

Basic Research Review for Clinicians: The cellular and molecular mechanisms of CTLA-4 and PD-1 function, and implications for tumor immunotherapy.
Keywords: tumor, receptor, inhibitory, coinhibitory, costimulatory, costimulation, PD-L1, ipilimumab
Abstract
Tumors can avoid immune surveillance by stimulating immune inhibitory receptors that function to turn off established immune responses. By blocking the ability of tumors to stimulate inhibitory receptors on T cells, sustained, anti-tumor immune responses can be generated in animals. Thus, therapeutic blockade of immune inhibitory checkpoints provides a potential method to boost anti-tumor immunity. The CTLA-4 and PD-1Rs represent two T cell-inhibitory receptors with independent mechanisms of action. Preclinical investigations revealed that CTLA-4 enforces an activation threshold and attenuates proliferation of tumor-specific T lymphocytes. In contrast, PD-1 functions primarily as a stop signal that limits T cell effector function within a tumor. The unique mechanisms and sites of action of CTLA-4 and PD-1 suggest that although blockade of either has the potential to promote anti-tumor immune responses, combined blockade of both might offer even more potent anti-tumor activity. See related review At the Bedside: CTLA-4 and PD-1 blocking antibodies in cancer immunotherapy.
Introduction
T lymphocytes play essential roles as orchestrators and effectors of the immune system. The power of T cells to defend against invading pathogens must be counterbalanced by mechanisms that prevent destructive immune responses targeted against self-tissues. Although thymic selection eliminates the majority of self-reactive T cells, a fraction escapes this process of central tolerance and retains the potential to inflict destructive autoimmune pathology against the self. The immune system evolved numerous strategies to restrain autoreactive T cells and maintain peripheral tolerance. In particular, a complex network of stimulatory and inhibitory receptors and ligands delivers cell-to-cell signals that dictate the outcome of T cell encounters with cognate antigen. Moreover, Tregs exert extrinsic control over autoreactive T cells, which is mediated, at least in part, by expression of many of the same stimulatory and inhibitory receptors.
The potential of T cells to attack and destroy tumors has long been recognized [1]. Moreover, recent experimental evidence has demonstrated that T cell surveillance throughout the life of an organism functions to prevent tumor development [2–4]. Nonetheless, T cells face an uphill battle in their attempts to recognize and eliminate tumors, as many of the same mechanisms that prevent autoimmunity also impair T cell responses to the “altered self” of tumors. In addition, it has become apparent that tumors are capable of selectively co-opting many of the inhibitory mechanisms that prevent sustained T cell responses to self-tissue. Selective blockade of these natural inhibitory checkpoints might provide a means of releasing the brakes on T cell activation and promoting potent anti-tumor responses. Indeed, antibodies that block the inhibitory receptors CTLA-4 and PD-1 have demonstrated an ability to improve outcomes for patients with at least some cancers [5–8].
CTLA-4 and PD-1 represent the two immune inhibitory receptors for which blocking agents have progressed furthest through clinical development as anti-cancer therapeutics. This review will highlight the distinctive biology of CTLA-4 and PD-1, including their unique molecular structures, expression patterns of receptors and ligands, and mechanisms of cell-intrinsic and cell-extrinsic activity. The distinct molecular and cellular mechanisms whereby CTLA-4 and PD-1 operate suggest that combined therapeutic manipulation of these pathways may be synergistic for cancer immunotherapy. The accompanying review by Callahan and Wolchok (At the bedside: CTLA-4 and PD-1 blocking antibodies in cancer immunotherapy)will focus on the clinical manipulation of CTLA-4 and PD-1, highlighting the future potential for combinatorial therapy in cancer patients.
CTLA-4
Discovery
Decades of research resulted in elucidation of the mechanisms, whereby T cells encounter antigen and undergo activation. It became apparent that TCR engagement is not in itself sufficient to enact clonal expansion and differentiation. Stimulation through the TCR alone results in an unresponsive, anergic state or activation-induced cell death, which are hypothesized to represent distinct mechanisms of peripheral self-tolerance [9, 10]. Indeed, theories of self/nonself-discrimination, put forward by Bretchser and Cohn [11] and Lafferty and Cunningham [12], had predicted the theoretical requirement for a second signal to drive lymphocyte clonal expansion. It was hypothesized that the first signal through the antigen receptor should provide specificity to the response, whereas an undefined, second signal should confirm that the specificity is directed against a nonself-antigen.
