

Abstract
The misassembly of soluble proteins into toxic aggregates, including amyloid fibrils, underlies a large number of human degenerative diseases. Cardiac amyloidoses, which are most commonly caused by aggregation of Ig light chains or transthyretin (TTR) in the cardiac interstitium and conducting system, represent an important and often underdiagnosed cause of heart failure. Two types of TTR-associated amyloid cardiomyopathies are clinically important. The Val122Ile (V122I) mutation, which alters the kinetic stability of TTR and affects 3% to 4% of African American subjects, can lead to development of familial amyloid cardiomyopathy. In addition, aggregation of WT TTR in individuals older than age 65 y causes senile systemic amyloidosis. TTR-mediated amyloid cardiomyopathies are chronic and progressive conditions that lead to arrhythmias, biventricular heart failure, and death. As no Food and Drug Administration-approved drugs are currently available for treatment of these diseases, the development of therapeutic agents that prevent TTR-mediated cardiotoxicity is desired. Here, we report the development of AG10, a potent and selective kinetic stabilizer of TTR. AG10 prevents dissociation of V122I-TTR in serum samples obtained from patients with familial amyloid cardiomyopathy. In contrast to other TTR stabilizers currently in clinical trials, AG10 stabilizes V122I- and WT-TTR equally well and also exceeds their efficacy to stabilize WT and mutant TTR in whole serum. Crystallographic studies of AG10 bound to V122I-TTR give valuable insights into how AG10 achieves such effective kinetic stabilization of TTR, which will also aid in designing better TTR stabilizers. The oral bioavailability of AG10, combined with additional desirable drug-like features, makes it a very promising candidate to treat TTR amyloid cardiomyopathy.
Keywords: drug design, crystal structure
The systemic amyloidoses are a group of diseases that are caused by protein aggregation, including amyloid fibril formation, in soft tissue, nervous system, and solid organs (1, 2). Transthyretin (TTR or prealbumin) is one of more than 30 proteins whose aggregation can cause disease by a gain-of-toxic function mechanism (3–5). Native tetrameric TTR consists of 127-aa, β-sheet–rich subunits primarily synthesized by the liver and secreted into the blood, where it acts as a backup carrier of thyroxine (T4) and the primary carrier of holo retinol-binding protein (RBP) (6, 7). The two T4-binding sites are formed by the weaker dimer–dimer interface (8) and, because of the presence of two other T4 transport proteins in blood, these sites remain largely unoccupied in humans (<1% T4 bound) (9). Dissociation of the TTR-tetramer at the T4-binding interface, which generates dimers that rapidly dissociate into amyloidogenic monomers, is the rate-limiting step during TTR misfolding and amyloid formation (Fig. 1) (3, 8, 10).
Fig. 1.

The TTR amyloidogenesis cascade and a table summarizing TTR-mediated amyloidoses. TTR amyloidoses require rate-limiting tetramer dissociation to dimers, followed by dissociation into monomers before partial unfolding of monomers yields the aggregation-prone amyloidogenic intermediate. The amyloidogenic intermediate can misassemble to form a variety of toxic aggregates, including amyloid fibrils. Disease-associated destabilizing mutations can kinetically or thermodynamically destabilize TTR. Kinetic stabilization can be achieved through trans allelic suppression with the kinetically stabilized T119M-TTR, binding to T4 or other small molecules (Lower Left).
There are more than 100 known amyloidogenic mutations in TTR, which segregate into ethnic and geographic groupings (5). Point mutations in TTR promote amyloidogenesis by lowering its thermodynamic stability and/or decreasing the kinetic barrier for tetramer dissociation (Fig. 1 and SI Appendix, Fig. S1) (11). These mutations lead to hereditary TTR amyloidoses such as familial amyloid polyneuropathy (FAP) (12, 13) and familial amyloid cardiomyopathy (FAC) (14, 15), which are autosomal-dominant conditions with varying ages of onset and penetrance depending on the TTR mutation and ethnic background of the carriers (summarized in Fig. 1). One of the clinically most important FAP-causing TTR mutations is the thermodynamically destabilized Val30Met TTR (V30M) mutation (16). The most common TTR variant with almost exclusive cardiac involvement is the kinetically destabilized V122I mutation (11, 17). Although the V122I-TTR monomer has similar stability to the WT-TTR monomer, the tetramer dissociates threefold faster under physiological conditions (Fig. 1) (17, 18). This allele occurs in 3% to 4% of African-American subjects (∼1.3 million people) and is hypothesized to contribute to the increased prevalence of heart failure among African Americans (14, 15, 17, 19). WT-TTR aggregation underlies the development of senile systemic amyloidosis (SSA), a condition that affects as much as 10% to 20% of the population older than age 65 y and, in some patients, leads to progressive congestive heart failure (20, 21). FAC and SSA are clinically important diseases that might be underappreciated and misdiagnosed as hypertensive heart disease (19).
