to fibroblasts, no decrease in cell speed was observed (Figure 9, F and G). Of interest, RNAi of Vav2 produced similar results (Figure 9, F and G) and also resulted in a decrease in directionality in NIH 3T3 cells (Figure 9H) but had no effect on cell speed (Figure 9I), suggesting that PKL-dependent activation of Vav2 is required to facilitate the role of PKL in establishing cellular polarity and maintaining directionality during cell migration.
DISCUSSION
This study outlines an important role for the interaction between tyrosine-phosphorylated PKL and Vav2 in regulating Vav2 activity during cell spreading and migration (summarized in Figure 10). We demonstrated that coexpression of GFP-PKL, but not the closely related GIT1, with Vav2 in CHO-K1 cells results in the formation of an “active” Vav2 phenotype by which cells display a nonpolarized, pancake-like morphology, as well as robust stress fibers, when spread on fibronectin. This “active” Vav2 phenotype is also observed in cells expressing a constitutively active form of Vav2 that was previously shown to promote the activity of Rac1, RhoA, and Cdc42 (Liu and Burridge, 2000). Indeed, coexpression of PKL with Vav2 promoted the GEF activity of Vav2 toward both Rac1 and RhoA, as demonstrated by pull-down assays using GST fusion proteins of nucleotide binding–defective mutants that selectively bind the activated form of GEFs (Garcia-Mata et al., 2006; Garrett et al., 2007). Formation of this “active” Vav2 phenotype and stimulation of Vav2 GEF activity are suppressed in cells expressing nonphosphorylatable GFP-PKL-3YF, and this mutant is unable to bind Vav2, demonstrating that PKL phosphorylation and binding of Vav2 via the SH2 domain are required for PKL-dependent activation of Vav2 activity. In previous studies, the carboxyl-terminus of Vav2, which contains the SH2 and SH3 domains, was suggested to play an autoinhibitory role in regulating Vav2 activity, acting as a dominant negative for EGF-induced Rac1 activation (Liu and Burridge, 2000). In addition, expression of Vav2 that either contained point mutations in the SH2 domain or lacked the carboxyl-terminal region resulted in a loss of tyrosine phosphorylation in response to EGF (Tamas et al., 2001). Therefore SH2 binding of tyrosine-phosphorylated proteins appears to be required for Vav2 phosphorylation and activation. As such, phospho-PKL binding to Vav2-SH2 might contribute to suppressing autoinhibition by the carboxyl-terminus of Vav2 and thereby promote Vav2 phosphorylation and activation.
FIGURE 10:

Schematic representation of findings outlined in this study. Phosphorylated PKL binds Vav2 via the Vav2 SH2 domain in response to EGF and FN signaling, resulting in increased Vav2 phosphorylation and stimulation of its GEF activity. These phosphorylation events are likely mediated by Src tyrosine kinase activity (data not shown; Servitja et al., 2003; Tu et al., 2003; Brown et al., 2005; Yu et al., 2009)). Active Vav2 promotes increased recruitment of PKL and β-PIX to paxillin-positive adhesions in an active Cdc42- and Rac1-dependent manner. In addition, Vav2 is required for efficient polarization of cells during directed cell migration and the corresponding distribution of PKL and β-PIX to the leading edge. In addition to the data outlined here, PKL may also facilitate the switching of Vav2 activity toward RhoA at later spreading time points and during migration to regulate adhesion maturation and disassembly.
Activation of Vav2 also requires membrane binding via its pleckstrin homology (PH) domain. Introduction of a point mutation into the Vav2 PH domain or treatment with the phosphoinositol-3-kinase (PI3K) inhibitor LY294002 inhibits Vav2 activity (Tamas et al., 2003), suggesting a key role for PI3K and the generation of phosphatidylinositol (3,4,5)-triphosphate (PIP3) in regulating Vav2 activity. Of interest, neutrophils isolated from PKL-null mice are unable to efficiently activate PI3K in order to produce and accumulate PIP3 at the leading edge of migrating cells (Mazaki et al., 2006). Therefore it is possible that PKL plays a multifaceted role in regulating Vav2 activity by influencing Vav2 membrane binding and activation via the regulation of PI3K activity and the generation of PIP3 in addition to promoting Vav2 phosphorylation via binding of the Vav2-SH2 domain.