TCR: Each T cell expresses on its cell surface a TCR with unique specificity. Engagement of the TCR with cognate peptide presented in the context of MHC molecules provides the initial signal (“signal one”) for T cell activation.
The two-signal theory of T cell activation received molecular confirmation by the discovery of stimulatory coreceptors, of which CD28 represented the founding member [13]. Coligation of the TCR, along with CD28, promoted T cell activation, proliferation, survival, and activation of effector function (Table 1)[14, 15]. In the absence of CD28 or its B7 ligands, T cell proliferation in response to TCR ligation was severely impaired [16, 17]. Important functions of CD28 costimulation were ultimately determined to include amplification of TCR signaling, stimulation of IL-2 production via stabilization of IL-2 mRNA, induction of the antiapoptotic molecule Bcl-XL, and activation of glucose uptake via signaling through the PI3K/Akt pathway, thereby providing the nutrients necessary for cell growth and proliferation [18–21].
Table 1. Summary of CD28, CTLA-4, and PD-1 functions on T cells.
| Receptor | Expression pattern | Ligand | Expression pattern | Functional outcome |
|---|---|---|---|---|
| CD28 | Constitutive expression on T cells | B7-2 (CD86) | Constitutive expression on APCs | Promotes: |
| Enhanced by APC activation | ∙ Productive activation | |||
| B7-1 (CD80) | Activated APCs | ∙ Clonal expansion | ||
| ∙ Acquisition of effector function | ||||
| CTLA-4 | Activated T cells | B7-1 (CD80) | Activated APCs | Raises activation threshold |
| Memory T cells | Attenuates clonal expansion | |||
| B7-2 (CD86) | Constitutive expression on APCs | |||
| Tregs | ||||
| Enhanced by APC activation | ||||
| PD-1 | Activated T cells | PD-L1 (B7-H1) | Constitutive expression on APCs | Restrains effector T cell function, primarily in nonlymphoid organs |
| “Exhausted” T cells | B and T cells | |||
| Nonhematopoietic cells | ||||
| Enhanced by inflammatory cytokines | ||||
| PD-L2 (B7-DC) | Constitutive expression on APCs | Restrains effector T cell function, primarily in lymphoid organs | ||
| Enhanced by APC activation |
Top row (CD28) highlights stimulatory, and middle (CTLA-4) and bottom (PD-1) rows highlight inhibitory receptor:ligand interactions, respectively. APCs include DCs, macrophages, and mast cells.
CD28: Stimulatory coreceptor expressed on the surface of T cells. Interactions between CD28 and B7 ligands expressed on the surface of APCs provide a second signal (“signal two”) required for productive T cell activation, clonal expansion, and acquisition of effector functions.
After the discovery and characterization of CD28, research efforts focused on identifying additional stimulatory coreceptors that could contribute to the activation of T cells. CTLA-4, also known at CD152, was identified initially as a gene expressed by activated cytotoxic T cells [22]. CTLA-4 shared significant homology with CD28 and bound to the same B7 ligands [23, 24]. Initial functional studies suggested that antibodies directed against CTLA-4 could enhance T cell activation in concert with antibodies directed against CD28, prompting the conclusion that CTLA-4 functioned as a costimulatory molecule [25]. However, it was later discovered that CTLA-4 was actually being inhibited under the conditions used in these experiments.
CTLA-4: Inhibitory coreceptor expressed on the surface of activated T cells. Interactions between CTLA-4 and B7 ligands expressed on the surface of APCs counteract CD28-mediated costimulatory signals and impair the activation of T cells.
Further investigation established that CTLA-4 functioned as an inhibitor of T cell activation [16, 26, 27]. Optimal cross-linking of the CTLA-4 receptor in the presence of TCR and CD28 stimulation resulted in suppression of IL-2 production and proliferative arrest, without induction of apoptosis [28, 29]. Engagement of CTLA-4 could induce an anergic phenotype similar to that observed in response to TCR stimulation alone [30], establishing the notion that CTLA-4 might function to counteract CD28-mediated costimulation.
The phenotype of CTLA-4-deficient mice confirmed its function as a crucial inhibitor of T cell activation. Mice with germline deletion of Ctla4 developed a rapidly progressive, fatal lymphoproliferative disease, characterized by multiorgan T cell infiltration and death by 3–4 weeks of age [31, 32]. The autoimmune phenotype was not attributable to abnormal T cell development in the thymus [33, 34]; however, it was strictly dependent on antigen-specific stimulation through the TCR [34] and costimulatory signals delivered through CD28:B7 receptor:ligand interactions [35–37]. Thus, CTLA-4 emerged as a key negative regulator of T lymphocyte activation and enforcer of peripheral tolerance, appearing to operate primarily via antagonism of CD28-mediated costimulation.
