

Abstract
The liver is a central organ for the synthesis and storage of nutrients, production of serum proteins and hormones, and breakdown of toxins and metabolites. Because the liver is susceptible to toxin- or pathogen-mediated injury, it maintains a remarkable capacity to regenerate by compensatory growth. Specifically, in response to injury, quiescent hepatocytes enter the cell cycle and undergo DNA replication to promote liver regrowth. Despite the elucidation of a number of regenerative factors, the mechanisms by which liver injury triggers hepatocyte proliferation are incompletely understood. We demonstrate here that eosinophils stimulate liver regeneration after partial hepatectomy and toxin-mediated injury. Liver injury results in rapid recruitment of eosinophils, which secrete IL-4 to promote the proliferation of quiescent hepatocytes. Surprisingly, signaling via the IL-4Rα in macrophages, which have been implicated in tissue repair, is dispensable for hepatocyte proliferation and liver regrowth after injury. Instead, IL-4 exerts its proliferative actions via IL-4Rα in hepatocytes. Our findings thus provide a unique mechanism by which eosinophil-derived IL-4 stimulates hepatocyte proliferation in regenerating liver.
Keywords: type 2 immunity, tissue injury and repair, inflammation, parasites
Liver is a highly regenerative organ in mammals. As such, injury to liver results in stimulation of quiescent hepatocytes to reenter the cell cycle, resulting in restoration of liver function (1, 2). Since the advent of partial hepatectomy in 1931 (3), a number of signaling pathways have been identified in rodents that stimulate hepatocyte proliferation. For instance, Kupffer cell-derived cytokines such as IL-6 and TNF-α, stellate cell-derived hepatocyte growth factor (HGF), and platelet-derived serotonin have been implicated in the activation and promotion of hepatocyte proliferation after injury (2, 4). Consistent with this, mice lacking IL-6, serotonin, or Met (the receptor for HGF in hepatocytes) exhibit impairment in liver regeneration after partial hepatectomy. In a similar manner, partial hepatectomy increases circulating levels of bile acids, which stimulate hepatocyte proliferation via activation of the nuclear receptor farnesoid X receptor (5).
In all vertebrate species, tissue injury results in activation of the innate immune system, suggesting its participation in the repair of tissues (6). Recent studies suggest that injury to epithelial surfaces, such as skin, lung, and colon, results in rapid activation of a type 2 immunity, which promotes the repair of damaged barrier surfaces by inducing the wound-healing program (7, 8). In each of these paradigms of tissue damage and repair, a central role has been ascribed to macrophages, which, in response to the locally produced type 2 cytokines IL-4 and IL-13, undergo alternative activation. However, it remains unknown whether the regenerative actions of type 2 immunity and alternatively activated macrophages are limited to replacement of barrier surfaces or whether they can also participate in regeneration of organs, such as the liver.
Another type 2 innate immune cell that has been implicated in tissue damage and repair is the eosinophil. Traditionally, tissue eosinophilia has been associated with pathogenesis of type 2 inflammatory pathologies, such as those associated with parasitic infections, allergies, and gastrointestinal disorders (9, 10). In these settings, activation of eosinophils results in the secretion of cytotoxic granule cationic proteins, such as major basic protein, eosinophil cationic protein, and eosinophil peroxidase, which in aggregate promote tissue damage and dysfunction. However, recent studies have highlighted a potential role for eosinophils in tissue homeostasis and repair (9, 11). For instance, in the model of surgical incision and closure, eosinophil number and alternative macrophage activation increased transiently after injury, suggesting that both cell types might be involved in the tissue repair response (12). Furthermore, in the context of liver regeneration, nodular regenerative hyperplasia of the liver has been observed in patients with idiopathic hypereosinophilic syndrome (13). Despite these advances, the functions of eosinophils and type 2 cytokine signaling have not been systematically investigated in the experimental paradigms of liver regeneration.
In this article, we present evidence that type 2 immune responses involving eosinophils and IL-4/IL-13 stimulate liver regeneration after partial hepatectomy and toxin-induced injury. Interestingly, signaling via the IL-4Rα in myeloid cells is not required for mediating the regenerative actions of type 2 cytokines in injured liver. Rather, type 2 cytokines stimulate liver regeneration, in part, via activation of the IL-4Rα on hepatocytes.
Results
Eosinophils Are Recruited to the Liver After Hepatic Injury.