Consistent with previous studies, we demonstrated that Vav2 is not phosphorylated in response to FN spreading alone (Liu and Burridge, 2000) but that in HT1080 cells Vav2 is phosphorylated after costimulation with EGF and FN. Vav2 was reported to undergo tyrosine phosphorylation in response to EGF, PDGF, and VEGF (Liu and Burridge, 2000; Marcoux and Vuori, 2003; Garrett et al., 2007) and to associate with the EGF receptor (Pandey et al., 2000; Tamas et al., 2003). Here we show that EGF-stimulated phosphorylation of Vav2 and therefore Vav2 GEF activity are suppressed after knockdown of PKL. Therefore PKL may be acting as a signaling intermediate to facilitate Vav2 phosphorylation and activity downstream of EGF stimulation. EGF-stimulated phosphorylation of Vav2 is primarily mediated by Src tyrosine kinase (Servitja et al., 2003; Tu et al., 2003), and Src also binds to and phosphorylates PKL (Brown et al., 2005; Yu et al., 2009). Therefore, PKL binding of Vav2 may also act to localize Vav2 to Src kinase to promote Src-dependent phosphorylation of Vav2 downstream of EGF stimulation.
Several reports have implicated a role for Vav2 signaling in cell spreading, scattering, and migration (Schuebel et al., 1998; Liu and Burridge, 2000; Marignani and Carpenter, 2001; Garrett et al., 2007). However, the molecular mechanisms by which Vav2 influences these aspects of cell behavior are poorly defined. Here we showed that under the conditions in which PKL-dependent phosphorylation of Vav2 occurs, EGF stimulation also promotes the recruitment of PKL and its binding partner β-PIX, another Cdc42/Rac1 GEF, to paxillin-positive focal adhesions. This recruitment can be stimulated by expression of CA-Vav2 in the absence of EGF and suppressed by expression of dominant-negative RS-Vav2 or knockdown of Vav2 by RNAi. These data suggest a potential feedforward loop by which PKL phosphorylation-dependent activation of Vav2 may locally activate Rac1 to promote the recruitment of the PKL and β-PIX (Turner et al., 1999) to focal adhesions. Redistribution of PKL and β-PIX to focal adhesions can be promoted via expression of either active-Rac1 or active Cdc42, but not active-RhoA, and requires the activation of PAK (Brown et al., 2002). Expression of CA-Vav2 (Liu and Burridge, 2000) and overexpression of Vav2 in EGF-stimulated cells (Tu et al., 2003) increase the activity of Cdc42, as well as of Rac1. Consistent with these observations, we demonstrate that the activity of Cdc42, as well as of Rac1, is required to facilitate Vav2-stimulated PKL recruitment to focal adhesions, whereas activity of RhoA is not required. It remains to be determined, however, whether EGF stimulation promotes endogenous Vav2 GEF activity toward Cdc42 and whether this stimulation is PKL dependent. In addition, Vav2 was also reported to demonstrate GEF activity toward RhoA (Abe et al., 2000), We were unable to detect endogenous Vav2-GEF activity toward RhoA under the conditions used to promote Vav2-dependent recruitment of the PKL to adhesions (data not shown). However, pull-down assays using the Rho-G17A mutant suggest that Vav2 exchange activity toward RhoA is enhanced in CHO-K1 cells coexpressing PKL and Vav2. The possibility that the PKL/Vav2 axis may be regulating RhoA activity is also supported by the presence of robust stress fibers in the “active” Vav2 phenotype and the observation that knockdown of either PKL or Vav2 in HT1080 cells results in cells with elongated tails typical of suppressed RhoA activity (Worthylake et al., 2001) when spread on FN in the presence of EGF for 2 h (data not shown). These data suggest that, at later time points, PKL could play a role in facilitating the switch of Vav2 activity toward RhoA to regulate focal adhesion maturation and disassembly.