Kinetic stabilization of the native tetrameric structure of TTR by interallelic trans suppression in compound heterozygote patients, who carry the destabilizing V30M mutation as well as a second, disease-suppressing mutation (T119M) (22), or by small molecule occupancy of the T4-binding sites (3, 23, 24), increases the dissociative transition state energy and prevents amyloidosis (Fig. 1). Recently, clinical trials of tafamidis, a TTR kinetic stabilizer, indicated that it slows the progression of early-stage neuropathy in patients with FAP (25). The nonsteroidal anti-inflammatory drug diflunisal is another drug undergoing clinical trials for patients with FAP (26), but its use may be contraindicated in patients with FAC as a result of the inhibition of cyclooxygenase (COX) enzymes (27, 28). At present, no Food and Drug Administration-approved drugs for prevention or treatment of FAC or SSA are available, and the therapy for most patients is confined to symptomatic relief. Liver transplantation, which removes the source of the mutated protein, has been the treatment of choice for hereditary TTR amyloidoses (29). Combined liver and heart transplantation is performed as a palliative measure for a subset of patients with FAC (30). The risk and cost of organ transplantation is substantial and transplant recipients require lifelong use of immune-suppressive therapy. Thus, there is an urgent need for a pharmacologic treatment of TTR cardiomyopathy.
Previously we reported a high-throughput screen for TTR ligands that enabled the identification of a variety of potent and structurally diverse TTR kinetic stabilizers (23). We used these compounds as a starting point for structure–activity relationship studies and synthesized a series of analogues of compound 1, which was one of best hits from this high-throughput screen (Fig. 2A and SI Appendix, Fig. S2) (23). In comparison with the clinical candidates tafamidis and diflunisal, we find that one of the analogues of 1, AG10 (Fig. 2A), is a highly effective and selective stabilizer of WT and V122I-TTR. AG10 also prevents the dissociation of V122I-TTR in serum obtained from patients with FAC and protects human cardiomyocytes from TTR amyloid toxicity very effectively. X-ray crystallography reveals important insights into the mechanism of how AG10 is able to bind V122I-TTR with high affinity and to kinetically stabilize the tetramer. Regarding treatment of TTR cardiac amyloidoses in humans, AG10 represents a lead structure that combines high selectivity and efficacy with encouraging pharmacokinetic properties.
Fig. 2.

AG10 binds to TTR and stabilizes it in buffer. (A) Chemical structures of TTR kinetic stabilizers. (B) Evaluation of ligand binding to TTR in buffer by FP. Competition of FP-probe 2 from TTR by increasing concentrations (0.006–12.5 µM) of AG10 (Kapp = 193 nM; R2 = 0.994) and tafamidis (Kapp = 247 nM; R2 = 0.990). Each point shows the mean (SD) of three replicates. (C) Assessment of the binding affinity of AG10 to TTR by ITC. Calorimetric titration of AG10 against TTR (Kd1 = 4.8 nM, Kd2 = 314 nM). Raw data (Upper) and integrated heats (Lower) from the titration of TTR (2 µM) with AG10 (25 µM). (D) Inhibition of WT- and V122I-TTR (4 µM) fibril formation by test compounds (4 and 2 µM) under acidic conditions after 24 h. Aggregation in the presence of solvent alone (DMSO) was defined as 100% fibril formation. Each bar shows the mean ± SD of three technical replicates.
Results
Design and Synthesis of AG10.
Our previously reported crystal structure of WT-TTR in complex with 1 showed that the 3,5-dimethyl-1H-pyrazole ring sat deep within the inner cavity of the T4-binding site and formed hydrogen bonds with S117 and S117′ (SI Appendix, Fig. S2) (23). The 2-fluorophenyl ring of 1 occupied the outer binding cavity, placing a portion of the aryl ring into the halogen binding pocket (HBP) 1 or 1′. With this observation and additional structure–activity relationship data obtained from other ligands (23, 24, 31), we predicted that introducing a carboxylic acid to the 2-fluorophenyl ring of 1 would allow the molecule to make additional electrostatic interactions with K15 and K15′ at the periphery of the pocket. We hypothesized that the formation of hydrogen bonds and electrostatic interactions with TTR within the T4 binding site would result in higher binding affinity and better TTR kinetic stabilization. We synthesized a series of analogues of 1 that probed the optimal position of the carboxylic acid moiety at the 2-fluorophenyl ring. Among these analogues, AG10 was the most potent compound (Fig. 2A and SI Appendix, Fig. S3 and Scheme S1).
Characterization of AG10 Binding Energetics to TTR.
