

Abstract
Identifying new targeted therapies that kill tumor cells while sparing normal tissue is a major challenge of cancer research. Using a high-throughput chemical synthetic lethal screen, we sought to identify compounds that exploit the loss of the von Hippel–Lindau (VHL) tumor suppressor gene, which occurs in about 80% of renal cell carcinomas (RCCs). RCCs, like many other cancers, are dependent on aerobic glycolysis for ATP production, a phenomenon known as the Warburg effect. The dependence of RCCs on glycolysis is in part a result of induction of glucose transporter 1 (GLUT1). Here, we report the identification of a class of compounds, the 3-series, exemplified by STF-31, which selectively kills RCCs by specifically targeting glucose uptake through GLUT1 and exploiting the unique dependence of these cells on GLUT1 for survival. Treatment with these agents inhibits the growth of RCCs by binding GLUT1 directly and impeding glucose uptake in vivo without toxicity to normal tissue. Activity of STF-31 in these experimental renal tumors can be monitored by [18F]fluorodeoxyglucose uptake by micro–positron emission tomography imaging, and therefore, these agents may be readily tested clinically in human tumors. Our results show that the Warburg effect confers distinct characteristics on tumor cells that can be selectively targeted for therapy.
INTRODUCTION
Conventional chemotherapeutic agents were identified by their ability to kill rapidly proliferating cells and therefore do not distinguish between healthy and cancer cells. Standard agents have low therapeutic indices and often show severe toxicity in normal tissue. Synthetic lethality is a therapeutically advantageous approach to drug discovery and is particularly suited to developing targeted therapeutics to treat cancers. Synthetic lethality is the genetic interaction whereby the simultaneous combination of mutation and/or inhibition of two genes leads to cell death (1). If only one of the two genes is altered, there are no deleterious effects and the cells are viable. In chemical synthetic lethality, the first mutation is essential for the development and survival of the cancer cells, whereas inhibition of a second gene or pathway by a small molecule results in cytotoxic cell death. This approach is appealing because it should kill tumor cells while not affecting normal, noncancerous tissue.
Renal cell carcinoma (RCC), the most common type of kidney cancer, is a particularly challenging, intractable disease, resistant to both radiation therapy and standard systemic chemotherapies (2). Less than 10% of metastatic RCC patients respond to immunotherapy (3). The targeting of receptor tyrosine kinases by sunitinib (Sutent) and sorafenib (Nexavar) has become the standard of care for advanced RCC (4), although these agents are not curative. RCC is uniquely suited for exploitation by synthetic lethal screening. In most RCCs, the von Hippel–Lindau (VHL) tumor suppressor gene is inactivated, driving tumor development. VHL deficiency imparts distinct characteristics on tumor cells, such as reliance on the high-affinity glucose transporter 1 (GLUT1) and aerobic glycolysis, also known as the Warburg effect (5). To specifically target RCC, we have used a synthetic lethal approach, seeking to identify compounds that exhibit selective cytotoxicity to cells that have lost functional VHL. In this report, we identify a class of compounds that impair glucose transport, resulting in specific killing of renal carcinoma cells.
RESULTS
A chemical synthetic lethal agent that targets loss of VHL in renal carcinoma
To discover compounds that selectively target RCC, we screened ~64,000 small molecules to identify those that functioned in a synthetic lethal manner to the loss of VHL. An RCC cell line with a naturally occurring VHL mutation and a genetically matched cell line with reintroduced wild-type VHL (fig. S1A) were labeled with yellow fluorescent protein and treated with a small-molecule library at a concentration of 10 to 20 μM. Fluorescence was read on day 4 as a surrogate marker for viability and growth. From this fluorescence-based cell assay, two classes of drugs exhibited toxicity for cells lacking VHL, but were relatively nontoxic to cells with functional VHL. One class, the pyridyl anilino thiazoles (for example, STF-62247), has previously been shown to induce autophagic cell death in VHL-deficient RCCs (6, 7) and is in preclinical development. Here, we characterize the selective cytotoxicity of a second class of compounds, the 3-series, typified by STF-31 (fig. S1B). We used both short-term viability and long-term survival assays to validate the primary screen (fig. S1C and Fig. 1, A and B). RCC4 cells without VHL showed reduced viability in the presence of drug in a concentration-dependent manner when compared to their wild-type counterparts (RCC4/VHL) as measured by XTT {2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2H-tetrazolium hydroxide} reduction after 4 days of treatment with STF-31 (fig. S1C). Clonogenic survival confirmed that STF-31 was specifically toxic to RCC4 cells, whereas RCC4/VHL cells were relatively unaffected (Fig. 1, A and B). RCC4/VHL cells treated with STF-31 largely recovered, whereas RCC4 cells under the same conditions did not (Fig. 1C). To corroborate the VHL dependence of STF-31 resistance, we tested ACHN cells, a renal carcinoma cell line with functional VHL, for STF-31 sensitivity (fig. S1A). Only ACHN cells with short hairpin RNA (shRNA) to VHL were sensitive to STF-31 (Fig. 1D). Thus, our chemical synthetic lethal screen identified compounds specifically cytotoxic to cells with impaired VHL function.
Fig. 1.

Chemical synthetic lethal screen identifies compounds that specifically target loss of VHL in renal carcinoma. (A) Effect of STF-31 on clonogenic survival of RCC4 with and without VHL (10 days) (*P < 0.00005). (B) Representative plates of clonogenic survival in RCC4 and RCC4/VHL cells (5 μM STF-31; 10 days). (C) Time course of STF-31 (5 μM) effect on cells (*P < 0.0005). (D) Effect of STF-31 on clonogenic survival of ACHN cells with and without shRNA to VHL (*P < 0.0001). (E) Effect of STF-31 (5 μM) on cell death in RCC4 and RCC4/VHL cells treated for 3 days. Cell death was measured with trypan blue staining (*P < 0.01). (F) Effect of STF-31 on RCC4, RCC4/VHL, or RCC4/VHL cell clones overexpressing HIF-2α (*P < 0.005). All error bars represent the SEM (n = 3).