In migrating HT1080 cells, Vav2 knockdown results in a reduction in cell polarity and directionality during cell migration, similar to observations made in NIH 3T3 cells expressing nonphosphorylatable GFP-PKL-3YF (Yu et al., 2009). A consequence of this loss of polarity is the inability to localize PKL/GIT1 and β-PIX to the leading edge of migrating cells. Therefore the effects of expressing GFP-PKL-3YF on cell migration might occur, at least in part, as a result of suppressed Vav2 activation. Previous reports demonstrated β-PIX recruitment to nascent adhesions at the leading edge of migrating cells (Loo et al., 2004; Nayal et al., 2006; ten Klooster et al., 2006; Kuo et al., 2011) and that β-PIX recruits Rac1 to adhesions via a direct interaction (ten Klooster et al., 2006). Activation of Rac1 by β-PIX promotes lamellipodial protrusion and nascent adhesion turnover (Nayal et al., 2006; Kuo et al., 2011), resulting in enhanced cell migration (Kuo et al., 2011). Of interest, β-PIX, like Vav2, is tyrosine phosphorylated downstream of EGF and Src activation, leading to its activation and increased adhesion turnover (Feng et al., 2010). In contrast to β-PIX, Vav2 has not been reported to localize to focal adhesions, nor have we observed it at adhesions in this study (data not shown), and it is therefore unlikely to be regulating Rac1 activity directly at this location. It is therefore possible that PKL plays a dual role in coordinating the spatiotemporal regulation of both Vav2 and β-PIX activities to coordinate Rho GTPase signaling during cell spreading and migration.
Of importance, the disregulation of Rho-family GTPase activity has been linked to numerous pathologies, including tumor growth and metastasis (Vega and Ridley, 2008). However, Rho-family GTPases are rarely mutated in human cancers. Instead, their abnormal activity most frequently results from overexpression or disregulation of their GEFs and GAPs. For instance, Vav proteins have been linked to the development of leukemias, melanoma, and pancreatic tumors (Billadeau, 2002; Bartolome et al., 2006; Kim et al., 2006; Prieto-Sanchez et al., 2006). In particular, Vav2 promotes invasion in melanoma (Bartolome et al., 2006), head and neck squamous cell carcinoma (Patel et al., 2007), and oral squamous cell carcinoma (Lai et al., 2008). Furthermore, Vav2 signaling regulates endothelial cell migration and angiogenesis, suggesting a possible role in tumor vascularization (Hunter et al., 2006; Garrett et al., 2007). Therefore, the identification of PKL as an upstream activator of Vav2 suggests that tumors in which expression of both PKL and Vav2 is elevated or phosphorylation of PKL is enhanced might have elevated Vav2 activity and thus an increased potential for invasion and metastasis.