To identify potential immune cells that participate in liver regeneration, we examined the immune cell repertoire of regenerating livers. Injury induced by partial hepatectomy or carbon tetrachloride (CCl4) led to a dramatic increase in the recruitment of innate immune cells, including eosinophils, neutrophils, macrophages, and dendritic cells (Figs. S1 A and B, and S2), which coincided with the onset of hepatocyte proliferation. In contrast, the content of adaptive immune cells did not change significantly in these models of liver injury (Figs. S1 C and D and S2). Because tissue eosinophilia has been implicated in tissue damage/repair and liver growth (10, 12–14), we quantified eosinophil numbers in the livers of uninjured and injured mice. In both models, eosinophil recruitment into injured livers was increased: A ∼2.4-fold increase was observed after partial hepatectomy, whereas injury by CCl4 resulted in a ∼6.9-fold increase in the number of eosinophils recovered from the liver (Fig. 1 A and B). This increased migration of eosinophils into injured livers was accompanied by higher expression of eotaxin-1 (Ccl11) (Fig. S1 E and F) and could be blocked by antibodies directed against the α4 and αL integrins, which bind vascular cell adhesion molecule 1 and intercellular cell adhesion molecule 1 to promote the migration of eosinophils into tissues (Fig. 1 A and B) (10, 15). In aggregate, these results suggest that injury to liver is a potent stimulus for eosinophil recruitment, potentially implicating eosinophils in liver’s regenerative response.
Fig. 1.

Impaired liver regeneration in mice lacking eosinophils. (A and B) Eosinophil numbers in livers of mice subjected to partial hepatectomy (PH) or toxin-induced injury (CCl4) were enumerated using flow cytometry. Migration of eosinophils into injured livers is integrin-dependent and can be blocked by antibodies against α4 and αL integrins (Ab). Isotype (Iso) immunoglobulins (IgG2a and IgG2b) were administered to control mice (n = 4–12 mice per group). (C) PH was performed in WT and ΔdblGATA mice, which lack eosinophils, and BrdU incorporation was examined at the indicated times to assess hepatocyte proliferation (n = 4–7 mice per genotype per time). (D) Remnant liver sections were stained for BrdU 36 h after partial hepatectomy. Representative images of WT and ΔdblGATA are shown. (E) Percentage of Ki67+ hepatocytes in WT and ΔdblGATA mice 2 d after administration of CCl4 (n = 3–5 mice per genotype). (F) Quantification of necrotic area in WT and ΔdblGATA mice 2 d after administration of CCl4 (n = 4–5 mice per genotype). *P < 0.05, **P < 0.01, ***P < 0.001. All data are presented as mean ± SEM.
Eosinophils Regulate Liver Regeneration.
To address this issue, we performed 70% hepatectomy in congenic wild-type (WT) and ΔdblGATA mice, with the latter lacking eosinophils because of a mutation in the promoter region of the Gata1 gene (16). In response to partial hepatectomy (3, 17), hepatocyte proliferation increased rapidly in WT mice, as assessed by the incorporation of 5-bromo-2′-deoxyuridine (BrdU) at 36, 48, and 72 h after hepatectomy (Fig. 1 C and D). This proliferative response of hepatocytes was reduced by ∼44–52% in ΔdblGATA mice 36, 48, and 72 h after hepatectomy (Fig. 1 C and D). Notably, the peak time for BrdU labeling was similar in WT and ΔdblGATA mice (Fig. 1C), indicating that the observed decrease in BrdU incorporation does not result from a temporal shift in hepatocyte proliferation. Congruent with this observation, liver/body weight was ∼20% lower in ΔdblGATA mice 72 h after partial hepatectomy (Fig. S3A).
To ascertain whether eosinophils were also required for liver regeneration after toxin-mediated injury, we treated WT and ΔdblGATA mice with a single dose of the liver toxin CCl4 (18). Although the extent of initial liver injury was similar in both strains of mice (Fig. S3B), their regenerative responses were quite different. WT mice rapidly underwent hepatocyte proliferation and regenerated their damaged livers, whereas ΔdblGATA mice exhibited ∼50% reduction in hepatocyte proliferation (quantified by staining for the cellular proliferation marker Ki67), which was associated with persistence of necrotic debris (Fig. 1 E and F and Fig. S3C). Moreover, this impaired regenerative response of ΔdblGATA mice to partial hepatectomy or CCl4-mediated injury was associated with complete absence of liver eosinophils (Fig. S4 A and B). These results highlight a previously unappreciated role for eosinophils in inducing hepatocyte proliferation after injury and implicate eosinophil-derived factors in the orchestration of liver regeneration.
Eosinophils Recruited to Injured Liver Secrete IL-4.