The binding affinity of AG10 and tafamidis to TTR at physiological pH was evaluated by using our fluorescence polarization (FP) assay (FP-probe 2; SI Appendix, Fig. S4 and Fig. S5) (23). The binding affinity of AG10 to WT-TTR [apparent binding constant (Kapp) = 193 nM; R2 = 0.994] was slightly better than that of tafamidis (Kapp = 247 nM; R2 = 0.990; Fig. 2B). Many ligands, including tafamidis (Kd1 = 4.4 nM, Kd2 = 280 nM, the dissociation constants for the first and second binding sites of TTR; SI Appendix, Figs. S6 and S7), bind TTR with negative cooperativity (24). We used isothermal titration calorimetry (ITC) to determine the binding constants of AG10 and tafamidis to WT-TTR and also to evaluate cooperativity between the two TTR T4 sites. Analysis of the free energies associated with AG10 binding to TTR shows high binding affinity, and the dissociation constants indicate that AG10 binds TTR with negative cooperativity (Kd1 = 4.8 nM, Kd2 = 314 nM; Fig. 2C). Despite the similar binding affinities of AG10 and tafamidis (i.e., similar Gibbs free energy, ΔG1 values; ΔG1 for tafamidis = −11.39 kcal/mol and ΔG1 for AG10 = −11.34 kcal/mol), the nature of binding for both compounds to TTR is very different. Whereas AG10 binding is almost entirely enthalpically driven (enthalpy change, ΔH1 = −13.60 kcal/mol, entropy change, TΔS1 = −2.26 kcal/mol), tafamidis binding is approximately 50% entropy and 50% enthalpy (ΔH1 = −5.00 kcal/mol and TΔS1 = 6.39 kcal/mol). This fact is also reflected in the differing modes for which these compounds bind to TTR as seen by X-ray crystallography (as described later). The negative cooperativity of AG10 binding to V122I-TTR was also corroborated by subunit exchange assays that yielded a Kd1 of 6.2 ± 2.0 nM and a Kd2 of 139 ± 80 nM (SI Appendix, Fig. S8 and Supplementary Text). For tafamidis, the fit yielded a Kd1 of 8.1 ± 0.8 nM, but Kd2 was too high to be accurately fit (estimated to be ∼1 μM; SI Appendix, Fig. S8).
AG10 Kinetically Stabilizes Native Tetrameric TTR in Vitro.
We then examined the ability of AG10 to kinetically stabilize TTR and prevent its aggregation. TTR forms amyloid fibrils in vitro under denaturing conditions that lower the pH, which promotes tetramer dissociation and partial monomer denaturation. Purified WT and V122I-TTR homotetramers (4 µM) were incubated with AG10 or tafamidis at TTR binding sites:test compound concentration ratios of 1:1 and 1:0.5. TTR amyloidogenesis, induced by lowering to pH 4.4, was assessed by measuring the turbidity at 400 nm (32). At 1:1 stoichiometry, AG10 and tafamidis were effective in stabilizing WT and V122-TTR. However, at substoichiometric ratio (1:0.5), AG10 was significantly (P < 0.0001) better than tafamidis in inhibiting the amyloidogenesis of WT and V122I-TTR (Fig. 2D).
AG10 Binds Selectively to WT-TTR in Human Serum.
To stabilize TTR tetramers in patients, small molecules must selectively bind to TTR in the presence of more than 4,000 other serum proteins. We examined the selectivity of AG10 for TTR in human serum by using a ligand competition assay using covalent probe 3 (SI Appendix, Figs. S4 and S9) (33). Probe 3 binds selectively to TTR in serum and then covalently modifies K15, creating a fluorescent conjugate (33). Ligands that bind selectively to TTR in serum decrease the binding of probe 3 to TTR, decreasing the fluorescence. In addition to selectivity, this assay depends also on the relative affinity of probe 3 and the test compound for the TTR binding sites, especially the second binding site (given that generally Kd2 > Kd1). Empirically, the results of the probe 3 competition assay at 3 h (33) correlate well with other direct measures of selectivity, such as the co-immunoprecipitatiob (co-IP) assay with TTR from biological fluids) (9).
Compounds (10 µM) were incubated with human serum (WT-TTR concentration ∼5 µM) or buffer with added WT-TTR (concentration = 3.6 μM) in the presence of probe 3 (3.6 µM). The rate of TTR covalent conjugate formation in serum and buffer was highest for diflunisal, which is a weak TTR ligand (>95% probe binding relative to control), (10) (Fig. 3 A and B and SI Appendix, Fig. S10). AG10 performs better in this assay in serum (3.1 ± 2.9% probe binding after 6 h; Fig. 3A and 3B) than tafamidis and T4 (70.5 ± 1.4% and 49.5 ± 2.6% probe binding after 6 h, respectively). Comparable results were obtained in buffer (SI Appendix, Fig. S10). It should be noted that AG10 performing better than tafamidis in this assay in buffer suggests that its affinity for TTR (especially the second binding site) is higher than that of tafamidis, and may have been underestimated by ITC.
Fig. 3.

AG10 binds selectively to TTR in human serum. (A) Fluorescence change caused by modification of TTR in human serum by covalent probe 3 monitored for 6 h in the presence of probe alone (black circles) or probe and TTR ligands (colors). (B) Percentage of covalent probe binding to TTR in the presence of ligands measured after 6 h of incubation relative to probe alone. (The lower the binding of the probe, the higher the binding selectivity of the ligand to TTR.) Each bar shows the mean (SD) of three replicates. (C and D) Stabilization of WT-TTR in serum against acid-mediated denaturation in the presence of AG10 and tafamidis. Serum samples were incubated (with DMSO, AG10, or tafamidis) in acetate buffer (pH 4.0) for the desired time period (0 and 72 h) before cross-linking and immunoblotting. The intensity of TTR bands (TTR tetramer, arrowhead; TTR bound to RBP, solid arrow) was quantified by using an Odyssey IR imaging system (LI-COR Bioscience) and reported as percentage of TTR tetramer, calculated as 100 × [(tetramer and tetramer + RBP density, 72 h)/(tetramer and tetramer + RBP density of DMSO, 0 h)]. (C) The shown blot is representative of four independent blots. (D) Error bars indicate SEM (n = 4).
