

Significance
The beneficial health effects of dietary phytochemicals make them promising candidates for treatment and prevention of multiple diseases. However, cellular targets for dietary components remain largely unknown. By combining phage display with high-throughput sequencing, we identified 160 human targets of apigenin, a flavonoid abundant in fruits and vegetables. The apigenin targets include hnRNPA2, a factor associated with numerous cellular malignancies and involved in mRNA metabolism/splicing. We show that, by inhibiting hnRNPA2 dimerization, apigenin affects the alternative splicing of key mRNAs. These findings provide a perspective on how dietary phytochemicals function and what distinguishes their action from pharmaceutical drugs.
Keywords: nanosensor, FRET, cancer, inflammation
Abstract
Flavonoids constitute the largest class of dietary phytochemicals, adding essential health value to our diet, and are emerging as key nutraceuticals. Cellular targets for dietary phytochemicals remain largely unknown, posing significant challenges for the regulation of dietary supplements and the understanding of how nutraceuticals provide health value. Here, we describe the identification of human cellular targets of apigenin, a flavonoid abundantly present in fruits and vegetables, using an innovative high-throughput approach that combines phage display with second generation sequencing. The 160 identified high-confidence candidate apigenin targets are significantly enriched in three main functional categories: GTPase activation, membrane transport, and mRNA metabolism/alternative splicing. This last category includes the heterogeneous nuclear ribonucleoprotein A2 (hnRNPA2), a factor involved in splicing regulation, mRNA stability, and mRNA transport. Apigenin binds to the C-terminal glycine-rich domain of hnRNPA2, preventing hnRNPA2 from forming homodimers, and therefore, it perturbs the alternative splicing of several human hnRNPA2 targets. Our results provide a framework to understand how dietary phytochemicals exert their actions by binding to many functionally diverse cellular targets. In turn, some of them may modulate the activity of a large number of downstream genes, which is exemplified here by the effects of apigenin on the alternative splicing activity of hnRNPA2. Hence, in contrast to small-molecule pharmaceuticals designed for defined target specificity, dietary phytochemicals affect a large number of cellular targets with varied affinities that, combined, result in their recognized health benefits.
Diet is at the cross-roads of many human chronic disease conditions. Dietary components influence all aspects of cellular function and also impact offspring development and gene expression (1). As life expectancy increases, age-related pathologies, including cardiovascular and neurological diseases, obesity, and cancer, inflict an immense pressure on healthcare costs and quality of life. Thus, there is an increasing interest in understanding the mechanisms of action of active nutritional compounds with health benefits (termed nutraceuticals). Lipids, proteins, and carbohydrates are the main dietary components that yield energy, but a healthy and balanced diet also provides all necessary micronutrients, which include minerals, vitamins, and a growing class of compounds generally known as dietary phytochemicals (2). The poorly understood mechanism of action of the vast majority of phytochemicals has made regulation by governmental agencies practically impossible, despite a continuously expanding market of dietary supplements and herbal medicines.
Phytochemicals accumulate as part of the adaptation of plants to particular ecological settings, providing plants with increased protection to biotic or abiotic stress conditions. Phytochemicals can be classified into broad classes that include the alkaloids, the terpenoids, and the phenylpropanoids, and they number more than 100,000 (3). Flavonoids, derived from the general phenylpropanoid pathway, are one subclass of phenolic compounds characterized by a C6-C3-C6 structure (Fig. S1A), and they constitute the largest class of dietary nutraceuticals. Depending on the organization and modifications of the three rings, flavonoids are classified into subclasses that include the flavanones, the flavones, the flavonols, the isoflavones, the anthocyanins, and the proanthocyanidins or condensed tannins. The health benefits of flavonoids are a consequence of a number of biological activities ascribed to them, including antiallergic, antimicrobial, antitumor, antiviral, and antiinflammatory functions (4–9). At least two features of flavonoids are important for their biological activities: their antioxidant properties and their ability to interact with proteins. The health benefits of flavonoids can be explained also by their function as signal molecules (8, 10), which bind to specific proteins and impede or enhance signal transduction pathways. The ability of estrogen receptors to recognize isoflavones and other phenolic molecules provides one of the best-described examples. These phytoestrogens have significant potential as hormone replacements for the treatment of hormone-dependent breast and prostate cancers as well as the prevention of the onset of cardiovascular diseases, and numerous efforts are underway to improve the phytoestrogenic potential of plants (11–14). However, for the vast majority of dietary components, cellular targets remain unknown (15). In fact, it is unclear whether dietary phytochemicals exert their beneficial effects either by significantly affecting the activity of just a few targets or through additive gains from modest effects on a large number of cellular targets. Identifying the proteins recognized by important components of our diet is challenging but an essential step to understanding how to improve human health through nutrition.
Similar to other flavones, apigenin is an important component of fruit- and vegetable-rich human diets (5). Apigenin derives from the flavanone naringenin by forming a double bond in ring C of the flavonoid structure (Fig. S1A). Despite their structural similarity, naringenin and apigenin elicit significantly different biological activities. Apigenin, but not naringenin (16), induces apoptosis of various cancer cell lines (17–19) and has potent antiinflammatory activity by inhibiting a pathway resulting in NF-κB activation (20). Apigenin is an abundant component in the Mediterranean diet, associated with lower prevalence of hypertension, cardiovascular disease, obesity, cancer, and diabetes (21–23). In addition, large population-based studies correlated the presence of apigenin with reduced risk of ovarian cancer (24). Apigenin is also the active compound in a number of popular dietary supplements. Hence, apigenin provides an attractive candidate compound from which to identify the comprehensive set of human targets as a first step to understanding how dietary phytochemicals confer health benefits.
Here, we describe the identification of 160 human cellular targets for apigenin using phage display coupled with second generation sequencing (PD-Seq), a method that permits the high-throughput discovery of small molecule–protein interactions. The identified apigenin targets are significantly enriched in three main functional categories corresponding to GTPase activation, membrane transport, and mRNA metabolism/alternative splicing. This last category includes the heterogeneous nuclear ribonucleoprotein A2 (hnRNPA2), an important factor in the progression of tumorigenesis by the regulation of splicing, mRNA stability, and mRNA transport. Using a combination of FRET and spectrophotometric methods, we show that the interaction between hnRNPA2 displays a high level of specificity and that the affinity of the apigenin–hnRNPA2 interaction is in the micromolar range, consistent with apigenin concentrations shown to exert biological responses. We established that apigenin interacts with the C-terminal domain of hnRNPA2 and affects hnRNPA2 multimerization, which is key for its activity. As a consequence of this effect, we show that apigenin modulates in vivo the alternative splicing of several hnRNPA2 substrates. Our results provide a comprehensive example of how a dietary flavonoid interacts with multiple targets and how at least one of these interactions, the interaction with hnRNPA2, cascades into quantitative effects on splicing for many more genes, helping explain the broad effect of dietary phytochemicals.
Results and Discussion
PD-Seq Identifies Candidate Cellular Targets for Apigenin.
For the comprehensive identification of apigenin cellular targets, we coupled apigenin to amino polyethyleneglycol-polyacrylamide copolymer beads (PEGA beads) after activation with 4-nitrophenyl bromoacetate (Fig. 1A). This method resulted in apigenin being coupled to the beads through either one of two –OH groups in the A-ring or through the single –OH group in the B-ring (Fig. 1A and Fig. S1A), exposing different faces of this flavone to proteins. The apigenin-loaded beads (A-beads) or the unloaded control beads (C-beads) were used to screen, in parallel, a commercially available human breast tumor phage display cDNA library (Fig. 1B). Three rounds of selection with C- and A-beads were performed in parallel, and phage DNA from each of the fractions was collected (Fig. 1C). Indexed libraries for sequencing by Illumina GAII were generated by amplifying inserts with primers in the phage arms (Table S1) and attaching indexed adapters to allow multiplexed sequencing (Fig. 1D).
Fig. 1.

Synthesis of apigenin beads and PD-Seq strategy outline. (A) Scheme for the chemical synthesis of A- (orange) and C-beads (blue). The coupling of apigenin to the beads occurred at the end of a polyethylene glycol linker (PEGA beads). Depending on the apigenin –OH group participating in the coupling to the phenyl bromoacetate group, A-beads consist of a combination of three products. (B) Schematic representation of the biopanning steps in the screening of a phage display cDNA library generated from human breast tumor cells mRNA. Three rounds of biopanning (3×), each including binding to the beads, washing, elution, and amplification, were performed in parallel using A- or C-beads. (C) Schematic representation of the fractions used to make the libraries for Illumina GAII sequencing. The preclearing and washing steps were skipped here for simplicity. The original library was an aliquot of a single amplified library purchased from Novagen. Original library (Ori-lib) and input and elution fractions (referred as E) obtained from the first and second rounds of biopanning using A- and C-beads (A-E1 and A-E2 and C-E1 and C-E2, respectively) were used to generate libraries for sequencing. (D) Schematic representation of Illumina GAII libraries preparation. PCR primers (indicated by arrows) at the cDNA insert and vector boundaries were used to amplify the cDNA-containing region, and they were subsequently ligated to Illumina adapters (gray areas) and indexed sequences (red area). (E) Heat map of nICPGs for each biopanning step was generated based on hierarchical clustering (Fig. S2). Cluster I, shown here, consisting of 160 genes is significantly enriched in the A-E2 fraction.
