

Abstract
Most high Mr thioredoxin reductases (TRs) have the unusual feature of utilizing a vicinal disulfide bond (Cys1-Cys2) which form an eight-membered ring during the catalytic cycle. Many eukaryotic TRs have replaced the Cys2 position of the dyad with the rare amino acid selenocysteine (Sec). Here we demonstrate that Cys- and Sec-containing TRs are distinguished by the importance each class of enzymes places on the 8-membered ring structure in the catalytic cycle. This hypothesis was explored by studying the truncated enzyme missing the C-terminal ring structure in conjunction with oxidized peptide substrates to investigate the reduction and opening of this dyad. The peptide substrates were identical in sequence to the missing part of the enzyme, containing either a disulfide or selenylsulfide linkage, but were differentiated by the presence (cyclic) and absence (acyclic) of the ring structure. The ratio of these turnover rates informs that the ring is only of modest importance for the truncated mouse mitochondrial Sec-TR (ring/no ring = 32), while the ring structure is highly important for the truncated Cys-TRs from D. melanogaster and C. elegans (ring/no ring > 1000). All three enzymes exhibit a similar dependence upon leaving group pKa as shown by the use of the acyclic peptides as substrates. These two factors can be reconciled for Cys-TRs if the ring functions to simultaneously allow for attack by a nearby thiolate while correctly positioning the leaving group sulfur atom to accept a proton from the enzymic general acid. For Sec-TRs the ring is unimportant because the lower pKa of the selenol relative to a thiol obviates its need to be protonated upon S-Se bond scission and permits physical separation of the selenol and the general acid. Further study of the biochemical properties of the truncated Cys and Sec TR enzymes demonstrates that the chemical advantage conferred on the eukaryotic enzyme by a selenol is the ability to function at acidic pH.
The thioredoxin system, comprised of thioredoxin reductase (TR), thioredoxin (Trx), and NADPH, is essential for maintaining redox balance in eukaryotic cells (1-2). Most eukaryotes contain a high Mr TR that is very similar in structure and mechanism to glutathione reductase (GR) (3). Both enzymes use NADPH as a redox cofactor to reduce a conserved disulfide redox center (…CVNVGC…). TR can be distinguished from GR by the presence of a C-terminal extension of 16 amino acids that contains an additional redox center that is functionally equivalent to oxidized glutathione (GSSG), the disulfide substrate for GR. In the case of GR, the reduced product (GSH) acts as a small molecule shuttle to carry reducing equivalents to many targets, especially glutaredoxin. TR however, utilizes the C-terminal redox center as an internal substrate shuttle (as part of the polypeptide chain) to convey reducing equivalents directly to the target substrate (Trx).
The C-terminal redox center of many high Mr TRs are unique in that they contain a conserved tetrapeptide motif of Xaa-Cys1-Cys2-Xaa (where Xaa is usually Gly or Ser) where Cys1 and Cys2 form a rare vicinal disulfide bond during the catalytic cycle (4). This type of disulfide bond has only been found in ~50 protein structures deposited in the Protein Data Bank (PDB) (5). The [Cys-Cys]ox dyad is part of a two residue turn that can fit the molecular constraints of types I, II, VIa, VIb, and VIII beta turns (5). In a few instances, it is thought to be part of a regulatory conformational switch as this motif can undergo a cis/trans isomerization of the peptide bond between Cys1 and Cys2 that would dramatically alter the protein topology where it is found (6).
Mammalian and other higher eukaryotic TRs have the interesting and unique feature of replacing Cys2 with a selenocysteine (Sec, U) residue (7). This Sec residue is essential to the function of the mammalian enzyme since mutation of Sec to Cys causes a large drop in kcat (8), however the presence of a selenium atom is not chemically required to catalyze the reduction of the disulfide bond of Trx (9). This point is further illustrated by the data in Table 1, which summarizes kinetic data for Sec-containing enzymes from mammals contrasted with Cys-containing TRs from D. melanogaster (DmTR), C. elegans (CeTR2), and A. gambiae (AgTR). These three Cys-TRs have a conventional Cys residue in the Cys2 position of the tetrapeptide motif and DmTR and AgTR catalyze the reduction of their respective cognate substrates with comparable catalytic efficiencies to the mammalian enzyme with only slightly reduced kcat values. The value of kcat for CeTR is substantial, although E. coli Trx was used as the substrate. In contrast, mutation of Sec to Cys in the mammalian enzyme results in losses of 175-fold and 540-fold in kcat for the cytosolic rat enzyme (rTR) and mitochondrial mouse enzyme (mTR3), respectively2 (8,10).
Table 1.
Kinetic Constants of Various TRs for the Reduction of Thioredoxin.
| Enzyme-tetrapeptide | Substrate | kcat (min−1) | Km (μM) | kcat/Km (min−1•M−1) |
|---|---|---|---|---|
| rTR-GCUGa | H. sapiens Trx | 2500 | 3.3 | 7.6 ×108 |
| rTR-GCCGa | H. sapiens Trx | 14.3 | 0.4 | 3.6 ×107 |
| mTR-GCUGb | E. coli Trx | 2220 | 70 | 3.3 ×107 |
| mTR-GCCGb | E. coli Trx | 4.1 | 49 | 8.4 ×104 |
| CeTR2-GCCGc | E. coli Trx | 610 | 610 | 1 ×106 |
| DmTR-SCCSd | D. m. Trx | 1320 | 7.1 | 1.9 ×108 |
| DmTR-SCCSb | E. coli Trx | 300 | 170 | 1.7 ×106 |
| AgTR-TCCSe | A. g. Trx | 924 | 8.5 | 1.13 ×108 |
Why is the activity of the mammalian enzyme so greatly affected by the loss of a selenium atom when the data in Table 1 shows that a sulfur atom can replace it in other eukaryotes without a dramatic loss in activity? As shown in Figure 1A, the presumptive nucleophile for the reduction of the disulfide bond of Trx is the residue that occupies the Cys2 position (X represents either S or Se in Figure 1) of the Cys1-Cys2 dyad. A reasonable assumption (though not explicitly stated in the literature) is that the loss of nucleophilicity that results from Sec to Cys mutation is responsible for the decrease in rate. The nucleophilicity of the Cys2 residue could be increased by lowering its pKa and low pKa thiolates in enzymes are certainly known (13). A mechanism for lowering the pKa of Cys2 (the presumptive nucleophile – Cys490’) in DmTR has been proposed (14-15).
Figure 1.

Comparison of the catalytic mechanisms of full-length TR (A), truncated TR utilizing a peptide substrate (B), GR utilizing oxidized GSSG as a substrate (C), and truncated TR utilizing the highly reactive disulfide DTNB as a substrate (D). In each case, the oxidoreductase accepts reducing equivalents from NADPH, which transfers a hydride to a non-covalently bound FAD cofactor. The N-terminal redox active disulfide is then reduced to form a charge transfer complex (thiolate-flavinCT), while the liberated interchange cysteine (SHIC) then either attacks the C-terminal redox center of the neighboring subunit (designated with a prime symbol and shown in red), which we refer to as the “ring opening step,” or attacks the substrate (also shown in red). Upon ring opening in the case of full-length TR, the nucleophile (Nuc’X either S from Cys or Se from Sec) forms a mixed disulfide with Trx, which is subsequently resolved by the vicinal cysteine (Res’SH) to reform the eight member ring and release the product. As can be seen in A and B, GR is essentially a truncated TR missing its C-terminal sixteen residues with GSSG (red) functionally equivalent to the TR C-terminal redox center. Oxidized cyclic and acyclic peptides (red – see Figure 2) that correspond to the C-terminal sequence of the missing TR can be reduced by the truncated enzyme. The arrow shows the direction of electron flow from NADPH to the substrate.