These results from the probe 3 assay indicate that AG10 is highly selective for TTR in biological fluids. However, the larger difference between AG10 and tafamidis in this assay corresponds to a smaller difference in other measures of selectivity, such as stabilization of serum TTR following acid-mediated denaturation (Fig. 3 and 4). By using the previously established linear relationship between the probe 3 assay and the co-IP–based selectivity assays, we can estimate the selectivity of AG10 for TTR (in the co-IP assay, selectivity values range from 0 to 2 equivalents of small molecule per TTR tetramer, with 0 equivalents indicating no selectivity and 2 equivalents indicating perfect selectivity for TTR; SI Appendix, Supplementary Text). The fluorescence from probe 3 assay for AG10 (<1% of control; Fig. 3A) and tafamidis (44.2 ± 3% of control; Fig. 3A) correspond to co-IP selectivity values of 1.55 ± 0.16 for AG10 and 0.7 ± 0.16 for tafamidis (which is close to the measured value of 0.81 for tafamidis; errors are single prediction errors; SI Appendix, Fig. S11) (24). The dramatic difference of AG10 and tafamidis in the probe 3 assay (0 ± 2.5% vs. 44.2 ± 3% probe 3 binding after 3 h, respectively) corresponds to a somewhat smaller difference in calculated selectivity (∼1.55 vs. 0.81). Nevertheless, it is clear that AG10 binds more selectively to TTR in serum than tafamidis. The higher selectivity of AG10 is also demonstrated through serum stabilization assays, as indicated in the Western blot data shown in Figs. 3 and 4.
Fig. 4.

Stabilization of V122I-TTR in serum from two patients with FAC against acid-mediated denaturation in the presence of AG10 and tafamidis. Stabilization effect of tafamidis and AG10 on serum from a heterozygous patient with FAC (heterozygous V122I/WT-TTR, African American, male, age 56 y) at 0 h (A and B) and after 72 h (C and D). (E and F) Stabilization effect of tafamidis and AG10 on serum from a homozygous patient with FAC (homozygous V122I-TTR, Caucasian, female, age 62 y). Sample processing and quantification was performed as described in Fig. 3. IgG was analyzed by using IRdye800 goat anti-human IgG. Error bars represent SEM of three replicates.
AG10 Increases Stability of WT-TTR in Human Serum Against Acid-Mediated Dissociation That Accelerates Amyloidogenesis.
We next tested AG10 and tafamidis for their ability to stabilize WT-TTR in human serum (Fig. 3 C and D) (26, 34). AG10 and tafamidis (concentration range between 0.1 and 10 µM) were preincubated with human serum (TTR ∼5 µM), and the pH was lowered to pH 4.0 to induce dissociation of TTR tetramers. Aliquots, taken at 0 and 72 h, were treated with glutaraldehyde to cross-link TTR tetramers in the serum sample. Western blot analysis was used to measure the amount of intact TTR tetramer after 72 h of acid treatment in the presence and absence of test compounds (Fig. 3 C and D). By using this previously described technique (23, 26, 34), we detected the TTR tetramer (Fig. 3C, arrowhead) as well as tetrameric TTR bound to RBP (Fig. 3C, solid arrow). We also observed bands with higher molecular weight than TTR-RBP, which might be a result of cross-linking of multiple TTR tetramers/dimers. To quantify the degree of stabilization of the tetrameric form of TTR by the two compounds, we measured the intensity of TTR and TTR-RBP bands by using quantitative IR Western blots. At all concentrations tested, AG10 was significantly more effective than tafamidis in stabilizing TTR. Increasing the concentration of tafamidis from 5 to 10 µM resulted in an increase in TTR stabilization after 72 h (at 5 µM, tetramer 37.8 ± 3.3%; at 10 µM, tetramer 49.4 ± 4.3%; Fig. 3 C and D). In contrast, AG10 was a very potent ligand that, even at 5 µM, stabilized most of the TTR (tetramer 74.9 ± 11.5%). Increasing the concentration of AG10 to 10 µM resulted in stabilization of almost all of TTR in serum (tetramer 97.6 ± 9.4%; Fig. 3 C and D). As TTR dimers and tetramers in serum are in equilibrium with each other, one can also observe more dimers in blots of samples that contain high tetramer levels (Fig. 3C, lanes 1, 5, and 6).
AG10 Increases Stability of Mutant V122I-TTR in Serum from Patients with FAC.