MATERIALS AND METHODS
Cell culture and reagents
HT1080 cells were cultured in MEM, 10% fetal bovine serum (FBS), 50 U/ml penicillin, and 50 μg/ml streptomycin. CHO.K1 cells were cultured in modified Ham's F12, 10% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cells were maintained at 37ºC and 5% CO2. The following antibodies were used for immunofluorescence and Western blotting: anti-GM130, anti-PKL/GIT1, anti-paxillin clone 165 (BD Transduction Laboratories, Lexington, KY), anti-paxillin clone H114, anti-GFP, anti–phospho Vav2 Y172 (Santa Cruz Biotechnology, Santa Cruz, CA), anti–α-actinin (Sigma-Aldrich, St. Louis, MO), anti-HA (Covance, Berkeley, CA), anti-Vav2 (Abcam, Cambridge, MA), anti-PKL (Cell Signaling Technology, Beverly, MA), and anti–β-PIX (Millipore, Billerica, MA). pEGFP.C1 PKL wild-type (PKL), PKL Y286F/Y392F/Y592F (PKL-3YF), and PKL-ΔPBS2 were described previously (Brown et al., 2005). HA-tagged Vav1, Vav2, and Vav3 cDNAs were purchased from ABgene (Epsom, United Kingdom). HA-constitutively active (CA)-Vav2 (consisting of only the DH, PH, and cysteine-rich domains of Vav2) and T7-tagged dominant-negative L342R/L343S Vav2 (RS)-Vav2 cDNA were generous gifts from Christopher Carpenter (Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA).
Transfections
CHO-K1 cells were transfected using Fugene6 (Roche, Basel, Switzerland) at a 3:1 ratio as per manufacturer's instructions and expression allowed to occur over 24 h. For HT1080 siRNA transfections, cells were transfected at 30–50% confluency using Lipofectamine-2000 (Invitrogen, Carlsbad, CA) according to manufacturer's instructions. Sequences for siRNAs (Ambion, Austin, TX) used are as follows: human PKL, 5′-CAACTTCTTTCATCCTGAATT-3′ and 5′-CAATGGTGCTAACTCTATATT-3′; human Vav2, 5′-TCACAGAGGCCAAGAAATT-3′; control, 5′-ACUCUAUCUGCACGCUGACUU-3′. Cells were used at 72 h posttransfection.
GST-binding assays
CHO.K1 cells transfected with GFP-PKL, GFP-PKL-3YF, or empty vector control, with or without HA-Vav2, were spread on 10 μg/ml FN for 2 h. Cells were lysed in 1% Triton lysis buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 10 mM MgCl2, 0.2 mM Na3VO4, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 10 mg/ml leupeptin). Clarified lysates were incubated end over end with GST-Vav2 SH2, GST-RacG15A, or GST-RhoG17A fusion proteins as indicated at 4ºC for 1 h and then washed three times in lysis buffer. Samples were solubilized in 2× SDS-sample buffer and evaluated by Western blotting.
Immunoprecipitation and Western blotting
CHO.K1 cells cotransfected with GFP-PKL, GFP-PKL-3YF, or empty vector control along with HA-Vav2 were spread on FN for 2 h and then lysed in 1% Triton lysis buffer. For assessment of endogenous Vav2 phosphorylation in HT1080 cells, cells were lysed in 1× nondenaturing lysis buffer (75 mM Tris, pH 7.6, 200 mM NaCl, 15 mM NaF, 1.5 mM Na3VO4, 7.5 mM EDTA, 7.5 mM ethylene glycol tetraacetic acid, 1% Triton X-100, 0.75% NP-40, 1 mM PMSF, and 10 mg/ml leupeptin). Clarified lysates were incubated end over end for 2 h at 4ºC with primary antibody and then an additional hour with protein A/G beads (Santa Cruz Biotechnology) before washing three times with lysis buffer. Immunoprecipitates were solubilized in sample buffer and analyzed by Western blot. For blotting, samples were run on 10% SDS–PAGE and transferred to nitrocellulose. Primary antibodies were incubated for 2 h, followed by 1-h incubation on secondary horseradish peroxidase–conjugated antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) at room temperature. Western blots were visualized by chemiluminescence using ECL (GE Healthcare Bio-Sciences, Piscataway, NJ) on a Bio-Rad (Hercules, CA) imaging system.