Molecular structure
CTLA-4 is a type 1 transmembrane glycoprotein of the Ig superfamily, comprised of four domains, including a signal peptide, an extracellular cellular ligand-binding domain, a transmembrane domain, and a short cytoplasmic tail [22, 38–40]. CTLA-4 forms a covalently linked heterodimer that binds to oligomerized B7-1 (CD80) and B7-2 (CD86) ligands with higher affinity and avidity than CD28 [24, 41–45]. Although the cytoplasmic domain lacks any intrinsic enzymatic activity, it recruits various molecules involved in signaling and intracellular trafficking.
Multiple splice variants of CTLA-4 exist [23], including a soluble form in humans and a ligand-independent form in mice. Polymorphisms in the soluble version of CTLA-4 have been implicated in human autoimmune disorders, including Grave's disease, Hashimoto's thyroiditis, and type I diabetes [46]. Likewise, polymorphisms in the ligand-independent form of CTLA-4 may play a role in the pathogenesis of diabetes in the NOD mouse model [46, 47]. The ligand-independent isoform of CTLA-4 appears to suppress self-reactive T cells by producing tonic inhibitory signals that increase the threshold required for T cell activation [47]. The specific contributions of each of these splice isoforms to the overall biologic function of CTLA-4 remain unknown.
Clinical Questions: Do CTLA-4 splice variants impair productive anti-tumor immunes responses in humans? Does CTLA-4 blockade mediate its anti-tumor effects, in part, by counteracting the functions of CTLA-4 splice variants?
Expression pattern
Expression of CTLA-4 is primarily restricted to T cells (Table 1) [22], although expression on B cells and other cell types has been described [48]. In contrast to CD28, which is expressed on the surface of resting and activated T cells, CTLA-4 exhibits minimal expression in resting T cells (Fig. 1). CTLA-4 is induced at the mRNA and protein level in response to TCR activation [43]. Expression of CTLA-4 is enhanced by costimulation through CD28 and/or IL-2 [49]. Protein expression of CTLA-4 peaks at 24–48 h post-TCR stimulation and requires entry into the cell cycle [49, 50]. Antigen-experienced memory CD4+ and CD8+ T cells, as well as CD4+ Tregs, maintain constitutive expression of CTLA-4 [51–54].
Figure 1. Unique spatiotemporal regulation of CTLA-4 and PD-1.
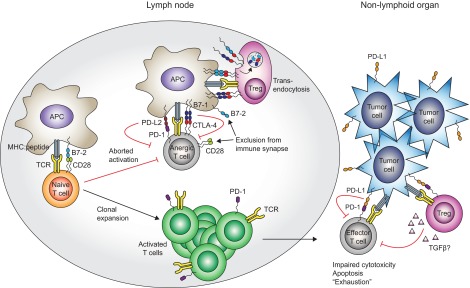
Activation of a naïve T cell requires TCR-mediated signals and costimulatory signals, generated by CD28:B7 ligand interactions. Upon activation, T cells induce expression of the inhibitory receptors CTLA-4 and PD-1, and the relative balance of stimulatory and inhibitory signaling can dictate the outcome of the T cell response. When CTLA-4- and PD-1-mediated inhibitory signals dominate, T cell activation is aborted, resulting in an unresponsive anergic state. Tregs can tip the balance toward inhibitory signals by removing B7 ligands from the APC surface via transendocytosis, thus favoring B7 ligand sequestration by the higher-affinity CTLA-4 receptor. When TCR- and CD28-mediated stimulatory signals dominate, T cells undergo clonal expansion, acquisition of effector function, and trafficking through nonlymphoid tissues. Effector T cell function can be limited by PD-1 interaction, with PD-L1 expressed on the surface of nonhematopoietic cells, including many different tumors. Moreover, PD-1:PD-L1 interactions can enhance Treg function, resulting in an additional layer of effector T cell inhibition.