While attenuating the nucleophilicity of the thiolate in a Cys-TR is one possible mechanistic explanation for the high catalytic efficiencies of Cys-TRs relative to Sec-TRs, other possibilities do exist. Figure 1 shows the mechanistic similarity of TR and GR, with TR containing an extra thiol-disulfide exchange step before electrons are transferred to the substrate. We refer to the additional thiol-disulfide exchange step of TR as the “ring opening” step because it involves reduction and opening of the eight-membered ring formed by the Cys1-Cys2/Sec2 dyad (16). We have previously investigated this step in the mechanism because we noted that the truncated enzyme, although mechanistically similar to GR, could only utilize a highly reactive disulfide such as 5,5′dithio-bis(2-nitrobenzoic acid) (DTNB) as a substrate, but not other simple disulfides such as cystine or GSSG. This observation led us to investigate the requirement for Sec in the mammalian enzyme by using the truncated enzyme and peptide disulfide substrates to study the ring opening reaction (16-17).
Here we test the hypothesis that the difference between Cys-containing TRs and Sec-containing TRs is the dependence each type of enzyme has on the unique eight-membered ring structure in the ring opening step of the catalytic cycle. Our approach was to synthesize peptide substrates with an eight-membered ring structure formed by a vicinal disulfide/selenylsulfide (cyclic peptide), or peptides substrates containing an interchain disulfide/selenylsulfide linking two peptides together (acyclic peptide). The acyclic peptide had the same sequence as the cyclic peptide except that the peptide bond of the Cys1-Cys2/Sec2 dyad is “broken” in comparison to the cyclic peptide (Figure 2). A second type of acyclic peptide was also synthesized that replaced the Cys2/Sec2 position with a 5-thio-2-nitrobenzoate group. Lastly, in order to differentiate the function of each position of the dyad, we also synthesized peptides in which Sec was placed in the Cys1 position. Our results using these unique substrates provide a mechanistic explanation for the presence of Sec in the mammalian enzyme.
Figure 2.

Peptide substrates constructed for this study: A) Cyclic, oxidized octamer; B) Acyclic, oxidized octamer; C) Acyclic hexamer in which Cys1 forms a mixed disulfide with the thiol of TNB, referred to as Pep-TNB in the text; and D) Cyclic octamer “switch” peptide in which Sec and Cys and have switched positions in the dyad. In all cases X = S or Se.
MATERIALS AND METHODS
Materials
DTNB, NADPH, were purchased from Sigma-Aldrich (St. Louis, MO). All restriction endonucleases, Vent DNA polymerase, and T4 DNA ligase were purchased from New England Biolabs (Ipswich, MA) and used with the supplied buffers according to the manufacturer’s guidelines. DNA primers were purchased from IDT (Coralville, IA). Yeast GR was purchased from Roche Diagnostics Corp. (Mannheim, Germany). Fmoc-protected amino acids and HBTU were purchased from Synbiosci Corporation (Livermore, CA). Resins for solid phase peptide synthesis were purchased from Novabiochem (San Diego, CA) and Applied Biosystems (Foster City, CA). Boc-Cys(5-Npys)-OH was purchased from Bachem (King of Prussia, PA). Fmoc-Sec(Mob)-OH was manufactured using a procedure previously reported (18-19). Solvents for peptide synthesis and all other required reagents were purchased from Fisher Scientific (Pittsburgh, PA) and were of reagent grade or better.
Cloning of Truncated TRs
Previously we reported the cloning of full-length wild-type (WT) DmTR as well as mutant C-terminal variants (10,16). Following identical cloning conditions from our previous reports, we constructed a truncated DmTR missing the final eight amino acids (PTPASCCS) from the C-terminus and we term this construct DmTRΔ8. For the cloning of this construct, upstream and downstream primers having respective sequences of 5′-AACAGACATATGGCGCCCGTGCAAGG-3′ and 5′-ACAGCCGGTACCTTAGTCCAGTCCGGAGCGCTTGGTGAT-3′ were used to construct the truncated DmTR for production as a recombinant protein in E. coli cells.
For construction of a truncated CeTR2 missing the C-terminal eight amino acids (PRTQGCCG) termed CeTR2Δ8, a clone incorporating a hexa-histidine tag at the N-terminus was constructed using an upstream primer of sequence 5′-ACAGCCCCGGATCCCTTCTCATCAAATAAATTTGATCTG-3′ and downstream primer of sequence 5′-ACAGCCAAGCTTGTCGACTCAGTCTTGTCCAGATCGTTTTGTAAT-3′ using identical amplification conditions to those above and reported previously (11). The construction of a truncated mTR3 missing the C-terminal eight amino acids (PTVTGCUG) termed mTR3Δ8 has been reported elsewhere (17).
Purification of Truncated TRs
The purification of mTR3Δ8 has been described in detail previously (17). For the production of recombinant enzymes CeTRΔ8 and DmTRΔ8, the constructed plasmids were transformed into E. coli ER2566 cells and grown overnight on LB plates containing 200μg/mL ampicillin. A single isolated colony was inoculated in 100 mL LB media containing 200μg/mL ampicillin and incubated overnight at 37 °C. The overnight inoculum was pelleted by centrifugation and resuspended in 30 mL of fresh TB (Terrific broth) media. Three, one-liter pyrex Fernbach flasks, each containing TB media containing a final concentration of 200μg/mL ampicillin and supplemented with 20 mg/L of niacinamide, riboflavin and pyridoxine, were inoculated with 10 mL of resuspended transformed cells. The bacterial growth was incubated at 37 °C, until the A600 reached 1.0 at which point the cells were incubated on ice until the temperature of the culture reached 20 °C. Protein production was induced by addition of IPTG to a final concentration of 0.5 mM. Cells were grown overnight at 20 °C and then harvested by centrifugation in a Beckman J21B centrifuge (7000 × g, 10 min, 4 °C) and stored frozen at −20 °C.
For the purification of recombinant enzyme DmTRΔ8, the frozen cells were thawed on ice and resuspended in 200 mL of 10 mM potassium phosphate, 10 mM NaCl, pH 8.0 and incubated with lysozyme (1 mg/mL) at 4 °C for 30 min and followed by brief sonication with a Branson 350 Sonifier (Danbury, CT) to break open any unlysed cells. The cellular debris was cleared by centrifugation in a Beckman J21B centrifuge (JA-14 rotor) at 7000 × g for 90 min at 4 °C. The clarified supernatant was gravity loaded onto DEAE column (2.5 × 16.5 cm, 80 mL) equilibrated with 10 mM potassium phosphate, 10 mM NaCl, pH 8.0 and washed until the A280 was ~0.05. Protein elution was achieved using a linear NaCl gradient of 10 – 400 mM (500 mL total volume) in the column buffer. All fractions were analyzed by a Cary 50 UV-Vis spectrophotometer from Varian (Walnut Creek, CA) to determine A280 and A460. Peak fractions were analyzed by 12% reducing SDS-PAGE and fractions exhibiting A460 and an apparent MW of 55 kDa were pooled and dialyzed into TE (10 mM Tris, 1mM EDTA, pH 8.0) buffer containing 10 mM NaCl. The dialyzed protein was incubated on ice in the presence of 20 mM βME for 30 min and gravity loaded onto a column (1.5 × 15 cm, 20 mL) containing 2′,5′-ADP Sepharose 4B (Amersham Biosciences Pharmacia Biotech, Uppsala, Sweden) equilibrated with TE buffer, pH 8.0 with 10 mM NaCl and 20 mM βME. The loaded column was then washed with buffer until the A280 was below 0.05 and the protein was liberated from the column with TE buffer containing 3.0 M NaCl. Peak fractions were analyzed and pooled based on their A460 and presence of a 55 kDa band observed on 12% reducing SDS-PAGE. Pooled fractions were gravity loaded onto a Fast Flow 6 Low Substitution Phenyl Sepharose (Amersham Biosciences Pharmacia Biotech) column (2.5 × 13 cm, 60 mL) equilibrated in TE buffer, pH 8.0 with 3.0 M NaCl. Following washing with column buffer, elution was carried out using a 500 mL linear gradient of 3.0 – 0.0 M NaCl. Fractions were analyzed for A460 content and visualized by 12% reducing SDS-PAGE to judge purity.