Among the factors secreted by eosinophils, IL-4 has been implicated in both wound repair and liver regeneration (8, 12, 19–21). We thus evaluated the competence of eosinophils for IL-4 secretion in injured liver. After CCl4-induced liver injury, eosinophils, which can be distinguished by their expression of sialic acid-binding Ig receptor (Siglec-F) (15), were the predominant cell type competent for IL-4 secretion (Fig. 2A and Fig. S5B) in IL-4 reporter mice (4get mice), a knock-in strain expressing the green fluorescent protein (GFP) from the IL-4 locus (22). Similarly, partial hepatectomy in 4get mice demonstrated that ∼81–84% of GFP+ cells were eosinophils (Fig. 2B and Fig. S5A), with the remainder being mast cells and T helper type 2 (Th2) cells (Figs. S5 A and B and S6 A and B). In contrast to a recent report (23), GFP+ cells did not express the pan-natural killer/natural killer T (NK/NKT) cell marker CD49b (Fig. S5 A and B). This difference likely stems from the increased sensitivity and specificity of 4get mice for detecting cells competent for IL-4 secretion. To further verify whether eosinophils were capable of secreting IL-4 in injured liver, we used knockin human CD2 (KN2) reporter mice in which cell surface expression of human CD2 (huCD2) faithfully reports the secretion of IL-4 protein (24). Congruent with the GFP positivity of eosinophils in 4get mice, eosinophils infiltrating injured liver expressed huCD2 on the cell surface, indicative of recent IL-4 secretion (Fig. 2 C and D). Furthermore, in both models of liver injury, cells competent for IL-4 secretion, as marked by GFP+ staining, colocalized with Ki67+ hepatocytes (Fig. 2E), suggesting a potential role for IL-4 in promoting hepatocyte proliferation.
Fig. 2.
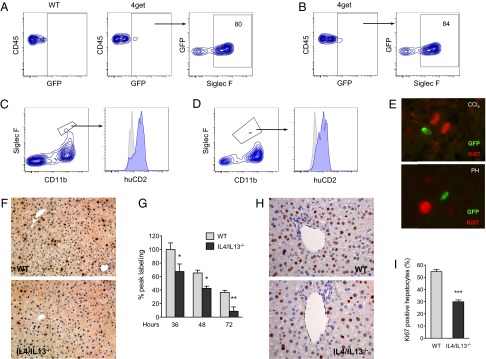
IL-4/IL-13 promote liver regeneration. (A and B) Identification of eosinophils as the major cell population competent for IL-4 secretion after CCl4-mediated injury (A) and PH (B). Two days after administration of CCl4 or PH in the IL-4 reporter mice (4get mice), the nonhepatocyte fraction was isolated and analyzed for expression of GFP and markers of eosinophils. Representative data from 3 independent experiments are shown in A and B. (C and D) Eosinophils produce IL-4 in injured livers. Expression of huCD2 on eosinophils (Siglec F+CD11b+) 2 d after PH (C) or CCl4-induced injury (D) in KN2 mice. Isotype, gray histogram; huCD2, blue histogram. (E) GFP+ cells localize in proximity to proliferating hepatocytes (Ki67+) 2 d after injury with CCl4 (Upper) or PH (Lower). (F) Liver sections stained for BrdU 36 h after partial hepatectomy. Representative images of WT and IL-4/IL-13−/− are shown. (G) Liver regeneration in WT and IL-4/IL-13−/− mice was assessed by BrdU incorporation at indicated times after PH (n = 4–9 mice per genotype per time). (H) Representative liver sections from WT and IL-4/IL-13−/− mice were stained for Ki67 2 d after administration of CCl4. (I) Percentage of Ki67+ hepatocytes in WT and IL-4/IL-13−/− mice 2 d after CCl4 treatment (n = 5 mice per genotype). *P < 0.05, **P < 0.01, ***P < 0.001. All data are presented as mean ± SEM.
Activation of IL-4/IL-13 Signaling Facilitates Liver Regeneration.
Because of the known functional redundancy between IL-4 and IL-13 (25, 26), we quantified the regenerative capacity of liver in IL-4/IL-13−/− mice. After 70% hepatectomy, BrdU incorporation was reduced by ∼35–75% at 36, 48, and 72 h in IL-4/IL-13−/− mice (Fig. 2 F and G). Consistent with our findings that hepatocyte proliferation is impaired in IL-4/IL-13−/− mice, the temporal profile of BrdU incorporation was similar in both genotypes (Fig. 2G), and liver/body weight was reduced by ∼30% in IL-4/IL-13−/− mice 72 h after partial hepatectomy (Fig. S7A). Furthermore, the regenerative response of IL-4/IL-13−/− mice to CCl4-induced liver injury was blunted, as evidenced by a ∼45% reduction in the number of Ki67+ hepatocytes (Fig. 2 H and I) and a ∼35% increase in necrotic debris area (Fig. S7 B and C).