Effect of STF-31 on autophagy, apoptosis, and DNA damage
We sought to determine whether STF-31 killed VHL-deficient cells through autophagic cell death or a different pathway. STF-31 did not induce any morphologic or biochemical features of autophagy, such as LC3-II lipidation (fig. S1D). There was no increase in nuclear condensation (fig. S1E), propidium iodide/annexin V staining (fig. S1F), and total p53 or phospho-p53 levels (fig. S1G) in RCC cells with or without VHL in response to STF-31, suggesting that STF-31 is not inducing apoptosis or DNA damage. However, in contrast to RCC4/VHL cells, RCCs without VHL underwent necrotic cell death in response to STF-31, as measured by the ability of the cells to exclude trypan blue, an indicator of cell membrane integrity (Fig. 1E). Together, these results indicate that STF-31 is synthetic lethal to the loss of VHL by causing a necrotic cell death.
Hypoxia-inducible factor overexpression and sensitivity to STF-31
VHL inactivation increases protein half-life of the hypoxia-inducible factor (HIF) family of transcription factors (8, 9). A nondegradable, constitutively active HIF was overexpressed in two clones of RCC4/VHL cells (10) and tested to determine whether sensitivity to STF-31 was HIF-dependent. Ectopic expression of HIF in cells with wild-type VHL sensitized these cells to STF-31 treatment, suggesting that deregulated HIF expression in VHL-deficient cells is at least partially responsible for the selective cytotoxicity of STF-31 (Fig. 1F). Therefore, STF-31 seems to function in a synthetic lethal manner to VHL mutation, preferentially targeting VHL-deficient cells and with cytotoxicity directly linked to the aberrant up-regulation of HIF.
Synthetic lethal interaction between glucose metabolism and VHL deficiency
HIF is important to adapting to low oxygen conditions through the transcription of many genes, including those involved in glucose metabolism (fig. S2A) (11). We hypothesized that STF-31 might inhibit metabolic pathways, leading to necrotic cell death. To examine whether this compound alters glycolysis, we measured intracellular lactate, which is rapidly converted from the glycolysis end product pyruvate, and extracellular acidification. Baseline levels of glycolysis were lower in wild-type VHL cells compared to VHL-deficient cells (Fig. 2, A and B), likely a result of the constitutive expression of HIF and subsequent overexpression of glucose transporters and glycolytic enzymes in the latter (12). Treatment with STF-31 significantly inhibited lactate production and extracellular acidification in VHL-deficient cells by about 60% compared to control-treated cells (Fig. 2, A and B). Treatment with STF-31 did not alter glycolysis in cells with wild-type VHL cells (Fig. 2, A and B). These results suggest that the selective toxicity of STF-31 is a consequence of the dependence of RCCs on glycolysis and/or a preferential targeting of pathways needed for glycolysis.
Fig. 2.

STF-31 inhibits glucose metabolism in VHL-deficient cells. (A) Lactate (μmol/cell), which is converted from pyruvate, the end product of glycolysis, in RCC4 and RCC4/VHL cells treated with either vehicle or STF-31 (5 μM) (*P < 0.01). (B) Relative extracellular acidification rate (ECAR) (mpH/min) of VHL-deficient cells and cells with wild-type VHL in response to STF-31 (5 μM) for 48 hours. Cells were stained with crystal violet and absorbance was measured for normalization (*P < 0.0005). (C) Relative glucose uptake after treatment with STF-31 (5 μM). Counts are normalized to cell number (*P < 0.000005). (D) Effect of STF-31 concentration on glucose uptake (*P < 0.00005). (E) Effect of STF-31 on hexokinase activity in whole-cell lysates after STF-31 treatment (5 μM). (F) Effect of STF-31 (5 μM) on glucose uptake in RCC4 cells transfected with small interfering RNA (siRNA) to HIF-1β (*P < 0.05). (G) Effect of STF-31 (5 μM) on oxygen consumption (nmol/min/106 cells). (H) Effect of STF-31 (5 μM) on relative ATP levels in cells with and without VHL (*P < 0.005). (I) Effect of STF-31 on ATP levels in cells with and without VHL (*P < 0.01). (J) Effect of STF-31 (5 μM) on glucose uptake and clonogenic cell survival in cells without VHL up to 72 hours. (K) Effect of STF-31 (5 μM) on glucose uptake and clonogenic cell survival in cells with VHL up to 72 hours. All error bars represent the SEM (n = 3).
STF-31 inhibits glucose uptake
We then examined whether this decrease in glycolysis in response to the 3-series was a result of a decrease in glucose uptake by using 2-deoxy-D-[3H]glucose, a nonhydrolyzable, radioactive glucose analog. STF-31 impaired glucose uptake in RCC4 cells (Fig. 2C) in a dose-dependent manner (Fig. 2D). RCC4/VHL cells had lower baseline glucose uptake compared to RCC4 cells (Fig. 2C) that was unaffected by increasing concentrations of STF-31 (Fig. 2D). Because the phosphorylation of glucose to glucose-6-phosphate by hexokinase is important for preventing glucose efflux from the cell, we asked whether STF-31 inhibits hexokinase. Hexokinase activity was inhibited by STF-31 only after 3 days of treatment in VHL-deficient RCC4 cells but was unchanged in RCC4/VHL cells with wild-type VHL (Fig. 2E). Again, baseline hexokinase activity was higher in RCC4 cells, consistent with VHL-deficient RCCs having higher rates of glycolysis and the hexokinase gene being a HIF target (Fig. 2, A, B, and E, and fig. S2A). The decrease in hexokinase activity occurred subsequent to changes in glucose uptake, indicating that inhibition of hexokinase was not directly responsible for the differential cytotoxicity of STF-31 in cells with and without VHL. Furthermore, inhibitors of glycolysis, non-specific glucose transport inhibitors, or hexokinase did not result in selective cytotoxicity to VHL-deficient cells (fig. S2, B to F). These data indicate that STF-31 decreases glycolysis by decreasing glucose transport and not by inhibiting a particular glycolytic step or enzyme.