For the purification of recombinant enzyme CeTR2Δ8, induced cells were thawed and then resuspended in 50 mM potassium phosphate, 500 mM NaCl, 10% glycerol, and 10 mM imidazole at pH 8.0 and incubated with lysozyme (1 mg/mL) at 4 °C for 30 min and then sonicated briefly. The cell lysate was cleared via centrifugation in a Beckman J21B centrifuge (JA-14 rotor) at 7000 × g for 90 min at 4 °C. The cleared supernatant was incubated with 20 mM βME for 30 min and then gravity loaded onto Ni-NTA agarose (Qiagen, Valencia, CA) column (2.5 × 5.0 cm, 20 mL) equilibrated with 50 mM potassium phosphate, 500 mM NaCl, 10% glycerol, and 10 mM imidazole, pH 8.0 and washed with 5 column volumes of buffer. The column was washed a second time with an additional 5 column volumes with column buffer containing 20 mM imidazole. Elution of the protein was achieved by addition of buffer containing 250 mM imidazole. The pooled fractions were then dialyzed against 50 mM potassium phosphate, 500 mM NaCl, 10% glycerol, and 1 mM dithiothreitol (DTT) pH 8. The dialysate was concentrated by ultrafiltration using an Amicon Ultra 30000 MWCO (Millipore) concentrator to 1 mL and loaded onto a PD10 desalting column.
Cloning and production of full-length WT mTR3 (20), DmTR (16), and CeTR2 (11) have been described previously. In this report the full-length enzymes are respectively abbreviated as mTR3-GCUG, DmTR-SCCS, and CeTR2-GCCG to indicate the sequence of their C-terminal redox active tetrapeptides.
Synthesis of Peptide Substrates
The amino acid sequence of cyclic and acyclic octamer peptides were chosen based upon the sequence of the C-terminus of each respective TR used in this study; PTVTGCUG for mTR3, PTPASCCS for DmTR, and PRTQGCCG for CeTR2. The procedure for the synthesis of the cyclic and acyclic versions of the C-terminal peptide for mTR3 has been recently published (17) and the corresponding peptides for DmTR and CeTR2 were synthesized using the same procedure. Here we also report on the synthesis of peptide fragments of mTR3, DmTR, and CeTR2 containing a mixed disulfide between Cys1 of the peptide and 5-thio-2-nitrobenzoate that we term as Pep-TNB (Figure 2C). These hexamer sequences were assembled on resin using standard Fmoc protocol and cleaved as previously described. Following Et2O trituration, the resulting crude peptide isolate was dissolved in 2 mL H2O. In a separate reaction vessel, 2 eq. DTNB was dissolved 2 mL 0.1M NH4HCO3. This mixture was added dropwise to the aqueous peptide solution over 5 min with constant stirring. Immediately following this addition, the reaction was acidified with a few drops of TFA, filtered through a 40 micron frit to remove excess precipitated DTNB, and injected in its entirety onto a preparative HPLC column. The fraction corresponding to the TNB conjugate was collected and lyophilized yielding a light yellow, crystalline product. The third type of peptide synthesized was of the sequence AA1-AA2-AA3-AA4-Sec1-Cys2-AA7-AA8 that we term the “switch peptide” because the position of the Cys and Sec residues have been exchanged. A switch peptide was made for mTR3 and DmTR using the native sequence of the respective enzyme for the remaining residues in the octapeptide. The procedures for the synthesis of the switch peptide, HPLC purification, characterization by MALDI-TOF mass spectrometry were identical to our previous report (17).
DTNB Reductase Activity
The TRΔ8 constructs were assayed for activity toward DTNB as described by Arner (21). All assays were performed on a Cary 50 UV-Vis spectrophotometer at 25 °C and pH 7.0 and were initiated by addition of enzyme. The concentration of homodimeric TR was determined using the flavin extinction coefficient of 22.6 mM−1•cm−1 (21). Activity was monitored over 2 min with Vo determined from the linear fit. Plots of Vo/ET versus substrate concentration were fit by the Michaelis-Menten equation using KaleidaGraph 4.02 from Synergy Software (Reading, PA) and activities are reported as moles of NADPH consumed per minute per mole of TR dimer.
The DTNB assay contained 0.2 mM NADPH and 10 mM EDTA in 100 mM potassium phosphate. At each concentration of DTNB, activity was corrected for background by addition of buffer only. Activity was measured by the increase in A412, calculated using the extinction coefficient for TNB-S− (5-thio-2-nitrobenzoate, 13.6 mM−1•cm−1), and divided by 2 to account for the production of two TNB-S− molecules per NADPH consumed. The concentrations of enzyme used in the assay were: 2 nM for mTR3Δ8, 5 nM for DmTRΔ8, and 5 nM for CeTR2Δ8.
pH Optima of DTNB Reduction
Activity toward DTNB was tested as a function of pH for constructs mTR3-GCUG, mTR3Δ8, DmTR-SCCS, DmTRΔ8, and yeast GR. Each assay contained buffer with a final concentration of 100 mM sodium acetate, MES, Tris, and dibasic potassium phosphate adjusted from pH 5.0 – 10.0 in intervals of 0.5 pH units. Each assay contained 150μM NADPH and 1 mM EDTA in a final volume of 500μL. The concentrations of enzyme and DTNB for each of the assays were as follows: 4 nM mTR3-GCUG/2.5 mM DTNB, 3 nM mTR3Δ8/2.5 mM DTNB, 4 nM DmTR-SCCS/1.0 mM DTNB, 5 nM DmTRΔ8/5.0 mM DTNB, and 50 nM yeast GR/0.1 mM DTNB. The activity was measured at A412 and background corrected, collected in triplicate, normalized to the percent of maximal activity for each given construct, and plotted as percent maximal activity versus pH.
We also assayed the truncated TRs and yeast GR using the mixed disulfide bond substrate denoted as Pep-TNB (Figure 2) as a function of pH. The assay conditions used were identical to those above and the following enzyme/substrate concentrations were used: 15 nM mTR3Δ8/0.5 mM PTVTGC-TNB, 25 nM DmTRΔ8/0.5 mM PTPASC-TNB, and 500 nM yeast GR/0.1mM PTPASC-TNB. Activity was monitored for changes in A340/min or A412/min as described above. Each pH point was assayed in triplicate and were background corrected against the spontaneous reduction of NADPH at acidic pH.
Assays using Peptide Disulfides as Substrates
All peptides were lyophilized, weighed and resuspended in a minimal amount of 500 mM potassium phosphate, pH 7.0, to create a working stock in the range of 30 – 60 mM. The peptide assay contained 0 – 25 mM peptide, 150μM NADPH, 1 mM EDTA, in 50 mM potassium phosphate at pH 7.0 in a volume of 500μL. Background activity was corrected for each concentration of peptide by performing an assay in which buffer was added instead of the peptide solution. A second control was performed in the presence of enzyme with peptide missing from the assay. Activity was measured by the decrease in A340 for the consumption of NADPH and calculated using an extinction coefficient of 6200 M−1•cm−1. The concentrations of TRΔ8 and corresponding peptides used in the assay are listed in Table S2 (Supporting Information). The concentration of each truncated TR was adjusted to achieve a signal to noise ratio that allowed a range of peptide concentrations to be assayed. When Pep-TNB was used as a substrate the assay conditions were identical to those discussed above except that A412 was used to follow the release of TNB-S− over time.