Immunofluorescence
Glass coverslips were coated with FN at 10 μg/ml at 4°C in phosphate-buffered saline (PBS) containing magnesium and calcium overnight. Coverslips were washed with PBS and blocked with 1% bovine serum albumin (BSA) in PBS. CHO.K1 and HT1080 cells were seeded at 4 × 104 cells/ml and allowed to adhere in the conditions indicated. Cells were subsequently fixed and permeabilized simultaneously using 4% paraformaldehyde with 0.5% Triton X-100 in PBS, quenched with 0.1 M glycine in PBS, and blocked overnight at 4°C with 3% (wt/vol) BSA. Fixed cells were stained with primary antibodies at 1:200 in 3% BSA in PBS for 2 h at room temperature. Rhodamine–phalloidin (1:1000) (Invitrogen) was used to visualize F-actin. Secondary antibodies (Jackson ImmunoResearch Laboratories) were used at 1:300 for 1 h at room temperature as indicated. PBS with 0.05% Tween-20 was used for subsequent washes. Cells were imaged using the Leica SP5 scanning confocal with a HCX PL APO 63×/1.40–0.60 Oil λ BL objective (Leica, Bannockburn, IL), and image analysis was performed using ImageJ (National Institutes of Health, Bethesda, MD).
Quantification of focal adhesion–localized proteins
Confocal immunofluorescence images were acquired using consistent acquisition settings per experiment and then background subtracted using rolling ball subtraction. After this, images of paxillin staining were thresholded to define focal adhesions, and masks were generated. Using a size cutoff of 0.1 μm, we then used these masks to acquire intensity values for paxillin and PKL/GIT1 or β-PIX at each adhesion within the cell. The ratio of PKL/GIT1 and PIX intensity to paxillin intensity at focal adhesions was subsequently determined such that the mean intensity of PKL/GIT1 and PIX at adhesions per cell was expressed relative to paxillin staining of the same cell. Pearson's r was determined using paxillin masks to define adhesions and background-subtracted images of the GFP-tagged protein of interest and the JACOP plug-in in ImageJ.
Wound healing
For wound-healing assays, cells were plated to confluence on FN-coated 24-well tissue culture dishes or glass coverslips. The monolayer was scraped using a pipette tip and washed four times with PBS. Time-lapse images were acquired every 3 min over a time course of 16 h on a Nikon Eclipse Ti microscope using a 10×/0.30 PL FLUOR Nikon objective and equipped with a Hamamatsu Orca R2 camera (Hamamatsu, Hamamatsu City, Japan) and Nikon NS-Elements software (Nikon, Melville, NY). Individual cells were tracked using ImageJ to acquire values for directionality and velocity. For immunofluorescence imaging, coverslips were fixed 2 hr postwounding and stained with primary antibodies as indicated. Cells at the wound edge with Golgi polarized to the front-facing 120° sector were scored as positive for polarization. Polarization of PKL/GIT1 and β-PIX in migrating cells was determined by counting the proportion of cells at the wound edge that demonstrated focal adhesion staining of PKL/GIT1 and β-PIX at the leading edge.
Statistical analysis
Values were calculated from at least three independent experiments and were compared by Student's t test, and p < 0.05 was considered statistically significant. Error bars represent SEM.
Supplementary Material
Acknowledgments
We thank members of the Turner lab, along with Patrick Caswell and Mark Morgan, for their insights and helpful discussions. We also gratefully acknowledge Chris Carpenter and Keith Burridge for providing reagents. This work was supported by National Institutes of Health Grants RO1 GM47607 (C.E.T.) and CA82258 (B.J.M.).
Abbreviations used:
- EGF
epidermal growth factor
- FN
fibronectin
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- PH
pleckstrin homology, PI3K, phosphoinositol-3-kinase
- PIP3
phosphatidylinositol (3,4,5)-triphosphate
- PKL
paxillin kinase linker
- SH2
Src homology 2
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-09-0654) on April 24, 2013.
*Present address: Wellcome Trust Center for Cell Matrix Research, Faculty of Life Sciences, University of Manchester, Manchester M13 9PT, United Kingdom.
