

Abstract
Repeated exposure to nicotine and other psychostimulant drugs produces persistent increases in their psychomotor and physiological effects (sensitization), a phenomenon related to the drugs' reinforcing properties and abuse potential. Here we examined the role of peripheral actions of nicotine in nicotine-induced sensitization of centrally mediated physiological parameters (brain, muscle, and skin temperatures), cortical and VTA EEG, neck EMG activity, and locomotion in freely moving rats. Repeated injections of intravenous nicotine (30 μg/kg) induced sensitization of the drug's effects on all these measures. In contrast, repeated injections of the peripherally acting analog of nicotine, nicotine pyrrolidine methiodide (nicotinePM, 30 μg/kg, i.v.) resulted in habituation (tolerance) of the same physiological, neuronal, and behavioral measures. However, after repeated nicotine exposure, acute nicotinePM injections induced nicotine-like physiological responses: powerful cortical and VTA EEG desynchronization, EMG activation, a large brain temperature increase, but weaker hyperlocomotion. Additionally, both the acute locomotor response to nicotine and nicotine-induced locomotor sensitization were attenuated by blockade of peripheral nicotinic receptors by hexamethonium (3 mg/kg, i.v.). These data suggest that the peripheral actions of nicotine, which precede its direct central actions, serve as a conditioned interoceptive cue capable of eliciting nicotine-like physiological and neural responses after repeated nicotine exposure. Thus, by providing a neural signal to the CNS that is repeatedly paired with the direct central effects of nicotine, the drug's peripheral actions play a critical role in the development of nicotine-induced physiological, neural, and behavioral sensitization.
Introduction
Nicotine, the primary active component of tobacco, is considered to be responsible for the development of tobacco addiction (Stolerman and Jarvis, 1995; Pontieri et al., 1996). In rats, repeated intermittent injections of nicotine typically cause sensitization or enhancement of the drug's locomotor activating effects (Clark and Kumar, 1983; Benwell and Balfour, 1992; Vezina et al., 2007). However, locomotor sensitization is not readily observed after continuous or high-dose nicotine exposure, or with short interinjection intervals (Ksir et al., 1987; Stolerman, 1990; Malin, 2001). Although various mechanisms are involved in drug-induced locomotor sensitization, classical conditioning is thought to play an important role (Kalivas and Stewart, 1991; Robinson and Berridge, 1993; Di Chiara, 2000). In the addiction field, conditioning typically refers to temporal pairings between exteroceptive environmental stimuli and the drug (O'Brien et al., 1992; Stewart, 1992). However, conditioning could also involve pairing between peripheral interoceptive effects of drugs and their central effects (Siegel, 1999; Paolone et al., 2004; Caprioli et al., 2007; Bevins et al., 2012).
Although nicotine rapidly reaches the brain after systemic administration (Berridge et al., 2010; Rose et al., 2010), this direct central action is always preceded by a transient neural activation due to stimulation of peripheral nicotinic receptors on the afferents of sensory nerves at the administration sites (lung alveoli, nasal and oral cavities) and within the circulatory system (Juan, 1982; Engberg and Hajos, 1994; Anand, 1996; Liu and Simon, 1996; Walker et al., 1996; Lenoir and Kiyatkin, 2011). After repeated nicotine exposure, these two pharmacological actions could interact in the CNS based on principles of Pavlovian conditioning, and the peripheral actions of nicotine could act as conditioned interoceptive cues, thus contributing to sensitization of nicotine's neural, physiological, and behavioral effects.
We tested this hypothesis at the physiological, neural, and behavioral levels. First, we examined how the effects of intravenous nicotine at a low, behaviorally active dose change after repeated injections. In addition to conventional locomotion, we monitored temperatures in NAcc, temporal muscle, and skin to provide measures of drug-induced metabolic neural activation and peripheral vasoconstriction (Kiyatkin, 2010). To characterize neural effects, we monitored electrical activity (EEG) in the cortex and VTA, and neck EMG activity. The NAcc and VTA play critical roles in mediating nicotine reward (Picciotto and Corrigal, 2002) and psychomotor sensitization (Balfour et al., 1998; Mao and McGehee, 2010). Second, we examined the effects of repeated exposure to nicotine pyrrolidine methiodide (nicotinePM), a peripherally acting nicotinic agonist (Aceto et al., 1983; Oldendorf et al., 1993), on the same behavioral, physiological, and electrophysiological measures. Third, we also determined how the effects of nicotinePM on the above measures are altered after repeated nicotine exposure. Finally, we examined the impact of the blockade of peripheral nicotinic receptors by hexamethonium on the acute locomotor effects of nicotine and their changes after its repeated exposure. Our data suggest that the peripheral actions of nicotine act as a conditioned interoceptive cue in drug-experienced rats, thus contributing to sensitization of nicotine's physiological, neuronal, and behavioral responses.
Materials and Methods
Subjects.
A total of 61 Long–Evans male rats (440 ± 20 g at the time of surgery, Charles River) were used as subjects. All rats were housed individually in a temperature-, humidity-, and light-controlled room (12 h/12 h light/dark cycle, lights on at 07:00) with free access to food and water. Procedures followed the guidelines outlined in the Guide for the Care and Use of Laboratory Animals (eighth edition; http://grants.nih.gov/grants/olaw/Guide-for-the-Care-and-Use-of-Laboratory-Animals.pdf) and were approved by the National Institute on Drug Abuse–Intramural Research Program Animal Care and Use Committee.
Overview of the study.
Our study consisted of three primary experiments. In Experiment 1, we examined the effects of repeated injections of nicotine and nicotinePM on NAcc, muscle, and skin temperatures and locomotion. We also examined the effect of acute nicotinePM injections on these measures after repeated nicotine exposure. In Experiment 2, we assessed the effects of the same drug treatments on cortical and VTA EEG, and neck EMG activity. In Experiment 3, we evaluated the impact the blockade of peripheral nicotinic receptors by hexamethonium has on the acute locomotor effects of nicotine and its sensitized locomotor responses, which develop after repeated injections.
In all experiments, nicotine ([−]nicotine hydrogen tartrate; Sigma) was injected intravenously at a dose of 30 μg/kg (calculated as drug base and dissolved in saline, pH ∼4.2). This dose is optimal for maintaining intravenous self-administration in rats (Cox et al., 1984; Donny et al., 1995; Matta et al., 2007) and comparable to that delivered during smoking of one to two cigarettes in humans (Rose and Corrigall, 1997; Berridge et al., 2010; Rose et al., 2010). To selectively activate or block peripheral nicotinic receptors, we used nicotinePM (Toronto Research Chemicals) or hexamethonium bromide (HEXA; Sigma), respectively. Both drugs have very limited blood–brain barrier (BBB) permeability (see the following paragraph). NicotinePM was delivered intravenously at the same dose as regular nicotine (30 μg/kg, calculated as drug base; dissolved in saline, pH ∼4.8). Because of the short duration of action of HEXA (Byck, 1961; Toyama et al., 1975), the drug was administered intravenously (3 mg/kg or ∼8 μm, dissolved in saline, pH ∼6.2) 4 min before intravenous nicotine injections. Under these conditions, HEXA is able to transiently block nicotinic receptors on the afferents of sensory nerves innervating heart and blood vessels (Byck, 1961; Ginzel, 1975). In all experiments, saline was used as a drug vehicle.
Drugs used in the study.
Nicotine (hydrogen tartrate salt) easily crosses the BBB, thus acting on both peripherally and centrally located nicotinic receptors, but nicotinePM resulting from its charge has a limited ability to enter the brain (Barlow and Dobson, 1955; Aceto et al., 1983; Oldendorf et al., 1993) and affects only the peripheral pool of nicotinic receptors. Although Aceto et al. (1983) have detected a small amount of nicotine in the brain after systemic injections of nicotinePM, the entry ratio was very small (1:140 vs regular nicotine) and the drug used systemically was fully inactive in the tail-flick test (1:513 vs nicotine); this drug was even slightly more active than regular nicotine with intraventricular injection. Taking into account this entry ratio and the low dose of nicotinePM used in this study (30 μg/kg), its dose equipotential to nicotine may be <0.2 μg/kg, below the minimal effective doses of nicotine found in physiological experiments (∼0.5–1.0 μg/kg) (Byck, 1961; Ginzel, 1975). As a charged molecule, nicotinePM may differ from regular nicotine in its receptor affinity, but based on existing evidence, it is thought that the nicotine-like physiological effects of nicotinePM are mediated via interaction with the same pool of peripheral nicotinic receptors activated by nicotine (Barlow and Hamilton, 1962; Aceto et al., 1983; Zuo et al., 2011). Because brains in the study of Aceto et al. (1983) were taken without saline perfusion, it is possible that brain samples were contaminated by drug residues contained in cerebral blood vessels. Thus, we conducted an additional experiment, using a highly sensitive mass spectrometry technique to verify that, under our experimental conditions, nicotinePM does not reach the brain (Experiment 4).