To further investigate the relationship between HIF and STF-31 toxicity, we silenced HIF-1β, the constitutively expressed binding partner of HIF-1α and HIF-2α, in RCC4 cells (fig. S3A). Transiently inhibiting HIF-1β rendered glucose uptake of RCC4 cells insensitive to STF-31 (Fig. 2F), consistent with the idea that HIF-dependent glucose uptake is responsible for the differential toxicity of STF-31 in VHL-deficient renal carcinomas.
Effect of inhibition of glucose uptake on both glycolysis and production of ATP
We next explored how a decrease in glycolysis could lead to selective necrotic cell death. One possibility is that STF-31 may inhibit oxidative phosphorylation and the conversion of pyruvate to acetyl coenzyme A. Although there was a difference in oxygen consumption, a marker of oxidative phosphorylation, between VHL-deficient and wild-type VHL cells (13, 14), there was no difference in oxygen consumption between cells treated with STF-31 and those that were not treated (Fig. 2G). Thus, the mitochondria and the oxidative pathway were unaffected by STF-31.
However, the decrease in glucose uptake in response to treatment with STF-31 in VHL-deficient cells resulted in a 75% decrease in adenosine 5′-triphosphate (ATP) levels (Fig. 2H). Inhibition of ATP production in response to STF-31 treatment was dose-dependent (Fig. 2I). Glucose inhibition preceded cell death in VHL-deficient cells but not in cells with wild-type VHL (Fig. 2, J and K). Together, these data suggest that loss of VHL is associated with reduced oxidative phosphorylation and greater dependence on glycolysis for ATP production. By disrupting glycolysis, STF-31 functions in a synthetic lethal manner to VHL mutation, ultimately killing VHL-deficient cells by inhibiting their primary mechanism of energy production.
Dependence of VHL-deficient cells on glycolysis
These data suggest that renal cells with defective VHL, like a range of other cancers, are highly dependent on aerobic glycolysis for energy production (5, 15, 16). We further examined this conditional genetic interaction of glucose dependence and VHL interaction by depriving RCCs of glucose in a growth curve assay. The growth of renal carcinoma cells lacking functional VHL was inhibited by glucose deprivation, whereas RCC4 cells with wild-type VHL continued to grow (fig. S1A and Fig. 3, A and B). The absence of pyruvate had little influence on cells with or without VHL, and the addition of pyruvate was unable to overcome the lack of glucose (Fig. 3, A and B). These results suggested that VHL-deficient cells are more sensitive than cells with VHL to changes in glucose. Together, these data demonstrate that VHL-deficient cells are unable to use oxidative phosphorylation to overcome their dependence on glycolysis for energy production.
Fig. 3.

VHL-deficient renal carcinomas are more sensitive to glucose deprivation and have higher GLUT1 levels than RCCs with wild-type VHL. (A) Effect of lack of glucose and/or pyruvate for 6 days on the number of RCC4 and RCC4/VHL cells (*P < 0.005). (B) Effect of lack of glucose and/or pyruvate for 6 days on the number of 786-O and 786/VHL cells (*P < 0.005). (C) Expression of GLUT1, GLUT2, GLUT3, and GLUT4 in a renal cancer data set containing normal renal tissue (Normal) and renal clear cell carcinomas (RCC) (17). (D) Relative mRNA expression of GLUT1 and GLUT2 in RCC4 and RCC4/VHL as determined by quantitative real-time PCR [normalized to TATA box binding protein (TBP)]. (E) Correlation of GLUT1 and GLUT2 expression in a renal cancer data set (17). All error bars represent the SEM (n = 3). (F) VHL status, HIF-1, HIF-2, GLUT1, and GLUT2 expression, and sensitivity to STF-31 in a panel of RCC cell lines.
Overexpression of GLUT1 in renal carcinomas
We next considered the glucose uptake and subsequent survival of RCCs with and without VHL treated with STF-31. We mined a published microarray data set in which clear cell renal carcinoma tumors and normal tissue were assessed (17). GLUT2, GLUT3, and GLUT4 were all expressed at low levels in RCC (Fig. 3C). GLUT1 was the most abundant glucose transporter in RCC (Fig. 3C). The two main glucose transporters are GLUT1, the inducible, high-affinity transporter, and GLUT2, which is responsible for basal glucose uptake (18). A further examination of glucose transporters by quantitative real-time polymerase chain reaction (PCR) and immunofluorescence revealed that GLUT1 was highly expressed in cells lacking VHL, whereas cells with VHL had very low levels of GLUT1 (Fig. 3D and fig. S3B). In contrast, GLUT2 was highly expressed in cells with wild-type VHL, whereas VHL-deficient cells had levels that could barely be detected (Fig. 3D and fig. S3B). Our analysis of the relative expression levels of GLUT1 and GLUT2 from the microarray data set demonstrates a significant inverse correlation between the two genes in clinical samples (Fig. 3E). GLUT1 levels were high in renal carcinomas, whereas GLUT2 levels were high in normal renal cells (Fig. 3E). A more detailed analysis of a panel of renal carcinoma cell lines revealed that sensitivity to STF-31 compounds is generally, but not exclusively, related to VHL status. It is more consistently linked with relatively high GLUT1 and low GLUT2 expression (Fig. 3F). STF-31 was toxic to RCCs that had GLUT1 but little or no GLUT2. High GLUT2 expression was able to rescue cells from cytotoxic effects of STF-31. The expression pattern of GLUT1 and GLUT2 indicates that STF-31 inhibits the higher-affinity GLUT1 glucose transporter, depriving RCCs of glucose and, consequently, energy needed to sustain the cells and maintain viability.