REFERENCES
- Abe K, Rossman KL, Liu B, Ritola KD, Chiang D, Campbell SL, Burridge K, Der CJ. Vav2 is an activator of Cdc42, Rac1, and RhoA. J Biol Chem. 2000;275:10141–10149. doi: 10.1074/jbc.275.14.10141. [DOI] [PubMed] [Google Scholar]
- Bartolome RA, Molina-Ortiz I, Samaniego R, Sanchez-Mateos P, Bustelo XR, Teixido J. Activation of Vav/Rho GTPase signaling by CXCL12 controls membrane-type matrix metalloproteinase-dependent melanoma cell invasion. Cancer Res. 2006;66:248–258. doi: 10.1158/0008-5472.CAN-05-2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billadeau DD. Cell growth and metastasis in pancreatic cancer: is Vav the Rho'd to activation. Int J Gastrointest Cancer. 2002;31:5–13. doi: 10.1385/IJGC:31:1-3:5. [DOI] [PubMed] [Google Scholar]
- Booden MA, Campbell SL, Der CJ. Critical but distinct roles for the pleckstrin homology and cysteine-rich domains as positive modulators of Vav2 signaling and transformation. Mol Cell Biol. 2002;22:2487–2497. doi: 10.1128/MCB.22.8.2487-2497.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Broussard JA, Webb DJ, Kaverina I. Asymmetric focal adhesion disassembly in motile cells. Curr Opin Cell Biol. 2008;20:85–90. doi: 10.1016/j.ceb.2007.10.009. [DOI] [PubMed] [Google Scholar]
- Brown MC, Cary LA, Jamieson JS, Cooper JA, Turner CE. Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol Biol Cell. 2005;16:4316–4328. doi: 10.1091/mbc.E05-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, West KA, Turner CE. Paxillin-dependent paxillin kinase linker and p21-activated kinase localization to focal adhesions involves a multistep activation pathway. Mol Biol Cell. 2002;13:1550–1565. doi: 10.1091/mbc.02-02-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Baird D, Yoo S, Antonyak M, Cerione RA. Phosphorylation of the cool-1/beta-Pix protein serves as a regulatory signal for the migration and invasive activity of Src-transformed cells. J Biol Chem. 2010;285:18806–18816. doi: 10.1074/jbc.M109.098079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 2006;406:425–437. doi: 10.1016/S0076-6879(06)06031-9. [DOI] [PubMed] [Google Scholar]
- Garrett TA, Van Buul JD, Burridge K. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp Cell Res. 2007;313:3285–3297. doi: 10.1016/j.yexcr.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- Hunter SG, Zhuang G, Brantley-Sieders D, Swat W, Cowan CW, Chen J. Essential role of Vav family guanine nucleotide exchange factors in EphA receptor-mediated angiogenesis. Mol Cell Biol. 2006;26:4830–4842. doi: 10.1128/MCB.02215-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011;3:a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Nozu F, Kusama K, Imawari M. Cholecystokinin stimulates the recruitment of the Src-RhoA-phosphoinositide 3-kinase pathway by Vav-2 downstream of G(alpha13) in pancreatic acini. Biochem Biophys Res Commun. 2006;339:271–276. doi: 10.1016/j.bbrc.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Kuo JC, Han X, Hsiao CT, Yates JR, 3rd, Waterman CM. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol. 2011;13:383–393. doi: 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai SY, Ziober AF, Lee MN, Cohen NA, Falls EM, Ziober BL. Activated Vav2 modulates cellular invasion through Rac1 and Cdc42 in oral squamous cell carcinoma. Oral Oncol. 2008;44:683–688. doi: 10.1016/j.oraloncology.2007.08.017. [DOI] [PubMed] [Google Scholar]