From a total of ∼8.3 million indexed 35-bp-long reads corresponding to the seven different libraries obtained (original library, Input1, C-E1, A-E1, Input2, C-E2, and A-E2) (Fig. 1C), 64% aligned with ORFs in the human genome (Table S2). To discover putative apigenin targets, the following criteria to the identified sequences were applied. (i) Inserts had to be in frame with the phage capsid, (ii) sequences had to be enriched in A- compared with C-beads (Fig. S2 A and B), and (iii) reads had to match one or multiple sequences in the human genome. In those cases where reads aligned to multiple coding sequences, weighted counts were used to obtain normalized in frame-aligned counts per gene models (nICPGs) (Fig. S1B). From a minimum of 15,568 genes represented in the original phage display cDNA library, which was established from combining sequence information from the original and selected fractions, 160 genes meeting these three criteria were identified (Fig. 1E), considered here the candidate apigenin targets (Table 1).
Table 1.
Identified apigenin targets
| ENSG number | Gene name | P value* | GO† | ENSG number | Gene name | P value* | GO† |
| ENSG00000122566 | HNRNPA2B1 | <1E-300 | c | ENSG00000110975 | SYT10 | 2.84E-09 | b |
| ENSG00000125351 | UPF3B | <1E-300 | c, d | ENSG00000123178 | C13orf1 | 3.73E-09 | c |
| ENSG00000121236 | TRIM34 | <1E-300 | c | ENSG00000138442 | WDR12 | 3.73E-09 | d |
| ENSG00000109906 | ZBTB16 | <1E-300 | c | ENSG00000182077 | PTCHD3 | 3.73E-09 | b |
| ENSG00000143476 | DTL | <1E-300 | b,c,d | ENSG00000157193 | LRP8 | 7.45E-09 | b,c,d |
| ENSG00000109132 | PHOX2B | 1.51E-273 | d | ENSG00000175471 | MCTP1 | 1.49E-08 | b,c |
| ENSG00000197768 | KRT17 | 3.81E-271 | d | ENSG00000105245 | NUMBL | 7.45E-09 | d |
| ENSG00000168038 | ULK4 | 3.89E-261 | d | ENSG00000112273 | HDGFL1 | 4.88E-04 | d |
| ENSG00000116544 | DLGAP3 | 1.83E-245 | b,d | ENSG00000147454 | SLC25A37 | 1.49E-08 | b,c,d |
| ENSG00000105643 | ARRDC2 | 1.88E-183 | c | ENSG00000213380 | COG8 | 1.49E-08 | b,d |
| ENSG00000137766 | UNC13C | 7.04E-133 | b,d | ENSG00000146950 | SHROOM2 | 2.98E-08 | b,d |
| ENSG00000214773 | AC112512.6 | 1.51E-123 | d | ENSG00000122034 | GTF3A | 2.98E-08 | c,d |
| ENSG00000213689 | TREX1 | 6.34E-117 | c,d | ENSG00000106484 | MEST | 1.19E-07 | b,c |
| ENSG00000187546 | TMEM195 | 2.30E-110 | b,d | ENSG00000119812 | FAM98A | 1.19E-07 | d |
| ENSG00000076928 | ARHGEF1 | 2.92E-107 | a,b,c,d | ENSG00000112701 | SENP6 | 2.38E-07 | c,d |
| ENSG00000170832 | USP32 | 2.15E-92 | b,d | ENSG00000102452 | VGCNL1 | 4.63E-07 | b,c |
| ENSG00000134668 | SPOCD1 | 3.14E-89 | c | ENSG00000145934 | ODZ2 | 4.77E-07 | b,d |
| ENSG00000140279 | DUOX2 | 3.14E-89 | b | ENSG00000144354 | CDCA7 | 4.77E-07 | c |
| ENSG00000137857 | DUOX1 | 3.14E-89 | b | ENSG00000165185 | KIAA1958 | 4.77E-07 | c,d |
| ENSG00000169126 | ARMC4 | 3.98E-89 | d | ENSG00000159784 | FAM131B | 4.77E-07 | d |
| ENSG00000103313 | MEFV | 6.28E-89 | c | ENSG00000203943 | SAMD13 | 3.81E-06 | c |
| ENSG00000182175 | RGMA | 8.43E-81 | b,c | ENSG00000166845 | C18orf54 | 3.81E-06 | c |
| ENSG00000008197 | TFAP2D | 2.16E-78 | d | ENSG00000127951 | FGL2 | 3.81E-06 | d |
| ENSG00000090920 | FCGBP | 2.76E-76 | b | ENSG00000215811 | BTNL10 | 3.81E-06 | b |
| ENSG00000159200 | DSCR1 | 1.77E-74 | c | ENSG00000155984 | TMEM185A | 7.63E-06 | b |
| ENSG00000203989 | RHOXF2B | 7.37E-70 | d | ENSG00000150938 | CRIM1 | 7.63E-06 | b,d |
| ENSG00000131721 | RHOXF2 | 7.37E-70 | d | ENSG00000026036 | TNFRSF6B | 1.53E-05 | c,d |
| ENSG00000204620 | AF196972.1 | 1.16E-69 | d | ENSG00000138735 | PDE5A | 3.05E-05 | c,d |
| ENSG00000058668 | ATP2B4 | 1.11E-55 | b,c,d | ENSG00000087087 | SRTT | 3.05E-05 | d |
| ENSG00000080031 | PTPRH | 1.42E-38 | b,c,d | ENSG00000168495 | POLR3D | 3.05E-05 | d |
| ENSG00000186517 | ARHGAP30 | 4.46E-38 | a,c,d | ENSG00000134490 | TMEM241 | 3.05E-05 | b,c |
| ENSG00000179542 | SLITRK4 | 3.76E-37 | b | ENSG00000165125 | TRPV6 | 3.05E-05 | b,c,d |
| ENSG00000107897 | ACBD5 | 4.93E-32 | b,c,d | ENSG00000160293 | VAV2 | 3.05E-05 | a,c,d |
| ENSG00000117385 | LEPRE1 | 2.64E-30 | c | ENSG00000101096 | NFATC2 | 3.05E-05 | c,d |
| ENSG00000035928 | RFC1 | 2.52E-29 | c,d | ENSG00000166471 | TMEM41B | 3.05E-05 | b,c,d |
| ENSG00000181295 | AL031289.1 | 8.08E-28 | d | ENSG00000182261 | NLRP10 | 4.08E-05 | d |
| ENSG00000119929 | CUTC | 2.58E-26 | d | ENSG00000101220 | C20orf27 | 6.10E-05 | d |
| ENSG00000154914 | USP43 | 5.17E-26 | c,d | ENSG00000126768 | TIMM17B | 6.10E-05 | b,d |
| ENSG00000116991 | SIPA1L2 | 8.27E-25 | a,c,d | ENSG00000010704 | HFE | 2.44E-04 | b,c,d |
| ENSG00000160007 | GRLF1 | 6.62E-24 | a,c | ENSG00000113448 | PDE4D | 2.44E-04 | b,c,d |
| ENSG00000103197 | TSC2 | 1.32E-23 | a,b,c,d | ENSG00000115290 | GRB14 | 2.44E-04 | b,d |
| ENSG00000196440 | ARMCX4 | 1.32E-23 | d | ENSG00000126803 | HSPA2 | 2.44E-04 | b |
| ENSG00000074621 | SLC24A1 | 3.43E-23 | b,c,d | ENSG00000184650 | ODF4 | 2.44E-04 | b |
| ENSG00000139223 | ANP32D | 1.06E-22 | d | ENSG00000115275 | MOGS | 2.44E-04 | b |
| ENSG00000107937 | GTPBP4 | 4.02E-21 | d | ENSG00000196792 | STRN3 | 2.44E-04 | b,c,d |
| ENSG00000086200 | IPO11 | 2.50E-20 | d | ENSG00000183783 | KCTD8 | 2.44E-04 | d |
| ENSG00000118096 | IFT46 | 3.09E-20 | d | ENSG00000196381 | ZNF781 | 2.44E-04 | c |
| ENSG00000033327 | GAB2 | 9.43E-20 | b,c,d | ENSG00000187621 | TCL6 | 2.44E-04 | d |
| ENSG00000092820 | VIL2 | 1.20E-19 | b | ENSG00000170734 | POLH | 2.44E-04 | c,d |
| ENSG00000145861 | C1QTNF2 | 4.34E-19 | b | ENSG00000105613 | MAST1 | 2.44E-04 | b,d |
| ENSG00000149187 | CUGBP1 | 4.34E-19 | c | ENSG00000153944 | MSI2 | 2.44E-04 | c,d |
| ENSG00000139842 | CUL4A | 2.17E-18 | c,d | ENSG00000124260 | MAGEA10 | 4.88E-04 | d |
| ENSG00000141424 | SLC39A6 | 3.77E-18 | b,c,d | ENSG00000198216 | CACNA1E | 9.77E-04 | b,c,d |
| ENSG00000181381 | DDX60L | 1.39E-17 | c | ENSG00000205334 | AC074091.13 | 9.77E-04 | d |
| ENSG00000166341 | DCHS1 | 2.78E-17 | b | ENSG00000002016 | RAD52 | 9.77E-04 | c,d |
| ENSG00000109814 | UGDH | 1.11E-16 | d | ENSG00000088756 | ARHGAP28 | 9.77E-04 | a,c,d |
| ENSG00000153902 | LGI4 | 2.22E-16 | c | ENSG00000072182 | ACCN4 | 9.77E-04 | b,c,d |
| ENSG00000115850 | LCT | 8.88E-16 | b | ENSG00000151693 | DDEF2 | 1.95E-03 | a,b,c |
| ENSG00000107821 | KAZALD1 | 3.55E-15 | c | ENSG00000120251 | GRIA2 | 1.95E-03 | b,c,d |
| ENSG00000056736 | IL17RB | 7.11E-15 | b,c | ENSG00000102858 | MGRN1 | 1.95E-03 | c,d |
| ENSG00000187800 | PEAR1 | 5.68E-14 | b,d | ENSG00000067057 | PFKP | 1.95E-03 | d |
| ENSG00000212882 | AL139010.29 | 5.68E-14 | b,d | ENSG00000178952 | TUFM | 1.95E-03 | d |
| ENSG00000178996 | SNX18 | 1.14E-13 | b,c,d | ENSG00000181036 | FCRL6 | 1.95E-03 | b,c,d |
| ENSG00000085433 | WDR47 | 2.12E-13 | c | ENSG00000177034 | MTX3 | 1.95E-03 | b,c,d |
| ENSG00000151303 | AGAP11 | 2.27E-13 | a | ENSG00000171160 | MORN4 | 1.95E-03 | c |
| ENSG00000184935 | AC090510.4 | 9.09E-13 | d | ENSG00000092847 | EIF2C1 | 1.95E-03 | c |
| ENSG00000116176 | TPSG1 | 9.09E-13 | b | ENSG00000162981 | FAM84A | 1.95E-03 | a,c |
| ENSG00000176009 | ASCL3 | 9.09E-13 | b | ENSG00000185686 | PRAME | 1.95E-03 | b |
| ENSG00000142319 | SLC6A3 | 9.09E-13 | b,d | ENSG00000152894 | PTPRK | 1.95E-03 | b,c,d |
| ENSG00000101489 | BRUNOL4 | 1.82E-12 | c | ENSG00000124279 | FASTKD3 | 1.95E-03 | d |
| ENSG00000008311 | AASS | 1.82E-12 | b | ENSG00000112112 | COL11A2 | 3.91E-03 | c |
| ENSG00000185499 | MUC1 | 3.64E-12 | b,c,d | ENSG00000067829 | IDH3G | 7.39E-03 | d |
| ENSG00000020256 | ZFP64 | 1.64E-11 | c,d | ENSG00000151989 | C2orf21 | 7.81E-03 | b,c |
| ENSG00000107262 | BAG1 | 5.82E-11 | c | ENSG00000157551 | KCNJ15 | 7.81E-03 | b,d |
| ENSG00000166948 | TGM6 | 1.16E-10 | c | ENSG00000174307 | PHLDA3 | 7.81E-03 | b |