The cyclic versions of each peptide and Pep-TNB were also uses as substrates for yeast GR and assayed in triplicate. The assay solution contained 50 mM potassium phosphate pH 7.0, 1 mM EDTA, 0.15 mM NADPH, and 1 mM peptide. For cyclic, oxidized peptides the decrease in A340 was monitored, while for the TNB labeled peptide the increase in A412 was monitored.
Spectral Characteristics as a Function of pH
Purified protein was dialyzed and concentrated to ~30 mg/mL (275 μM) in 10 mM potassium phosphate containing 300 mM NaCl, 1 mM EDTA at pH 7.4. A reaction mixture (1 mL) was prepared in the cuvette containing 3.5 μM homodimeric TR (7 μM subunits) with the addition of 250 μM NADPH after the initial (oxidized TR) scan. Each reaction contained 150 mM potassium phosphate, 150 mM citrate, 300 mM NaCl, and 1 mM EDTA. The reaction was monitored between 240 nm and 700 nm for 5 min. Spectral scans were converted to extinction coefficient by determining the concentration of TR in each assay using the A462 and the extinction coefficient of monomeric TR, 11.3 mM−1•cm−1. Difference spectra were calculated to determine the peak wavelength for the thiolate-flavin charge-transfer complex by subtracting the absorbance at each wavelength of the oxidized enzyme from the absorbance of the reduced form of the enzyme. After a 5 min incubation with NADPH, 50 μM DTNB was added to the cuvette to evaluate the thiol redox state. DTNB was added to the oxidized enzyme as a control.
RESULTS AND DISCUSSION
Purification of Enzymes
mTRΔ8 and DmTRΔ8 enzymes were purified to homogeneity using three different chromatographic steps as judge by 12% SDS-PAGE as shown in Figure S1 (Supporting Information). Protein yields for mTRΔ8 and DmTRΔ8 were 32 mg/L and 156 mg/L of culture, respectively. The clone for CeTR2Δ8 contained a hexa-histidine tag, which allowed for single step purification by an affinity column and had a similar purity as the other TRs in this study as judged by SDS-PAGE (Figure S1). The protein yield for CeTR2Δ8 was 35 mg/L of cell culture.
DTNB Reductase Activity
A summary of the DTNB reductase activity for the truncated TRs missing their final eight amino acids is presented in Table 2. All truncated enzymes maintained their ability to reduce DTNB at rates comparable to their corresponding full-length WT enzymes. In the case of mTR3Δ8, its kcat increased 2.3-fold, while Km also increased 1.8-fold, so that the truncated enzyme has nearly an identical catalytic efficiency as the full-length enzyme. DmTRΔ8 followed a similar pattern; its kcat increased 8.2-fold, while the Km increased 18.6-fold, resulting in a decrease in catalytic efficiency of 2.3-fold. The kinetic constants for CeTRΔ8 were essentially the same as its full-length enzyme.
Table 2.
Kinetic Constants for the Reduction of DTNB
| Enzyme | kcat (min−1) | Km (mM−1) | kcat/Km (min−1•mM−1) |
|---|---|---|---|
| mTR3-GCUGa | 1251 ± 71 | 0.45 ± 0.09 | 2720 |
| mTR3-Δ8 | 2905 ± 232 | 0.83 ± 0.27 | 3500 |
| DmTR-SCCSa | 157 ± 12 | 0.22 ± 0.07 | 713 |
| DmTR-Δ8 | 1294 ± 277 | 4.1 ± 2.1 | 316 |
| CeTR2-GCCGa | 134 ± 7 | 0.41 ± 0.09 | 327 |
| CeTR2-Δ8 | 152 ± 14 | 0.53 ± 0.19 | 287 |
Previously reported (10).
pH Optima of DTNB Reduction
To evaluate the pH dependence of the reduction of DTNB in the truncated enzymes and their full-length WT counterparts, we performed pH rate profiles. The truncated mouse enzyme shows a pH optimum for the reduction of DTNB at pH 6.5, while truncated DmTR has a pH optimum at one pH unit higher as shown in Figure 3A. The advantage in using the truncated enzyme with a highly reactive, disulfide substrate in this experiment is that it allows for an estimation of enzymic titratable pKa’s without complications arising from the C-terminal redox center or interference from leaving group effects of the substrate, since in the case of DTNB the pKa of the leaving group thiol is 4.75 (22), eliminating its need to be protonated upon S-S bond cleavage. From this experiment, we can estimate two apparent, macroscopic pKa’s. For the mammalian enzyme (mTR3), pK1 = 5.8 and pK2 = 7.8, while for DmTR, pK1 = 6.5 and pK2 = 9.1. Recently Huang and coworkers performed a similar experiment with WT DmTR and H464’Q DmTR using Trx as the substrate (23). Similar to our results, they observe an apparent, macroscopic pKa of 6.4 (pK1), which they assign to either Cys57 or Cys490’. Since we have eliminated the C-terminal redox center containing Cys490’, a likely interpretation of our experiment is that the pKa of 6.4 can be ascribed to Cys57 in DmTR.
Figure 3.

Activity of the various forms of TR in this study toward DTNB as a function of pH. A) Truncated mTR3Δ8 (closed squares) and truncated DmTRΔ8 (open circles). B) Full length WT mouse enzyme – mTR3-GCUG (closed squares), full length WT Drosophila enzyme – DmTR-SCCS (open circles). The raw data used to compose the plots is given in Table S1 of the Supporting Information.
Extrapolation of this interpretation to mTR3 means that Cys52 in mTR3 has a pKa of 5.8, which is the attacking thiolate in this thiol-disulfide exchange reaction (labeled as CysIC in Figure 1). Also based on the results presented by Huang and coworkers, we can assign pK2 to that of an imidazolium ion (His464’ in DmTR and His463’ in mTR3) (23). Further support for this assignment is the mechanistic relationship of GR to TR, where pK2 has also been assigned to an imidazolium ion, which exists as an ion pair with a thiolate ion (24). The pH profiles for both truncated enzymes using Pep-TNB as a substrate are very similar to the profiles when DTNB is used as the substrate, as might be expected due to the similarity of both substrates (data not shown). The results of the experiments with DTNB and Pep-TNB clearly demonstrate that the two truncated enzymes exhibit very different apparent macroscopic pKa’s that are independent of the presence of the C-terminal redox center. As will be demonstrated and discussed further in this report the difference between the two enzymes is not merely confined to the presence or absence of Sec in the C-terminal redox center.
For comparison, the pH rate profiles for the full-length enzymes are shown in Figure 3B. Here the profiles are similar, but somewhat broadened in comparison with mTR3 showing a pH optimum of 7 – 7.5 and DmTR having an optimum of 8 – 8.5. This effect is most likely due to the fact that both the N-terminal and C-terminal disulfide redox centers can interact with DTNB. We also evaluated yeast GR for its ability to catalyze the reduction of DTNB at varying pHs because of its structural and mechanistic similarity to our truncated TRs and the results can be seen in Figure S2 of the Supporting Information. The results of this experiment show that pK2 is ~ 8.7, close to the reported value of 9.1 for His467’ (24), thus validating our approach.
Assays using Peptide Substrates
During the catalytic cycle electron pairs are transferred from the N-terminal disulfide redox center to the C-terminal disulfide/selenylsulfide redox center. Truncated TRs are capable of reducing peptide disulfides/selenylsulfides that correspond to their missing C-terminal sequence (ring opening step – Figure 1) (16-17). Using this property of TR we were able to evaluate the relative importance that the unique eight-membered ring makes to the catalytic rate by comparing the turnover rate of cyclic and acyclic peptide substrates. In each instance the native peptide was oxidized, creating a disulfide/selenylsulfide linkage, resulting in the formation of a cyclic intrachain peptide containing a strained eight-membered ring, or an unstrained, acyclic interchain peptide (Figure 2).