Of note, although CD4+ and CD8+ T cells express CTLA-4, the inhibitory functions of CTLA-4 on CD4+ T cells appear to be relatively more important for the prevention of autoimmune pathology. CTLA-4-deficient CD8+ T cells are incapable of inducing autoimmune pathology in the absence of CTLA-4-deficient CD4+ T cells [35, 55]. This may be attributable, at least in part, to CD4+ Tregs, which constitutively express high levels of CTLA-4 and depend on CTLA-4 for their suppressive functions [53, 54, 56]. Nonetheless, CTLA-4 does exert inhibitory activities on CD8+ T cells and may be particularly important as a regulator of secondary responses by effector/memory CD8+ T cells [51, 52, 57].
Clinical Question: What are the relative contributions of effector and memory CD4+ and CD8+ T cells to the anti-tumor activity of CTLA-4 blockade in humans?
Cellular localization
One of the most remarkable features of CTLA-4 biology is its pattern of intracellular localization and trafficking (Fig. 2). The majority of CTLA-4 resides within intracellular vesicles of the trans-Golgi network and endosomal compartments [49, 58, 59]. In resting T cells, a small amount of CTLA-4 protein cycles continuously from the Golgi apparatus to the cell surface, followed by rapid endocytosis and lysosomal degradation [49]. This intracellular trafficking pattern is mediated by association of the cytoplasmic tail of CTLA-4 with the clathrin-associated adaptor proteins AP-1 and AP-2/AP50 [60–64]. In addition, the interaction of CTLA-4 with a protein—TCR-interacting molecule (TRIM)—appears to be important for its proper intracellular localization and trafficking [65].
Figure 2. Distinct mechanisms of intracellular signaling by CTLA-4 and PD-1.
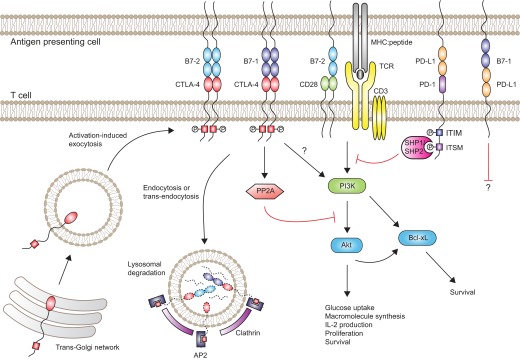
Upon activation, T cells synthesize CTLA-4, and intracellular vesicles containing CTLA-4 undergo transport to the immune synapse. Phosphorylation of CTLA-4 at the YKVM motif (red box) promotes sustained expression of CTLA-4 at the cell surface by preventing AP2- and clathrin-mediated endocytosis. At the cell surface, CTLA-4 competes with CD28 for access to B7 ligands. CTLA-4-mediated transendocytosis of B7 ligands can further limit availability of B7 ligands for CD28. Ligation of CTLA-4 by B7-1 or B7-2 blocks TCR/CD28-mediated activation of Akt, in part, through CTLA-4-mediated activation of the serine/threonine phosphatase PP2A. However, CTLA-4 ligation does not interfere with PI3K-mediated induction of the antiapoptotic molecule Bcl-xL. In contrast, ligation of PD-1 by PD-L1 or PD-L2 (not shown) results in phosphorylation of ITSM (purple box) and ITIM (blue box) motifs within the cytoplasmic tail of PD-1. The phosphorylated ITSM motif recruits the tyrosine phosphatases SHP-1 and SHP-2, which in turn, block TCR/CD28-mediated activation of PI3K. Signals delivered via B7-1:PD-L1 interactions may also inhibit T cell function, although the signaling pathways have not been elucidated.
Clinical Questions: Do CTLA-4 blocking antibodies undergo endocytosis and lysosomal degradation? If so, would inhibition of these processes enhance the anti-tumor efficacy of CTLA-4 blockade?
Upon TCR engagement, CTLA-4 expression is induced, and intracellular vesicles containing CTLA-4 undergo relocalization to the immune synapse [58]. TCR-induced kinases Lck and ZAP70 phosphorylate the cytoplasmic tail of CTLA-4 at Y165, resulting in disruption of the CTLA-4:AP-2 interaction and retention of CTLA-4 at the cell surface in the immune synapse [62, 64, 66]. Of note, the stronger the TCR signal, the more CTLA-4 accumulates at the immune synapse, thus providing a dynamic and tunable inhibitory signal [67].