To gain an understanding of how IL-4/IL-13 regulate liver regeneration, we performed genome-wide transcriptional analysis with microarrays on livers of WT and IL-4/IL-13−/− mice treated with CCl4. Gene set enrichment analysis with Database for Annotation, Visualization and Integrated Discovery (DAVID) (27) revealed that gene ontology (GO) terms associated with the cell cycle, nuclear division, and mitosis were overrepresented within the differentially expressed genes (Fig. 3A and Table S1). This suggested that IL-4/IL-13 stimulate the reentry of quiescent hepatocytes into the cell cycle. In support of this, genes required for entry into and progression through the cell cycle, such as cyclin B1 (Ccnb1), cell division cycle 20 (Cdc20), early growth response protein-1 (Egr1), forkhead box protein M1 (Foxm1), were expressed at lower levels in livers of IL-4/IL-13−/− mice after injury (Fig. 3 B and C). Moreover, expression of genes encoded by alpha-fetoprotein (Afp), H19 and insulin-like growth factor 2 (Igf2), which are dynamically regulated in fetal (expressed), adult (repressed), and regenerating (expressed) liver, was also reduced in the livers of IL-4/IL-13−/− mice (Fig. 3C). These changes in gene expression were verified by quantitative RT-PCR analysis of liver mRNAs of WT and IL-4/IL-13−/− mice (Fig. 3C). Importantly, expression of FoxM1 protein, a master regulator of hepatocyte proliferation and liver regeneration (28), and its target gene cyclin b1 (Ccnb1) was markedly lower in mice lacking IL-4/IL-13 (Fig. 3 C and D). A similar decrease in expression of genes required for hepatocyte proliferation was observed in the ΔdblGATA mice, which lack eosinophils, after partial hepatectomy (Fig. 3E).
Fig. 3.

IL-4/IL-13 signaling promotes cell cycle progression after liver injury. (A) Global gene expression analysis of WT and IL-4/IL-13−/− livers was performed 2 d after injury with CCl4. Gene ontology (GO) terms associated with cell cycle and mitosis are enriched among the differentially expressed genes. (B) Heat map presentation of differentially expressed genes in livers of WT and IL-4/IL-13−/− mice 2 d after injury with CCl4 (red, high; green, low). (C) Quantitative RT-PCR analysis of genes associated with hepatocyte proliferation and liver regeneration. mRNAs from WT and IL-4/IL-13−/− mice were analyzed 2 d after injury with CCl4 (n = 3–5 per genotype and treatment). (D) Western blot analysis of FoxM1 in WT and IL-4/IL-13−/− livers. (E) Quantitative RT-PCR analysis of genes associated with hepatocyte proliferation in WT and ΔdblGATA mice was performed 2 d after PH (n = 5–8 per genotype and treatment). *P < 0.05, **P < 0.01, ***P < 0.001. All data are presented as mean ± SEM.
IL-4Rα Signaling in Hepatocytes, Not Myeloid Cells, Regulates Liver Regeneration.
Previous studies have implicated an IL-4- and IL-13-driven program of alternative macrophage activation in tissue repair (12, 21, 29), prompting us to examine the role of these cells in liver regeneration. For these studies, we used mice harboring deletion of IL-4Rα in myeloid cells [IL4RαL/LLysMCre (IL4Rα floxed allele crossed with Cre recombinase driven by Lysozyme 2 promoter); Fig. S8 A and B], which are impaired in alternative macrophage activation (30, 31). Surprisingly, genetic disruption of IL-4/IL-13 signaling in myeloid cells did not decrease the rate of BrdU incorporation in hepatocytes after partial hepatectomy (Fig. 4A). Consistent with these findings, the hepatic regenerative response was similar in control (IL4RαL/L) and IL4RαL/LLysMCre mice after CCl4-mediated injury (Fig. 4B and Fig. S8C), implying that IL-4/IL-13 might act directly on hepatocytes to promote their proliferation.
Fig. 4.
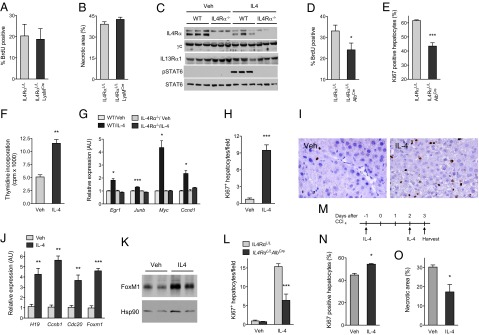
IL-4 stimulates hepatocyte proliferation in a cell-autonomous manner. (A and B) IL4Rα signaling in myeloid cells is not required for liver regeneration. (A) Liver regeneration in IL4RαL/L and IL4RαL/LLysMCre mice was assessed by BrdU incorporation at 36 h after PH (n = 3–5 mice per genotype). (B) Quantification of necrotic area in IL4RαL/L and IL4RαL/LLysMCre mice 2 d after administration of CCl4 (n = 4–5 per genotype). (C) Immunoblot analyses of IL-4/IL-13 signaling receptors in primary hepatocytes. (D and E) IL4Rα signaling in hepatocytes is required for liver regeneration after injury. (D) BrdU incorporation in IL4RαL/L and IL4RαL/LAlbCre mice was assessed 36 h after PH (n = 6–7 mice per genotype). (E) Quantification of Ki67+ hepatocytes in IL4RαL/L and IL4RαL/LAlbCre mice 2 d after administration of CCl4 (n = 5–7 mice per genotype). (F–K) Stimulation with IL-4 promotes hepatocyte proliferation in vitro and in vivo. (F) 3H-thymidine incorporation in primary hepatocytes after in vitro stimulation with IL-4. Data are representative of 2 independent experiments. (G) Quantitative RT-PCR analysis of genes associated with hepatocyte proliferation in primary hepatocytes stimulated with Veh or IL-4 for 48 h (n = 3 per genotype and treatment). (H) WT C57BL/6J mice were injected with Veh or IL-4 for 5 d, and hepatocyte proliferation was quantified by Ki67 positivity (n = 5–6 per treatment group). (I) Representative sections showing staining for Ki67 in livers of mice treated with Veh or IL-4. (J) Quantitative RT-PCR analysis of genes associated with hepatocyte proliferation and liver growth in WT mice treated with Veh and IL-4. (K) Immunoblot analysis of FoxM1 expression in livers of mice treated with Veh or IL-4. (L) Mitogenic actions of IL-4 in IL4RαL/L and IL4RαL/LAlbCre mice. Ki67+ hepatocytes were quantified in Veh or IL-4-treated mice (n = 5 mice per genotype and treatment). (M–O) Treatment with IL-4 protects mice from CCl4-induced liver injury. (M) Schematic for dosing of IL-4 to mice administered CCl4. (N) Percentage of Ki67+ hepatocytes in Veh and IL-4-treated mice 3 d after CCl4 administration (n = 4 per treatment group). (O) Quantification of necrotic area in Veh and IL-4-treated mice 3 d after CCl4-induced injury (n = 4–5 mice per treatment group). *P < 0.05, **P < 0.01, ***P < 0.001. All data are presented as mean ± SEM.
To determine whether IL-4 can directly signal in hepatocytes, we examined the patency of the IL-4 signaling pathway in purified primary hepatocytes. WT murine hepatocytes expressed IL-4Rα, IL-13Rα1, and the common γc chain, which mediated the signaling response to IL-4 (Fig. 4C) (26). On the basis of these findings, we tested whether deletion of IL-4Rα in hepatocytes, as in IL4RαL/LAlbCre (Fig. S8 D and E) might impair liver’s regenerative response to injury. Similar to IL-4/IL-13−/− mice (Fig. 2F), BrdU incorporation was reduced by ∼30% after partial hepatectomy in IL4RαL/LAlbCre (IL4Rα floxed allele crossed with Cre recombinase driven by Albumin promoter) (Fig. 4D and Fig. S9A). Congruent with this observation, hepatocyte proliferation, as quantified by Ki67 staining, was also decreased by ∼30% in IL4RαL/LAlbCre after injury with CCl4 (Fig. 4E and Fig. S9B). Taken together, these results indicate that signaling via the hepatocyte IL4Rα mediates liver regeneration after injury.
IL-4 Stimulates Liver Growth.
We next examined whether IL-4 signaling could enhance hepatocyte proliferation in the absence of injury. In primary hepatocytes, stimulation with IL-4 increased thymidine incorporation by ∼230% (Fig. 4F) and induced expression of genes associated with hepatocyte proliferation (Fig. 4G). Similarly, IL-4 administration to WT mice for 5 d enhanced hepatocyte proliferation, as demonstrated by a ∼10-fold increase in staining for the cellular proliferation marker Ki67 (Fig. 4 H and I). To probe the molecular mechanism by which IL-4 promotes hepatocyte proliferation, we analyzed the expression of genes that were down-regulated in livers of IL-4/IL-13−/− mice. Treatment with IL-4 increased expression of FoxM1 mRNA and protein about five- and between ∼1.5- and twofold, respectively (Fig. 4 J and K), and also induced markers of hepatocyte proliferation, such as the mRNAs encoded by Ccnb1, Cdc20, FoxM1, and H19 (Fig. 4J). Thus, in the absence of injury, treatment with IL-4 is sufficient to drive quiescent hepatocytes into cell cycle and promote hepatocyte DNA replication both in vitro and in vivo.
A previous study has suggested an indirect mechanism by which IL-4 stimulates hepatocyte proliferation. Specifically, IL-4 was postulated to induce the expression of the potent hepatocyte mitogen IL-6 via deposition of complement (23). To determine whether direct or indirect mechanisms contribute to IL-4-induced hepatocyte proliferation, we injected uninjured IL4RαL/L and IL4RαL/LAlbCre mice with IL-4. As expected, treatment of IL4RαL/L mice with IL-4 enhanced DNA synthesis in hepatocytes (Fig. 4L). Notably, IL-4 driven hepatocyte proliferation was decreased by ∼60% in IL4RαL/LAlbCre mice (Fig. 4L), indicating that the proliferative actions of IL-4 are, in part, mediated via its direct effects on hepatocytes. The residual increase in hepatocyte proliferation might result from the ability of IL-4 to enhance IL-6 secretion from nonparenchymal cells, such as macrophages and stellate cells (23).