The second order rate constants for these peptide turnover rates are given in Table 3. In order to compare the importance of the ring structure to each class of enzyme, the important factor to consider is whether the native peptide contains Sec or Cys. Thus for the mammalian enzyme we must compare the turnover rates for the cyclic Se-peptide and the acyclic Se-peptide. The data in Table 3 shows that the mammalian enzyme prefers the cyclic peptide over that of the acyclic peptide by a factor of 32. The other two enzymes in Table 3 are Cys-TRs so that the relevant rates for comparison are the cyclic S-peptide and the acyclic S-peptide. In these two cases, a much different result is obtained. Comparison of these rates for the truncated versions of DmTR and CeTR2 reveals that the ring is much more important to catalysis, contributing a factor of 1025 and 2267, respectively. This result strongly implies a different mechanism for the ring opening step in each class of enzyme. This is a result of a difference in ring structure in the two types of enzymes (P.B. Deker and R.J. Hondal, unpublished) and also of the difference in structure of the tetrapeptide binding site in each type of enzyme (16).
Table 3.
Rate Data for Truncated TRs using Peptide Disulfides as Substrates.
| Substrate | Rate (min−1•mM−1) |
||
|---|---|---|---|
| Enzyme (octapeptide) |
|||
| mTR3Δ8 (PTVTGCUG) |
DmTRΔ8 (PTPASCCS) |
CeTR2Δ8 (PRTQGCCG) |
|
| Cyclic Se-peptide | 260 ± 16a | 499 ± 24 | 49 ± 4 |
| Acyclic Se-peptide | 8.2 ± 1.4b | 31 ± 0.4 | 19 ± 3.3 |
| Cyclic S-peptide | 0.03 ± 0.002a | 41 ± 1.5 | 136 ± 34 |
| Acyclic S-peptide | 0.02 ± 0.001a | 0.04 ± 0.001 | 0.06 ± 0.03 |
| Switch mutant | 0.23 ± 0.012 | 4.6 ± 0.04 | NDc |
| DTNB | 3500 ± 230a | 316 ± 79 | 287 ± 37 |
| Peptide-TNB | 220 ± 62 | 21 ± 0.45 | 32 ± 9 |
We have previously presented the hypothesis that protonation of the leaving group atom in this ring-opening step is partially rate limiting (16). This contention is supported by the data in Table 3 if we compare the ratio of turnover rates for the acyclic Se-peptide to the acyclic S-peptide. This ratio allows for a determination of the importance of leaving group pKa on the reaction since these acyclic peptides differ by only one atom with differing pKa values (5.2 for Se and 8.3 for S) (25). These ratios, summarized in Table 4, are very similar for all three enzymes in this study (in the range of 300 to 800). Our interpretation of this result, consistent with our previous studies, is that leaving group pKa is an extremely important facet of this reaction. This can be rationalized by the representation of the ring opening reaction shown in Figure 4A. In the case of the cyclic peptide, the ring can adopt a conformation to allow for attack by the thiolate of CysIC, while simultaneously positioning the leaving group atom in a position to accept a proton from the general acid/base catalyst (His464’).
Table 4.
Ratio of Turnover Rates of Peptide Substrates.
| Enzyme | Acyclic-Se/ Acyclic-S |
Cyclic /Acyclica |
|---|---|---|
| mTR3Δ8 | 410 | 32 |
| DmTRΔ8 | 775 | 1025 |
| CeTR2Δ8 | 317 | 2267 |
For mTrR3Δ8, the cyclic and acyclic peptides both contained Se at the leaving group position, while for DmTRΔ8 and CeTRΔ8 both peptides contained S at the leaving group position.
Figure 4.

Mechanism of truncated TR toward cyclic and acyclic peptide substrates. The peptide substrate is positioned in the active site such that nucleophilic CysIC thiolate can attack the Cys1 position, and cause the release of X (magenta). A) In the case of the cyclic peptide, when X = S, the ring is critical for positioning X near the conserved His, to receive a proton to stabilize the leaving group. When X = Se, the position of the ring near the His matters less due to the much lower pKa of a selenolate in comparison to that of a thiolate. B) In the case of the flexible, acyclic peptide, which has more degrees of motion in comparison the cyclic peptide, protonation of X is much less likely to occur due to the “floppy” nature of the peptide. However, when X = Se, protonation does not need to occur for transition-state stabilization due to its lower pKa. Experimentally we observe that when X = S, the acyclic peptides are extremely poor substrates.
As illustrated in Figure 4B, binding of the very flexible, acyclic peptide in the active site most likely results in a physical disconnection of the leaving group from the general acid. This assertion is supported by the data in Tables 3 and 4. For example, DmTR turns over the acyclic Se-peptide at nearly the same rate as the cyclic S-peptide (compare 31 to 41 in Table 3), while the acyclic S-peptide is turned over 1000 fold slower. The reason that the acyclic Se-peptide is such a good substrate in comparison to the acyclic S-peptide is that the low pKa of the selenolate leaving group overcomes the physical disconnection to the general acid. This same leaving group effect is also apparent in the case of the substrates that have the TNB-S− anion (pKa = 4.75) as the leaving group (Pep-TNB and DTNB in Table 3). The disconnection between the leaving group and the general acid, caused by disruption of the ring structure, explains the experimental results regarding the importance of the ring in both classes of TRs and the observed leaving group effect. The ring is much more important in the Cys-containing TRs because the exact positioning of the leaving group thiol is required in order to facilitate proton transfer from the general acid (irrespective of the final cis/trans conformation of the ring in the holoenzyme). The ring becomes relatively unimportant for Sec-TRs because the much lower pKa of the selenolate allows it to leave without receiving a proton from His463’. This result implies that the mammalian enzyme binds the ring in a different conformation so that the two types of TRs are differentiated mechanistically in the ring opening step.
Additionally, cyclic, “switch” mutant peptides were constructed containing a selenylsulfide bond where the positions of Sec and Cys were switched so that in the case of the mammalian enzyme Sec now occupies the Cys1 position of the dyad, with Cys occupying the Cys2 position of the dyad. The mammalian enzyme had an activity of 0.23 min−1•mM−1 using this peptide substrate, which is 1130-fold lower than the cyclic peptide in which Sec is in the second position of the dyad (260 min−1•mM−1). By contrast, DmTR had an activity of 4.6 min−1•mM−1 with this switch peptide substrate (Sec1-Cys2), which is only 9-fold lower than the native, Cys1-Cys2 peptide substrate (41 min−1•mM). The lower magnitude of this switch effect on the Cys-containing TRs in comparison to the mammalian Sec-TR can be rationalized by the fact that the sulfur atom of CysIC must attack a more electronegative atom during the ring opening step, resulting in a lower rate. The fact that the effect is far less dramatic in the Cys-TR suggests that the rate reducing effect of the Se atom in the Cys1 position is largely overcome by the correct juxtaposition of Cys2 for protonation by His464’. The effect of this mutation is illustrated in Figure 5.
Figure 5.

Importance of the position of the Sec residue in the Cys1-Sec2 dyad (forming an eight-membered ring) in the ring opening step. In the case in which Sec occupies the 2nd position of the dyad (A), the mammalian enzyme has 1130 fold higher activity than when Sec is in the 1st position of the dyad as in (B). DmTR also has a preference for the Sec residue in the 2nd position of the dyad as it has 12-fold higher activity in comparison to its native peptide sequence. In the case in which Sec occupies the 1st position of the dyad (B), DmTR only suffers a 9-fold loss in activity compared to its native peptide sequence, while the effect is severe for the mammalian enzyme as already noted. In the case of DmTR for the situation shown in (B), the sulfur atom is still in position to accept a proton from His, so the loss in activity is due to the effect of the thiolate attacking a more electronegative atom.