Specific binding of STF-31 to GLUT1
We determined whether STF-31 directly inhibits GLUT1. Treatment with STF-31 did not affect GLUT1 protein levels in a time- or concentration-dependent manner (fig. S3C), nor did STF-31 change soluble GLUT1 protein levels (fig. S3D). To explore potential interactions between STF-31 and GLUT1, we structurally modeled STF-31 with GLUT1 (Fig. 4A). This model predicts docking of the drug within the central channel of GLUT1 with potential interactions to two residues, Arg126 and Trp412, of GLUT1. Both residues are essential for optimal glucose transport because mutation of either inhibits glucose transport activity (19, 20). Furthermore, mutated Arg126 was identified in several patients who showed diminished glucose transporter activity clinically (19). Fasentin, a fas-dependent glucose transport inhibitor, which was not toxic to either VHL-deficient or wild-type VHL cells (fig. S2B), docked toward the extracellular end of the channel, not in the central pocket (21). Thus, in a structural model of GLUT1, STF-31 is predicted to interact within the central pore of the solute carrier.
Fig. 4.

Inhibition of GLUT1 leads to cell death in VHL-deficient cells. (A) Schematic representation of STF-31 docking within the solute channel of GLUT1. (B) Cell lysates of RCC4 and RCC4/VHL were incubated with Affi-Gel–immobilized STF-31 derivatized with a linker, and then eluted with urea buffer. Elutions were probed with antibodies to GLUT1, GLUT2, or GLUT3. (C) Relative mRNA levels of GLUT1 from RCC4 and RCC4/VHL cells carrying a stable, inducible shRNAmir to GLUT1. Cells were treated with doxycycline (DOX) (500 ng/ml) for 5 days. (D) Cell viability of RCC4 and RCC4/VHL after induction of the shRNAmir to GLUT1. All error bars represent the SEM (n = 3). (E) Fluorescence-activated cell sorting analysis of RCC4 and RCC4/VHL cells expressing an inducible shRNAmir to GLUT1. Cells were treated with doxycycline (500 ng/ml) to induce the shRNAmir, which also turns on the expression of TurboRFP. After 4 days, cells were collected, stained with a marker of cell necrosis, and subjected to flow cytometry.
To confirm the specificity of binding, we examined direct interaction of STF-31 to GLUT1, GLUT2, and GLUT3. Total cell extracts from RCC4 and RCC4/VHL were incubated with STF-31 immobilized to a column with a polyethylene glycol linker (fig. S1B). The affinity columns were eluted with urea, and the eluate was immunoblotted for the three glucose transporters. GLUT1 from RCC4 cells bound to the column (Fig. 4B). RCC4/VHL cell lysates did not bind to the immobilized drug, likely due to low expression of GLUT1 in these cells. Notably, the immobilized drug did not bind GLUT2 from RCC4/VHL extracts, demonstrating that STF-31 can bind specifically to GLUT1 and that it does not bind to other glucose transporters.
Inhibition of GLUT1 and cell death
To further explore the relationship between GLUT1-mediated glucose transport and selective toxicity of STF-31, we examined the relative toxicities of a number of compounds that can influence glucose metabolism. Simply inhibiting glycolysis with 2-deoxy-d-glucose, a non-degradable glucose analog, was not sufficient to confer differential toxicity between RCC cells with and without VHL in either short-term or long-term survival assays (fig. S3, E and F). Fasentin, a glucose transporter inhibitor, was nontoxic (fig. S2B), whereas a different nonspecific glucose transporter inhibitor (phloretin) and the hexokinase inhibitors (bromopyruvate, clotrimazole, and lonidamine) were all toxic to both RCC4 and RCC4/VHL cells (fig. S2, C to F). In vitro testing of a broad range of 50 different kinases demonstrated no significant decrease in any of the kinases examined in response to STF-31 (table S1). Together, these data suggest that the preferential toxicity to RCC4 cells depends on the selective inhibition of GLUT1.
From these observations, we hypothesized that specific, genetic inhibition of GLUT1 would mimic STF-31 cytotoxicity. Using a doxycycline-inducible shRNAmir, we achieved about 50% reduction in GLUT1 levels in both RCC4 and RCC4/VHL cells (Fig. 4C), which translated into a 40% reduction in cell viability of cells with mutant VHL (Fig. 4D). This inducible promoter also drives expression of a TurboRFP reporter, allowing GLUT1 silencing to be monitored via flow cytometry. Cells were stained with a fluorescent necrosis marker, permitting simultaneous detection of GLUT1 inhibition and cell death. Induction of GLUT1 inhibition resulted in a marked increase in red fluorescence in RCC4, as well as a concomitant increase in necrosis, particularly among those cells with the strongest TurboRFP induction (Fig. 4E). In contrast, induction of the GLUT1 shRNA in RCC4/VHL cells had little effect on necrosis. These results indicate directly that genetic GLUT1 inhibition causes death in mutant VHL cells. Specific inhibition of GLUT1, either pharmacologically with STF-31 or genetically with RNA interference, leads to necrosis of cells lacking VHL.