HEXA is a quaternary ammonium compound that, after systemic injections, affects only peripheral nicotinic receptors because the drug's chemical structure prevents it (or at least substantially limits it) from crossing the BBB (Wassermann, 1972; Ginzel, 1973, 1975; Woods et al., 2008). Whereas in an early study small amounts of HEXA were detected in the brain after its systemic injections at a high dose (Asghar and Roth, 1971), a more recent study revealed virtually no entry (4–5 m−18 /g after 0.7 mg/kg i.p. injection; Petrali et al., 1984). Because the brains in the Asghar and Roth (1971) study were also taken by decapitation without saline perfusion, their data may have been affected by drug residues contained in cerebral vessels and on the luminal, blood side of the BBB. Consistent with the preferentially peripheral action of HEXA, Malin et al. (1997) reported that intraventricular injections of HEXA at a low dose (12 ng/kg) precipitated opiate withdrawal symptoms (a centrally mediated effect), whereas systemic injections at a high dose (10 mg/kg, s.c.) did not. By this functional measure, HEXA was 195,000-fold more effective after central than peripheral administration.
We did not normalize the pH of injected solutions because pH could alter drug–receptor interactions, which are especially important for the immediate, local effects of highly changed nicotinePM and HEXA. However, because of rapid mixing with blood, which is a highly efficient buffer, the slightly acidic pH of the injected solutions is unlikely to affect the pH of arterial blood that reaches the brain some time after drug injections. Therefore, at the time of possible interaction with cellular elements of the BBB, the pH of arterial blood containing administered substances is presumably unchanged (∼7.4) and drugs are apparently ionized at the levels corresponding to this normal pH.
Surgical procedures.
In all experiments, rats were implanted under general anesthesia (Equithesin, 3.3 ml/kg) with a chronic intravenous catheter in the right jugular vein. The catheter was fed subcutaneously to the head mount and connected to an injection port secured on the skull with dental cement. Catheters were flushed daily with 0.2 ml of sterile saline to maintain catheter patency.
In Experiment 1, the rats (n = 23) were also implanted with three copper–constantan thermocouple electrodes as previously described (Kiyatkin and Brown, 2005). Four holes were drilled through the skull: three for securing screws and one for the electrode to be inserted stereotaxically in the NAcc shell (anteroposterior [AP], 1.2 mm; mediolateral [ML], 0.8 mm; dorsoventral [DV], 7.4 mm below the scull surface) using the coordinates of Paxinos and Watson (1998). A second, thermocouple electrode was implanted subcutaneously along the nasal ridge with the tip ∼15 mm anterior to bregma. This electrode was used to monitor skin temperature and provide a measure of peripheral vasoconstriction, a centrally mediated response elicited by various arousing stimuli (Altschule, 1951; Baker et al., 1976) and nicotine (Ginzel at al., 1975; Zuo et al., 2011). The third electrode was inserted in the deep temporal muscle (musculus temporalis), a nonmotor head muscle that receives the same arterial blood supply as the brain from the carotid artery. Parallel temperature recordings from the brain site and this location allowed us to determine brain-temporal muscle temperature differential, an important measure of drug-induced metabolic brain activation (Kiyatkin, 2010). All probes were secured with dental cement to the three stainless steel screws threaded into the skull.
In Experiment 2, the rats (n = 14) were also surgically prepared for chronic EEG and EMG recordings as previously described (Kiyatkin and Smirnov, 2010; Lenoir and Kiyatkin, 2011). Rats were implanted with three stainless steel screws threaded into the skull (two active screws on the left side: AP, 1.0 mm; ML, 1.5 mm; AP, −4.0 mm; ML, 1.5 mm; and a ground screw on the right side: AP, 2.0 mm; ML, 2.0 mm (Paxinos and Watson, 1998) and two stainless steel EMG electrodes (custom-made from four insulated 50 μm wires) implanted bilaterally in deep neck muscle. In addition, an insulated stainless steel electrode (375 μm in diameter, with 0.5 mm open active area at the tip) was implanted in the VTA (AP, −5.5 mm; ML, 2.0 mm; DV, 8.4 mm with 10° angle). After implantation, all six connectors were inserted into a plastic socket and fixed with dental acrylic as a head mount. For cortical recordings, we used two active screws threaded into the skull on the left side and a ground screw implanted on the right side. With these electrode locations, the electrical signal reflected the activity of the entire cortical mantle. For VTA recordings, we used an electrode implanted in the VTA and the most frontal screw on the left side. The same ground screw was also used as a reference electrode for differential EMG recording, with two active EMG electrodes implanted bilaterally in deep neck muscle. During testing, electrical activity from EEG and EMG electrodes passed through an amplifier (Pinnacle Technology) incorporated inside of an extension cord and electrical swivel to the main amplifiers (P15-D for EEG, P55 for EMG; Grass Electronics), which were used for additional signal amplification and filtering. EEG and EMG signals were bandpass-filtered from 1 to 100 Hz and 100 to 1000 Hz, respectively. The filtered signals were then passed to a Micro 1401 MK2 interface (Cambridge Electronic Design), allowing their acquisition, recording, and analysis using a Spike-2 interface (Cambridge Electronic Design).
Experimental protocols
Experiments 1 and 2: locomotor activity, temperatures, EEG, and EMG.
Recordings of locomotion and temperature (Experiment 1) were conducted inside a Plexiglas chamber (32 × 32 × 32 cm) equipped with four infrared motion detectors (Med Associates) and placed inside a sound attenuation chamber. Rats were attached via a flexible cord and channel electrical commutator to thermal recording hardware (Thermes 16, Physitemp). Room temperature was maintained at 23–24°C and controlled by another thermocouple sensor located in the recording chamber. Electrophysiological recordings (Experiment 2) were conducted in an electrically insulated cage (38 × 47 × 47 cm) placed inside a sound- and light-attenuated box (60 × 56 × 70 cm) with electromagnetic insulation.
A similar protocol was used in both experiments (Fig. 1). After a 3-day presurgery habituation to the experimental chamber (at least 6 h) and a 4–5 d after surgery recovery period, recording sessions (∼8 h each) were performed daily (9:30–17:30) over the next 5 to 12 d. After placement in the cage, the sockets on the rat's head were connected to the recording cable and a plastic catheter extension. This catheter extension was connected to a liquid swivel and an additional catheter extension, allowing stress-free drug delivery from outside of the cage and minimizing possible detection of the injection procedure by the rat. Because rats are sensitive to extraneous stimuli, special care was taken to exclude all possible stimuli immediately before and during drug injections. The first two recording sessions served as additional habituation sessions, during which the rat was observed and received at least three intravenous saline injections (0.15 ml over 15 s). Drug testing began after an ∼2 h habituation period. During this period, the rat returned to a quiet state after the transfer from the animal facility and the connection of recording cables, and both locomotor activity and brain and muscle temperatures gradually decreased to relatively stable levels maintained during the rest of the recording session. A similar habituation period was also used in electrophysiological experiments where drug injections were initiated when the rat was inactive and baseline electrical activity showed high-amplitude fluctuations typical of a sleep-like state.
Figure 1.

Experimental procedures of Experiments 1–3. Each square represents a daily session, and arrows of different color represent intravenous injections of different drugs (red, nicotine; blue, NicotinePM; green, saline; and brown, HEXA). Time intervals between single (Experiment 1–3) and paired (Experiment 3; two arrows connected by a blue line) drug injections were 120 min. Time intervals between two injections in a pair (Experiment 3) were 4 min. For other details, see Materials and Methods.
All rats were divided into two groups, depending upon the drug tested (nicotine or nicotinePM; Fig. 1). During two initial test sessions (treatment days 1 and 2), rats received three injections of either nicotine or nicotinePM at the same low dose (30 μg/kg, i.v., 0.15 ml over 15 s). These injections were spaced at least 2 h apart to avoid the influence of previous drug effects and maintain the same conditions before each injection. All drug injections were made when the rats were in quiet wakefulness or drowsiness/sleep state with no overt movements during the 120 s preceding each drug or saline injection. During these periods, baseline EEG activity showed high-magnitude fluctuations (synchronization) and EMG activity was low and stable; this condition was essential to exclude spontaneous neural activation associated with awakening and behavioral activity, which can mask drug-induced neural activation. After one day without drug treatment, rats of each group were tested again with the same drug during two daily sessions (treatment days 3 and 4). During each session, they received three injections of either nicotine or nicotinePM under the same experimental conditions. Rats that received nicotine treatment were tested in two more sessions under identical conditions to examine the effects of nicotinePM (three i.v. injections per session) after nicotine exposure in the previous sessions. During the last recording session, rats with different histories of drug treatment received at least three saline injections to test the influence of treatment on any possible nonspecific effects of the injection procedure.
When recording was completed, the rats were deeply anesthetized and decapitated. The brains were removed for sectioning, and all locations of cerebral implants were histologically verified. The thermocouple probes in Experiment 1 were located within the medial portion of ventral striatum (NAcc shell) (Paxinos and Watson, 1998).
Experiment 3: locomotor activity: HEXA and nicotine.