Finally, we tested whether administration of IL-4 could protect WT mice from toxin (CCl4)-mediated liver injury (Fig. 4M). Although the initial extent of liver damage was similar in both groups of mice (Fig. S10A), hepatocyte proliferation, as quantified by nuclear staining for Ki67 (Fig. S10B), was increased by ∼22% in IL-4-treated animals (Fig. 4N). Furthermore, this increase in hepatocyte proliferation was associated with a ∼43% decrease in necrotic cell area (Fig. 4O and Fig. S10C), suggesting a potential therapeutic benefit of IL-4 in acute liver injury.
Discussion
The regenerative capacity of the liver in part reflects its susceptibility to damage by ingested toxins and pathogenic infections. As such, multiple homeostatic mechanisms are activated on injury to initiate liver regeneration, each contributing only partially to the restoration of liver function (1, 2). Here, we have demonstrated a role for eosinophils in orchestrating the regrowth of liver after injury. Moreover, our work has elucidated a direct mechanism by which eosinophil-derived IL-4 signals via the hepatocyte IL-4Rα to stimulate liver regeneration. In broad terms, these findings suggest that activation of type 2 immunity is integrally linked to reparative and regenerative programs in tissues, thereby exemplifying the host’s protective strategy of tolerance against tissue damage (8, 32). In support of this idea, we recently reported that eosinophil-derived IL-4 facilitates muscle regeneration (11). Last, the ability of IL-4 to induce cell cycle progression in hepatocytes and protect against toxin-mediated injury has potential clinical implications for augmenting liver growth and regeneration in patients with liver disease.
Our studies differ in a number of important ways from a recent report that demonstrated a role for IL-4 in liver regeneration (23). First, using homozygous KN2 mice that lack IL-4 (24), we failed to detect any differences in liver’s regenerative response to injury. A substantial defect in liver regeneration was observed in IL-4/IL-13 knockout mice, which suggests that functional redundancy between IL-4 and IL-13 accounts for these discrepant results. Indeed, a large number of studies indicate that these two type 2 cytokines regulate an overlapping set of biological programs, including those in host defense, allergic inflammation, and metabolism (25, 26, 31). Second, using the highly sensitive and specific 4get reporter mice, we failed to detect GFP expression in NKT cells, which previously had been implicated as a source of IL-4 in regenerating livers. A potential reason for this difference might stem from the methods used to identify IL-4 producing cells in regenerating livers. Although we used knock-in 4get and KN2 reporter mice to demonstrate eosinophils as the cellular source of IL-4, DeAngelis et al. (23) identified NKT cells as the cellular source of IL-4, using intracellular staining. Because intracellular staining protocols require restimulation of cells ex vivo and the subsequent trapping of the produced cytokines in the Golgi complex, they tend to give false-positive results and do not faithfully report the endogenous production of cytokines in vivo (22). In addition, rather than an indirect mechanism involving IL-4, complement, and IL-6, we provide genetic evidence for a direct mechanism in which activation of the IL-4Rα in hepatocytes stimulates hepatocyte proliferation.
Our current work also raises some interesting questions for future investigations. For instance, although treatment of uninjured mice with IL-4 is sufficient to drive hepatocyte proliferation, deletion of IL-4Rα in hepatocytes does not completely abrogate this response. This suggests that additional pathways are activated by IL-4 that can stimulate hepatocyte proliferation. Indeed, in various primary cell types and tissues, treatment with IL-4 can rapidly induce the expression of IL-6, a known activator of hepatocyte proliferation. Thus, in the future, it will be important to identify other IL-4Rα+ cells in liver that participate in liver’s regenerative response after injury.
Materials and Methods
Animal Studies.
Male mice aged 8–16 wk were used in the studies. Colonies of WT, ΔdblGATA, IL-4/IL-13−/−, 4get, 4get ΔdblGATA, IL4RαL/L, and IL4RαL/LLysMCre mice (all on the BALB/cJ background) and IL4RαL/L and IL4RαL/LAlbCre mice on the C57BL/6J background were maintained in the Cardiovascular Research Institute’s pathogen-free barrier facility according to institutional guidelines. Partial hepatectomy, which surgically removes 70% of the liver, was performed under Isoflurane anesthesia, as described previously (3, 17). Briefly, after a midline incision, the left lateral and median lobes were exposed, ligated, and completely excised, leaving behind the gall bladder and right and caudate lobes. For the sham-operated controls, the liver was exteriorized through a midline incision and then subsequently put back into the peritoneal cavity. Acute liver injury was performed by a single i.p. injection of CCl4 (0.4 mg/g, Sigma) suspended in olive oil. To block integrin-mediated migration of eosinophils, mice were given an i.v. injection of isotype control antibody (100 μg) or anti-αL (100 μg, M17/4, UCSF Monoclonal Antibody Core) and anti-α4 (100 μg, PS/2) 24 h before initiation of injury and every 24 h thereafter until harvest. To quantify the number of proliferating hepatocytes, animals were given an i.p. injection of BrdU (100 mg/kg, Sigma) 2 h before euthanasia. For IL-4 treatment studies, mice were injected intraperitoneally for 5 consecutive days with 2 μg IL-4 (Peprotech) complexed with 10 μg anti-IL-4 antibody (BD Biosciences, BV-1B11). To evaluate the protective effects of IL-4 on CCl4-induced liver injury, mice were administered 2 μg IL-4 complexed with 10 μg anti-IL-4 antibody 1 d before and 2 d after injection of CCl4. Hepatic regenerative response were quantified 3 d after administration of CCl4. All animal care and procedures were performed in accordance with Stanford University’s Administrative Panel on Laboratory Animal Care and the University of California San Francisco’s Institutional Animal Care and Use Committee guidelines.