| ENSG00000063761 | ADCK1 | 1.16E-10 | c,d | ENSG00000117984 | CTSD | 7.81E-03 | d |
| ENSG00000011451 | WIZ | 4.66E-10 | c,d | ENSG00000157093 | LYZL4 | 7.81E-03 | d |
| ENSG00000100441 | KHNYN | 4.66E-10 | d | ENSG00000176700 | SCAND2 | 7.81E-03 | c |
| ENSG00000114859 | CLCN2 | 4.66E-10 | b,c,d | ENSG00000145242 | EPHA5 | 7.81E-03 | b,c,d |
| ENSG00000110057 | UNC93B1 | 9.31E-10 | b,d | ENSG00000155975 | VPS37A | 1.56E-02 | b,c,d |
Shading indicates validated apigenin targets. ENSG, Ensembl gene; UPF3B, up-frame shift suppressor 3 homolog B.
P value of log10(nICPGInput2) compared with log10(nICPGA-E2).
GO: a, GTPase activation; b, membrane; c, alternative splicing; d, others.
Simultaneously to PD-Seq, we characterized clones recovered after the third round of panning by the traditional phage display approach by performing bacterial infections and individual plaque analysis (Fig. S3 A and B). As is usually the case (25, 26), this approach yielded a very small number of clones. We analyzed a total of 16 plaques by PCR. Three of them showed a small fragment, which on sequencing, proved to be identical to each other. They corresponded to what we have named the MKET clone, which we found to be highly enriched in all fractions; however, it may possibly be an artifact of library preparation (Fig. S3 B–F). The remaining 13 clones had similarly sized inserts, and sequencing all of them determined that they corresponded to the C-terminal region (residues 264–341) (Fig. S3C) of hnRNPA2, also identified as the top candidate by PD-Seq (Table 1).
HnRNPA2 and its splice variant hnRNPB1 play fundamental roles in the progression of tumorigenesis by regulating splicing, mRNA stability, and mRNA transport (27). Consistent with its presence in the phage display library used here, hnRNPA2/B1 higher expression has been reported in several human cancers, including breast (28), and hnRNPA2 expression is recognized as a marker of glioblastoma and lung cancer (29–31).
The hnRNPA2 C-terminal region (hnRNPA2C) contains three of six YGGG repeats that characterize the protein–protein interaction glycine-rich domain (hnRNPA2GRD) (Fig. 2B) (32). As a first step to determine the specificity of hnRNPA2C for different flavonoids, we performed A-bead pull-down assays of hnRNPA2C phage (ϕ-hnRNPA2C) suspensions in the presence of apigenin or naringenin. Apigenin, but not naringenin, significantly competed the binding of ϕ-hnRNPA2C to A-beads (Fig. S4A).
Fig. 2.

hnRNPA2 directly binds apigenin through the GRD. (A) Lysates from HeLa cells expressing full-length hnRNPA2-GFP or GFP alone were pulled down with A- or C-beads. Pull-down assays were resolved by SDS/PAGE and analyzed by Western blot using anti-GFP antibodies. (B) Schematic representation of GST-hnRNPA2 clones used in the pull-down assays with C- or A-beads. RRM, RNA recognition motifs of hnRNPA2. hnRNPA2C corresponds to the C-terminal 78-aa fragment present in ϕ-hnRNPA2C identified by conventional phage display screening. hnRNPA2∆C corresponds to a clone in which this C-terminal 78-aa fragment was deleted. hnRNPA2∆GRD corresponds to a clone in which the GRD domain was deleted. GST-hnRNPA2∆C corresponds to hnRNPA2 lacking the C-terminal region. All forms of hnRNPA2 were GST-tagged, E. coli-expressed, and affinity-purified forms. (C) Different versions of recombinant affinity-purified GST-hnRNPA2 proteins were pulled down with A- or C-beads (indicated as A or C, respectively). Pull-down assays (bound) and supernatants fractions were resolved by SDS/PAGE and analyzed by Western blot using anti-GST antibodies. Arrows indicate the correctly sized products; smaller bands present in some of the lanes correspond to degradation products.
The identification of just hnRNPA2 by conventional phage display screening underscores one of its main limitations, namely the retrieval of very few candidates, likely because many bona fide targets fail to be properly amplified or selected through multiple biopanning rounds (33). The results here presented show that PD-Seq overcomes this shortcoming, providing an opportunity for the high-throughput identification of small-molecule targets.
Apigenin Targets Are Enriched in Three Main Categories.
A comparison of the sequence of 160 putative apigenin targets failed to reveal any obvious common protein domains or stretches of conserved amino acids. However, analyses based on known functional annotations and gene ontology (GO) showed that three main categories (GTPase activation, membrane transport, and mRNA metabolism/alternative splicing) are highly significantly (P < 0.01) overrepresented among the identified 160 apigenin targets (Table 1 and Fig. S2). The GTPase activation functional category contains proteins such as Rho-guanine nucleotide exchange factor 1 (ARHGEF1) involved in the activation of Rho-GTPases (34), a family of proteins regulating cell polarity and cell migration (processes previously shown to be modulated by apigenin) (35, 36). The membrane transport category includes sodium, zinc, and calcium ion transporters as well as mitochondrial membrane transport proteins, consistent with the mitochondrial envelope being a major apigenin accumulation site in sensitive cells (16). The third very significantly enriched category corresponds to factors involved in alternative splicing and mRNA metabolism, with hnRNPA2 and up-frame shift suppressor 3 homolog B (UPF3B) showing the highest fold enrichment from all identified apigenin targets (Table 1). Together, these three GO functional classes correspond to 121 of 160 candidate apigenin targets identified.
To evaluate the reliability of PD-Seq for the identification of small-molecule binding proteins, we combined three different validation approaches. The first approach consisted of searching the literature for described flavonoid binding activities for these proteins. Four putative apigenin targets [hnRNPA2, UDP-glucose dehydrogenase (UGDH), lactase (LCT), and Mucin 1 (MUC1)] (Table 1) had been previously shown to interact with different flavonoids (37–40). HnRNPA2/B1 was recently identified as a proanthocyanidin-interacting protein responsible for the antiviral activity of blueberry leaf extracts against hepatitis C virus (37). UGDH activity is inhibited by the flavonol quercetin, resulting in decreased proliferation of breast cancer cells (38). LCT binds and hydrolyzes a range of flavonol and isoflavone glycosides (39). Apigenin interacts with the cytoplasmic domain of MUC1, inhibiting its dimerization in breast cancer cells (40).
The second approach consisted of testing identified candidate apigenin targets that displayed enzymatic activity for apigenin effects. As control, we used naringenin, which displays none of the biological activities of apigenin (16). Two candidate apigenin targets were tested in human MDA-MB-231 breast cancer cells treated with 50 μM apigenin, naringenin, or DMSO diluent control. Isocitrate dehydrogenases 3 (IDH3) corresponds to one of three human isocitrate dehydrogenases, which catalyses the NAD+-dependent oxidative decarboxylation of isocitrate to α-ketoglutarate (41), whereas UGDH catalyzes the NAD+-dependent oxidation of UDP-glucose to UDP-glucoronate (42). In enzymatic assays conducted to investigate the effects of apigenin on these enzymes, IDH3 activity was inhibited by more than 60% in isolated mitochondria from MDA-MB-231 breast cancer cells treated with apigenin, whereas naringenin had no effect (Fig. S4B). Similarly, apigenin, but not naringenin, significantly (P < 0.05) decreased the UGDH enzymatic activity in breast cancer cell extracts (Fig. S4C). Our results show that the inhibition of UGDH by flavonols can be extended to flavones.