We collected serum samples from two patients with FAC harboring the V122I-TTR mutations. One of these patients is heterozygous, which will result in expression of TTR heterotetramers consisting of V122I and WT subunits, whereas the other patient is homozygous for the V122I-TTR mutation. AG10 and tafamidis (10 µM) were evaluated for their effect on stabilization of V122I-TTR following acid denaturation of the patient’s serum samples (Fig. 4). Because of the increased instability of the mutant samples at pH 4 (compared with WT-TTR), we observed that, relative to AG10-treated samples, samples treated with only DMSO already showed a small reduction of TTR tetramer levels at 0 h (Fig. 4 A and B). There was no significant difference in the stabilizing effect of AG10 toward heterozygous (at 10 µM, tetramer 108.3 ± 5.2%) and homozygous V122I-TTR (at 10 µM, tetramer 114.4 ± 15.8%; Fig. 4) compared with its effect toward WT-TTR (at 10 µM, tetramer 97.6 ± 9.4%; Fig. 3 C and D). Stabilization of the V122I/WT-TTR heterotetramer by tafamidis (at 10 µM, tetramer: 57.2 ± 2.8%; Fig. 4 C and D) was also similar to its effect toward WT-TTR (at 10 µM, tetramer 49.4 ± 4.3%; Fig. 3 C and D). However, the stabilizing effect of tafamidis on homozygous V122I mutant TTR (at 10 µM, tetramer 31.1 ± 4.8%) was significantly lower than its effect on WT-TTR or heterozygous V122I/WT-TTR (Fig. 4 E and F). In addition, based on the probe 3 assay described earlier, binding of AG10 to V122I-TTR in serum from a patient with FAC was also very selective (6.9 ± 1.6% probe binding; SI Appendix, Fig. S12). At 8 μM, AG10 and tafamidis protected cardiomyocytes from the proteotoxicity of amyloidogenic V122I-TTR in vitro (SI Appendix, Fig. S13 and Supplementary Text).
AG10 Has No Significant Interactions with a Number of Receptors and Enzymes.
COX inhibitors such as diflunisal can produce significant gastrointestinal and cardiovascular side effects (27, 28). In addition, many previously known TTR ligands, designed based on the T4 structure, bind to thyroid hormone nuclear receptor. AG10 shows no significant inhibition of COX-1 or COX-2 and no significant binding to thyroid hormone nuclear receptor (IC50 > 10 µM; SI Appendix, Table S1). In addition, AG10 has very minimal inhibition of two common off-targets in drug discovery, the potassium ion channel hERG (human ether-a-go-go related gene) (IC50 > 100 µM) and a number of cytochrome P450 isozymes (IC50 > 50 µM; SI Appendix, Table S1).
AG10 Is Orally Bioavailable in Rodents and Shows No Toxicity.
Pharmacokinetic evaluation showed that AG10 is orally bioavailable in rodents and has excellent chemical and metabolic stability (SI Appendix, Figs S14 and S15 and Table S2). A more detailed account of pharmacokinetic properties of AG10 is provided in SI Appendix. The effect of AG10 and tafamidis on viability and proliferation of four cell lines was evaluated (SI Appendix, Fig. S16). AG10 showed no cytotoxic effects toward any of the cell lines that were tested, whereas tafamidis had only minor cytotoxic effects against Michigan cancer foundation-7 (MCF7) cells at high concentrations. We then investigated the in vivo toxicity of AG10 in Wistar rats. In a subacute (28-d) toxicity study, rats received a daily dose of AG10 (50 mg/kg/d by oral gavage; plasma Cmax ∼40 µM). No signs of toxicity or mortality were observed and no macroscopic changes were noted at necropsy. After 28 d of dosing, comprehensive evaluations of the health status of AG10-treated and untreated control animals were performed. This evaluation included a clinical chemistry panel of 23 variables and a complete blood count (SI Appendix). None of the assayed parameters showed significant differences between the treated and untreated groups. All animals gained the expected body weight throughout the whole study period (SI Appendix, Fig. S17). Histopathological evaluation of liver, kidney, heart, spleen, thymus, and lung showed no signs of pathologic processes in the AG10-treated animals (SI Appendix, Fig. S18).
Crystal Structures of AG10 and Tafamidis in Complex with V122I-TTR.
We determined the high-resolution cocrystal structures of AG10 and tafamidis in complex with V122I-TTR at 1.18-Å and 1.20-Å resolution, respectively (Fig. 5 and SI Appendix, Table S3). The 3,5-dimethyl-1H-pyrazole ring of AG10 sits deep within the inner cavity of the T4-binding site, and forms two hydrogen bonds with the S117 and S117′ of adjacent subunits (Fig. 5B). The methyl groups of this ring are placed into the HBP3 and 3′, where they make hydrophobic interactions. The carboxylic acid moiety on the para-fluoro-aryl ring makes additional electrostatic interactions directly with the ε-amino groups of K15 and K15′ at the periphery of the T4-binding site, and the fluorine atom is placed into HBP1. These two salt bridge interactions close the T4 pocket around AG10 and are partially shielded from the solvent, resulting in high-affinity binding of AG10 to TTR (Fig. 5B and Fig. S19A). In the case of tafamidis, there is no hydrogen bonding at the base of the T4 pocket; instead, the chlorine atoms of the 3,5-dichloro ring are also placed into HBP3 and 3′, where they bridge the subunits through predominantly hydrophobic interactions (Fig. 5C). Although AG10 and tafamidis possess a carboxylate group, the manner in which they interact with the ε-amino group of K15/15′ differs greatly. A closer look at the relative positions of the carboxylate groups for both molecules reveals these to be in different conformations (SI Appendix, Fig. S20). The aliphatic ether linker in AG10 between the 3,5-dimethyl-1H-pyrazole and phenyl ring affords the carboxylate group greater conformational freedom, whereas the more rigid benzoxazole scaffold of tafamidis limits the conformations this carboxylate can occupy. In doing so, it appears the carboxylate of tafamidis is unable to optimally bind to ε-amino group of K15 or K15′ directly. Instead, these groups are bridged through water molecules, which are likely weaker interactions by comparison, and leave the pocket more open with the ligand more exposed to the solvent (Fig. S19B).