In vivo efficacy of 3-series drugs
We next wanted to evaluate whether STF-31 might be clinically useful. Because GLUT1 is the predominant glucose transporter in human erythrocytes, we tested the effect of a more soluble analog of STF-31 on glucose uptake in human red blood cells. This soluble analog of STF-31 significantly impaired glucose uptake in red blood cells (Fig. 5A), but did not cause hemolysis (Fig. 5B). These studies suggested that the 3-series drugs should be further evaluated in vivo.
Fig. 5.

Efficacy of 3-series drugs can be monitored in vivo. (A) Glucose uptake measured in human erythrocytes after treatment with a soluble STF-31 analog (5 μM) (*P < 0.001) (n = 3). (B) Representative photos of human red blood cells treated with either red blood cell (RBC) lysis buffer or an STF-31 analog (5 μM). (C) FDG-PET imaging of 786-O cells, an RCC with a naturally occurring VHL mutation, implanted subcutaneously into the flanks of CD-1 nude mice. Representative axial cross section of a mouse before treatment (left) and after three daily intraperitoneal injections of a soluble analog of STF-31 (11.6 mg/kg) (right), overlaid with CT scan. (D) Quantification of FDG-PET inhibition as determined by the 90th percentile region of interest for percent injected dose per gram (ID/g) (*P < 0.01). (E) (a) Kidney from vehicle-treated mouse. (b) Kidney from STF-31 analog–treated mouse. (c) Spleen from vehicle-treated mouse. (d) Spleen from STF-31 analog–treated mouse. (e) Liver from vehicle-treated mouse. (f) Liver from STF-31 analog–treated mouse. (g) Heart from vehicle-treated mouse. (h) Heart from STF-31 analog–treated mouse. (i) Salivary gland from vehicle-treated mouse. (j) Salivary gland from STF-31 analog–treated mouse. (k) Brain from vehicle-treated mouse. (l) Brain from STF-31 analog–treated mouse. Animals were treated for 10 days with vehicle or drug (11.6 mg/kg for the first 3 days, followed by 7.8 mg/kg for the next week). Scale bar, 100 μm. (F) 786-O tumor-bearing mice were treated once or twice a day with vehicle (11 animals) or a soluble STF-31 analog (12 animals) (11.6 mg/kg for the first 3 days, followed by 7.8 mg/kg for the next week) (*P < 0.005). (G) ACHN cells expressing a short hairpin RNA to VHL were implanted subcutaneously into the flanks of immunocompromised mice (five mice per group). Once tumors reached an average of >60 mm3, mice were treated daily with STF-31 analog or vehicle (*P < 0.01). All error bars represent the SEM.
The high utilization of glucose by cancer cells compared to normal cells is the basis of fluorodeoxyglucose positron emission tomography (FDG-PET) in the diagnosis of cancer. We hypothesized that if STF-31 was functioning by inhibiting glucose uptake, we could monitor its effects by FDG-PET (22). Pretreatment scans of animals inoculated with subcutaneous VHL-deficient human RCCs revealed high glucose uptake within the tumors (Fig. 5C). After three daily doses of a soluble analog of STF-31, there was a marked decrease in tumor glucose uptake (Fig. 5, C and D). Despite variation in initial tumor FDG uptake, treatment consistently decreased FDG uptake, suggesting that the inhibition of glucose uptake by the 3-series may lead to tumor inhibition (Fig. 5D). These results demonstrate that the effectiveness of STF-31 can be directly monitored in vivo by FDG-PET.
Animals treated with a soluble analog of STF-31 for 14 days exhibited no toxicity in normal tissues (Fig. 5E), immunosuppression (table S2), or apparent seizures. Mouse erythrocytes, which primarily express GLUT4 (23), did not undergo hemolysis in response to treatment (fig. S3G). Brain glucose utilization monitored by FDG-PET was relatively unaffected in treated mice this STF-31 analog (fig. S3H), and they had normal levels of red blood cells, hemoglobin, and hematocrit (table S2). Together, these data suggest that the soluble analog of STF-31 that we tested might have a high therapeutic index.
We next tested whether the 3-series compounds are effective in treating RCC tumor xenografts. Daily systemic treatment of mice with VHL-deficient xenografts with the soluble analog of STF-31 for 10 to 14 days markedly delayed tumor growth in two RCC model systems: 786-O with a naturally occurring VHL mutation and ACHN expressing shRNA to VHL (Fig. 5, F and G). In both of these models, drug treatment delayed tumor growth compared to tumors treated with vehicle alone. In contrast, STF-31 analog treatment did not affect the growth rate of ACHN tumors with wild-type VHL (fig. S3I). Together, these findings suggest that in vivo, the 3-series compounds may effectively and specifically target GLUT1-dependent cancer cells, such as renal carcinomas that have become dependent on glycolysis as a consequence of VHL deficiency.
DISCUSSION
STF-31 represents the second class of small molecules that we have identified that selectively kill RCCs (6). This class of drug has a distinct mechanism of killing than the previously described STF-62247. Whereas STF-62247 selectively induces autophagic cell death in a HIF-independent manner, the 3-series class acts by disrupting glucose uptake and utilization. The GLUT1-selective cytotoxicity induced by these drugs provides direct evidence that many cancer cell types are dependent on glycolysis, including most of RCCs (24). RCCs are selectively sensitive to STF-31 because aberrant HIF stabilization (either HIF-1 or HIF-2) results in diminished mitochondrial activity, causing these cells to become highly dependent on glucose uptake for glycolysis and ATP production (Fig. 6). By inhibiting glucose uptake, STF-31 specifically targets the Achilles’ heel of RCCs. Normal kidney cells are not strictly dependent on glycolysis or GLUT1 for viability and use other glucose transporters, such as GLUT2, and are therefore insensitive to STF-31 toxicity. Our findings indicate that the differential metabolism of cancer cells can be exploited for the preferential targeting of these cells by small molecules.