The third approach consisted of evaluating the interaction of the identified targets expressed as GFP-tagged with apigenin in human epithelial HeLa cells. Pull-down assays of lysates from HeLa cells expressing hnRNPA2-GFP with either A- or C-beads and immunoblotted with anti-GFP showed that hnRNPA2-GFP specifically binds the A- but not C-beads (Fig. 2A, line 2). Similar results were obtained for ARHGEF1-GFP and GFP–BCL2-associated athanogene (BAG1), an antiapoptotic protein that enhances the activity of B-cell lymphoma 2 (BCL2) (43) (Fig. S4 D, line 2 and E, line 2).
Collectively, our validation results underscore the value of PD-Seq for the identification of apigenin targets. The finding that most of the apigenin targets correspond to one of three main categories (GTPase activation, membrane transport, and mRNA metabolism/alternative splicing) has several interesting implications. First, despite being structurally similar, apigenin, but not naringenin, shows specific biochemical effects (e.g., inhibiting enzymatic activity, which is shown in Fig. S4 B and C). Second, kinases and other proteins with ATP binding proteins have been generally believed to be main targets for flavonoids (44, 45). Interestingly, our screen identified only five kinases [aarF domain containing kinase 1 (ADCK1), ephrin type-A receptor 5 (EPHA5), microtubule associated serine/threonine kinase 1 (MAST1), phosphofructokinase platelet (PFKP), and unc-51-like kinase 4 (ULK4)] (Table 1) as candidate apigenin targets, despite more than 560 kinases being represented in the library. Third, it is tempting to speculate that the three main classes of apigenin targets reflect different temporal effects of this compound, because it is well-documented that dietary components frequently elicit short- and long-term biological responses (46). If so, these targets may reveal cellular functions associated with the use of flavonoids for both preventive (long-term) and therapeutic (short-term) interventions.
Specific and High-Affinity Interaction of Apigenin and Other Flavonoids with the Dimerization Region of hnRNPA2.
The identification of hnRNPA2 as an apigenin target by both PD-Seq and conventional biopanning and the importance of this protein in cancer make the apigenin–hnRNPA2 interaction an ideal candidate for additional characterization. To determine the regions in hnRNPA2 that bind apigenin, we generated fragments that represented the full-length protein, the protein lacking the last 78 aa, the protein lacking the GRD, and just the last 78 aa fused to GST (hnRNPA2, hnRNPA2∆C, hnRNPA2∆GRD, and hnRNPA2C, respectively) (Fig. 2B). Each of these proteins was then tested for its ability to interact with A- or C-beads in pull-down experiments, which were analyzed by Western blot using antibodies against GST (Fig. 2C). The interaction of hnRNPA2 with apigenin likely involves multiple sites in the GRD, because GST-hnRNPA2∆C still binds to A-beads (Fig. 2C). Showing that the interaction with apigenin requires the GRD, the deletion of this domain (hnRNPA2∆GRD) abolished A-bead binding (Fig. 2C). These results confirm the interaction of apigenin with hnRNPA2 and identify the GRD as the hnRNPA2 protein domain with flavones binding capacity.
To determine the affinity and specificity of the interaction between flavonoids and hnRNPA2, two strategies were pursued. First, we took advantage of the ability of apigenin and other flavones to absorb light at 310 and 370 nm (Fig. 3A). The light absorption of apigenin increases when incubated with GST-hnRNPA2 but not GST alone (Fig. 3A). Similar changes in absorption of flavonoids have been reported as a consequence of their interactions with proteins (47–49). The increased absorption at 370 nm was used to determine the apparent dissociation constant (KD) of GST-hnRNPA2 for apigenin, which was estimated at 2.66 ± 1.09 μM (Fig. 3 B and C). Comparable results were obtained if we used the absorption change at 310 nm. The strength of the interaction of hnRNPA2 for apigenin is, therefore, comparable, if not higher, than described for other protein–flavonoid interactions. For example, the KD of quercetin for collagen was determined to be at ∼12 μM (48).
Fig. 3.

Binding affinity of the interaction of hnRNPA2 with apigenin. (A) Apigenin (10 μM) was titrated with increasing concentrations of purified GST-hnRNPA2 (0, 0.2, 0.5, 1, 2, 5, 10, and 20 μM) or GST (Inset). Changes in absorption across the UV-visible spectrum were determined over the 250- to 450-nm range. (B) Changes in absorbance of apigenin at 370 nm as determined in A. (C) Dissociation constant (KD) of the apigenin–hnRNPA2 complex calculated using the Benesi–Hilderbrand method as described in Materials and Methods. Data represent the mean ± SEM (n = 3). (D) hnRNPA2C (Fig. 2B) was cloned into the pFLIP2 vector in frame between the regions coding for the N-terminal CFP and C-terminal YFP. The affinity-purified FLIP2-3-hnRNPA2C protein was incubated with increasing concentrations of apigenin (0, 1, 5, 10, 25, 50, and 100 μM) for 3 h at 37 °C. Relative fluorescence units (RFUs) were determined by spectrofluorometry (λext = 405 nm; λemi = 460–600 nm) and represented as emission spectra. The absence of an isosbestic point in the nanosensor spectra is likely a consequence of flavonoids, particularly flavones, absorbing light at a wavelength that partially overlaps with the CFP excitation spectrum (Fig. S6). YFP fluorescence is not affected by the various flavonoids over the broad concentration range tested (Fig. S6). (E) The calculated YFP/CFP fluorescent ratios (530/480 nm) are represented over the 0- to 100-μM concentration range for each flavonoid. (F) Flavonoid-dependent changes in YFP/CFP ratios were transformed into saturation curves as described in Materials and Methods. KD value was determined by using nonlinear regression. Data represent the mean ± SEM (n = 3).
The second strategy consisted of developing a genetically encoded flavonoid nanosensor based on FRET. To this objective, we cloned hnRNPA2C (Fig. 3D) in a collection of vectors for expression as translational fusions between fluorescent proteins with distinct excitation/emission spectra (50). Of eight constructs tested, FRET was observed in four (Figs. S5 and S6), but in only one instance, p-fluorescent indicator protein 2-3-hnRNPA2C (pFLIP2-3-hnRNPA2C) (Fig. 3D), was the energy transfer increased by apigenin in a concentration-dependent manner (Fig. 3E). This construct was, therefore, used to estimate the apparent KD of apigenin for this fragment, which was determined to be 22.99 ± 7.70 μM (Fig. 3F).
The lower affinity (higher apparent KD) determined by FRET most likely reflects the differences between the two KD determination methods and the absence of additional apigenin binding sites in hnRNPA2C (Fig. 2 B and C). However, one advantage of the FRET-based method over the spectrophotometric approach is that it permits us to investigate the specificity and relative affinity of the interaction between hnRNPA2 and other flavonoids. Luteolin differs from apigenin by an additional –OH in ring B and also interacts with hnRNPA2, although with lower affinity (Table 2 and Fig. S7A). However, a methyl group in the additional B-ring –OH, which is present in chrysoeriol, a common flavone in many medicinal plants, results in the complete inhibition of the interaction (Table 2 and Fig. S7A). Apigenin is often found in the diet as C- or O-glucosides (51). Revealing the important role of the 7-O group in the interaction with hnRNPA2, apigenin 7-O-glucoside shows little, if any, biological activity (52). In contrast, highlighting the importance of the 7-O group itself in the interaction with hnRNPA2 rather than a steric hindrance by the glucosyl group, apigenin 6-C-glucoside (isovitexin) significantly binds to hnRNPA2 with lower affinity than aglycone apigenin but higher affinity than luteolin (Table 2 and Fig. S7B). Consistent with ϕ-hnRNPA2C recognizing apigenin but not naringenin (Table 2 and Fig. S4A), neither naringenin nor the related flavanone eriodictyol affect the FRET of the nanosensor (Table 2 and Fig. S7D). Suggesting that the binding to hnRNPA2 is not limited to flavones and consistent with the fact that many biological activities of flavones being shared by flavonols (5), quercetin and kaempferol also show a significant interaction with hnRNPA2 but with lower affinity than apigenin (Table 2 and Fig. S7E). No binding of hnRNPA2 to flavopiridol or the isoflavone genistein was detected, suggesting that these biologically active compounds function through mechanisms distinct from apigenin (Fig. S7 C and F).
Table 2.
Binding affinities of different flavonoids to the FRET nanosensor
| Flavonoid | P value* (one-way ANOVA) | R2 | KD (μM) |
| Apigenin | 0.00012 | 0.873 | 22.99 ± 7.70 |
| Luteolin | 0.00031 | 0.880 | 131 ± 78.89 |
| Quercetin | 0.00002 | 0.916 | 126.6 ± 60.18 |
| Kaempferol | 0.01873 | 0.815 | 27.13 ± 12.24 |
| Apigenin 6-C-glucoside | 0.91532 | 0.897 | 60.88 ± 24.14 |
| Apigenin 7-O-glucoside | 0.73034 | 0.468 | N.B.† |
| Chrysoeriol | 0.10702 | 0.213 | N.B. |
| Naringenin | 0.10737 | 0.120 | N.B. |
| Eriodyctiol | 0.96653 | 0.233 | N.B. |
| Genistein | 0.03077 | 0.037 | N.B. |
| Flavopiridol | 0.99484 | 0.503 | N.B. |
Data represent mean ± SEM (n = 3). KD, dissociation constant; R2, coefficient of determination of saturation curves fitted by nonlinear regression (Materials and Methods).