Monitoring of locomotor activity was conducted in Plexiglas chambers (42 × 42 × 30 cm; AccuScan Instruments) equipped with 16 beams located in both the horizontal and vertical planes, with beams separated from each other by 2.4 cm. Locomotor detection chambers were operated simultaneously via a PC interface, and experiments were controlled using VersaMax software (AccuScan Instruments). Before surgeries, rats were placed in the chambers for two consecutive days (at least 6 h per day) for environmental habituation (Fig. 1 for a schematic of experimental protocol). After a 5 day postsurgery recovery, rats received daily treatment sessions of 7 h duration. During the first session, rats were additionally habituated to the chamber for 2 h and then received 3 intravenous saline injections (0.2 ml over 20 s) separated by 2 h interinjection intervals. Then, the rats were divided into four equal groups (n = 6) using a 2 (nicotine dose, 0, 30 μg/kg) × 2 (HEXA dose, 0, 3 mg/kg) factorial design. On days 1 and 2 (pretreatment), each group received three pairs of injections according to their assigned group (saline–nicotine, saline–saline, HEXA–nicotine, HEXA–saline) with a within-pair injection interval of 4 min and between-pair interval of 120 min; the injection volume was 0.20 ml and the injection was given over 20 s. On day 3 (nicotine sensitization test), after a 2 h habituation to the recording chamber, rats of all groups received three single intravenous injections of nicotine at 2 h interinjection intervals. Unlike Experiments 1 and 2, where all injections were given via catheter extensions to minimize any extraneous influences, in Experiment 3, due to logistical limitations, drugs or saline were injected directly into the injection ports located on the rat's head. Although this procedure is minimally stressful for the rats, the saline injection procedure induced locomotor activation, which was absent when saline was injected via a catheter extension in Experiments 1 and 2 (see Results).
Experiment 4: measurements of brain nicotine.
Rats (n = 7) were prepared with chronic intravenous catheters and, after 5 d recovery, they were injected with either nicotine (100 μg/kg, n = 2), nicotinePM (100 μg/kg, n = 2 and 200 μg/kg, n = 1), or saline (n = 2). The doses were higher than those used in Experiments 1–3 to ensure that nicotine can be reliably detected in the brain as a reference point for nicotinePM at the detection sensitivity of the mass spectrometry instrument. All injections were conducted after 3 h habituation under quiet resting conditions and in equal volume (0.4 ml). Four minutes after the injections, rats were injected with Equithesin (1.0 ml over 1 min, i.v.) and perfused with cold saline for 15 min. Brains were rapidly removed and frozen in chilled isopentane for subsequent analysis. Brains were homogenized in distilled water at a concentration of 50 mg brain tissue/ml and extracts were desalted using SPEC18 columns. The extracts were diluted 1:1 in MeOH: 0.1% formic acid for mass analysis on an Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) with a static nanospray source in positive ion mode.
Data analysis.
The data obtained in thermorecording and electrophysiological experiments were grouped according to the injected drug (nicotine, nicotinePM, and saline) and the pharmacological history of the rat (naive, injections 1–3 on day 1; experienced, injections 7–12 on days 3–4 for each individual drug, and for nicotinePM after prior nicotine exposure). Temperatures were recorded with a time resolution of 10 s and movement was recorded as the number of infrared beam breaks per 1 min. Temperatures and locomotor activity were analyzed as slow changes (1- or 2-min bins for 60 min after injection), and temperature data were also analyzed as rapid changes (10-s bins for the first 10 min after injection). All temperature data are reported as changes from the preinjection, quiet-rest baselines. In addition to individual temperatures (NAcc, temporal muscle, and facial skin), we also analyzed brain–muscle and skin–muscle temperature differentials (i.e., relative differences in temperature change between the two locations). Because temperatures in each body location are determined by two primary variables (heat production resulting from metabolism and heat loss/gain resulting from circulation), these two parameters serve as valuable measures of metabolic brain activation and peripheral vasoconstriction/vasodilatation, respectively (Kiyatkin, 2010). One-way repeated-measures (RM) ANOVA was used as a primary tool for evaluating the effects of individual drugs and saline on locomotion and temperatures, and two-way RM ANOVA was used for statistical evaluation of between-group differences in mean temperature and locomotion. Significant main and interaction effects were followed by Fisher's PLSD post hoc test.
Because of their rapid, second-scale dynamics, electrophysiological parameters were analyzed with a 5 s time resolution from 2 min before to 10 min after each drug/saline injection. As we have shown previously, the 5 s temporal resolution of data analysis is optimal for detecting rapid changes in electrophysiological parameters, and the 10-min postinjection interval covers the time when nicotine has its primary physiological effects (Lenoir and Kiyatkin, 2011). For each drug test, we determined changes in EEG and EMG total powers (filtered within 2–58 Hz and 58–1000 Hz, respectively) and changes in power of individual EEG frequencies: δ (2–4 Hz), θ (4–8 Hz), α (8–15 Hz), β (15–29 Hz), and γ (29–58 Hz). Data on drug-induced alterations in individual EEG waves are not shown. Because EEG and EMG signals differ in their magnitude between individual rats, absolute values of total power were transformed into relative changes, taking a basal value (mean for 60 s preinjection) as 100%. EMG total power was analyzed statistically as natural logarithmic derivatives because of large fluctuations after drug administrations. One-way RM ANOVA (followed by Fisher's post hoc test) was used for evaluating statistical significance and duration of EEG and EMG changes induced by drug/saline injections. Between-group differences were quantified by comparing both the time course data and mean values of the immediate (5–30 s) and long-term (30–300 s) effects of drugs using a two-way RM ANOVA followed by post hoc PLSD Fisher tests (for point-to-point comparisons in time course) and Bonferroni test (for mean values).
Although several parameters of locomotor activity (i.e., total distance, vertical activity, time of movements, and number of movements) were recorded in Experiment 3, horizontal activity quantified as the number of beam breaks per minute was chosen as a primary parameter to evaluate drug-induced changes in locomotion. Two-way pairwise RM ANOVAs (followed by Fisher PLSD post hoc test) were used for time course analyses of drug-induced changes in locomotor activity (time within-session as a within-subject factor and pretreatment as a between-subject factor) and evaluation of between-group differences in locomotor responses on each treatment day. A mixed three-way ANOVA with pretreatment (HEXA vs saline) and drug (nicotine vs saline) as between-subject factors and day (day 1 vs day 2) as a within-subject factor was used for statistical evaluations of mean changes in locomotor activity. The different factors in the statistical analyses are provided in Results.
Results
Sensitization of locomotor, physiological, and neural effects of nicotine after repeated injections
When used in drug-naive conditions (day 1), intravenous nicotine induced weak locomotor activation (F(29,929) = 4.0; p < 0.01), a rapid and strong decrease in skin temperature (F(29,929) = 17.5; p < 0.01), and much weaker biphasic, up-down changes in NAcc and muscle temperatures (Fig. 2A, left). All these effects, except skin temperature, strongly increased (sensitized) after 2 d drug experience (six previous injections) (F(30,960) = 8.0, 17.3, and 14.8, respectively; p < 0.01; see Fig. 2A, right panel, B). Statistical analyses showed significant effects of drug experience on changes in locomotion and NAcc temperatures (F(1,59) = 14.5, F(1,55) = 25.1, respectively; both p < 0.001); the mean values increased more than twofold for locomotion and >7-fold for NAcc temperature (Fig. 2B). Nicotine-treated rats also showed larger increases in NAcc–muscle temperature differentials and larger decreases in skin–muscle differentials (Fig. 2A, middle), suggesting the enhancement of metabolic brain activation and peripheral vasoconstriction, respectively.
Figure 2.

Sensitization of locomotor and physiological effects of intravenous nicotine after repeated injections. A, Top, Mean (±SEM) relative changes in temperatures in NAcc, temporal muscle, and skin (°C), Middle, Changes in NAcc–muscle and skin–muscle temperature differentials (°C). Bottom, Locomotor activity (counts/min) induced by intravenous nicotine in drug-naive and nicotine-experienced conditions. n is the number of averaged tests per group. Vertical hatched line represents the start of injection. Values significantly different from the preinjection baseline (at least p < 0.05) are shown by horizontal lines of the respective colors. B, Mean (±SEM) changes in temperatures (°C) and locomotor activity calculated for 20 min after injection. Data are grouped based on the drug tested (nicotine, saline) and previous drug experience: (Naive), No experience; (Nicotine), 2 d of nicotine exposure. ○Significantly different from saline control (p < 0.05). #Significant differences between drug-naive and drug-experienced conditions (p < 0.05). Numbers of trials in each group are shown within bars.
In both drug-naive and drug-experienced conditions, nicotine induced rapid, strong, and prolonged EEG desynchronization equally evident in the cortex and VTA, as well as robust EMG activation (Fig. 3A,B). In both conditions, the total power of EEG signals dropped significantly (day 1, F(14,464) = 23.4 and 32.8; days 3–4, F(20,650) = 35.2 and 40.7 for cortex and VTA, respectively; all p < 0.001) within the injection duration (2.5–7.5 s) and slowly returned toward baseline thereafter. The EMG signal in both conditions strongly increased within the timing of drug injection, peaked at ∼30 s, and then slowly decreased toward baseline (F(14,464) = 12.2 and F(20,650) = 17.1 on day 1 and days 3–4, respectively, each p < 0.001). Although the pattern of EEG response was similar in both brain structures and in each drug condition, nicotine-induced decreases in total power were more prolonged after previous nicotine exposure (Fig. 3; Table 1). As shown in Table 1, the immediate effects (5–30 s) of nicotine on cortical and VTA total powers were identical in both groups, but the long-term effects (30–300 s) were stronger in nicotine-experienced conditions (p < 0.05 for cortex, p = 0.08 for VTA). This difference, however, was relatively small. In contrast, the increase in EMG total power was equally rapid and strong in both conditions, with no differences in either rapid or long-term components (Fig. 3A–C; Table 1).
Figure 3.