- LaLonde DP, Grubinger M, Lamarche-Vane N, Turner CE. CdGAP associates with actopaxin to regulate integrin-dependent changes in cell morphology and motility. Curr Biol. 2006;16:1375–1385. doi: 10.1016/j.cub.2006.05.057. [DOI] [PubMed] [Google Scholar]
- Lee J, et al. p85 beta-PIX is required for cell motility through phosphorylations of focal adhesion kinase and p38 MAP kinase. Exp Cell Res. 2005;307:315–328. doi: 10.1016/j.yexcr.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Liu BP, Burridge K. Vav2 activates Rac1, Cdc42, and RhoA downstream from growth factor receptors but not beta1 integrins. Mol Cell Biol. 2000;20:7160–7169. doi: 10.1128/mcb.20.19.7160-7169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo TH, Ng YW, Lim L, Manser E. GIT1 activates p21-activated kinase through a mechanism independent of p21 binding. Mol Cell Biol. 2004;24:3849–3859. doi: 10.1128/MCB.24.9.3849-3859.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida K, et al. High-throughput phosphotyrosine profiling using SH2 domains. Mol Cell. 2007;26:899–915. doi: 10.1016/j.molcel.2007.05.031. [DOI] [PubMed] [Google Scholar]
- Marcoux N, Vuori K. EGF receptor mediates adhesion-dependent activation of the Rac GTPase: a role for phosphatidylinositol 3-kinase and Vav2. Oncogene. 2003;22:6100–6106. doi: 10.1038/sj.onc.1206712. [DOI] [PubMed] [Google Scholar]
- Marignani PA, Carpenter CL. Vav2 is required for cell spreading. J Cell Biol. 2001;154:177–186. doi: 10.1083/jcb.200103134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazaki Y, et al. Neutrophil direction sensing and superoxide production linked by the GTPase-activating protein GIT2. Nat Immunol. 2006;7:724–731. doi: 10.1038/ni1349. [DOI] [PubMed] [Google Scholar]
- Nabi IR. The polarization of the motile cell. J Cell Sci. 1999;112(Pt 12):1803–1811. doi: 10.1242/jcs.112.12.1803. [DOI] [PubMed] [Google Scholar]
- Nayal A, Webb DJ, Brown CM, Schaefer EM, Vicente-Manzanares M, Horwitz AR. Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J Cell Biol. 2006;173:587–589. doi: 10.1083/jcb.200509075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiya N, Kiosses WB, Han J, Ginsberg MH. An alpha4 integrin-paxillin-Arf-GAP complex restricts Rac activation to the leading edge of migrating cells. Nat Cell Biol. 2005;7:343–352. doi: 10.1038/ncb1234. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A, Podtelejnikov AV, Blagoev B, Bustelo XR, Mann M, Lodish HF. Analysis of receptor signaling pathways by mass spectrometry: identification of vav-2 as a substrate of the epidermal and platelet-derived growth factor receptors. Proc Natl Acad Sci USA. 2000;97:179–184. doi: 10.1073/pnas.97.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V, Rosenfeldt HM, Lyons R, Servitja JM, Bustelo XR, Siroff M, Gutkind JS. Persistent activation of Rac1 in squamous carcinomas of the head and neck: evidence for an EGFR/Vav2 signaling axis involved in cell invasion. Carcinogenesis. 2007;28:1145–1152. doi: 10.1093/carcin/bgm008. [DOI] [PubMed] [Google Scholar]
- Pegtel DM, Ellenbroek SI, Mertens AE, van der Kammen RA, de Rooij J, Collard JG. The Par-Tiam1 complex controls persistent migration by stabilizing microtubule-dependent front-rear polarity. Curr Biol. 2007;17:1623–1634. doi: 10.1016/j.cub.2007.08.035. [DOI] [PubMed] [Google Scholar]