Fig. 5.
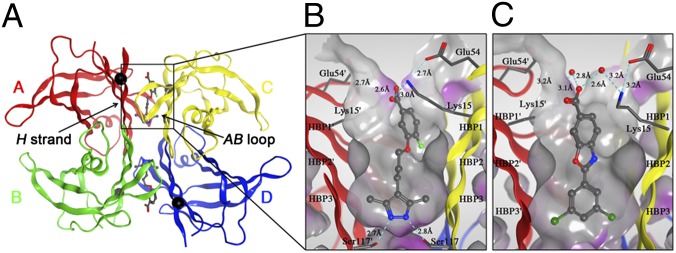
Crystal structures of V122I-TTR ligand complexes. (A) Quaternary structure of AG10 bound to V122I-TTR shown as a ribbon representation with monomers colored individually and positions of each of the V122I mutations shown as black spheres located on the H β-strand, which interacts with the adjacent AB-loop on the AC/BD interface. (B) AG10 in complex with V122I-TTR. (C) Tafamidis in complex with V122I-TTR. Close-up views of one of the two identical T4 binding sites with different colored ribbons for the two monomers of the tetramer composing the binding site. A Connolly molecular surface (40) was applied to residues within 10 Å of ligand in the T4 binding pocket and colored gray for hydrophobic and purple for polar residues.
Discussion
We hypothesized that the efficacy and selectivity of TTR stabilizers could be optimized if a compound were able to bind to the inner cavity and the periphery of the T4 binding site. This would be especially critical for compounds aimed at stabilization of disease-associated TTR variants. Guided by structural information on TTR ligands (23, 24, 31), we designed and synthesized AG10 and tested it for affinity, selectivity, and stabilization of TTR.
Our studies showed that AG10 exhibited remarkable selectivity for TTR binding in human serum in the presence of more than 4,000 other serum proteins (Fig. 3 A and B). The exceptional binding selectivity of AG10 to serum TTR surpasses that of TTR’s natural ligand, T4, as well as that of the clinical candidates, tafamidis and diflunisal. AG10 selectively stabilized serum WT-TTR in a dose-dependent manner and was significantly more effective than tafamidis at all concentrations tested (Fig. 3 C and D). The Cmax of tafamidis in humans, following a 20-mg daily dose, was estimated to be 7.4 μM (35). At 10 μM, tafamidis stabilized 49.4 ± 4.3% of TTR tetramers in serum. In comparison, at 10 μM, AG10 provided almost full stabilization (97.6 ± 9.4%) of serum WT-TTR (Fig. 3 C and D).
A key consideration for the development of kinetic stabilizers of TTR is that such ligands should stabilize mutant and WT-TTR equally well. This is especially critical in light of protein sequencing data, which compared the composition of soluble plasma TTR to TTR amyloid deposits in affected tissues of heterozygous patients with TTR amyloidoses. It was found that in plasma, mutant TTR accounts for one third of TTR, whereas, in amyloid fibrils in tissue, it accounts for two thirds of TTR present. This inverse 2:1 WT/mutant peptide ratio in plasma and 1:2 WT/mutant ratio in amyloid deposits suggested that mutant TTR contributes disproportionally to amyloid formation in heterozygous patients, probably because tetramers containing at least two mutant subunits are less stable and deposit preferentially in tissues (36). AG10 stabilizes the kinetically destabilized, mutant V122I-TTR tetramers equally well as WT-TTR tetramers. This can be seen most clearly when comparing the ability of AG10 and tafamidis to stabilize WT- and V122I-TTR in human serum. There is a statistically significant difference (P < 0.02) between stabilization of TTR tetramers composed of WT-TTR (49.4 ± 4.3% stabilization) and V122I-TTR (31.1 ± 2.7% stabilization) monomers by tafamidis (at 10 μM). Conversely, AG10 stabilizes TTR from WT control serum and serum from a V122I homozygous patient with FAC serum equally well (Figs. 3 C and D and 4F). Similarly, there is a statistically significant difference (P < 0.01) in prevention of WT- vs. V122I-TTR amyloid fibril formation by tafamidis, whereas there is no significant difference between WT- and V122I-TTR seen for AG10 (Fig. 2D). These data suggest that the nature of interactions that AG10 forms within the T4 binding site might be critical for targeting the kinetically destabilized V122I-TTR as effectively as WT-TTR. Our crystallographic studies of AG10 bound to the mutant V122I-TTR revealed valuable insights into how AG10 achieves such potent kinetic stabilization of V122I-TTR (Fig. 5).