Sensitization of electrophysiological effects of nicotine and tolerance (habituation) of electrophysiological effects of nicotinePM after repeated drug exposure. Time course of changes (mean ± SEM for 5 s intervals) in total power of EEG desynchronization in the cortex (A,D) and VTA (B,E) and EMG activation (C,F) induced by intravenous injections of nicotine (A–C) or nicotinePM (D–F) in drug-naive (blue) and drug-experienced (red) conditions. The effects of intravenous saline injections are shown as black lines. Filled symbols represent values significantly different (p < 0.05) from baseline (60 s preinjection). Green bold lines within each graph represent significant between-group differences (p < 0.05). Two vertical hatched lines represent the timing of drug injection (0–15 s). EMG data were analyzed as ln derivatives and shown in ln scale. Data are grouped based on the drug tested (nicotine or nicotinePM) and previous drug experience: (naive), No experience; (Nicotine) or (NicotinePM), 2 d of nicotine or nicotinePM injections. n represents the number of analyzed tests per group.
Table 1.
Mean changes in cortical and VTA EEG and EMG total powers induced by intravenous nicotine, nicotinePM, and saline in drug-naive conditions and after drug experiencea
| Group | Immediate effect (5–30 s) |
Long-term effect (30–300 s) |
||||
|---|---|---|---|---|---|---|
| Cortex | VTA | EMG | Cortex | VTA | EMG | |
| Nicotine (naive) | 32.47 ± 2.99 | 26.23 ± 2.11 | 8.38 ± 0.30 | 36.00 ± 2.50 | 27.43 ± 2.13 | 8.11 ± 0.34 |
| Nicotine (Nicotine) | 30.39 ± 2.25 | 24.51 ± 1.99 | 7.90 ± 0.27 | 29.85 ± 1.66* | 23.00 ± 1.76 | 7.65 ± 0.33 |
| NicotinePM (naive) | 38.85 ± 2.28 | 31.06 ± 4.23 | 6.58 ± 0.11 | 74.94 ± 3.14 | 68.02 ± 5.12 | 4.94 ± 0.51 |
| NicotinePM (NicotinePM) | 63.48 ± 3.86*** | 51.96 ± 5.08*** | 5.87 ± 0.09*** | 93.25 ± 4.42*** | 84.43 ± 4.25* | 4.15 ± 0.37 |
| NicotinePM (Nicotine) | 36.04 ± 1.43†† | 32.71 ± 2.04†† | 7.11 ± 0.19†† | 59.06 ± 6.38*,†† | 48.90 ± 4.98*,†† | 6.99 ± 0.49*,††† |
| Saline (naive) | 53.68 ± 3.14 | 46.16 ± 4.17 | 5.68 ± 0.25 | 87.63 ± 3.18 | 80.10 ± 3.52 | 4.64 ± 0.25 |
| Saline (NIC) | 41.99 ± 5.00 | 35.25 ± 4.12 | 6.73 ± 0.28* | 80.73 ± 4.14 | 72.17 ± 4.09 | 4.71 ± 0.26 |
aData are grouped depending upon the drug tested (nicotine, nicotinePM, or saline) and previous drug experience: (naive), No experience, (Nicotine) and (NicotinePM), 2 d of previous nicotine or nicotinePM treatment, respectively. EEG total power is represented in percentages versus preinjection baseline (100%). Because of large-magnitude changes, EMG total power is shown as ln(%).
Significant differences in each drug group (saline, nicotine, and nicotinePM) versus “naive” conditions (*p < 0.05; ***p < 0.001; Student's t test). Significant differences between the effects of nicotinePM after previous nicotinePM and nicotine experience (†p < 0.05; ††p < 0.01; †††p < 0.001).
Habituation of locomotor, physiological, and neural effects of nicotinePM after repeated injections
In contrast to nicotine, its peripherally acting analog induced weak locomotor activation, weak increases in NAcc temperatures (F(25,545) = 1.6 and 1.6, respectively; p < 0.05), and equally rapid but smaller decreases in skin temperatures (F(25,545) = 8.4, p < 0.001) in drug-naive conditions (Fig. 4A, left). All effects, except the skin temperature response, significantly decreased (habituated) after prior nicotinePM injections (at least p < 0.05; Fig. 4A, right, B) to the levels induced by saline injections. NicotinePM in both conditions did not affect NAcc–muscle temperature differentials but slightly decreased skin–muscle differentials, and the latter effect in nicotinePM-treated rats was similar to that induced by saline.
Figure 4.

Habituation (tolerance) of the behavioral and physiological effects of nicotinePM. A, Top, Relative changes in NAcc, temporal muscle, and facial skin temperatures (°C). Middle, Changes in NAcc–muscle and skin-muscle temperature differentials (°C). Bottom, Locomotor activity (counts/min) induced by intravenous injections of nicotinePM in drug-naive conditions (left) and after 2 d of previous drug exposure (right). n is the number of averaged tests. Vertical hatched line shows the start of injection. Horizontal lines of respective colors represent values significantly different from preinjection baseline (p < 0.05). B, Mean (±SEM) changes in temperatures (°C) and locomotor activity (counts/min) calculated for 20 min after injection. Data are grouped depending on the drug tested (saline, nicotinePM) and previous drug experience: (Naive), No experience; (nicotinePM), 2 d of nicotinePM exposure. ○Significantly different from saline control (p < 0.05). #Significant differences in the effects of nicotinePM between drug-naive and drug-experienced conditions (p < 0.05). Numbers of trials in each group (n) are shown within bars.
Unlike regular nicotine, nicotinePM induced much weaker, transient EEG desynchronization and EMG activation in drug-naive conditions (Fig. 3D–F; F(15,495) = 7.1, 7.5, and 4.9 for cortical EEG, VTA, and EMG signals, respectively; each p < 0.001). These acute electrophysiological effects were significantly smaller than those of nicotine for both the immediate and long-term components, and all effects significantly decreased after repeated nicotinePM injections (Table 1; Fig. 3D–F). Although nicotinePM after previous injections still induced significant changes in EEG and EMG signals (F(22,712) = 3.0, 3.5, and 2.7; p < 0.01), these effects were similar to those observed after saline injections (Fig. 3D–F; Table 1).
Nicotine-like neural and physiological responses induced by nicotinePM after prior exposure to nicotine
In contrast to the weak, transient physiological effects of nicotinePM that habituated after repeated treatment, this drug at the same low dose strongly increased NAcc and muscle temperatures and strongly decreased skin temperature in animals with previous nicotine exposure (Fig. 5A; F(24,774) = 5.9, 5.0, 8.1, respectively, all p < 0.001). However, the increase in locomotor response was weak and did not reach statistical significance (F(24,774) = 1.4, p = 0.09). The effect of drug history (prior nicotine vs nicotinePM exposure) was significant for all temperature changes (F(10,520) = 9.2, 9.0, and 8.5 for NAcc, muscle, and skin, respectively; all p < 0.01) but only approached significance for locomotion (F(20,1040) = 1.4, p = 0.09). The effects of nicotinePM after nicotine exposure were ∼18-fold larger for NAcc temperature elevation and fourfold larger for skin temperature decrease than those observed with nicotinePM after nicotinePM exposure (Fig. 5B). In contrast, locomotor responses were equally weak and about the same in both conditions. After nicotine exposure, nicotinePM also strongly increased NAcc–muscle and strongly decreased skin–muscle, temperature differentials (F(24,774) = 6.0 and 7.8, respectively; p < 0.001; Fig. 5A, middle). These nicotine-like effects were either absent or very weak in drug-naive conditions (see above). The changes in both these parameters are also indicated by a significant drug history × time interaction (F(6,312) = 2.4, p < 0.05 and F(10,520) = 7.0, p < 0.001, respectively).
Figure 5.

Nicotine-like effects of nicotinePM after repeated nicotine exposure: temperature and locomotion. A, Top, Mean (±SEM) changes in NAcc, temporal muscle, and facial skin temperatures (°C). Middle, Changes in NAcc–muscle and skin–muscle temperature differentials (°C). Bottom, Locomotor activity (counts/min) induced by intravenous injections of nicotinePM after 2 d of nicotine exposure (°C). n is number of averaged tests. Vertical hatched line represents the start of injection. Horizontal lines of respective colors represent values significantly different from preinjection baseline (p < 0.05; Fisher test). B, Mean (±SEM) changes in NAcc and skin temperatures (°C) and locomotor activity (counts/min) calculated for 20 min after injection. Data are grouped depending upon the drug tested (nicotine and nicotinePM) and previous drug experience: (Naive), no experience (nicotine); (NicotinePM), 2 d of exposure to nicotine or nicotinePM treatment. ○Significant differences compared with nicotinePM in drug-naive conditions (p < 0.05). *Significant differences compared with nicotinePM after nicotinePM injections. #Differences in locomotor effects of nicotinePM and nicotine after nicotine exposure (red and yellow bars, respectively). Numbers of trials in each group are shown within bars. C, Rapid changes in NAcc (top) and skin (bottom) temperatures for 120 s before and 600 s after the onset of intravenous drug injections (vertical hatched line). Filled symbols represent values significantly different from preinjection baseline (p < 0.05). Bold horizontal green lines represent significant between-group differences (p < 0.05).
Strong changes in the effects of nicotinePM after prior nicotine exposure were particularly evident when NAcc and skin temperatures were analyzed at high temporal resolution (Fig. 5C). In contrast to the slow, weak changes in drug-naive conditions, nicotinePM after nicotine exposure induced a rapid and strong rise in NAcc temperature (F(24,774) = 12.2, p < 0.001) similar to that induced by nicotine. A similar potentiation was also observed for skin temperature, suggesting the appearance of a nicotine-like vasoconstrictive response of nicotinePM (Fig. 5C, bottom).