Receptor:ligand interactions
One of the primary mechanisms whereby CTLA-4 inhibits T cell function is via competition with CD28 for access to B7 ligands. CD28 and CTLA-4 share the ligands B7-1 (CD80) and B7-2 (CD86), whose expression is restricted to APCs, including DCs, B cells, and macrophages (Table 1) [24, 44, 45, 68–70]. Compared with CD28, CTLA-4 has higher affinity and avidity for B7 ligands, which has been attributed to homodimer formation by CTLA-4 that allows for bivalent binding of B7 molecules, in contrast to the monovalent binding of B7 ligands by CD28 [40, 71]. The stronger ligand-binding activity of CTLA-4 allows it to exert its inhibitory functions at a much lower surface density compared with that of CD28.
B7-1 (CD80): Ligand expressed on the surface of activated APCs. Although it can interact with CD28, B7-1 appears to be the primary ligand for CTLA-4, forming multivalent interactions that deliver inhibitory signals to T cells.
The expression kinetics of B7-2 and B7-1 parallel the expression kinetics of CD28 and CTLA-4, respectively. B7-2 is constitutively expressed on resting APCs with further induction after activation, reaching a peak within 24 h. In contrast, B7-1 does not exhibit significant expression on resting APCs but is maximally induced ∼48 h after APC activation [69, 70]. Thus, the current model suggests that CD28:B7-2 receptor:ligand interactions initiate T cell activation, whereas CTLA-4:B7-1 interactions can terminate the response in the later stages of activation [71]. Support for this model came from the observations that B7-2 stabilizes CD28 in the immune synapse, whereas B7-1 recruits CTLA-4 to the immune synapse at later stages of activation, resulting in the displacement of CD28 [72].
B7-2 (CD86): Ligand expressed constitutively on the surface of resting and activated APCs. Appears to be the primary ligand responsible for delivering costimulatory signals to T cells via monovalent interactions with CD28.
Recent observations have demonstrated an interesting connection between CTLA-4:B7 receptor:ligand interactions and the unique cellular trafficking of the CTLA-4 molecule. Activated T cells expressing CTLA-4 were shown to acquire the ligands B7-1 and B7-2 from APCs by a process of transendocytosis [73]. The B7 molecules were endocytosed and degraded in the lysosomes of T cells in a CTLA-4-dependent manner (Fig. 1). As a consequence, CTLA-4-expressing T cells caused significant depletion of the B7 ligands from the surface of APCs. This provided a molecular mechanism for prior observations that the conditioning of activated APCs with CTLA-4-expressing Tregs resulted in the selective loss of B7 ligands from the surface of the APCs in a CTLA-4-dependent manner, thereby impairing the ability of the conditioned APCs to activate T cells [74, 75]. Thus, CTLA-4:B7 transendocytosis provides a mechanistic explanation of how CTLA-4 might inhibit T cell function in cis and in trans.
Intracellular signaling
In addition to competition for B7 ligands, CTLA-4 inhibits T cell activation through cell-intrinsic signaling cascades [76]. The importance of intracellular signaling via the cytoplasmic tail of CTLA-4 is demonstrated by the inability of CTLA-4 constructs lacking the cytoplasmic domain to suppress lymphoproliferative disease in CTLA-4-deficient mice [77]. On the other hand, CTLA-4 constructs incapable of binding to B7 ligands are capable of partially suppressing the autoimmune phenotype induced by CTLA-4-deficient T cells [78]. Thus, simple sequestration of B7 ligands by the extracellular domain of CTLA-4 is insufficient to explain its mechanism of inhibitory function. However, the requirement of the cytoplasmic domain of CTLA-4 may also be explained partly by its role in vesicular trafficking and transendocytosis.
The cytoplasmic tail of CTLA-4 interacts with a number of signaling molecules that inhibit proximal signaling via the TCR and CD28 (Fig. 2). TCR/CD28 signaling results in activation of numerous kinases, including Lck, Fyn, Lyn, Rlk, and Jak2, which are capable of phosphorylating Y165 and Y182 of the cytoplasmic domain [79–82]. Initial studies demonstrated that phosphorylation at Y165 creates a docking site for the protein tyrosine phosphatase SHP-2, which subsequently inhibits proximal TCR signaling via dephosphorylation of the TCRζ chain, linker for activation of T cells, and the Ras regulator p52SHC [83, 84]. CTLA-4 interferes with the formation of lipid rafts, TCR:ZAP70 microclusters, and the central supramolecular activation complex, each of which plays important roles in T cell activation [85–88]. In addition, CTLA-4 stabilizes expression of the ubiquitin ligase Cbl-b, an important negative regulator of signaling via the TCR and CD28 [89].