To support our argument that the second position of the dyad is the leaving group position, we substituted Se for S in the second position of the dyad for cyclic peptide PTPASCUS(ox) and this peptide was turned over with a rate of 499 min−1•mM−1 by DmTR, which is 12-fold faster than with the native cyclic peptide (PTPASCCS(ox)). Since the lower pKa of Se makes it a better leaving group than S, this result makes sense if the second position of the dyad is in the leaving group position.
By examining the kinetics of this ring opening step, we are not focused on the nucleophilicity of selenium, but rather on its electrophilicity since a selenylsulfide bond, like a disulfide bond, is an electrophile that is attacked by a thiolate nucleophile in this step of the enzymatic mechanism. The overall electrophilicity of the selenylsulfide bond is not dependant on the position of the Sec residue in the dyad. Yet for the mammalian enzyme when Sec is placed in the first position of the dyad a very large loss in activity occurs, while only a small loss in activity occurs for the Cys-TR. The explanation for this is that for the mammalian enzyme, the ring structure is different than the ring structure for Cys-TRs and this difference in structure causes a disconnection between the general acid and the leaving group. This physical disconnection necessitates the need for a low pKa leaving group in the second position of the dyad and a selenolate fills that role. We have argued previously that the mammalian enzyme uses a trans conformation of the ring and it is this conformation that creates the barrier between His and Sec (16). This is opposite of the cis geometry that is shown for the ring in Figure 4A.
Our hypothesis regarding the dominance of the leaving group in the ring opening step was further tested using yeast GR. The substrate for GR is oxidized glutathione (GSSG – a simple disulfide), the reduction of which yields 2 molecules of GSH; the thiol groups of each have a pKa of 8.56 (26). In our assay using GSSG as a substrate, the specific activity is 4357 moles NADPH/min/mole of GR. However, the peptides with vicinal Cys-Cys disulfides that we used as substrates for the truncated versions of TR in this study were not substrates for yeast GR as can be see by the data in Table 5. Of course a major determinant in this reaction is that GR has evolved to bind GSSG as a substrate and not other types of disulfides, rendering these peptide disulfides poor substrates. However, it has been shown that proton transfer to the leaving group thiol of GSH2 is partially rate determining and GR accomplishes this proton transfer by making use of His467’, which is analogous to His464’ in DmTR and His463’ in mTR3. We also tested the vicinal, Cys-Sec-containing peptides as substrates for yeast GR and as can be seen in Table 5, the Sec-containing peptides are 17 to 38 fold better substrates than their Cys analogs. We also tested two different versions of Pep-TNB as substrates for yeast GR, and these mixed disulfide substrates are also turned over at rates > 190 fold higher than the vicinal disulfide peptide substrates. The results show that yeast GR also demonstrates a strong dependence on leaving group pKa, similar to the ring opening step in TR.
Table 5.
Peptide Disulfides as Substrates for Yeast GR.
| Substrate | Activity (moles NADPH/min/mole of enzyme)a |
|---|---|
| PTVTGCUG(ox)b | 1.89 ± 0.31 |
| PTVTGCCG(ox)b | 0.05 ± 0.005 |
| PTPASCUS(ox)b | 1.19 ± 0.023 |
| PTPASCCS(ox)b | 0.07 ± 0.018 |
| PTVTGC-TNBb | 9.54 ± 0.27 |
| PTPASC-TNBb | 16.07 ± 0.22 |
| GSSGc | 4357 ± 130 |
Activity is reported per monomer of GR
At 1 mM peptide in the assay cocktail.
At 250 μM GSSG and 1 nM yeast GR in the assay cocktail.
The effect of pH on the spectral properties of TR
It has been previously observed that crystals of mTR3 grown at pH 6.5 would change from a yellow color in the oxidized state to a red color upon reduction with NADPH (27) characteristic of formation of the thiolate-flavin charge-transfer complex observed between 525 nm and 540 nm for the pyridine nucleotide oxidoreductase family of proteins (28). The mTR3 C-terminal truncation mutant used in this study also crystallized at pH 6.5 while the truncated DmTR crystallized at pH 5.5. In performing NADPH soaking experiments, mTR3 crystals turned red as expected while DmTR crystals did not. Rather, the DmTR crystals became near colorless. This dramatic observation is shown in Figure 6, which compares crystals of DmTRΔ8 and mTR3Δ8 reduced by NADPH at pH 5.3 and 5.85.
Figure 6.

(A) Crystals of DmTRΔ8 soaked in crystallization solution containing 10 mM NADPH at pH 5.3 (top) and at pH 5.85 (bottom). (B) Crystals of mTR3Δ8 soaked in crystallization solution containing 10 mM NADPH at pH 5.3. The time points are as indicated in the figure.
We subsequently verified this observation spectrophotometrically between pH 4.9 – 9.0. Very weak, transient charge-transfer absorbance is observed below pH 5.5 for DmTRΔ8 upon incubation with NADPH, while there is the characteristic initial decrease at 460 nm followed by a shift to 440 nm (spectra at pH 5.1 is shown in Figure 7A). The situation is much different at pH 7.0 (shown in Figure 7B) where (as expected) significant charger-transfer absorbance is seen. In performing the same experiment for mTR3Δ8, very significant charger-transfer absorbance is observed at pH 5.1 (Figure 7C), and comparison of the spectra of the two enzymes at pH 5.1 presents a sharp contrast (compare Figure 7A to 7C). This indicates a less stable thiolate-flavin charge-transfer complex for DmTRΔ8 at low pH compared to that of mTR3Δ8. The spectrum of mTR3Δ8 at pH 7.0 is shown in Figure 7D for comparison.
Figure 7.

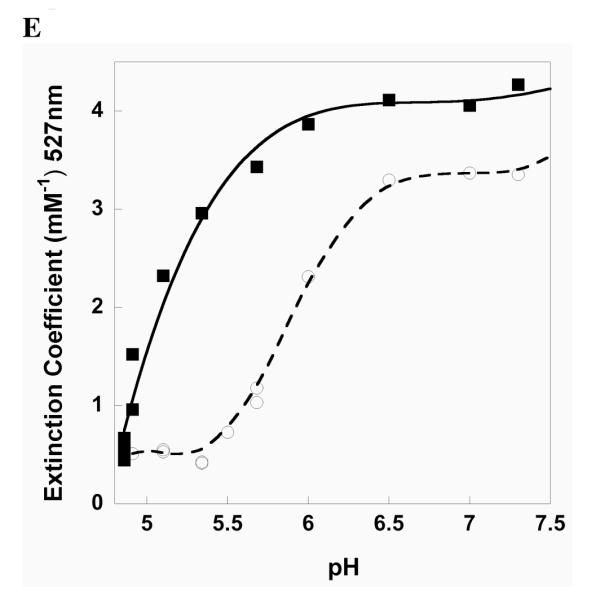
Spectral characteristics of thioredoxin reductase. Spectra are shown for NADPH-reduced DmTRΔ8 at pH 5.1(A) and pH 7.0 (B). The same spectra are shown for mTR3Δ8 at pH 5.1 (C) and pH 7.0 (D). For each figure, the oxidized spectrum is highlighted in gray and the 30 sec time point post-NADPH reduction has been shown in bold. The insets compare the time course at 527 nm at pH 5.1 (open circles) and pH 7.0 (closed squares) for the respective enzymes. (E) Charge-transfer as a function of pH measured at 527 nm for DmTRΔ8 (open circles) and mTR3Δ8 (closed squares).