Statistical significance of the YFP/CFP ratios over the tested flavonoid concentration range (Fig. 3E and Fig. S7).
N.B., no binding.
Apigenin Inhibits hnRNPA2 Multimerization and Splicing Activity.
In the cell, hnRNPA2 forms oligomers through the GRD, and multimerization is required for mRNA binding by the RNA recognition motif (RRM) domain (53–55). To determine if the binding of apigenin to the GRD affects hnRNPA2 dimerization, the amplified luminescent proximity homogenous assay (ALPHA) was used. This assay recapitulates the reported hnRNPA2 dimerization, which is reflected in the ability of 6xHis-hnRNPA2 to form heterodimers with GST-hnRNPA2 but not GST (Fig. 4A). The addition of apigenin (100 μM) to the GST-hnRNPA2/6xHis-hnRNPA2 heterodimers resulted in luminescence being reduced to less than one-half (Fig. 4B, Api), whereas no decrease in luminescence was observed with naringenin (100 μM) (Fig. 4B, Nar). These results show that binding of apigenin to the GRD inhibits hnRNPA2 dimerization.
Fig. 4.

Apigenin affects hnRNPA2 dimerization. (A) Purified 6xHis-hnRNPA2 (125 nM) protein was incubated with 125 nM native GST-hnRNPA2, 125 nM boiled GST-hnRNPA2, or 125 nM GST for 1 h at room temperature followed by the addition of GSH-acceptor and anti–His-donor beads for 6 h. (B) GST-hnRNPA2 (125 nM) was incubated with 125 nM 6xHis-hnRNPA2 for 1 h at room temperature followed by the addition of GSH-acceptor and anti–His-donor beads for 6 h at room temperature. Apigenin (Api; 100 μM), naringenin (Nar; 100 μM), or diluent control (DMSO) was added for 15 min at room temperature. Data represent the mean ± SEM (n = 4). *P < 0.05.
To determine the biological consequences of the hnRNPA2–apigenin interaction, the effect of apigenin on alternative splicing of known hnRNPA2 substrates (29) was evaluated. The splice isoforms of c-FLIP (c-FLIPL and c-FLIPS) (Fig. 5A) and caspase-9 (caspase-9a and -9b) (Fig. 5B) were compared in two human cell lines with contrasting hnRNPA2 expression. MDA-MB-231 breast cancer cells express high levels of hnRNPA2 (Fig. S8), whereas the MCF-10A immortalized noncarcinogenic breast epithelial cells have very low levels of hnRNPA2 mRNA and protein (Fig. S8). In MDA-MB-231 but not MCF-10A cells, apigenin reduced the levels of c-FLIPS (Fig. 5 A and D) and caspase-9b (Fig. 5 B and E) without affecting the splicing of baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5) (Fig. 5 C and F), an alternatively spliced non-hnRNPA2 substrate (56). In agreement with its ability to also bind hnRNPA2 (Table 2 and Fig. S7A), the flavone luteolin had a similar effect on splicing as apigenin (Fig. 5). In contrast, naringenin, a non-hnRNPA2 binding flavonoid, showed no effect on splicing (Fig. 5). Taken together, these results show that apigenin (and likely, other flavones) interacts with hnRNPA2, inhibiting its dimerization and altering the alternative splicing patterns of hnRNPA2 substrates.
Fig. 5.

Apigenin regulates alternative splicing of hnRNPA2 substrates in breast cancer cells. (A–C) MDA-MB-231 breast cancer cells were treated with 50 μM apigenin (Api), luteolin (Lut), or naringenin (Nar) or diluent DMSO for 48 h. Total RNA was isolated, and alternative splicing was analyzed by RT-PCR using specific primers for (A) c-FLIP, (B) caspase-9, and (C) BIRC5. GAPDH expression was used as the loading control. Reactions were resolved in 2% (wt/vol) agarose gels. Positions of the primers (arrows) and splicing variants (boxes) are represented schematically on the right. (D–F) Graphs represent the percent of the indicated splice isoform. Data represent the mean ± SEM (n = 3). *P < 0.05 compared with DMSO control samples.
Increased expression of hnRNPA2 has been reported in several cancers (28–31). Cancer cells are often resistant to apoptosis (57), which is normally triggered by the activation of signal transduction cascades that culminate in the activation of the caspases. The caspase-9a splice isoform, present in both the malignant and noncarcinogenic breast cancer cells (Fig. 5 B and E), encodes the functional apoptotic caspase-9, which is responsible for inducing cell death. In contrast, the splice isoform caspase-9b (lacking exons 3–6) (Fig. 5B), significantly increased in malignant cells and down-regulated by apigenin, encodes a caspase-9 protein that exhibits a dominant-negative activity and inhibits apoptosis (58). A similar situation happens with c-FLIP; c-FLIPS, inhibited by apigenin, has an alternate 3′ exon (exon 7 instead of 8) (Fig. 5 A and D), which encodes a c-FLIP protein that completely prevents the activation of specific death receptors (59). Thus, the addition of apigenin to malignant cells reverts the splicing of caspase-9 and c-FLIP, two key regulators of apoptosis, to the splice variants present in noncarcinogenic cells.
Conclusion
We describe here the comprehensive identification of the human cellular targets of the flavone apigenin, an abundant dietary phytochemical with anticarcinogenic activities, by PD-Seq, a high-throughput strategy that combines phage display with next generation sequencing. The identified apigenin candidate targets are not obviously enriched in ATP binding enzymes, which could have been expected based on previous studies (44, 45); also, they are not randomly distributed across multiple types of proteins. Rather, most (121/160) of the candidate targets fall into one of three categories: GTPase activation, membrane transport, and mRNA metabolism/alternative splicing. This specificity of apigenin for specific types of proteins is revealing in terms of how this flavone may exert very specific cellular responses (60, 61). Evidence of how phytochemicals perturb gene function is provided by the ability of apigenin to bind to the GRD of hnRNPA2 and inhibit its dimerization, which is essential for RNA binding and hence, important for the participation of hnRNPA2 in alternative splicing. Indeed, we show that apigenin affects the splicing of hnRNPA2 substrates in breast cancer cells. These results may help explain how apigenin exerts an anticarcinogenic activity by decreasing the inhibition of apoptosis, thereby increasing the efficacy of chemotherapeutic drugs. This study offers a fresh view on how dietary phytochemicals are likely to influence the systems network by impacting multiple (hundreds) cellular targets with relatively low (micromolar range) affinity, moderately affecting their activities (enzymatic or protein–protein interaction). Thus, in contrast to a pharmaceutical drug selected to have high affinity and specificity, the effect of a dietary phytochemical would be distributed across the entire network, resulting in a fine-tuning effect, with a consequent long-term impact on health.
Materials and Methods
Preparation of Apigenin-Immobilized PEGA Beads.
Apigenin was immobilized to amino PEGA beads (EMD Biosciences). These beads are referred throughout the text as A-beads. Acetylated PEGA beads were used as controls and are referred throughout the text as C-beads. C-beads were loaded with acetic anhydride (Ac2O) according to the procedure reported previously (62). Detailed information on the generation of beads can be found in SI Materials and Methods.
Phage Display Screening.
The phage display screening was performed using a T7 Select Human Breast Tumor cDNA phage library (EMD Biosciences). The original library was amplified by Plate Lysate Amplification according to the manufacturer’s protocol. Preclearing of the amplified library was done by incubating 2 mL T7 phage (109 pfu/mL) with 200 µL (10 mg/mL) C-beads at 4 °C overnight. The precleared phage suspension (1 mL) was incubated with 100 µL 10 mg/mL A- or C-beads at 4 °C overnight and washed 10 times with 1 mL buffer of 20 mM Tris, pH 8.0, 150 mM NaCl, and 0.05% Tween 20 followed by elution with 100 µL 1% SDS for 10 min at room temperature. Eluted fractions (5 µL) were inoculated into 3 mL Escherichia coli Rosetta-gami B5615 (EMD Bioscience) host bacteria cells and incubated for 3 h at 37 °C. Phage-infected cells were centrifuged at 800 × g for 5 min, and supernatants containing phage particles were used for the next biopanning step. Phage titers for each biopanning step were evaluated by counting pfu per milliliter according to the manufacturer’s protocol (EMD Biosciences).
Illumina GAII Second Generation Sequencing of cDNA Phage Libraries.
Phage DNA was isolated from the input and the elution fractions obtained in the first and second rounds of biopanning using either A- or C-beads (Fig. 1C) (referred as A–E or C–E, respectively) by phenol/chloroform extraction and amplified by PCR using T7 flanking insert primers (Fig. 1D and Table S1). The primers include three consecutive random nucleotides at the 5′ regions to help cluster recognition in Illumina GAII. Amplified PCR fragments were used for preparation of Illumina libraries (Illumina) according to the manufacturer’s protocol with some modifications. Briefly, 1.5 pmol PAGE-purified grade indexed adapter oligos were ligated with 100 ng amplified cDNA with Ultrapure T4 ligase (Enzymatics) for 15 min at room temperature. Ligation products were purified using a PCR purification kit (Qiagen) followed by amplification using Phusion Hot Polymerase (New England Biolabs) and sequenced by Illumina GAII.
Analysis of PD-Seq Data.