No structural information was available for any of the pathogenic TTR variants in complex with clinical candidates. This is of particular importance for the FAC variant, as the V122I mutation lies directly on the weaker dimer–dimer interface, leading to an increased rate of dissociation, so-called kinetic destabilization. Surprisingly, although the binding affinities for AG10 and tafamidis are very similar, the degree of stabilization of AG10 as determined through multiple assays is higher. In WT-TTR, V122 is located on the periphery of the H β-strand, which makes an antiparallel β-sheet interaction with another monomer, stabilizing the AC/BD dimer interface. The side chain of the mutated I122 packs against the side chains of F87 and Y114 of the neighboring subunit, and this packing is slightly altered relative to the WT V122-Y114 interaction. This subtle movement of the Y114 side chain in the V122I homotetramer alters its interactions with the so-called AB-loop of a second dimer at the interface (AB/CD) and is thought to be the mechanism by which the V122I mutation selectively destabilizes the tetrameric quaternary structure (37). Unlike tafamidis, the 3,5-dimethyl-1H-pyrazole ring of AG10 forms hydrogen bonds with S117 and 117′, which bridge the H strands of adjacent monomers, whereas the carboxylate of the para-fluoro-aryl ring directly forms a salt bridge interaction with K15 and K15′. Thus, binding of AG10 can compensate for the loss in stability at the AB/CD interface of V122I-TTR and increase the energy barrier for dissociation, thereby ameliorating the amyloid cascade as shown in our studies. The highly optimized binding of AG10 within the TTR T4 pocket is reflected in the favorable binding enthalpy associated with the formation of an extensive network of hydrogen bonds and salt bridges. The binding enthalpy is critical for the development of high-affinity drugs, and it is typically more difficult to optimize than entropy (38).
Despite the similar binding affinities of AG10 and tafamidis to TTR in buffer, their ability to stabilize TTR in buffer and serum are different. Our studies show that, because of the nature of interaction with the target, molecules with similar binding affinities could have very different efficacy in stabilizing amyloidogenic proteins. Because of the kinetic instability of V122I-TTR, it appears to be advantageous for small molecules targeting the FAC-associated V122I-TTR to have enthalpically driven binding (by forming more hydrogen bonds and ionic interactions) as well as multiple interactions with different subunits of the protein. It has been proposed that, analogous to TTR, small-molecule stabilizers of the physiological tetramer of α-synuclein could reduce its pathogenicity in Parkinson disease (39). Our studies could provide insights for the development of better stabilizers for α-synuclein and potentially other amyloidogenic proteins.
In conclusion, the potency and selectivity of AG10 to TTR in human serum exceeds those of drugs currently in clinical trials. The oral bioavailability, lack of toxicity in rodents, and additional favorable pharmacokinetic properties make AG10 a very promising candidate compound for treatment of TTR amyloidoses. Importantly, our biophysical and structural data of AG10 bound to the V122I-TTR reveal critical insights into the mechanisms by which compounds are able to achieve not only high binding affinity, but more importantly high efficacy and selectivity for TTR in plasma. The ability of AG10 to stabilize mutant and WT-TTR equally well might prove to be critical in its use as a potential therapeutic agent to prevent aggregation of mutant TTR, and will be valuable in the design of future kinetic stabilizers of TTR.
Methods
The procedures for AG10 synthesis and characterization (1H spectroscopy, mass spectra) are described in SI Appendix, as are the methods for X-ray crystallography and structure determination and for assaying AG10-mediated TTR stabilization in buffer and serum.
Supplementary Material
Acknowledgments
The authors acknowledge Dr. M. M. Davidson (Columbia University) for providing the AC16 cells. This work was supported by New Investigator Award from the American Association of Colleges of Pharmacy; a gift from Bill and Ann Nutting (to M.M.A.); a grant from the Stanford Translational Research Program (to M.L. and I.A.G.); a grant from the SPARK program (to I.A.G.), and National Institutes of Health Grants AI42266 (to I.A.W.) and AG 032285 (to N.R.).
Footnotes
Conflict of interest statement: E.T.P. and J.W.K. have a financial interest in the regulatory agency-approved drug tafamidis.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4HIQ [AG10:V122I-TTR] and 4HIS [tafamidis:V122-TTR]).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1300761110/-/DCSupplemental.