The difference spectrum calculated between the reduced and oxidized forms for each TR show a maxima at 527 nm for the charge-transfer complex. The extinction at 527 nm over the course of the assay at pH 5.1 is compared to pH 7.0 in the insets of Figure 7B and 7D and the stable charge-transfer plotted as a function of pH in Figure 7E. The data demonstrates a stronger charge-transfer for mTR3Δ8 than DmTRΔ8 and that the charge-transfer is significantly more stable for mTR3Δ8 as pH is decreased. The data in Figure 7E allows for calculation of the pKa of the charge-transfer Cys residue (Cys57 in mTR3 and Cys62 in DmTR – CysCT). This data can be fit to a hyperbolic curve by normalizing the signal at 527 nm so that all values of the normalized signal fell between 0 and 1 and then plotting this normalized signal against H+ concentration. The analytically determined pKa of CysCT for mTR3 and DmTR was thus determined to be 5.02 ± 0.27 and 5.95 ± 0.085, respectively. Reaction of DmTRΔ8 (reduced by NADPH) with DTNB produced 0.83 to 1.04 equivalents of TNB-S− per subunit of TR below pH 5.5, while complete reduction of DTNB to 100 μM TNB-S− was observed at neutral pH. The stable shift to 440 nm upon reduction at low pH and only one equivalent of TNB-S− produced indicates reduction of the enzyme and likely protonation of the CysCT.
CONCLUSION
The notion that the selenium atom is required by mammalian TR due only to its superior nucleophilicity is largely dispelled by the data in Table 1. While the difference in nucleophilicities between the two classes of TRs is not known, model studies show that the difference in nucleophilicity between selenium and sulfur is in the range of 8-10 fold (29). However, there is a marked difference in reactivity between selenium and sulfur at low pH (18).
The data presented in this study is the culmination of a series of papers that have focused on the ring opening step in the catalytic mechanism that we have argued distinguishes Cys-TRs from Sec-TRs (10, 16-17). Here we have determined the relative importance of the ring structure in the ring opening step by synthesizing peptide substrates (each containing either a disulfide or selenylsulfide) that are identical in sequence, but were differentiated by the presence and absence of the ring structure (Figure 2). The ratio of these turnover rates by the respective truncated enzymes tells us that the ring is only of modest importance for the truncated Sec-TR (ring/no ring = 32 – Table 4), while the ring structure is highly important for the two truncated Cys-TRs (ring/no ring > 1000 – Table 4). However, the magnitude of the leaving group effect for both classes of enzymes was very similar (300 – 800 fold), indicating that leaving group pKa is of comparable importance to the ring opening step.
These experiments imply why only a special type of disulfide, a vicinal disulfide with cis amide geometry, can be a substrate for the truncated Cys-TR because only this type of disulfide has the right geometry to allow for thiolate attack by CysIC while at the same time position the leaving group sulfur atom correctly to accept a proton from the general acid (shown in Figure 4A). Absent of this type of geometry, acyclic disulfides such as DTNB can be substrates because of superior leaving group ability. We also conclude that Sec-TRs are as dependent upon leaving group pKa as Cys-TRs because of a difference in conformation of the eight-membered ring and this different conformation separates the general acid from the selenolate, otherwise substitution of Cys for Sec would not have an effect in the ring opening step in the mammalian enzyme. From a mechanistic viewpoint, this physical separation explains the need for the utilization of Sec by the mammalian enzyme because of the need for a low pKa leaving group in the ring opening step. We note that other groups led by Williams (30) and Barycki (27) have come to the opposite conclusion regarding this step in the mechanism. While the complete mechanistic details of Cys-TRs still remain unknown, this study clearly shows that the Cys and Sec enzymes can be distinguished by the ring opening step of the mechanism.
Irrespective of the C-terminal redox center, this study also indicates that the entire catalytic machineries of the two classes of enzymes are different. This is evident by the different pKa values for CysIC of mTR3 and DmTR, which are 5.8 and 6.5, respectively. The reason for this difference may be that the mammalian enzyme needs a more fully ionized thiolate to attack the selenylsulfide bond of the ring, which has a lower redox potential compared to that of a disulfide (31). Correspondingly, the pKa of CysCT must be lower in order to resolve the mixed disulfide (compare 5.02 vs. 5.95 for the pKa of CysCT of mTR3 and DmTR, respectively).
Thus from a mechanistic perspective the biochemical differences between the two classes of enzymes demonstrate that the mammalian enzyme has evolved to take advantage of the superior chemical properties conferred by a selenium atom at acidic pH (also discussed in 32), allowing for Sec-TRs to function at significantly lower pH compared to Cys-TRs. While this conclusion is drawn from studying the differing mechanistic properties of the two classes of enzymes in vitro, the biological significance of this finding is not yet apparent. The finding that a very significant difference between Cys-TRs and Sec-TRs is the difference in the mechanism of the ring opening step implies that: (i) substitution of Se for S in the ring affects the structure of the ring in the mammalian enzyme; (ii) the structure of the tetrapeptide binding pockets of the two classes of enzymes are different; and (iii) the mammalian enzyme may utilize other substrates that interact directly with the N-terminal disulfide redox center.
Supplementary Material
ACKNOWLEDGEMENT
We wish to thank Dr. Stephen J. Everse for providing a critical reading of the manuscript.
ABBREVIATIONS
- A280
absorbance at 280nm
- A460
absorbance at 460nm
- ADP
adenosine diphosphate
- βME
β-mercaptoethanol
- C. elegans
Caenorhabditis elegans
- CeTR2
mitochondrial TR from Caenorhabditis elegans
- Cys
cysteine
- CysCT
charge-transfer cysteine
- CysIC
interchange cysteine
- D. melanogaster
Drosophila melanogaster
- DEAE
diethylaminoethyl
- DmTR
TR from Drosophila melanogaster
- DTNB
5,5′dithio-bis(2-nitrobenzoic acid)
- DTT
dithiothreitol
- E. coli
Escherichia coli
- EDTA
ethylaminediamine tetraacetic acid
- Gly
glycine
- GR
glutathione reductase
- GSH
glutathione, reduced
- GSSG
glutathione, oxidized
- HEPES
N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid
- IPTG
isopropyl-β-D-thiogalactopyranoside
- LB
Luria-Bertani media
- MDS
mixed disulfide
- MES
2-(4-morpholino)-ethane sulfonic acid
- Mr
molecular ratio
- mTR3
mitochondrial TR from mouse
- MW
molecular weight
- MWCO
molecular weight cut off
- NaCl
sodium chloride
- NADPH
β-nicotinamide adenine dinucleotide phosphate, reduced
- Ni-NTA
nickel nitrilotriacetic acid
- O.D.
optical density
- PDB
Protein Data Bank
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- Sec
selenocysteine
- Ser
serine
- TB
terrific broth
- TE
Tris EDTA buffer
- TNB-S−
5-thio-2-nitrobenzoate anion
- TR
thioredoxin reductase
- Tris
tris-(hydroxymethyl)aminomethane
- Trx
thioredoxin
- U
the one letter code for Sec
- UGA
sense codon for Sec
- WT
wild-type
Footnotes
These studies were supported by National Institutes of Health Grant GM070742 to RJH.
While the value of kcat for the Sec => Cys mutant of mTR3 drops 555 fold using E. coli Trx as substrate, the drop in kcat could be smaller if we had used the cognate Trx as a substrate instead, similar to the 175 fold decrease in kcat with the same mutant of the rat enzyme.