Sequences corresponding to empty clones, named multicloning site clones, and a contaminant clone referred as MKET were subtracted from the total number of sequences (Fig. S3D). Remaining reads were aligned to human coding sequences using SeqMap (63) allowing two mismatches. nICPGs were calculated as follows. The numbers of aligned sequences per gene model were counted, and in cases where a read aligned to multiple coding sequences, the number of reads was divided by the number of aligned coding sequences. For instance, a read that aligns to two coding sequences, A and B, results in 0.5 as a weighted count. If a second read matches to A but not B, then the total ICPG for A will be 1.5, and the total ICPG for B will be 0.5 (Fig. S1B). Thus, by using weighted counts, the total count number is identical to total number of reads. To determine the normalized ICPG for a given gene A, ICPGA was divided by the sum of all ICPGs and multiplied by 106 for better handling. All genes appearing in at least one library were used for additional analysis. A binomial test was used to predict significant changes between two samples. Log10(nICPG) was used for clustering, and log10(nICPG) was arbitrarily converted to zero if nICPG was zero for a particular gene. Clustering and heat map representations were calculated by applying the hierarchical clustering using MeV software (64) with average linkage clustering. Enrichment of functional categories was analyzed using the Database for Annotation, Visualization, and Integrated Discovery (65). The total number of genes appearing in the heat map (15,568 genes) was used as a background dataset to obtain enrichment of functional categories.
Plasmid Construction.
Detailed information of all clones can be found in SI Materials and Methods.
Recombinant Protein Expression and Production.
Different GST-tagged hnRNPA2 proteins were obtained as follows. BLR(DE)LysS cells transformed with plasmid Destination (pDEST) 15-hnRNPA2, pDEST15-hnRNPA2∆C, pDEST15-hnRNPA2∆GRD, pDEST15-hnRNPA2GRD, or pDEST15-hnRNPA2C clones were grown in LB, and protein expression was induced with 1 mM isopropyl-β-D-thiogalactoside for 2 h at 37 °C. After induction, cells were harvested by centrifugation, resuspended in lysis buffer (PBS, pH 7.4, 1 mM DTT, 0.1 mM PMSF, 2 μg/mL protease inhibitors chymostatin, pepstatin, antipain, and leupeptin, 1% Tween 20, 10 mg/mL lysozyme), and sonicated using a Branson Sonifier 450 (output control: 8; duty cycle: 80%; 5 cycles, 10 pulses each cycle; Branson Ultrasonics). Proteins were purified by glutathione binding affinity chromatography (EMD Biosciences) in batches (150 μL beads/10 mL bacteria lysates) at 4 °C for 2 h. Beads were washed three times in a column with 1 mL PBS, pH 7.4 and eluted with 1 mL elution buffer (10 mM reduced glutathione, 50 mM Tris, pH 8.0). GST-tagged proteins were dialyzed in 20 mM Tris, pH 7.6, NaCl 150 mM, 1 mM DTT, and 0.1 mM PMSF (TBS buffer) for 6 h at 4 °C (1:500 dilution). 6xHis-hnRNPA2 and 6xHis-FLIP-hnRNPA2 proteins were obtained as previously described (66). Briefly, BLR(DE)LysS cells were grown in LB, and protein expression was induced with 1 mM IPTG for 2 h at 30 °C. After induction, cells were collected and lysed by sonication with buffer containing 20 mM Tris, pH 8.0, 1 mM DTT, 0.1 mM PMSF, and 2 μg/mL protease inhibitor mixture containing chymostatin, pepstatin, antipain, and leupeptin using the above-mentioned conditions. 6xHis-tagged proteins were purified by His binding affinity chromatography (EMD Biosciences). Binding to the beads was performed in batches (125 μL beads/10 mL bacteria lysates) at 4 °C for 2 h, and beads were washed with three 1-mL aliquots of 20 mM Tris⋅HCl and eluted with discontinuous imidazole gradient (20–50 mM imidazole in 20 mM, pH 8.0). Elution fractions containing the 6xHis-hnRNPA2 and 6xHis-FLIP-hnRNPA2 proteins were dialyzed in 20 mM Tris buffer, pH 8.0, for 6 h at 4 °C (1:500 dilution).
Pull-Down Assays.
GST–pull-down assays were carried out by incubating 100 nM purified recombinant GST-hnRNPA2 proteins or GST alone with 150 μg A- or C-beads in 100 μL TBS buffer, pH 7.6, containing 1 mM DTT, 0.1 mM PMSF, and 2 μg/mL protease inhibitor mixture for 12 h at 4 °C. The beads were spun down and washed three times with TBS buffer. Supernatants were recovered and kept at −70 °C. The bound proteins were eluted with 2% (wt/vol) SDS. Both bound and supernatant fractions were analyzed by Western blot using anti-GST antibodies (Thermo Scientific). For pull-down assays using cellular apigenin candidate targets, cell lysates from HeLa cells transiently expressing hnRNPA2-GFP, ARHGEF1-GFP, GFP-BAG1, or GFP alone were incubated with 300 μg A- or C-beads in 100 μL TBS buffer, pH 7.6, containing 1 mM DTT, 0.1 mM PMSF, and 2 μg/mL protease inhibitor mixture for 12 h at 4 °C. The beads were spun down and washed three times with TBS buffer. The bound proteins were eluted with 2% (wt/vol) SDS, and both eluted bound fractions and supernatants were kept at −70 °C. The bound proteins and supernatant (0.25 volumes total supernatant) fractions were analyzed by Western blot using anti-GFP antibodies (Abcam).
Spectrophotometric Analyses.
The change in the UV-visible absorption of apigenin was determined by a spectrophotometric method. Free apigenin (10 μM) was incubated with increasing concentrations of GST-hnRNPA2 or GST alone (0.2, 0.5, 1, 2, 5, 10, and 20 μM) in 150 μL TBS buffer, pH 8.0, containing protease inhibitor mixture for 15 min at 37 °C using a 96-well plate (SensoPlate 96W Sterile; Greiner). Absorption spectra ranging from 260 to 450 nm were measured using a spectrofluorometer plate reader (FlexStation3; Molecular Devices). The dissociation constant of the complex, KD, was calculated with the Benesi–Hilderbrand method (49, 67):
where CApigenin and ChnRNPA2 are concentrations of apigenin and GST-hnRNPA2, respectively, ΔA is the change in absorbance, ε is the extinction coefficient of the complex, l is the path length (0.4 cm for this system), and K is the association constant corresponding 1/KD. KD was, therefore, determined by plotting (lCApigenin/ΔA) vs. (1/ChnRNPA2).
FRET.
Emission spectra of purified FLIP-hnRNPA2C proteins were determined using a spectrofluorometer plate reader (FlexStation3; Molecular Devices) by exciting CFP at 405 nm and recording emission over the range of 460–600 nm. CFP shows a maximum peak at 480 nm, whereas YFP shows a maximum peak at 530 nm. FRET was determined as the intensity of fluorescence at 530 nm divided by the intensity of fluorescence at 480 nm (YFP/CFP ratio). Binding of flavonoids to FLIP-hnRNPA2C was assessed by incubating 1 μM purified FLIP-hnRNPA2C with increasing concentrations of apigenin, luteolin, chrysoeriol, naringenin, eriodictyol, quercetin, kaempferol, flavopiridol, genistein, apigenin 7-O-glucoside, apigenin 6-C-glucoside (ranging from 1 to 100 μM), or diluent DMSO as control in 200 μL 20 mM Tris, pH 8.0, at 37 °C for 3 h.
The dissociation constants of the complexes, KD, were calculated by fitting the YFP/CFP ratio curves to the equation for the binding of a ligand to a protein:
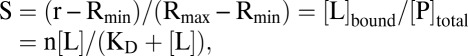 |
where S is saturation, [L] is ligand concentration, [L]bound is concentration of bound ligand, n is the number of equal binding sites, [P]total is the total concentration of FLIP nanosensor, r is the ratio, Rmin is the minimum ratio in the absence of ligand, and Rmax is the maximum ratio at saturation with ligand (66). Saturation curves were obtained, and KD was determined by nonlinear regression using the GraphPad Prism. Levels of statistical significance between means in FRET experiments were determined by one-way ANOVA.
ALPHA.
Dimerization of hnRNPA2 was determined by ALPHA using GSH-acceptor and anti–His-donor beads according to the manufacturer’s instructions (Perkin-Elmer). Briefly, 6xHis-hnRNPA2 (125 nM) was incubated with 125 nM GST-hnRNPA2 for 1 h at room temperature followed by the addition of 20 μg/mL GSH-acceptor and 20 μg/mL anti–His-donor beads in 20 μL TBS buffer, pH 7.6, containing 1 mM DTT, 0.1 mM PMSF, and 2 μg/mL each chymostatin, pepstatin, antipain, and leupeptin and incubated for an additional 6 h at room temperature. Apigenin (100 μM), naringenin (100 μM), or diluent control (DMSO) was added for 15 min at room temperature. Arbitrary fluorescence units were determined using the EnSpire multimode plate reader with ALPHA technology (Perkin-Elmer) and expressed divided by 1,000. Statistical significance was determined by one-way ANOVA using the GraphPad Prism software.
Cell Culture.
Detailed information is in SI Materials and Methods.
Enzymatic Assays.
To determine UGDH activity, MDA-MB-231 cells were treated with 50 μM apigenin, naringenin, or diluent DMSO for 3 h. Cells were homogenized by douncing (10 strokes) in lysis buffer (10 mM Tris⋅HCl, pH 8.7, 50 mM KCl, 1.5 mM MgCl2, 0.1 mM PMSF, 2 μg/mL each chymostatin, pepstatin, antipain, and leupeptin) and centrifuged at 20,000 × g for 15 min at 4 °C. Cell lysates (250 μg protein) were incubated in 200 μL buffer containing 1 mM UDP-glucose, 100 mM sodium glycine, pH 8.7, and 1 mM NAD+ at room temperature. Activity was determined by assessing the change in NAD+ absorbance at 340 nm for 30 min using the EnSpire multimode plate spectrophotometer reader (Perkin-Elmer).