Immunohistochemistry and Immunoblotting.
For immunohistochemical studies, livers were fixed in neutral-buffered formalin and methacarn and embedded in paraffin. Tissues were sectioned at 5 μm, stained with H&E or anti-BrdU (1:40; Dako M0744). To determine the percentage of BrdU+ hepatocytes (labeling index), 2,000–2,500 nuclei were counted per animal in 10 random fields. For Ki67 staining, antigens were retrieved with Dako universal antigen retrieval solution (at pH 6.0), and tissue sections were subsequently stained with Ki67 antibody (1:50; Dako M7249). Secondary biotinylated anti-rat (1:200; Dako) coupled with Streptavidin-horseradish peroxidase (HRP) was used for detection of Ki67. Immunoblotting for proteins was performed by standard methods, using the following antibodies: IL4Rα (Santa Cruz E-1), IL2Rγ (Santa Cruz H-300), IL13Rα1 (Abcam Ab79277), phosphorylated pSTAT6 (BD Biosciences), STAT6 (Santa Cruz M20), FoxM1 (1:100; Santa Cruz K-19), β-actin (Sigma AC-40), and HSP90 (Santa Cruz H114).
Flow Cytometry.
Liver was minced and digested with Dispase (2.4 U/mL; Invitrogen), dispersed, and subjected to RBC lysis. Cell suspension was then filtered through 100-μm and 40-μm cell strainers sequentially. For flow cytometric analysis, cells were stained with the following reagents and antibodies: Live/Dead stain and human CD2 (Invitrogen); Fc block, CD45, CD3, CD4, CD8, CD11b, F4/80, CD11c, CD49b, CD117, B220, and Ly6G (all from Biolegend); mouse IL4Rα and Siglec-F (BD Biosciences); and IL13R and FcεRI (eBioscience).
Gene Expression Analyses.
TRIzol reagent (Invitrogen) was used to extract total RNA from tissues, which was then treated with DNase (Invitrogen) to remove genomic DNA. Reverse transcription was performed on 1 μg of tissue RNA using the first strand cDNA synthesis kit (Origene). As described previously (33), quantitative real-time PCR analyses were carried out with cDNA templates using SsoFast EvaGreen Supermix (Biorad). All primer sequences are available on request. Relative expression of target mRNAs was calculated by the ΔΔCT method after normalization to the housekeeping gene 36B4. For microarray studies, 25 μg total RNA was hybridized to Illumina MouseRef-8 v2.0 Expression BeadChip. After normalization, statistical analyses were performed with significance analysis of microarray (SAM), with a cutoff of > twofold and P value < 0.05. Functional annotation of differentially expressed genes was performed with DAVID. Complete linkage clustering of genes and arrays was performed using CLUSTER, and visualized with TREEVIEW. The complete microarray data have been deposited with the Gene Expression Omnibus and can be accessed at GSE45002.
Hepatocyte Culture.
As described previously, hepatocytes were isolated by perfusion of liver with digestion buffer containing collagenase, followed by isodensity purification on a Percoll gradient (34). Primary hepatocytes were grown in Hepatozyme serum-free media (Invitrogen) at a density of ∼2 × 104 per cm2 and stimulated with vehicle (Veh) or 10 ng/mL of IL-4 (Peprotech) for 36 h. Twenty-four hours before harvest, cells were pulsed with 2 μCi/mL 3H-thymidine to monitor proliferation.
Statistical Analysis.
All data are presented as mean ± SEM. Statistical significance was determined using the Student t test. A P value of <0.05 was considered to be statistically significant and is presented as *P < 0.05, **P < 0.01, or ***P < 0.001.
Supplementary Material
Acknowledgments
We thank members of the A.C. laboratory and A. Loh for comments on the manuscript. This work was supported by Grants DK076760 and DK081405 and National Institutes of Health Director’s Pioneer Award DP1AR064158 (to A.C.). Y.P.G. was supported by Agency for Science, Technology and Research National Science Scholarship, Stanford Graduate Fellowship supported K.D.N., and N.C.H. was supported by a Wellcome Trust Intermediate Clinical Fellowship (ref. 085187).
Footnotes
The authors declare no conflict of interest.