Nicotine exposure also changed the electrophysiological effects of nicotinePM, resulting in robust, prolonged increases in EEG desynchronization and much greater EMG activation (F(11,371) = 5.0, 2.8, and 3.2 for cortical, VTA EEG, and EMG signals, respectively; p < 0.01 in each case), greatly exceeding the effects of this drug in both drug-naive and nicotinePM-experienced conditions (Fig. 6A,B; Table 1). The effect of drug history was significant for all three parameters (F(1,33) = 7.7, 5.6, and 27.2, respectively; all p < 0.01), but drug history × time interaction was significant only for the EMG signal (F(120,3960) = 2.0, p < 0.01). The potentiating effect of nicotine exposure was equally strong and highly significant with respect to both the immediate and long-term EEG effects of the drug (Table 1). In contrast to the transient EMG activation and weak locomotor response seen in drug-naive conditions (Figs. 3F and 5A), nicotinePM given after nicotine exposure induced a strong and prolonged EMG activation, which remained evident for ∼8 min after injection interval (Fig. 6C; Table 1).
Figure 6.

Nicotine-like effects of nicotinePM after repeated nicotine exposure: cortical and VTA EEG and muscle EMG. A–C, Changes (mean ± SEM for 5 s time bins) in total powers of EEG desynhronization in the cortex (A) and VTA EEG (B) and EMG activation (C) induced by intravenous injections of nicotinePM after previous exposure to nicotinePM (blue) and nicotine (red). D–F, Changes in the same parameters induced by intravenous injections of saline after previous exposure to nicotine (red) and saline (blue). Filled symbols represent values significantly different from baseline. Green bold lines within each graph represent between-group differences in individual data points (p < 0.05). Two vertical hatched lines represent the timing of drug injection (0–15 s). EMG data were analyzed as ln derivatives and shown in ln-scale. Data are grouped depending upon the drug tested (nicotinePM or saline) and previous drug exposure: (NicotinePM) and (Nicotine), 2 d of nicotinePM or nicotine exposure; (saline) and (nicotine), 2 d of previous saline or nicotine exposure. n = number of averaged tests per group.
Electrophysiological effects of saline are also affected by nicotine exposure
Consistent with our previous observations (Kiyatkin and Lenoir, 2011), saline injections made during quiet resting conditions induced rapid, transient EEG desynchronization coupled with sharp and brief EMG activation (Fig. 6D–F; F(16,526) = 5.6, 5.9, and 5.4 for cortical, VTA, and EMG signals, respectively; p < 0.001). Compared to drug-naive conditions, these effects significantly increased after nicotine exposure (Table 1; Fig. 6C,D). The time × drug history interaction was significant for both cortical and VTA EEG and EMG signals (F(17,510) = 1.7, F(13,390) = 1.8, and F(7,210) = 2.2; p < 0.05) for 85, 65, and 15 s after injection onset, respectively). As shown in Table 1 and Figure 6C, D, the mean changes in EEG and EMG total powers induced by saline after prior nicotine exposure were significantly stronger than those in drug-naive controls from 22.5 to 57.5 s (cortical EEG) and 22.5 to 32.5 s (EMG) after the injection onset, with only a tendency toward an increase in the VTA.
Peripheral actions of nicotine are necessary for drug-induced locomotor activation and the development of locomotor sensitization
Recently, we showed that HEXA, a highly charged nicotinic antagonist with limited BBB permeability, strongly attenuates cortical EEG desynchronization and acute EMG activation induced by intravenous nicotine in drug-naive conditions (Lenoir and Kiyatkin, 2011). Therefore, we hypothesized that the blockade of peripheral nicotinic receptors could decrease the acute locomotor effects of nicotine and attenuate nicotine-induced locomotor sensitization. During two identical pretreatment days (days 1 and 2), rats received intravenous injections of either saline or HEXA, and 4 min later received intravenous injections of either saline or nicotine (a 2 × 2 factorial design). On test day 3, rats of all four groups (two primary and two control) received intravenous nicotine injections. In contrast to our major experiments, in which drugs were delivered via catheter extensions from distant locations, drug and saline in this experiment were directly injected into the head-mounted injection ports.
Under these conditions, injections of both nicotine and saline on day 1 induced similar locomotor activation (Fig. 7A,C). However, when nicotine injection was preceded by HEXA, the nicotine-induced locomotor response was strongly inhibited compared with the group pretreated with saline (significant time × pretreatment interaction for 13 min after injection; F(20,680) = 2.2, p < 0.01; Fig. 7A). Although saline-induced locomotor activation as a mean change was only marginally lower after HEXA injection (HEXA–saline group), this pretreatment significantly inhibited motor activity for ∼8 min after HEXA administration (F(9,316) = 5.1, p < 0.01; Fig. 7A). Similar to that on day 1, rats that received HEXA as the first injection showed a strongly attenuated locomotor response to nicotine on day 2 (significant time × pretreatment interactions for 13 min after injection; F(21,714) = 6.8, p < 0.01). Nicotine-induced locomotor activation on day 2 also increased versus day 1 (p < 0.05; red bars), saline-induced locomotion decreased (p < 0.01; black bars), and a significant difference emerged between the effects of nicotine and saline (p < 0.01). These results were confirmed using three-way ANOVA (drug, pretreatment, pretreatment day) for mean values of locomotion summed for 20 min after the second injection. In this case, both drug × pretreatment and drug × day interactions were significant (F(1,68) = 6.8 and 12.8, respectively; p < 0.01), suggesting that HEXA decreases nicotine-induced locomotion and has no significant effects on saline-induced locomotion on both days, and that effects of nicotine and saline change differently according to the day.
Figure 7.

Blockade of peripheral nicotine receptors attenuates nicotine-induced locomotion and prevents the development of nicotine locomotor sensitization. A–C, Development of locomotor sensitization. Time course (mean number of beam breaks/min ± SEM) and mean changes in horizontal locomotion (total number of beam breaks for 20 min after injection) induced by different pretreatments on day 1 and day 2. On both days, rats in each group (n = 6) received two consecutive intravenous injections (black arrows) at −4 and 0 min, respectively. The first injection was either saline or HEXA, and the second injection was either saline or nicotine. *Different from the nicotine–HEXA group (p < 0.05). D, E, Test for the expression of locomotor sensitization. Time course (mean number of beam breaks/min ± SEM) and mean changes in locomotion (total number of counts for 20 min after injection) induced by nicotine in rats with different pretreatment histories on days 1 and 2. E, ○Significant differences compared with saline–saline group. *Significant differences compared with HEXA–saline group. #Significant difference in effects of nicotine between saline–nicotine and HEXA–nicotine groups. n = number of rats(test) per group.
Nicotine-induced locomotor activation, assessed in animals of all groups on test day 3, differed depending upon previous drug experience (Fig. 7D,E). Rats that received previous nicotine treatment (saline–nicotine) showed a larger locomotor response to nicotine than those that received saline injections (saline–saline; time × pretreatment interaction: F(20,680) = 2.2; p < 0.01); this nicotine response was also larger than that induced by this drug on day 1 (p < 0.01). This sensitized locomotor response to nicotine was completely blocked in rats that received nicotine after HEXA pretreatment (significant time × pretreatment interaction: F(20,680) = 2.6, p < 0.001). In animals of this group, the attenuating effect was evident in both the initial, injection-related rise in locomotion, and the lower, tonic increase, which were almost absent relative to those in the saline–nicotine group. HEXA pretreatment (HEXA–saline) did not affect the nicotine-induced locomotor response seen after saline treatment (saline–saline) but unexpectedly, this response to nicotine after HEXA–nicotine pretreatment was significantly weaker than that in rats that received both saline–saline and HEXA–saline pretreatment (F(20,680) = 4.2 and 2.0, respectively; both p < 0.01; Fig. 7D,E). Possible reasons for this finding will be considered in the Discussion.
NicotinePM fails to enter the brain after intravenous injections
Because some of our observed effects of nicotinePM could be explained by its direct central action, we used mass spectrometry to verify whether this drug can enter the brain after intravenous administration. We first tested our extraction and desalting methods by spiking brain homogenates with nicotine, which was easily detected at a concentration of 250 pmol (10−12 mol) per milliliter of homogenate. During calibration of the mass spectrometer, both nicotine and nicotinePM standards were detected down to a concentration of 1 fmol (10−15 mol)/μl with a mass error of <2 ppm. By using electrospray ionization mass spectrometry, nicotine was detected in measurable amounts after nicotine injections but was undetectable after nicotinePM injections (Fig. 8). Additionally, we analyzed the brain samples in rats injected with nicotinePM at 200 μg/kg dose, and we were still unable to detect nicotine in brain homogenates. Based on the ion counts for the nicotine-injected brain compared with the spiked nicotine delivery, the amount of nicotine is most likely <1 nmol or lower per brain. To further confirm the assignment of nicotine, we conducted fragmentation (MS/MS) analysis on the nicotine ion. Based on the strong agreement in spectra for the nicotine standard and the nicotine peak in nicotine-injected brains, we confirmed our assignment of nicotine. Because no nicotine was detected in nicotinePM-injected brains, it is impossible to determine the entry ratio. However, it could be estimated to be at least two orders of magnitude lower than for regular nicotine based on the detection limit for the nicotinePM standard and the signal observed for the regular nicotine. Therefore, this experiment confirms the previous work showing that nicotinePM is virtually BBB-impermeable (Barlow and Hamilton, 1962; Aceto et al., 1983) and suggests that, with a proper brain perfusion, nicotine cannot be detected in the brain after a high-dose systemic injection of nicotinePM.