IDH3 activity was determined in mitochondria preparations from MDA-MB-231 cells treated with 50 μM apigenin, naringenin, or diluent DMSO as control for 3 h. Mitochondria were isolated from 2 × 107 cells by dounce homogenization (100 strokes) in 400 μL mitochondria isolation (MI) buffer containing 20 mM Tris, pH 7.2, 0.8 M sucrose, 40 mM KCl, 2 mM EGTA, 1 mg/mL BSA, 0.1 mM PMSF, and 2 μg/mL each chymostatin, pepstatin, antipain, and leupeptin and centrifuged at 1,500 × g for 10 min at 4 °C. Pellets were resuspended in 400 μL MI buffer and centrifuged at 17,000 × g for 30 min at 4 °C. Pellets containing mitochondrial fraction were resuspended in 200 μL MI buffer and lysed by three rounds of freeze and thaw. Purity of the isolated mitochondria was verified by Western blot using antibodies against cytochrome c, a specific mitochondrial marker, and GAPDH, a cytoplasmic marker. IDH3 activity was evaluated by incubating 250 μg mitochondrial protein in 200 μL buffer containing 100 mM K2HPO4, 100 mM KH2PO4, 8 mM MgCl2, 500 μM NAD+, and 2 mM sodium isocitrate, pH 7.6. Activity was determined by assessing the change in NAD+ absorbance at 340 nm for 30 min using the EnSpire multimode plate reader.
Enzymatic units were calculated using the following formula: enzymatic units = (ΔA340 × Vf × d.f.)/(ε × mg × l), where ΔA340 is the change in absorbance at 340 nm over time, Vf is the final reaction volume, d.f. is the dilution factor, ε is the extinction coefficient of NAD+ determined to be 6.22, mg is the amount of protein, and l is the light path estimated to be 0.68. Levels of statistical significance between treatments were determined by one-way ANOVA.
Analysis of Alternative Splicing by RT-PCR.
Total RNA from MDA-MB-231 and MCF-10A treated with 50 μM apigenin, luteolin, naringenin, or diluent DMSO for 48 h was obtained using TRIzol (Life Technologies) and reverse transcribed to cDNA using the ThermoScript RT-PCR System (Life Technologies) according to manufacturer’s instructions. A 20-µL mixture containing 1 µL cDNA (20 ng) template, 0.25 µM primers, 0.2 mM dNTPs, and 1 U Platinum Taq DNA Polymerase (Life Technologies) was run using the following conditions: 95 °C for 5 min and 40 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 45 s followed by 72 °C for 5 min. Primers used to amplify splice forms are listed in Table S1: caspase-9 [PrimersAndreaOhio (PAO)-462/PAO-463], cFLIPL (PAO-545/PAO-546), cFLIPS (PAO-547/PAO-548), BIRC5 (PAO-673/PAO-674), and GAPDH (PAO-230/PAO-231) as loading control. Splice variants were resolved in 2% (wt/vol) agarose gels. Isoform percent was calculated by densitometry as follow: 100 × (density of isoform X)/(density of all isoforms). Statistical significance between treatments was determined by one-way ANOVA.
Supplementary Material
Acknowledgments
The authors thank Dr. V. Gopalan for his insightful comments on the manuscript. We thank Dr. Wolf Frommer for the FLIP vectors and advisement on nanosensors. We also thank Drs. Lexie Friend, Adrian R. Krainer, Ann C. Williams, and Philip B. Wedegaertner for constructs. D.A. was supported by the Public Health Preparedness for Infectious Diseases (PHPID) predoctoral fellowship. A.Y. was supported by National Institutes of Health Training Fellowship 5 T32 CA106196-05. This work was supported by US Department of Agriculture National Institute of Food and Agriculture Agricultural and Food Research Initiative Competitive Grant 2010-65115-20408 (to E.G.) and National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (NHLBI) Grant R01HL075040-01 (to A.I.D.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.D. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303726110/-/DCSupplemental.
References
- 1.Choi SW, Friso S. Epigenetics: A new bridge between nutrition and health. Adv Nutr. 2010;1(1):8–16. doi: 10.3945/an.110.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Higdon J. An Evidence-Based Approach to Dietary Phytochemicals. New York: Thieme; 2007. [Google Scholar]
- 3.Verpoorte R. Pharmacognosy in the new millennium: Leadfinding and biotechnology. J Pharm Pharmacol. 2000;52(3):253–262. doi: 10.1211/0022357001773931. [DOI] [PubMed] [Google Scholar]
- 4.Clifford M, Brown JE. In: Flavonoids: Chemistry, Biochemistry and Applications. Andersen OM, Markham KR, editors. Boca Raton, FL: Taylor and Francis Group; 2006. pp. 320–370. [Google Scholar]
- 5.Crozier A, Jaganath IB, Clifford MN. Dietary phenolics: Chemistry, bioavailability and effects on health. Nat Prod Rep. 2009;26(8):1001–1043. doi: 10.1039/b802662a. [DOI] [PubMed] [Google Scholar]
- 6.Riemersma RA, Rice-Evans CA, Tyrrell RM, Clifford MN, Lean ME. Tea flavonoids and cardiovascular health. QJM. 2001;94(5):277–282. doi: 10.1093/qjmed/94.5.277. [DOI] [PubMed] [Google Scholar]
- 7.Carroll KK, Guthrie N, So FV, Chambers AF. In: Flavonoids in Health and Disease. Rice-Evans CA, Packer L, editors. New York: Marcel Dekker, Inc.; 1998. pp. 437–467. [Google Scholar]
- 8.Williams RJ, Spencer JP, Rice-Evans C. Flavonoids: Antioxidants or signalling molecules? Free Radic Biol Med. 2004;36(7):838–849. doi: 10.1016/j.freeradbiomed.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Prasad S, Phromnoi K, Yadav VR, Chaturvedi MM, Aggarwal BB. Targeting inflammatory pathways by flavonoids for prevention and treatment of cancer. Planta Med. 2010;76(11):1044–1063. doi: 10.1055/s-0030-1250111. [DOI] [PubMed] [Google Scholar]
- 10.Taylor LP, Grotewold E. Flavonoids as developmental regulators. Curr Opin Plant Biol. 2005;8(3):317–323. doi: 10.1016/j.pbi.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Du H, Huang Y, Tang Y. Genetic and metabolic engineering of isoflavonoid biosynthesis. Appl Microbiol Biotechnol. 2010;86(5):1293–1312. doi: 10.1007/s00253-010-2512-8. [DOI] [PubMed] [Google Scholar]
- 12.Trantas E, Panopoulos N, Ververidis F. Metabolic engineering of the complete pathway leading to heterologous biosynthesis of various flavonoids and stilbenoids in Saccharomyces cerevisiae. Metab Eng. 2009;11(6):355–366. doi: 10.1016/j.ymben.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Deavours BE, Dixon RA. Metabolic engineering of isoflavonoid biosynthesis in alfalfa. Plant Physiol. 2005;138(4):2245–2259. doi: 10.1104/pp.105.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dixon RA. Phytoestrogens. Annu Rev Plant Biol. 2004;55:225–261. doi: 10.1146/annurev.arplant.55.031903.141729. [DOI] [PubMed] [Google Scholar]
- 15.Panagiotou G, Nielsen J. Nutritional systems biology: Definitions and approaches. Annu Rev Nutr. 2009;29:329–339. doi: 10.1146/annurev-nutr-080508-141138. [DOI] [PubMed] [Google Scholar]
- 16.Vargo MA, et al. Apigenin-induced-apoptosis is mediated by the activation of PKCδ and caspases in leukemia cells. Biochem Pharmacol. 2006;72(6):681–692. doi: 10.1016/j.bcp.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 17.Kaur P, Shukla S, Gupta S. Plant flavonoid apigenin inactivates Akt to trigger apoptosis in human prostate cancer: An in vitro and in vivo study. Carcinogenesis. 2008;29(11):2210–2217. doi: 10.1093/carcin/bgn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi EJ, Kim GH. Apigenin causes G(2)/M arrest associated with the modulation of p21(Cip1) and Cdc2 and activates p53-dependent apoptosis pathway in human breast cancer SK-BR-3 cells. J Nutr Biochem. 2009;20(4):285–290. doi: 10.1016/j.jnutbio.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Das A, Banik NL, Ray SK. Mechanism of apoptosis with the involvement of calpain and caspase cascades in human malignant neuroblastoma SH-SY5Y cells exposed to flavonoids. Int J Cancer. 2006;119(11):2575–2585. doi: 10.1002/ijc.22228. [DOI] [PubMed] [Google Scholar]
- 20.Nicholas C, et al. Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-κB through the suppression of p65 phosphorylation. J Immunol. 2007;179(10):7121–7127. doi: 10.4049/jimmunol.179.10.7121. [DOI] [PubMed] [Google Scholar]
- 21.Buckland G, Bach A, Serra-Majem L. Obesity and the Mediterranean diet: A systematic review of observational and intervention studies. Obes Rev. 2008;9(6):582–593. doi: 10.1111/j.1467-789X.2008.00503.x. [DOI] [PubMed] [Google Scholar]
- 22.La Vecchia C. Association between Mediterranean dietary patterns and cancer risk. Nutr Rev. 2009;67(Suppl 1):S126–S129. doi: 10.1111/j.1753-4887.2009.00174.x. [DOI] [PubMed] [Google Scholar]
- 23.Lairon D. Intervention studies on Mediterranean diet and cardiovascular risk. Mol Nutr Food Res. 2007;51(10):1209–1214. doi: 10.1002/mnfr.200700097. [DOI] [PubMed] [Google Scholar]
- 24.Gates MA, et al. Flavonoid intake and ovarian cancer risk in a population-based case-control study. Int J Cancer. 2009;124(8):1918–1925. doi: 10.1002/ijc.24151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin Y, Yu J, Yu YG. Identification of hNopp140 as a binding partner for doxorubicin with a phage display cloning method. Chem Biol. 2002;9(2):157–162. doi: 10.1016/s1074-5521(02)00096-0. [DOI] [PubMed] [Google Scholar]