References
- 1.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426(6968):900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Johnson SM, et al. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: A focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38(12):911–921. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 4.Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337(13):898–909. doi: 10.1056/NEJM199709253371306. [DOI] [PubMed] [Google Scholar]
- 5.Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10(3):160–184. doi: 10.3109/13506120308998998. [DOI] [PubMed] [Google Scholar]
- 6.Blake CC, Geisow MJ, Oatley SJ, Rérat B, Rérat C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol. 1978;121(3):339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 7.Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science. 1995;268(5213):1039–1041. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- 8.Foss TR, Wiseman RL, Kelly JW. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry. 2005;44(47):15525–15533. doi: 10.1021/bi051608t. [DOI] [PubMed] [Google Scholar]
- 9.Purkey HE, Dorrell MI, Kelly JW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci USA. 2001;98(10):5566–5571. doi: 10.1073/pnas.091431798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammarström P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299(5607):713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 11.Sekijima Y, et al. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121(1):73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 12.Ando Y, Suhr OB. Autonomic dysfunction in familial amyloidotic polyneuropathy (FAP) Amyloid. 1998;5(4):288–300. doi: 10.3109/13506129809007303. [DOI] [PubMed] [Google Scholar]
- 13.Benson MD. Familial amyloidotic polyneuropathy. Trends Neurosci. 1989;12(3):88–92. doi: 10.1016/0166-2236(89)90162-8. [DOI] [PubMed] [Google Scholar]
- 14.Connors LH, et al. Cardiac amyloidosis in African Americans: Comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J. 2009;158(4):607–614. doi: 10.1016/j.ahj.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Buxbaum J, et al. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864–870. doi: 10.1016/j.ahj.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammarström P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci USA. 2002;99(suppl 4):16427–16432. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang X, Buxbaum JN, Kelly JW. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc Natl Acad Sci USA. 2001;98(26):14943–14948. doi: 10.1073/pnas.261419998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286–1300. doi: 10.1161/CIRCULATIONAHA.111.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Falk RH. Cardiac amyloidosis: A treatable disease, often overlooked. Circulation. 2011;124(9):1079–1085. doi: 10.1161/CIRCULATIONAHA.110.010447. [DOI] [PubMed] [Google Scholar]
- 20.Westermark P, Sletten K, Johansson B, Cornwell GG., 3rd Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci USA. 1990;87(7):2843–2845. doi: 10.1073/pnas.87.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rapezzi C, et al. Transthyretin-related amyloidoses and the heart: A clinical overview. Nat Rev Cardiol. 2010;7(7):398–408. doi: 10.1038/nrcardio.2010.67. [DOI] [PubMed] [Google Scholar]
- 22.Hammarström P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293(5539):2459–2462. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 23.Alhamadsheh MM, et al. Potent kinetic stabilizers that prevent transthyretin-mediated cardiomyocyte proteotoxicity. Sci Transl Med. 2011;3(97):97ra81. doi: 10.1126/scitranslmed.3002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bulawa CE, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA. 2012;109(24):9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coelho T, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology. 2012;79(8):785–792. doi: 10.1212/WNL.0b013e3182661eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13(4):236–249. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- 27.Page J, Henry D. Consumption of NSAIDs and the development of congestive heart failure in elderly patients: An underrecognized public health problem. Arch Intern Med. 2000;160(6):777–784. doi: 10.1001/archinte.160.6.777. [DOI] [PubMed] [Google Scholar]
- 28.Wallace JL. Pathogenesis of NSAID-induced gastroduodenal mucosal injury. Best Pract Res Clin Gastroenterol. 2001;15(5):691–703. doi: 10.1053/bega.2001.0229. [DOI] [PubMed] [Google Scholar]
- 29.Jonsén E, Suhr OB, Tashima K, Athlin E. Early liver transplantation is essential for familial amyloidotic polyneuropathy patients’ quality of life. Amyloid. 2001;8(1):52–57. doi: 10.3109/13506120108993814. [DOI] [PubMed] [Google Scholar]
- 30.Hamour IM, et al. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8(5):1056–1059. doi: 10.1111/j.1600-6143.2008.02162.x. [DOI] [PubMed] [Google Scholar]
- 31.Connelly S, Choi S, Johnson SM, Kelly JW, Wilson IA. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr Opin Struct Biol. 2010;20(1):54–62. doi: 10.1016/j.sbi.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bourgault S, et al. Mechanisms of transthyretin cardiomyocyte toxicity inhibition by resveratrol analogs. Biochem Biophys Res Commun. 2011;410(4):707–713. doi: 10.1016/j.bbrc.2011.04.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi S, Kelly JW. A competition assay to identify amyloidogenesis inhibitors by monitoring the fluorescence emitted by the covalent attachment of a stilbene derivative to transthyretin. Bioorg Med Chem. 2011;19(4):1505–1514. doi: 10.1016/j.bmc.2010.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tojo K, Sekijima Y, Kelly JW, Ikeda S. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res. 2006;56(4):441–449. doi: 10.1016/j.neures.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 35. European Medicines Agency Committee for Medicinal Products for Human Use (2011) Tafamidis Meglumine (Vyndaqel) assessment report, 22 September 2011. EMA/729083/2011. Procedure No. EMEA/H/C/002294.
- 36.Dwulet FE, Benson MD. Primary structure of an amyloid prealbumin and its plasma precursor in a heredofamilial polyneuropathy of Swedish origin. Proc Natl Acad Sci USA. 1984;81(3):694–698. doi: 10.1073/pnas.81.3.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Damas AM, Ribeiro S, Lamzin VS, Palha JA, Saraiva MJ. Structure of the Val122Ile variant transthyretin - a cardiomyopathic mutant. Acta Crystallogr D Biol Crystallogr. 1996;52(Pt 5):966–972. doi: 10.1107/S0907444996003307. [DOI] [PubMed] [Google Scholar]
- 38.Freire E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov Today. 2008;13(19-20):869–874. doi: 10.1016/j.drudis2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477(7362):107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Connolly ML. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983;221(4612):709–713. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.