Figure 8.
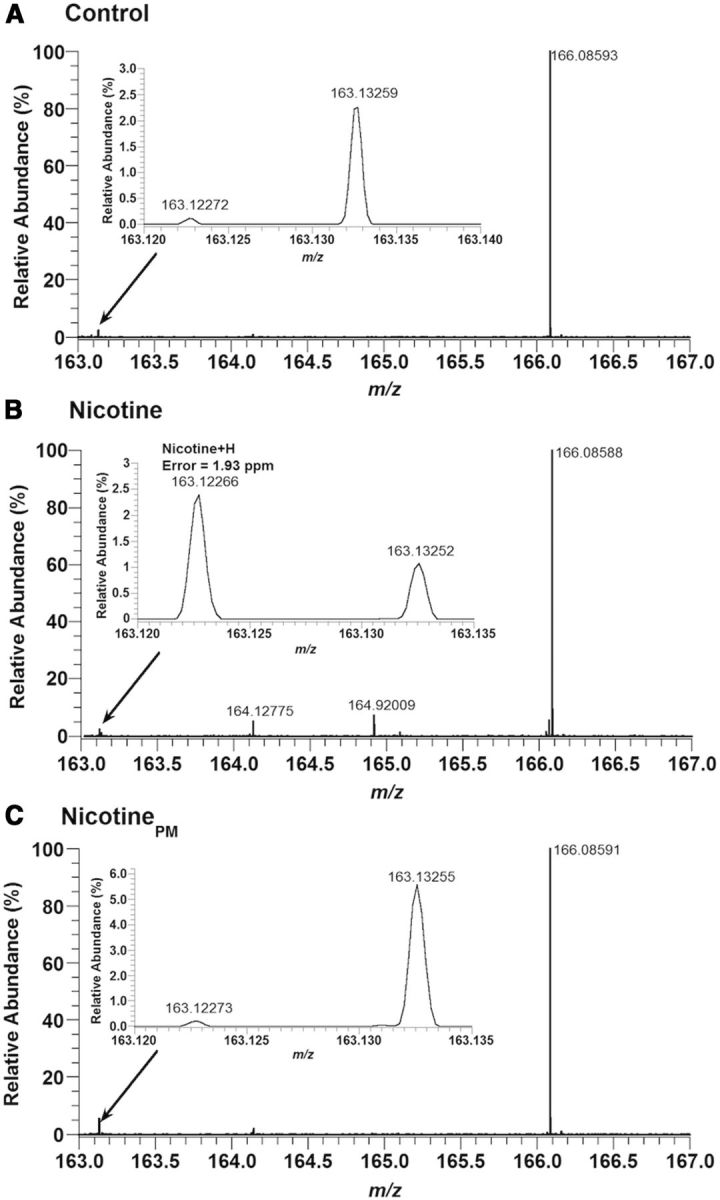
Nicotine is not detected in the brain after intravenous injections of nicotinePM. Electrospray mass spectra in the nicotine mass range in brain samples obtained from rats injected with saline (A, control), nicotine (B, 100 μg/kg), or nicotinePM (C, 100 μg/kg). The nicotine mass peak (M + H) was only detected above baseline for in the brains of nicotine-injected rats. This assignment was further confirmed by fragmentation (MS/MS) of this mass peak. The “nicotine peaks” in the brain were identical in rats injected with saline and nicotinePM.
Discussion
This study aimed to answer two major questions. First, by monitoring electrical activity in the cortex, VTA, and neck muscle, as well as temperatures in the NAcc, muscle, and skin, we examined how central and centrally mediated effects of intravenous nicotine at a low, behaviorally active dose change after repeated drug exposure and how they relate to changes in locomotor activity. Second, we explored the role of peripheral actions of nicotine in the sensitization of its physiological, neuronal, and behavioral effects.
Sensitization of physiological and neural effects of nicotine
Although the sensitization of nicotine's locomotor effects reported here agrees with previous data (Domino, 2001; Vezina et al., 2007; Mao and McGehee, 2010), the rapid experience-dependent enhancement of the physiological and neural effects of nicotine is a novel finding. Consistent with the literature (Rezvani and Levin, 2004; Ruskin et al., 2007), nicotine at this low dose in drug-naive conditions had weak effects on locomotion and slightly decreased NAcc and muscle temperatures but rapidly and strongly decreased skin temperature, reflecting the known centrally mediated vasoconstrictive effect of nicotine (Comroe, 1960; Ginzel, 1975). However, after relatively limited exposure, nicotine induced much stronger locomotor activation, modest elevations in brain and muscle temperatures, and a greater increase in NAcc–muscle temperature differential. The rise in the latter parameter suggests that transient metabolic brain activation induced by nicotine in drug-naive conditions becomes stronger and more prolonged after drug experience. In contrast, nicotine-induced EEG desynchronization in both the cortex and VTA remained equally rapid and strong but became more prolonged after nicotine exposure. Interestingly, in contrast to the large increases in locomotor activity, the nicotine-induced EMG response reflecting tonic and phasic changes in muscular tone was equally strong regardless of drug experience.
Critical role of peripheral actions of nicotine in the development of neural sensitization
Previous studies suggest that nicotine's actions on peripherally located neural substrates contribute significantly to the acute neural effects of nicotine (see Introduction). Here, we used two pharmacological tools, nicotinePM and HEXA, to clarify the role of these peripheral actions in the development of nicotine-induced sensitization. Both drugs are highly charged molecules and have very limited BBB permeability. Although these drugs after systemic administration could be detected in brain tissue in extremely low amounts (see Materials and Methods), we confirmed the inability of nicotinePM to enter the brain using a highly sensitive mass spectrometry method. With proper brain perfusion, which was often missing in early autoradiographic studies, nicotine was easily detected in brain tissue after intravenous injection of regular nicotine but remained undetectable after higher-dose nicotinePM injections. Therefore, based on existing knowledge and our measurements, we assume that the effects induced by these drugs are mediated primarily by their interaction with peripheral nicotinic receptors.
Unlike nicotine, which activates both peripherally and centrally located nicotinic receptors, nicotinePM, its peripherally acting analog, induced much weaker, transient responses in drug-naive conditions, mimicking the effects of mild somatosensory stimuli. Similar to sound (Kiyatkin and Smirnov, 2010) and saline injections (this study), nicotinePM induced brief EEG desynchronization equally evident in the cortex and VTA, and equally brief EMG activation. Similar to weak sensory stimuli, nicotinePM slightly increased locomotion, weakly elevated brain and body temperatures, and transiently decreased skin temperature. Consistent with the known habituation of neural effects induced by simple somatosensory stimuli (Hendry et al., 1999; Stancak, 2006; Sandler and Tsitolovsky, 2008), the electrophysiological effects of nicotinePM weaken after repeated exposure. Habituation was also evident at behavioral and physiological levels, suggesting that repeated stimulation of only peripheral nicotinic receptors quickly loses its arousing potential.
However, nicotinePM at the same, apparently “ineffective” dose induced strong physiological and neural effects when injected after nicotine experience. Under these conditions, nicotinePM strongly increased NAcc and muscle temperatures, decreased skin temperatures, and induced powerful and prolonged EEG desynchronization and EMG activation, suggesting that, after nicotine exposure, this drug acquired the properties of a conditioned, nicotine-related interoceptive cue. The enhancement of NAcc–muscle and skin–muscle temperature differentials induced by nicotinePM under these conditions (Fig. 5) suggests that it also induces nicotine-like metabolic brain activation and peripheral vasoconstriction, novel drug effects that were not evident before nicotine exposure. If selective stimulation of peripheral nicotinic receptors after nicotine experience is able to induce a powerful conditioned neural and physiological activation, this rapid effect may interact in the CNS with the later-occurring, more prolonged actions of nicotine in the brain, thus contributing to the enhanced (sensitized) physiological and neural effects of nicotine with repeated exposure. Therefore, the effects of nicotine after previous experience are always “conditioned drug effects” (Stewart, 1992), reflecting a combination of pharmacological and learning factors. Nicotine experience also slightly (but significantly) potentiated transient EEG desynchronization and EMG activation induced by saline injection (Fig. 6D–F). Although saline injection is usually viewed as a neutral stimulus, it is associated with transient changes in temperature and pressure, which are detected by multiple subtypes of ionic channels (Clapham, 2003), resulting in transient neural activation due to brief stimulation of visceral sensory afferents. Importantly, this test suggests that even weak, nonspecific stimulation of vascular sensory afferents, a pathway transmitting the nicotine sensory signal, could act in nicotine-experienced individuals as a weak, drug-related sensory cue. This mechanism could explain why habitual smokers often perceive similar immediate subjective and autonomic responses during smoking of nicotine-free cigarettes (Butschky et al., 1995; Westman et al., 1996).
The peripherally triggered conditioned effect of nicotinePM was strong at the level of neural activity (cortical and VTA EEG) and centrally organized physiological parameters (EMG, NAcc, body and skin temperatures) but unexpectedly weak at the level of locomotion. Although the postnicotine locomotor response to nicotinePM was significantly larger than that after nicotinePM experience, the difference in effect was no greater than the initial drug response in drug-naive conditions (Fig. 5B). Although this finding may seem surprising, it agrees with evidence that nicotine's action on central neurons (which are absent in nicotinePM) are essential to induce behavioral activation.