- 26.Rodi DJ, et al. Screening of a library of phage-displayed peptides identifies human bcl-2 as a taxol-binding protein. J Mol Biol. 1999;285(1):197–203. doi: 10.1006/jmbi.1998.2303. [DOI] [PubMed] [Google Scholar]
- 27.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136(4):777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou J, et al. Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2/B1 (hnRNP-A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res Treat. 2001;66(3):217–224. doi: 10.1023/a:1010631915831. [DOI] [PubMed] [Google Scholar]
- 29.Golan-Gerstl R, et al. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71(13):4464–4472. doi: 10.1158/0008-5472.CAN-10-4410. [DOI] [PubMed] [Google Scholar]
- 30.Sueoka E, et al. Detection of plasma hnRNP B1 mRNA, a new cancer biomarker, in lung cancer patients by quantitative real-time polymerase chain reaction. Lung Cancer. 2005;48(1):77–83. doi: 10.1016/j.lungcan.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Wu S, et al. hnRNP B1 protein may be a possible prognostic factor in squamous cell carcinoma of the lung. Lung Cancer. 2003;41(2):179–186. doi: 10.1016/s0169-5002(03)00226-5. [DOI] [PubMed] [Google Scholar]
- 32.Cartegni L, et al. hnRNP A1 selectively interacts through its Gly-rich domain with different RNA-binding proteins. J Mol Biol. 1996;259(3):337–348. doi: 10.1006/jmbi.1996.0324. [DOI] [PubMed] [Google Scholar]
- 33.Derda R, et al. Diversity of phage-displayed libraries of peptides during panning and amplification. Molecules. 2011;16(2):1776–1803. doi: 10.3390/molecules16021776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aittaleb M, Boguth CA, Tesmer JJ. Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol Pharmacol. 2010;77(2):111–125. doi: 10.1124/mol.109.061234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu XW, Meng D, Fang J. Apigenin inhibited migration and invasion of human ovarian cancer A2780 cells through focal adhesion kinase. Carcinogenesis. 2008;29(12):2369–2376. doi: 10.1093/carcin/bgn244. [DOI] [PubMed] [Google Scholar]
- 36.Lee WJ, Chen WK, Wang CJ, Lin WL, Tseng TH. Apigenin inhibits HGF-promoted invasive growth and metastasis involving blocking PI3K/Akt pathway and beta 4 integrin function in MDA-MB-231 breast cancer cells. Toxicol Appl Pharmacol. 2008;226(2):178–191. doi: 10.1016/j.taap.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 37.Takeshita M, et al. Proanthocyanidin from blueberry leaves suppresses expression of subgenomic hepatitis C virus RNA. J Biol Chem. 2009;284(32):21165–21176. doi: 10.1074/jbc.M109.004945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang EY, et al. Inhibitory effects of gallic acid and quercetin on UDP-glucose dehydrogenase activity. FEBS Lett. 2008;582(27):3793–3797. doi: 10.1016/j.febslet.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 39.Day AJ, et al. Dietary flavonoid and isoflavone glycosides are hydrolysed by the lactase site of lactase phlorizin hydrolase. FEBS Lett. 2000;468(2–3):166–170. doi: 10.1016/s0014-5793(00)01211-4. [DOI] [PubMed] [Google Scholar]
- 40.Zhou Y, Rajabi H, Kufe D. Mucin 1 C-terminal subunit oncoprotein is a target for small-molecule inhibitors. Mol Pharmacol. 2011;79(5):886–893. doi: 10.1124/mol.110.070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qi F, Chen X, Beard DA. Detailed kinetics and regulation of mammalian NAD-linked isocitrate dehydrogenase. Biochim Biophys Acta. 2008;1784(11):1641–1651. doi: 10.1016/j.bbapap.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egger S, et al. Structural and kinetic evidence that catalytic reaction of human UDP-glucose 6-dehydrogenase involves covalent thiohemiacetal and thioester enzyme intermediates. J Biol Chem. 2012;287(3):2119–2129. doi: 10.1074/jbc.M111.313015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aveic S, Pigazzi M, Basso G. BAG1: The guardian of anti-apoptotic proteins in acute myeloid leukemia. PLoS One. 2011;6(10):e26097. doi: 10.1371/journal.pone.0026097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hou DX, Kumamoto T. Flavonoids as protein kinase inhibitors for cancer chemoprevention: Direct binding and molecular modeling. Antioxid Redox Signal. 2010;13(5):691–719. doi: 10.1089/ars.2009.2816. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Revalde JL, Reid G, Paxton JW. Interactions of dietary phytochemicals with ABC transporters: Possible implications for drug disposition and multidrug resistance in cancer. Drug Metab Rev. 2010;42(4):590–611. doi: 10.3109/03602531003758690. [DOI] [PubMed] [Google Scholar]
- 46.Tuñón MJ, García-Mediavilla MV, Sánchez-Campos S, González-Gallego J. Potential of flavonoids as anti-inflammatory agents: Modulation of pro-inflammatory gene expression and signal transduction pathways. Curr Drug Metab. 2009;10(3):256–271. doi: 10.2174/138920009787846369. [DOI] [PubMed] [Google Scholar]
- 47.Huhta MS, Chen HP, Hemann C, Hille CR, Marsh EN. Protein-coenzyme interactions in adenosylcobalamin-dependent glutamate mutase. Biochem J. 2001;355(Pt 1):131–137. doi: 10.1042/0264-6021:3550131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang X, et al. Spectroscopy study on the interaction of quercetin with collagen. J Agric Food Chem. 2009;57(9):3431–3435. doi: 10.1021/jf803671s. [DOI] [PubMed] [Google Scholar]
- 49.Zhu M, et al. The flavonoid baicalein inhibits fibrillation of α-synuclein and disaggregates existing fibrils. J Biol Chem. 2004;279(26):26846–26857. doi: 10.1074/jbc.M403129200. [DOI] [PubMed] [Google Scholar]
- 50.Deuschle K, et al. Construction and optimization of a family of genetically encoded metabolite sensors by semirational protein engineering. Protein Sci. 2005;14(9):2304–2314. doi: 10.1110/ps.051508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iwashina T. The structure and distribution of the flavonoids in plants. J Plant Res. 2000;113(3):287–299. [Google Scholar]
- 52.Hostetler G, et al. Flavone deglycosylation increases their anti-inflammatory activity and absorption. Mol Nutr Food Res. 2012;56(4):558–569. doi: 10.1002/mnfr.201100596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barnett SF, Theiry TA, LeStourgeon WM. The core proteins A2 and B1 exist as (A2)3B1 tetramers in 40S nuclear ribonucleoprotein particles. Mol Cell Biol. 1991;11(2):864–871. doi: 10.1128/mcb.11.2.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carson JH, Blondin N, Korza G. Rules of engagement promote polarity in RNA trafficking. BMC Neurosci. 2006;7(Suppl 1):S3. doi: 10.1186/1471-2202-7-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carson JH, Barbarese E. Systems analysis of RNA trafficking in neural cells. Biol Cell. 2005;97(1):51–62. doi: 10.1042/BC20040083. [DOI] [PubMed] [Google Scholar]
- 56.Huelga SC, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012;1(2):167–178. doi: 10.1016/j.celrep.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Igney FH, Krammer PH. Death and anti-death: Tumour resistance to apoptosis. Nat Rev Cancer. 2002;2(4):277–288. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- 58.Seol DW, Billiar TR. A caspase-9 variant missing the catalytic site is an endogenous inhibitor of apoptosis. J Biol Chem. 1999;274(4):2072–2076. doi: 10.1074/jbc.274.4.2072. [DOI] [PubMed] [Google Scholar]
- 59.Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem. 2001;276(23):20633–20640. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- 60.Gonzalez-Mejia ME, Voss OH, Murnan EJ, Doseff AI. Apigenin-induced apoptosis of leukemia cells is mediated by a bimodal and differentially regulated residue-specific phosphorylation of heat-shock protein-27. Cell Death Dis. 2010;1:e64. doi: 10.1038/cddis.2010.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arango D, et al. Apigenin induces DNA damage through the PKCδ-dependent activation of ATM and H2AX causing down-regulation of genes involved in cell cycle control and DNA repair. Biochem Pharmacol. 2012;84(12):1571–1580. doi: 10.1016/j.bcp.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuramochi K, et al. Identification of small molecule binding molecules by affinity purification using a specific ligand immobilized on PEGA resin. Bioconjug Chem. 2008;19(12):2417–2426. doi: 10.1021/bc8002716. [DOI] [PubMed] [Google Scholar]
- 63.Jiang H, Wong WH. SeqMap: Mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008;24(20):2395–2396. doi: 10.1093/bioinformatics/btn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saeed AI, et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques. 2003;34(2):374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 65.Huang DW, Sherman BT, Lempick RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;44(1):44–59. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 66.Fehr M, Frommer WB, Lalonde S. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proc Natl Acad Sci USA. 2002;99(15):9846–9851. doi: 10.1073/pnas.142089199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benesi HA, Hildebrand JH. A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons. J Am Chem Soc. 1949;71(8):2703–2707. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.