SUPPORTING INFORMATION AVAILABLE There are two Tables and two Figures in the Supporting Information. Table S1 tabulates the raw data from Figure 3, while Table S2 tabulates the concentration of peptide and enzyme used in our complementation assays. Figure S1 is a 12% SDS-PAGE of the purified enzymes in this study while Figure S2 shows the pH rate profiles for yeast GR. This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- 1.Holmgren A. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- 2.Arnér ES, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 3.Arscott LD, Gromer S, Schirmer RH, Becker K, Williams CH., Jr. The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 1997;94:3621–3626. doi: 10.1073/pnas.94.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carugo O, Cemazar M, Sahariev S, Hudaky I, Gaspari Z, Perczel A, Pongor S. Vicinal disulfide turns. Protein Eng. 2003;16:637–639. doi: 10.1093/protein/gzg088. [DOI] [PubMed] [Google Scholar]
- 5.Hudaky I, Gaspari Z, Carugo O, Cemazar M, Pongor S, Perczel A. Vicinal disulfide bridge conformers by experimental methods and by ab initio and DFT molecular computations. Proteins. 2004;55:152–168. doi: 10.1002/prot.10581. [DOI] [PubMed] [Google Scholar]
- 6.Creighton CJ, Reynolds CH, Lee DH, Leo GC, Reitz AB. Conformational analysis of the eight-membered ring of the oxidized cysteinyl-cysteine unit implicated in nicotinic acetylcholine receptor ligand recognition. J. Am. Chem. Soc. 2001;123:12664–12669. doi: 10.1021/ja016505m. [DOI] [PubMed] [Google Scholar]
- 7.Gladyshev VN, Jeang KT, Stadtman TC. Selenocysteine identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase corresponds to TGA in the human placental gene. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6146–51. doi: 10.1073/pnas.93.12.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong L, Holmgren A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J. Biol. Chem. 2000;275:18121–18128. doi: 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]
- 9.Kanzok SM, Fechner A, Bauer H, Ulschmid JK, Muller HM, Botella-Munoz J, Schneuwly S, Schirmer R, Becker K. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science. 2001;291:643–646. doi: 10.1126/science.291.5504.643. [DOI] [PubMed] [Google Scholar]
- 10.Eckenroth BE, Lacey BM, Lothrop AP, Harris KM, Hondal RJ. Investigation of the C-terminal redox center of high Mr thioredoxin reductase by protein engineering and semisynthesis. Biochemistry. 2007;46:9472–9483. doi: 10.1021/bi7004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lacey BM, Hondal RJ. Characterization of mitochondrial thioredoxin reductase from C. elegans. Biochem. Biophys. Res. Commun. 2006;346:629–636. doi: 10.1016/j.bbrc.2006.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer H, Gromer S, Urbani A, Schnolzer M, Schirmer RH, Muller HM. Thioredoxin reductase from the malaria mosquito Anopheles gambiae. Eur. J. Biochem. 2003;270:4272–4281. doi: 10.1046/j.1432-1033.2003.03812.x. [DOI] [PubMed] [Google Scholar]
- 13.Jao SC, English Ospina SM, Berdis AJ, Starke DW, Post CB, Mieyal JJ. Computational and mutational analysis of human glutaredoxin (thioltransferase): probing the molecular basis of the low pKa of cysteine 22 and its role in catalysis. Biochemistry. 2006;45:4785–4796. doi: 10.1021/bi0516327. [DOI] [PubMed] [Google Scholar]
- 14.Gromer S, Johansson L, Bauer H, Arscott LD, Rauch S, Ballou DP, Williams CH, Jr., Schrimer RH, Arner ES. Active sites of thioredoxin reductases: why selenoproteins? Proc. Natl. Acad. Sci. U. S. A. 2003;100:12618–12623. doi: 10.1073/pnas.2134510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johansson L, Arscott LD, Ballou DP, Williams CH, Jr, Arnér ES. Studies of an active site mutant of the selenoprotein thioredoxin reductase: the Ser-Cys-Cys-Ser motif of the insect orthologue is not sufficient to replace the Cys-Sec dyad in the mammalian enzyme. Free Radic Biol Med. 2006;41:649–56. doi: 10.1016/j.freeradbiomed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Eckenroth BE, Rould MA, Hondal RJ, Everse SJ. Structural and biochemical studies reveal differences in the catalytic mechanisms of mammalian and Drosophila melanogaster thioredoxin reductases. Biochemistry. 2007;46:4694–4706. doi: 10.1021/bi602394p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flemer S, Jr., Lacey BM, Hondal RJ. Synthesis of peptide substrates for mammalian thioredoxin reductase. J. Pept. Sci. 2008;14:637–647. doi: 10.1002/psc.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hondal RJ, Nilsson BL, Raines RT. Selenocysteine in native chemical ligation and expressed protein ligation. J. Am. Chem. Soc. 2001;123:5140–5141. doi: 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]
- 19.Hondal RJ, Raines RT. Semisynthesis of proteins containing selenocysteine. Methods Enzymol. 2002;347:70–83. doi: 10.1016/s0076-6879(02)47009-7. [DOI] [PubMed] [Google Scholar]
- 20.Eckenroth B, Harris K, Turanov AA, Gladyshev VN, Raines RT, Hondal RJ. Semisynthesis and characterization of mammalian thioredoxin reductase. Biochemistry. 2006;45:5158–517. doi: 10.1021/bi0517887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arner ES, Zhong L, Holmgren A. Preparation and assay of mammalian thioredoxin reductase. Methods Enzymol. 1999;300:226–239. doi: 10.1016/s0076-6879(99)00129-9. [DOI] [PubMed] [Google Scholar]
- 22.Danehy JP, Elia VJ, Lavelle CJ. Alkaline decomposition of organic disulfides. IV. Limitation on the use of Ellman’s reagent. 2,2′Dinitro-5,5′dithiodibenzoic acid. J. Org. Chem. 1971;36:1003–1005. [Google Scholar]
- 23.Huang HH, Arscott LD, Ballou DP, Williams CH., Jr. Acid-base catalysis in the mechanism of thioredoxin reductase from Drosophila melanogaster. Biochemistry. 2008;47:1721–1731. doi: 10.1021/bi702040u. [DOI] [PubMed] [Google Scholar]
- 24.Sahlman L, Williams CH., Jr. Titration studies on the actives sites of pig heart lipoamide dehydrogenase and yeast glutathione reductase as monitored by the charge transfer absorbance. J. Biol. Chem. 1989;264:8033–3038. [PubMed] [Google Scholar]
- 25.Huber RE, Criddle RS. Comparison of the chemical properties of selenocysteine and selenocystine and their sulfur analogs. Arch. Biochem. Biophys. 1967;122:164–173. doi: 10.1016/0003-9861(67)90136-1. [DOI] [PubMed] [Google Scholar]
- 26.Danehy JP, Parameswaran KN. Acidic dissociation constants of thiols. J. Chem. Eng. Data. 1968;13:386–389. [Google Scholar]
- 27.Biterova EI, Turanov AA, Gladyshev VN, Barycki JJ. Crystal structures of oxidized and reduced mitochondrial thioredoxin reductase provide molecular details of the reaction mechanism. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15018–15023. doi: 10.1073/pnas.0504218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rietveld P, Arscott LD, Berry A, Scrutton NS, Deonarain MP, Perham RN, Williams CH., Jr. Reductive and oxidative half-reactions of glutathione reductase from Escherichia coli. Biochemistry. 1994;33:13888–13895. doi: 10.1021/bi00250a043. [DOI] [PubMed] [Google Scholar]
- 29.Pearson RG, Sobel H, Songstad J. Nucleophilic reactivity constants toward methyl iodide and trans-[Pt(py)2Cl2] J. Am. Chem. Soc. 1968;90:319–326. [Google Scholar]
- 30.Bauer H, Massey V, Arscott LD, Schirmer RH, Ballou DP, Williams CH., Jr. The mechanism of high Mr thioredoxin reductase from Drosophila melanogaster. J Biol Chem. 2003;278:33020–8. doi: 10.1074/jbc.M303762200. [DOI] [PubMed] [Google Scholar]
- 31.Besse D, Siedler F, Diercks T, Kessler H, Moroder L. The redox potentials of selenocystine in unconstrained cyclic peptides. Angew. Chem. Int. Ed. Eng. 1997;36:883–885. [Google Scholar]
- 32.Wessjohann LA, Schneider A, Abbas M, Brandt W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol Chem. 2007;388:997–1006. doi: 10.1515/BC.2007.138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.