- Prieto-Sanchez RM, Hernandez JA, Garcia JL, Gutierrez NC, San Miguel J, Bustelo XR, Hernandez JM. Overexpression of the VAV proto-oncogene product is associated with B-cell chronic lymphocytic leukaemia displaying loss on 13q. Br J Haematol. 2006;133:642–645. doi: 10.1111/j.1365-2141.2006.06094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuebel KE, Bustelo XR, Nielsen DA, Song BJ, Barbacid M, Goldman D, Lee IJ. Isolation and characterization of murine vav2, a member of the vav family of proto-oncogenes. Oncogene. 1996;13:363–371. [PubMed] [Google Scholar]
- Schuebel KE, Movilla N, Rosa JL, Bustelo XR. Phosphorylation-dependent and constitutive activation of Rho proteins by wild-type and oncogenic Vav-2. EMBO J. 1998;17:6608–6621. doi: 10.1093/emboj/17.22.6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servitja JM, Marinissen MJ, Sodhi A, Bustelo XR, Gutkind JS. Rac1 function is required for Src-induced transformation. Evidence of a role for Tiam1 and Vav2 in Rac activation by Src. J Biol Chem. 2003;278:34339–34346. doi: 10.1074/jbc.M302960200. [DOI] [PubMed] [Google Scholar]
- Tamas P, Solti Z, Bauer P, Illes A, Sipeki S, Bauer A, Farago A, Downward J, Buday L. Mechanism of epidermal growth factor regulation of Vav2, a guanine nucleotide exchange factor for Rac. J Biol Chem. 2003;278:5163–5171. doi: 10.1074/jbc.M207555200. [DOI] [PubMed] [Google Scholar]
- Tamas P, Solti Z, Buday L. Membrane-targeting is critical for the phosphorylation of Vav2 by activated EGF receptor. Cell Signal. 2001;13:475–481. doi: 10.1016/s0898-6568(01)00172-3. [DOI] [PubMed] [Google Scholar]
- ten Klooster JP, Jaffer ZM, Chernoff J, Hordijk PL. Targeting and activation of Rac1 are mediated by the exchange factor beta-Pix. J Cell Biol. 2006;172:759–769. doi: 10.1083/jcb.200509096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu S, Wu WJ, Wang J, Cerione RA. Epidermal growth factor-dependent regulation of Cdc42 is mediated by the Src tyrosine kinase. J Biol Chem. 2003;278:49293–49300. doi: 10.1074/jbc.M307021200. [DOI] [PubMed] [Google Scholar]
- Turner CE. Paxillin and focal adhesion signalling. Nat Cell Biol. 2000;2:E231–E236. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, Bagrodia S, Thomas S, Leventhal PS. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J Cell Biol. 1999;145:851–863. doi: 10.1083/jcb.145.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582:2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Webb DJ, Parsons JT, Horwitz AF. Adhesion assembly, disassembly and turnover in migrating cells—over and over and over again. Nat Cell Biol. 2002;4:E97–E100. doi: 10.1038/ncb0402-e97. [DOI] [PubMed] [Google Scholar]
- West KA, Zhang H, Brown MC, Nikolopoulos SN, Riedy MC, Horwitz AF, Turner CE. The LD4 motif of paxillin regulates cell spreading and motility through an interaction with paxillin kinase linker (PKL) J Cell Biol. 2001;154:161–176. doi: 10.1083/jcb.200101039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormer D, Deakin NO, Turner CE. CdGAP regulates cell migration and adhesion dynamics in two-and three-dimensional matrix environments. Cytoskeleton (Hoboken) 2012;69:644–658. doi: 10.1002/cm.21057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthylake RA, Lemoine S, Watson JM, Burridge K. RhoA is required for monocyte tail retraction during transendothelial migration. J Cell Biol. 2001;154:147–160. doi: 10.1083/jcb.200103048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JA, Deakin NO, Turner CE. Paxillin-kinase-linker tyrosine phosphorylation regulates directional cell migration. Mol Biol Cell. 2009;20:4706–4719. doi: 10.1091/mbc.E09-07-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.