Fig. 6.
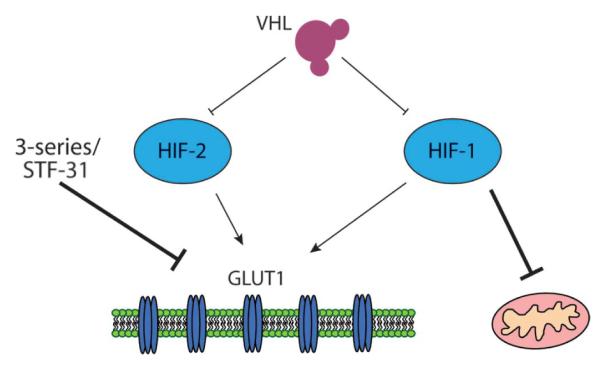
STF-31 is synthetically lethal to cells dependent on GLUT1 for aerobic glycolysis. VHL regulates both HIF-1 and HIF-2, which can increase GLUT1 expression. HIF-1 also inhibits the mitochondria, causing cells to switch to aerobic glycolysis. STF-31 compounds inhibit GLUT1 in RCCs with high levels of GLUT1. These RCCs are dependent on GLUT1 for glycolysis and cell viability.
Our results have a number of implications for the development of new cancer therapeutics. First, this method of screening for compounds that are synthetically lethal to the loss of VHL should be adaptable to other tumor types with distinct genotypes, such as the loss of function of tumor suppressor genes or gain of function of oncogenes. Second, the selective cytotoxicity of STF-31 is not restricted to VHL-deficient tumors. We initially sought to identify small molecules directly synthetically lethal to VHL mutation. However, subsequent experiments illustrate that STF-31 is generally synthetically lethal to cancer cells that have high GLUT1 levels and require glycolysis. It is likely that a number of other cancer types have additional genetic or epigenetic alterations that make them highly dependent on aerobic glycolysis for energy production and therefore sensitive to STF-31.
Note also that targeting GLUT1 in human renal cell cancers should be feasible because Glut1 heterozygous knockout mice are viable and recapitulate the human GLUT1 deficiency syndrome, which is effectively treated by a ketogenic diet (25, 26). We did not observe any toxicity in normal tissue, including brain, in these studies. Finally, our data show that the effectiveness of the 3-series compounds can be monitored by in vivo imaging. This property offers the potential advantages of enabling dosage optimization and identification of which kidney cancers will respond best to treatment in phase I clinical trials. Diagnosing and predicting the response of RCC by FDG-PET imaging will be aided by simultaneous computed tomography (CT), which is useful in evaluating responses to receptor tyrosine kinase inhibition (27). Furthermore, FDG-PET imaging of RCC will likely benefit patients with high-grade tumors or tumors that have metastasized beyond the kidney. Being able to track the response of a particular tumor is both cost-effective and lends itself to personalized medicine, which are two of the primary objectives of future cancer therapies.
MATERIALS AND METHODS
Cell culture and reagents
Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) + 10% fetal calf serum (FCS). HIF-overexpressing clones have been described (10). ON-TARGETplus SMART pools against HIF-1β/ARNT (Dharmacon) were transfected with DharmaFECT Reagent1 according to the manufacturer’s directions. Inducible shRNAmir to GLUT1 was from Open Biosystems. GLUT1, GLUT2, and GLUT3 antibodies were purchased from R&D Systems. Lactate levels and hexokinase activity were measured by fluorometric assay (BioVision and Sigma-Aldrich, respectively). ATP levels were measured by bioluminescence assay (Molecular Probes).
Cell viability assays
XTT and clonogenic survival assays were performed as in (10). Necrosis was determined by trypan blue exclusion.
Glucose uptake
One hundred thousand cells per well in a six-well plate were treated for the indicated time and concentration. Cells were washed twice and incubated in low-glucose medium (30 min), and [3H]2-deoxyglucose (0.5 μCi) was added in 1 ml of glucose-free medium (1 hour). Cells were washed twice and lysed (0.2 N NaOH, 0.2% SDS). Glucose uptake was quantified with a scintillation counter.
Extracellular acidification rate
Cells were treated with 5 μM STF-31 (24 hours), counted, seeded (8000 cells per well) in XF 96-well microplates, and incubated at 37°C (24 hours) with or without STF-31. Cells were rinsed and resuspended in unbuffered DMEM (25 mM glucose, 5% fetal bovine serum, phenol red, pH 7.4). Extracellular acidification rate (ECAR) was measured in real-time with a Seahorse Bioscience XF96 extracellular flux analyzer under basal conditions. The microplate was stained with crystal violet, and absorbance was measured at 570 nm for normalization.
Oxygen consumption
Treated cells were resuspended (5 × 106 cells/ml, DMEM + 10% FCS) and oxygen consumption was measured with an Oxytherm electrode unit (Hansatech).
Quantitative real-time reverse transcription–PCR
Relative mRNA levels were quantified as in (6).
Gene expression analysis
Expression data from a human clinical cancer study were extracted for all probe sets of GLUT1, GLUT2, GLUT3, and GLUT4 after robust multiarray normalization (17). The Jones study (GSE15641) included 49 RCC tumors, 20 non-RCC renal tumors, and 23 normal kidney samples.