Our experiments with HEXA confirmed the critical role of nicotine's action on peripheral nicotinic receptors in the development of locomotor sensitization. Consistent with the strong attenuation of nicotine-induced EEG desynchronization and EMG activation seen in drug-naive conditions (Lenoir and Kiyatkin, 2011), HEXA pretreatment strongly inhibited the acute locomotor effects of nicotine and fully blocked the sensitized locomotion after repeated nicotine exposure. Therefore, nicotine's action on peripheral nicotinic receptors appears to be critical for triggering the acute locomotor response to nicotine and may be essential for the development of behavioral sensitization.
Unexpectedly, the nicotine-induced locomotion in the HEXA–nicotine pretreatment group was even weaker than that in the saline–saline pretreatment group that never received nicotine (Fig. 7D,E). We hypothesize that the relative decrease in nicotine-induced locomotion on day 3 may be the result of a repeated attenuation of its acute neural effects by HEXA during pretreatment. This difference could also result from procedural factors (i.e., direct drug/saline administrations) via head-mounted injection ports instead of a catheter extensions used in the main experiments. Under these conditions, saline injection induced locomotor activation (Fig. 7A–C), which was otherwise absent under fully stress- and cue-free conditions (Fig. 2). This behavioral activation associated with the injection procedure revealed a transient inhibiting effect of HEXA on locomotion, which was also absent during cue- and stress-free injections (Lenoir and Kiyatkin, 2011), and masked some of the initial locomotor activating effects of nicotine observed in drug-naive rats in Experiments 1 and 2 (Fig. 2). Therefore, this difference in the effects of nicotine could also result from the relative increase in nicotine-induced locomotion in the saline–saline pretreatment group due to habituation to repeated injection procedure. Finally, despite much evidence suggesting the exclusively peripheral action of HEXA, we cannot exclude the possibility that the drug can enter the brain in small amounts via specific BBB-weak locations and affect central neurons.
In conclusion, we provide evidence for a critical role of nicotine's actions on peripheral nicotinic receptors in the development of sensitization of nicotine's physiological, neural, and behavioral effects. By stimulating these receptors, abundantly expressed on afferents of sensory nerves, nicotine administration produces neural signal, which rapidly reaches the brain and triggers neural activation. This effect interacts with the subsequent, slower and more prolonged direct drug effects in the brain, and during repeated administration, these initially independent drug actions may become interdependent according to principles of Pavlovian conditioning. Therefore, in drug-experienced individuals, the peripheral actions of nicotine may serve as powerful interoceptive conditioned cues and play an important role in human cigarette smoking. Consistent with this idea, blockade of sensory afferents of the upper respiratory tract by local anesthetics makes human smoking much less rewarding (Rose et al., 1985). Finally, based on our data, the human observations described above, and the evidence that a similar interoceptive conditioning mechanism may be applicable to other addictive drugs (Brown and Kiyatkin, 2008; Kiyatkin and Smirnov, 2010; Wise and Kiyatkin, 2011), we suggest that behavioral and pharmacological treatment strategies targeting interoceptive conditioned drug effects may be more efficacious in drug abusers than those that focus exclusively on devaluing the unconditioned rewarding effects of drugs in the brain.
Footnotes
This work was supported by the National Institute on Drug Abuse–Intramural Research Program. We thank Drs. Y. Shaham, S. Ahmed, C. Mejias-Aponte, K. Guillem, C. Lupica, P. Redgrave, E. Gardner, and K. Wakabayashi for helpful suggestions regarding this manuscript; Dr. S. Jackson for assistance in nicotine measurement experiment; and postbaccalaureate trainees S. Schwarzchild and S. Myal for technical assistance.
The authors declare no competing financial interests.
References
- Aceto MD, Awaya H, Martin BR, May EL. Antinociceptive action of nicotine and its methiodide derivatives in mice and rats. Br J Pharmacol. 1983;79:869–876. doi: 10.1111/j.1476-5381.1983.tb10531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschule MD. Emotion and the circulation. Circulation. 1951;3:444–454. doi: 10.1161/01.CIR.3.3.444. [DOI] [PubMed] [Google Scholar]
- Anand A. Role of aortic chemoreceptors in the hypertensive response to cigarette smoke. Respir Physiol. 1996;106:231–238. doi: 10.1016/S0034-5687(96)00087-4. [DOI] [PubMed] [Google Scholar]
- Asghar K, Roth LJ. Entry and distribution of hexamethonium in the central nervous system. Biochem Pharmacol. 1971;20:2787–2795. doi: 10.1016/0006-2952(71)90189-4. [DOI] [PubMed] [Google Scholar]
- Baker MA, Cronin MJ, Mountjoy DG. Variability of skin temperature in the waking monkey. Am J Physiol. 1976;230:449–455. doi: 10.1152/ajplegacy.1976.230.2.449. [DOI] [PubMed] [Google Scholar]
- Balfour DJ, Benwell ME, Birrell CE, Kelly RJ, Al-Aloul M. Sensitization of the mesoaccumbens dopamine response to nicotine. Pharmacol Biochem Behav. 1998;59:1021–1030. doi: 10.1016/S0091-3057(97)00537-6. [DOI] [PubMed] [Google Scholar]
- Barlow RB, Dobson NA. Nicotine monomethiodide. J Pharm Pharmacol. 1955;7:27–34. doi: 10.1111/j.2042-7158.1955.tb12000.x. [DOI] [PubMed] [Google Scholar]
- Barlow RB, Hamilton JT. Effects of some isomers and analogues of nicotine on junctional transmission. Br J Pharmacol Chemother. 1962;18:510–542. doi: 10.1111/j.1476-5381.1962.tb01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJ. The effects of acute and repeated nicotine treatment on nucleus accumbens dopamine and locomotor activity. Br J Pharmacol. 1992;105:849–856. doi: 10.1111/j.1476-5381.1992.tb09067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MS, Apana SM, Nagano KK, Berridge CE, Leisure GP, Boswell MV. Smoking produces rapid rise of [11C]nicotine in human brain. Psychopharmacology (Berl) 2010;209:383–394. doi: 10.1007/s00213-010-1809-8. [DOI] [PubMed] [Google Scholar]
- Bevins RA, Barrett ST, Polewan RJ, Pittenger ST, Swalve N, Charntikov S. Disentangling the nature of the nicotine stimulus. Behav Processes. 2012;90:28–33. doi: 10.1016/j.beproc.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PL, Kiyatkin EA. Sensory effects of intravenous cocaine on dopamine and non-dopamine ventral tegmental area neurons. Brain Res. 2008;1218:230–249. doi: 10.1016/j.brainres.2008.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butschky MF, Bailey D, Henningfield JE, Pickworth WB. Smoking without nicotine delivery decreases withdrawal in 12-hour abstinent smokers. Pharmacol Biochem Behav. 1995;50:91–96. doi: 10.1016/0091-3057(94)00269-O. [DOI] [PubMed] [Google Scholar]
- Byck R. The effect of hexamethonium on the carotid chemoreceptor response to nicotine and cyanide. Br J Pharmacol Chemother. 1961;16:15–22. doi: 10.1111/j.1476-5381.1961.tb00293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprioli D, Celentano M, Paolone G, Badiani A. Modeling the role of environment in addiction. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1639–1653. doi: 10.1016/j.pnpbp.2007.08.029. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Clarke PB, Kumar R. The effects of nicotine on locomotor activity in non-tolerant and tolerant rats. Br J Pharmacol. 1983;78:329–337. doi: 10.1111/j.1476-5381.1983.tb09398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comroe JH. The pharmacological actions of nicotine. Ann N Y Acad Sci. 1960;90:48–51. doi: 10.1111/j.1749-6632.1960.tb32616.x. [DOI] [PubMed] [Google Scholar]
- Cox BM, Goldstein A, Nelson WT. Nicotine self-administration in rats. Br J Pharmacol. 1984;83:49–55. doi: 10.1111/j.1476-5381.1984.tb10118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G. Role of dopamine in the behavioral actions of nicotine related to addiction. Eur. J Pharmacol. 2000;393:295–314. doi: 10.1016/S0014-2999(00)00122-9. [DOI] [PubMed] [Google Scholar]
- Domino EF. Nicotine induced behavioral locomotor sensitization. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:59–71. doi: 10.1016/S0278-5846(00)00148-2. [DOI] [PubMed] [Google Scholar]
- Donny EC, Caggiula AR, Knopf S, Brown C. Nicotine self-administration in rats. Psychopharmacology (Berl) 1995;122:390–394. doi: 10.1007/BF02246272. [DOI] [PubMed] [Google Scholar]
- Engberg G, Hajos M. Nicotine-induced activation of locus ceruleus neurons: an analysis of peripheral versus central induction. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:443–446. doi: 10.1007/BF00169131. [DOI] [PubMed] [Google Scholar]