†This Direct Submission article had a prearranged editor.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE45002).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304046110/-/DCSupplemental.
References
- 1.Michalopoulos GK. Liver regeneration after partial hepatectomy: critical analysis of mechanistic dilemmas. Am J Pathol. 2010;176(1):2–13. doi: 10.2353/ajpath.2010.090675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taub R. Liver regeneration: From myth to mechanism. Nat Rev Mol Cell Biol. 2004;5(10):836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 3.Higgins GM, Anderson RM. Experimental pathology of the liver. I. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol (Chic) 1931;12:186–202. [Google Scholar]
- 4.Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276(5309):60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 5.Huang W, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312(5771):233–236. doi: 10.1126/science.1121435. [DOI] [PubMed] [Google Scholar]
- 6.Stoick-Cooper CL, Moon RT, Weidinger G. Advances in signaling in vertebrate regeneration as a prelude to regenerative medicine. Genes Dev. 2007;21(11):1292–1315. doi: 10.1101/gad.1540507. [DOI] [PubMed] [Google Scholar]
- 7.Allen JE, Wynn TA. Evolution of Th2 immunity: a rapid repair response to tissue destructive pathogens. PLoS Pathog. 2011;7(5):e1002003. doi: 10.1371/journal.ppat.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palm NW, Rosenstein RK, Medzhitov R. Allergic host defences. Nature. 2012;484(7395):465–472. doi: 10.1038/nature11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg HF, Dyer KD, Foster PS. Eosinophils: Changing perspectives in health and disease. Nat Rev Immunol. 2013;13(1):9–22. doi: 10.1038/nri3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol. 2006;24:147–174. doi: 10.1146/annurev.immunol.24.021605.090720. [DOI] [PubMed] [Google Scholar]
- 11.Heredia JE, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153(2):376–388. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loke P, et al. Alternative activation is an innate response to injury that requires CD4+ T cells to be sustained during chronic infection. J Immunol. 2007;179(6):3926–3936. doi: 10.4049/jimmunol.179.6.3926. [DOI] [PubMed] [Google Scholar]
- 13.Baker BL, Axiotis C, Hurwitz ES, Leavitt R, Di Bisceglie AM. Nodular regenerative hyperplasia of the liver in idiopathic hypereosinophilic syndrome. J Clin Gastroenterol. 1991;13(4):452–456. doi: 10.1097/00004836-199108000-00018. [DOI] [PubMed] [Google Scholar]
- 14.Malyshev VV, Treshchuk LI, Popova NS. [Changes in the content of eosinophils, corticosterone and catecholamines during liver regeneration following resection] Biull Eksp Biol Med. 1978;86(12):663–664. [PubMed] [Google Scholar]
- 15.Wu D, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332(6026):243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu C, et al. Targeted deletion of a high-affinity GATA-binding site in the GATA-1 promoter leads to selective loss of the eosinophil lineage in vivo. J Exp Med. 2002;195(11):1387–1395. doi: 10.1084/jem.20020656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc. 2008;3(7):1167–1170. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- 18.Henderson NC, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci USA. 2006;103(13):5060–5065. doi: 10.1073/pnas.0511167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anthony RM, Rutitzky LI, Urban JF, Jr, Stadecker MJ, Gause WC. Protective immune mechanisms in helminth infection. Nat Rev Immunol. 2007;7(12):975–987. doi: 10.1038/nri2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen F, et al. An essential role for TH2-type responses in limiting acute tissue damage during experimental helminth infection. Nat Med. 2012;18(2):260–266. doi: 10.1038/nm.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohrs M, Shinkai K, Mohrs K, Locksley RM. Analysis of type 2 immunity in vivo with a bicistronic IL-4 reporter. Immunity. 2001;15(2):303–311. doi: 10.1016/s1074-7613(01)00186-8. [DOI] [PubMed] [Google Scholar]
- 23.DeAngelis RA, et al. A complement-IL-4 regulatory circuit controls liver regeneration. J Immunol. 2012;188(2):641–648. doi: 10.4049/jimmunol.1101925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohrs K, Wakil AE, Killeen N, Locksley RM, Mohrs M. A two-step process for cytokine production revealed by IL-4 dual-reporter mice. Immunity. 2005;23(4):419–429. doi: 10.1016/j.immuni.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finkelman FD, et al. Interleukin-4- and interleukin-13-mediated host protection against intestinal nematode parasites. Immunol Rev. 2004;201:139–155. doi: 10.1111/j.0105-2896.2004.00192.x. [DOI] [PubMed] [Google Scholar]
- 26.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 27.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Kiyokawa H, Dennewitz MB, Costa RH. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc Natl Acad Sci USA. 2002;99(26):16881–16886. doi: 10.1073/pnas.252570299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 30.Herbert DR, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20(5):623–635. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen KD, et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480(7375):104–108. doi: 10.1038/nature10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335(6071):936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Odegaard JI, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Odegaard JI, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7(6):496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.