Molecular modeling
The likely protonation state of STF-31 at pH 7.4 was predicted with Filter, and conformers were generated with OMEGA2 (both from OpenEye Scientific Software). GOLD (28) was then used to dock the lowest energy conformer into a 20-Å cavity that covered the internal channel of a GLUT1 homology model [Protein Data Bank entry 1SUK (29)]. Goldscore function was used at maximum search efficiency with a cutoff of 20 poses separated by a minimum root mean square deviation of 2.5 Å. All predicting binding poses were subsequently refined by energy minimization with SZYBKI with the MMFF94s force field and Poisson-Boltzmann implicit solvent model enabled. STF-31 and all protein atoms with 8 Å of the ligand were allowed to move.
Affinity columns
Affi-Gel 15 (Bio-Rad)–activated affinity media were coupled to an analog with a polyethylene glycol linker (fig. S1B) to generate immobilized affinity linkers. One microgram of total cell extract was incubated with 1 ml of immobilized drug. These resins were washed and eluted with 9 M urea and analyzed by Western blotting.
Flow cytometry
Single-cell suspensions were prepared in phosphate-buffered saline (PBS) (1 × 106 cells/ml) for fluorescence-activated cell sorting with a BD LSR II cytometer (BD Biosciences). Total cell viability was detected with LIVE/DEAD fixable dead cell stain (Invitrogen) as per the manufacturer’s suggested protocol. ArC amine-reactive compensation beads (Invitrogen) were used as a positive control. The induction of the shRNAmir to GLUT1 was detected with the PE channel (TurboRFP), whereas cell viability was detected with aqua amine (violet green) emission settings. Ten thousand events per sample were collected, and data were processed with FlowJo software (version 8.8.6, TreeStar Inc.).
In vivo studies
All experiments were approved by Stanford’s Administrative Panel on Laboratory Animal Care in accordance with institutional and national guidelines. Cells (5 × 106) were implanted subcutaneously into the flanks of nude mice (4 to 6 weeks old) (Charles River Laboratories). Tumors were measured with calipers. V = W2 × 0.5 L. When tumors reached >100 mm3, mice were randomized into vehicle (dimethyl sulfoxide in 16% cremophor EL/PBS) or treated groups. Mice were treated with the STF-31 soluble analog (11.6 mg/kg for 3 days, followed by 7.8 mg/kg for 7 to 9 days). Five-micrometer sections were stained with hematoxylin and eosin. For FDG-PET imaging, mice were fasted overnight, anesthetized (2% isoflurane), and injected intraperitoneally (250 μCi of FDG). One hour after injection, mice were imaged for 10 min (Rodent R4 MicroPET, Concorde Microsystems). Data were reconstructed into three-dimensional volumes with an ordered subset expectation maximization algorithm and were calibrated into units of percent injected dose per gram.
Red blood cell hemolysis
Blood was collected from mice and humans and centrifuged at 600g (4°C for 10 min). The plasma was aspirated and cells were washed twice in PBS + 10% FCS. Red blood cells were then resuspended in PBS and treated with the 3-series (2.5 or 5 μM) or Red Blood Cell Lysis solution (Sigma).
Statistical analyses
Student’s t tests were used to determine significance. All error bars represent the SEM.
Supplementary Material
Acknowledgments
We thank G. Thomas for ACHN cells and G.-O. Ahn, A. Krieg, and I. Papandreou for technical assistance.
Funding: This investigation was supported by grants from the Action to Cure Kidney Cancer/Cecile and Ken Youner Fund for Cancer Research, the Association for International Cancer Research 10-0042 (M.B.), the Maurice Wilkins Centre for Biodiscovery (M.P.H. and J.U.F.), the Skippy Frank Translational Medicine and Life Sciences Fund (A.J.G.), the Silicon Valley Foundation (A.J.G.), National Cancer Institute (NCI)-CA-458899 (D.A.C.), NCI-T32-CA-121940 (E.W.L.), NCI-CA-67166 (A.J.G.), and NCI-CA-082566 (M.P.H. and A.J.G.).
Footnotes
SUPPLEMENTARY MATERIAL www.sciencetranslationalmedicine.org/cgi/content/full/3/94/94ra70/DC1
Materials and Methods
Fig. S1. STF-31 does not induce autophagy, apoptosis, or DNA damage.
Fig. S2. Hexokinase and other glucose transporter inhibitors do not affect VHL-deficient cell survival.
Fig. S3. GLUT1 protein levels are unaffected by STF-31.
Table S1. STF-31 does not inhibit a broad range of kinases.
Table S2. Complete blood counts from control- and STF-31–treated mice (10 days). References
Author contributions: D.A.C., P.D.S., P.N., S.T., E.W.L., D.E.S.-C., M.P.H., and A.J.G. designed the experiments. D.A.C., P.D.S., P.N., S.T., E.W.L., A.B., G.E.R., M.B., J.U.F., M.P.H., and A.J.G. performed the experiments and analyzed the data. J.-T.C. performed the microarray analyses. J.W. and D.E.S.-C. assisted and provided technical advice for the high-throughput screening. Modeling was performed by J.U.F. D.M.B. contributed to the in vivo toxicity studies. E.E.G. provided technical advice and analysis of the imaging studies. D.A.C., W.A.D., M.P.H., and A.J.G. supervised these studies. D.A.C. and A.J.G. wrote most of the article.
Competing interests: Stanford University holds a patent “Preparation of heteroaryl benzamides useful in treating diseases” PCT Int. Appl. WO 2011011514, which is related to the work described in this paper, with P.D.S., S.T., D.A.C., W.A.D., M.P.H., and A.J.G. named as inventors. A.J.G. is a founder of and D.A.C. is a scientific consultant for Ruga, a company that discovers and develops targeted therapeutics in oncology and has licensed this patent. Drugs used in these studies are available from the author.