- Ginzel KH. Muscle relaxation by drugs which stimulate sensory nerve endings: II. The effects of nicotinic agents. Neuropharmacology. 1973;12:149–164. doi: 10.1016/0028-3908(73)90084-1. [DOI] [PubMed] [Google Scholar]
- Ginzel KH. The importance of sensory nerve endings as sites of drug action. Naunyn Schmiedebergs Arch Pharmacol. 1975;288:29–56. doi: 10.1007/BF00501812. [DOI] [PubMed] [Google Scholar]
- Hendry SC, Hsiao SS, Brown MC. Fundamentals of sensory systems. In: Zigmond MJ, Bloom FE, Landis SC, Roberts JL, Squire LR, editors. Fundamental neuroscience. San Diego: Academic; 1999. pp. 657–670. [Google Scholar]
- Juan H. Nicotinic nociceptors on perivascular sensory nerve endings. Pain. 1982;12:259–264. doi: 10.1016/0304-3959(82)90157-9. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-U. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA. Brain temperature homeostasis: physiological fluctuations and pathological shifts. Front Biosci. 2010;15:73–92. doi: 10.2741/3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyatkin EA, Brown PL. Brain and body temperature homeostasis during sodium pentobarbital anesthesia with and without body warming in rats. Physiol Behav. 2005;84:563–570. doi: 10.1016/j.physbeh.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Lenoir M. Intravenous saline injection as an interoceptive signal in rats. Psychopharmacology (Berl) 2011;217:387–396. doi: 10.1007/s00213-011-2294-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyatkin EA, Smirnov MS. Rapid EEG desynchronization and EMG activation induced by intravenous cocaine in freely moving rats: a peripheral, nondopamine neural triggering. Am J Physiol Regul Integr Comp Physiol. 2010;298:R285–R300. doi: 10.1152/ajpregu.00628.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksir C, Hakan RL, Kellar KJ. Chronic nicotine and locomotor activity: influences of exposure dose and test dose. Psychopharmacology. 1987;92:25–29. doi: 10.1007/BF00215474. [DOI] [PubMed] [Google Scholar]
- Lenoir M, Kiyatkin EA. Critical role of peripheral actions of intravenous nicotine in mediating its central effects. Neuropsychopharmacology. 2011;36:2125–2138. doi: 10.1038/npp.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Simon SA. Capsaicin and nicotine both activate a subset of rat trigeminal ganglion neurons. Am J Physiol. 1996;270:C1807–C1814. doi: 10.1152/ajpcell.1996.270.6.C1807. [DOI] [PubMed] [Google Scholar]
- Malin DH. Nicotine dependence: studies with a laboratory model. Pharmacol Biochem Behav. 2001;70:551–559. doi: 10.1016/S0091-3057(01)00699-2. [DOI] [PubMed] [Google Scholar]
- Malin DH, Lake JR, Schopen CK, Kirk JW, Sailer EE, Lawless BA, Upchurch TP, Shenoi M, Rajan N. Nicotine abstinence syndrome precipitated by central but not peripheral hexamethonium. Pharmacol Biochem Behav. 1997;58:695–699. doi: 10.1016/S0091-3057(97)90006-X. [DOI] [PubMed] [Google Scholar]
- Mao D, McGehee DS. Nicotine and behavioral sensitization. J Mol Neurosci. 2010;40:154–163. doi: 10.1007/s12031-009-9230-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology. 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- O'Brien CP, Childress AR, McLellan AT, Ehrman R. Classical conditioning in drug-dependent humans. Ann N Y Acad Sci. 1992;654:400–415. doi: 10.1111/j.1749-6632.1992.tb25984.x. [DOI] [PubMed] [Google Scholar]
- Oldendorf WH, Stoller BE, Harris FL. Blood–brain barrier penetration abolished by N-methyl quaternization of nicotine. Proc Natl Acad Sci U S A. 1993;90:307–311. doi: 10.1073/pnas.90.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolone G, Palopoli M, Marrone MC, Nencini P, Badiani A. Environmental modulation of the interoceptive effects of amphetamine in the rat. Behav Brain Res. 2004;152:149–155. doi: 10.1016/j.bbr.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Paxinos J, Watson C. The rat brain in stereotaxic coordinates. Sydney: Academic; 1998. [Google Scholar]
- Petrali JP, Maxwell DM, Lenz DE, Mills KR. A study of the effect of soman on the rat blood–brain barrier. Proceedings of the 4th Annual Chemical Defense Review (395–405); Aberdeen Proving Grounds, MD: US Army Medical Research Institute of Chemical Defense; 1984. [Google Scholar]
- Picciotto MR, Corrigall WA. Neuronal systems underlying behaviors related to nicotine addiction: neural circuits and molecular genetics. J Neurosci. 2002;22:3338–3341. doi: 10.1523/JNEUROSCI.22-09-03338.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontieri FE, Tanda G, Orzi F, Di Chiara G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature. 1996;382:255–257. doi: 10.1038/382255a0. [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Levin ED. Adolescent and adult rats respond differently to nicotine and alcohol: motor activity and body temperature. Int J Dev Neurosci. 2004;22:349–354. doi: 10.1016/j.ijdevneu.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-P. [DOI] [PubMed] [Google Scholar]
- Rose JE, Corrigall WA. Nicotine self-administration in animals and humans: similarities and differences. Psychopharmacology. 1997;130:28–40. doi: 10.1007/s002130050209. [DOI] [PubMed] [Google Scholar]
- Rose JE, Tashkin DP, Ertle A, Zinser MC, Lafer R. Sensory blockade of smoking satisfaction. Pharmacol Biochem Behav. 1985;23:289–293. doi: 10.1016/0091-3057(85)90572-6. [DOI] [PubMed] [Google Scholar]
- Rose JE, Mukhin AG, Lokitz SJ, Turkington TG, Herskovic J, Behm FM, Garg S, Garg PK. Kinetics of brain nicotine accumulation in dependent and nondependent smokers assessed with PET and cigarettes containing 11C-nicotine. Proc Natl Acad Sci U S A. 2010;107:5190–5195. doi: 10.1073/pnas.0909184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin DN, Anand R, LaHoste GJ. Menthol and nicotine oppositely modulate body temperature in the rat. Eur J Pharmacol. 2007;559:161–164. doi: 10.1016/j.ejphar.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Sandler U, Tsitolovsky L. Neural cell behavior and fuzzy logic. New York: Springer; 2008. [Google Scholar]
- Siegel S. Drug anticipation and drug addiction: the 1998 H. David Archibald Lecture. Addiction. 1999;94:1113–1124. doi: 10.1046/j.1360-0443.1999.94811132.x. [DOI] [PubMed] [Google Scholar]
- Stancák A. Cortical oscillatory changes occurring during somatosensory and thermal stimulation. Prog Brain Res. 2006;159:237–252. doi: 10.1016/S0079-6123(06)59016-8. [DOI] [PubMed] [Google Scholar]
- Stewart J. Neurobiology of conditioning to drugs of abuse. Ann N Y Acad Sci. 1992;654:335–346. doi: 10.1111/j.1749-6632.1992.tb25979.x. [DOI] [PubMed] [Google Scholar]
- Stolerman IP. Behavioural pharmacology of nicotine: implications for multiple brain nicotinic receptors. Ciba Found Symp. 1990;152:3–16. doi: 10.1002/9780470513965.ch2. discussion 16–22. [DOI] [PubMed] [Google Scholar]
- Stolerman IP, Jarvis MJ. The scientific case that nicotine is addictive. Psychopharmacology. 1995;117:2–10. doi: 10.1007/BF02245088. discussion 14–20. [DOI] [PubMed] [Google Scholar]
- Toyama T, Yokoyama I, Nishi K. Effects of hexamethonium and other ganglionic blocking agents on electrical activity of the esophagus induced by vagal stimulation in the dog. Eur J Pharmacol. 1975;31:63–71. doi: 10.1016/0014-2999(75)90079-5. [DOI] [PubMed] [Google Scholar]
- Vezina P, McGehee DS, Green WN. Exposure to nicotine and sensitization of nicotine-induced behaviors. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1625–1638. doi: 10.1016/j.pnpbp.2007.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JC, Kendal-Reed M, Keiger CJ, Bencherif M, Silver WL. Olfactory and trigeminal responses to nicotine. Drug Dev Res. 1996;38:160–168. doi: 10.1002/(SICI)1098-2299(199607/08)38:3/4<160::AID-DDR4>3.0.CO%3B2-K. [DOI] [Google Scholar]
- Wassermann O. Studies on the pharmacokinetics of bis-quaternary ammonium compounds: 3. Autoradiographic studies on the substituent-dependent distribution pattern of 3H-hexamethonium derivatives in mice. Naunyn Schmiedebergs Arch Pharmacol. 1972;275:251–261. doi: 10.1007/BF00500054. [DOI] [PubMed] [Google Scholar]
- Westman EC, Behm FM, Rose JE. Dissociating the nicotine and airway sensory effects of smoking. Pharmacol Biochem Behav. 1996;53:309–315. doi: 10.1016/0091-3057(95)02027-6. [DOI] [PubMed] [Google Scholar]
- Wise RA, Kiyatkin EA. Differentiating the rapid actions of cocaine. Nat Rev Neurosci. 2011;12:479–484. doi: 10.1038/nrn3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods AS, Moyer SC, Jackson SN. Amazing stability of phosphate-quaternary amine interactions. J Proteome Res. 2008;7:3423–3427. doi: 10.1021/pr8001595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Lu H, Vaupel DB, Zhang Y, Chefer SI, Rea WR, Moore AV, Yang Y, Stein EA. Acute nicotine-induced tachyphylaxis is differentially manifest in the limbic system. Neuropsychopharmacology. 2011;36:2498–2512. doi: 10.1038/npp.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]