REFERENCES AND NOTES
- 1.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–1068. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 2.Motzer RJ, Russo P. Systemic therapy for renal cell carcinoma. J. Urol. 2000;163:408–417. [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, White DE, Steinberg SM. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: Identification of the antigens mediating response. Ann. Surg. 1998;228:307–319. doi: 10.1097/00000658-199809000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rathmell WK, Martz CA, Rini BI. Renal cell carcinoma. Curr. Opin. Oncol. 2007;19:234–240. doi: 10.1097/CCO.0b013e3280ad4388. [DOI] [PubMed] [Google Scholar]
- 5.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 6.Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell. 2008;14:90–102. doi: 10.1016/j.ccr.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hay MP, Turcotte S, Flanagan JU, Bonnet M, Chan DA, Sutphin PD, Nguyen P, Giaccia AJ, Denny WA. 4-Pyridylanilinothiazoles that selectively target von Hippel–Lindau deficient renal cell carcinoma cells by inducing autophagic cell death. J. Med. Chem. 2010;53:787–797. doi: 10.1021/jm901457w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iliopoulos O, Kibel A, Gray S, Kaelin WG., Jr. Tumour suppression by the human von Hippel–Lindau gene product. Nat. Med. 1995;1:822–826. doi: 10.1038/nm0895-822. [DOI] [PubMed] [Google Scholar]
- 9.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 10.Sutphin PD, Chan DA, Li JM, Turcotte S, Krieg AJ, Giaccia AJ. Targeting the loss of the von Hippel-Lindau tumor suppressor gene in renal cell carcinoma cells. Cancer Res. 2007;67:5896–5905. doi: 10.1158/0008-5472.CAN-07-0604. [DOI] [PubMed] [Google Scholar]
- 11.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suganuma N, Segade F, Matsuzu K, Bowden DW. Differential expression of facilitative glucose transporters in normal and tumour kidney tissues. BJU Int. 2007;99:1143–1149. doi: 10.1111/j.1464-410X.2007.06765.x. [DOI] [PubMed] [Google Scholar]
- 13.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 15.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 16.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 17.Jones J, Otu H, Spentzos D, Kolia S, Inan M, Beecken WD, Fellbaum C, Gu X, Joseph M, Pantuck AJ, Jonas D, Libermann TA. Gene signatures of progression and metastasis in renal cell cancer. Clin. Cancer Res. 2005;11:5730–5739. doi: 10.1158/1078-0432.CCR-04-2225. [DOI] [PubMed] [Google Scholar]
- 18.Pajor AM, Randolph KM, Kerner SA, Smith CD. Inhibitor binding in the human renal low- and high-affinity Na+/glucose cotransporters. J. Pharmacol. Exp. Ther. 2008;324:985–991. doi: 10.1124/jpet.107.129825. [DOI] [PubMed] [Google Scholar]
- 19.Brockmann K, Wang D, Korenke CG, von Moers A, Ho YY, Pascual JM, Kuang K, Yang H, Ma L, Kranz-Eble P, Fischbarg J, Hanefeld F, De Vivo DC. Autosomal dominant Glut-1 deficiency syndrome and familial epilepsy. Ann. Neurol. 2001;50:476–485. doi: 10.1002/ana.1222. [DOI] [PubMed] [Google Scholar]
- 20.Garcia JC, Strube M, Leingang K, Keller K, Mueckler MM. Amino acid substitutions at tryptophan 388 and tryptophan 412 of the HepG2 (Glut1) glucose transporter inhibit transport activity and targeting to the plasma membrane in Xenopus oocytes. J. Biol. Chem. 1992;267:7770–7776. [PubMed] [Google Scholar]
- 21.Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, Gronda M, Eberhard Y, Minden MD, Bilan PJ, Klip A, Batey RA, Schimmer AD. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol. Cancer Ther. 2008;7:3546–3555. doi: 10.1158/1535-7163.MCT-08-0569. [DOI] [PubMed] [Google Scholar]
- 22.Larson SM, Schöder H. Advances in positron emission tomography applications for urologic cancers. Curr. Opin. Urol. 2008;18:65–70. doi: 10.1097/MOU.0b013e3282f19cde. [DOI] [PubMed] [Google Scholar]
- 23.Montel-Hagen A, Kinet S, Manel N, Mongellaz C, Prohaska R, Battini JL, Delaunay J, Sitbon M, Taylor N. Erythrocyte Glut1 triggers dehydroascorbic acid uptake in mammals unable to synthesize vitamin C. Cell. 2008;132:1039–1048. doi: 10.1016/j.cell.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 24.Kroemer G, Pouyssegur J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, Yoo J, Noebels JL, De Vivo DC. A mouse model for Glut-1 haploinsufficiency. Hum. Mol. Genet. 2006;15:1169–1179. doi: 10.1093/hmg/ddl032. [DOI] [PubMed] [Google Scholar]
- 26.Klepper J, Leiendecker B. GLUT1 deficiency syndrome—2007 update. Dev. Med. Child Neurol. 2007;49:707–716. doi: 10.1111/j.1469-8749.2007.00707.x. [DOI] [PubMed] [Google Scholar]
- 27.Cowey CL, Fielding JR, Rathmell WK. The loss of radiographic enhancement in primary renal cell carcinoma tumors following multitargeted receptor tyrosine kinase therapy is an additional indicator of response. Urology. 2010;75:1108–1113.e1. doi: 10.1016/j.urology.2009.06.105. [DOI] [PubMed] [Google Scholar]
- 28.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein–ligand docking using GOLD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 29.Salas-Burgos A, Iserovich P, Zuniga F, Vera JC, Fischbarg J. Predicting the three-dimensional structure of the human facilitative glucose transporter glut1 by a novel evolutionary homology strategy: Insights on the molecular mechanism of substrate migration, and binding sites for glucose and inhibitory molecules. Biophys. J. 2004;87:2990–2999. doi: 10.1529/biophysj.104.047886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.