Several studies indicated that an important CTLA-4-interacting partner is the serine/threonine phosphatase PP2A [90, 91]. CTLA-4-mediated recruitment of PP2A results in inhibition of Akt, thereby inhibiting CD28-mediated induction of glucose uptake [19]. Importantly, CTLA-4 ligation does not affect upstream PI3K activation by CD28. In fact, the cytoplasmic tail of CTLA-4 may also interact directly with and activate PI3K [92, 93]. Unimpeded PI3K activation permits induction of the antiapoptotic factor Bcl-xL, which facilitates survival of the anergic T cell after receiving inhibitory signals via CTLA-4 [93, 94]. However, T cell blastogenesis and proliferation cannot proceed in the absence of Akt-stimulated glucose uptake [93, 94].
PP2A: Intracellular serine/threonine phosphatase whose functions include inhibition of Akt upon recruitment to the cytoplasmic tail of CTLA-4.
Additional downstream consequences of CTLA-4 ligation include inhibition of IL-2 production and induction of cell cycle arrest [26–28]. The inhibition of IL-2 production appears to be a consequence of impaired nuclear accumulation of AP-1, NF of activated T cells (NFAT), and NF-κB [95, 96]. Cell cycle arrest results from CTLA-4-mediated inhibition of CDK4, CDK6, and cyclin D3, an effect that is partially independent of IL-2 [97].
Clinical Question: Could pharmacologic inhibition of intracellular signaling molecules downstream of CTLA-4, such as PP2A, enhance and/or recapitulate the anti-tumor effects of CTLA-4 blockade in humans?
Despite biochemical evidence that CTLA-4 might inhibit proximal TCR signaling, gene expression profiling suggests that the net effect of CTLA-4 ligation is blockade of CD28-mediated signals, without altering the number of transcripts activated downstream of the TCR [98]. The biologic function of CTLA-4 as a specific inhibitor of CD28 has been corroborated with numerous genetic approaches, most notably, with the demonstration that a single point mutation in CD28 is capable of completely abrogating the autoimmune phenotype in CTLA-4-deficient mice [37].
Role on Tregs
One striking observation regarding the phenotype of CTLA-4-deficient mice was that the defects observed were not cell-autonomous. T cell-deficient mice, reconstituted with a mixture of WT and CTLA-4-deficient T cells, remain healthy and do not develop the fatal autoimmune disorder [99, 100]. Moreover, CTLA-4-deficient T cells, present in mixed chimeric mice, respond normally to viral infection [101]. These findings do not exclude the presence of cell-autonomous mechanisms of CTLA-4 function but do suggest that they can be over-ridden by cell-extrinsic mechanisms.
Numerous lines of evidence suggest that CTLA-4-expressing Tregs are responsible for the cell-extrinsic inhibitory effects on conventional T cells. As mentioned above, in distinction to conventional T cells, Tregs exhibit constitutive expression of CTLA-4 [53, 54], and CTLA-4 is a direct target of Foxp3, the lineage-specifying transcription factor for Tregs [102–104]. Antibody-mediated CTLA-4 blockade on Tregs abrogates their suppressive function in vivo [105].
Treg: Subset of CD4+ T cells defined by expression of the transcription factor Foxp3. Tregs play a critical role in preventing destructive immune responses, which may, in part, be mediated by their expression of CTLA-4.
Elegant genetic studies demonstrated that expression of CTLA-4 by Tregs is critical for the cell-extrinsic control of conventional T cell activation. Conditional deletion of Ctla4, only in Tregs, resulted in fatal systemic autoimmune disease with similar features to that seen in mice with germline deletion of Ctla4, albeit with somewhat delayed kinetics [56]. The phenotype was not a result of impaired survival of Tregs but was associated with heightened expression of CD80 and CD86 on APCs in the mice with Treg-specific deletion of Ctla4. Thus, it was concluded that CTLA-4 expression by Tregs limits availability of CD80 and CD86 on APCs, thereby inhibiting conventional T cell activation in a cell-extrinsic fashion.
Clinical Question: To what degree does antagonism of CTLA-4 function on Tregs contribute to the anti-tumor effects of CTLA-4 blocking agents in humans?
Another mechanism whereby CTLA-4 might function on Tregs is via “reverse” B7 ligand-mediated signals transmitted to APCs [106]. Tregs can induce expression of the tryptophan-catabolizing enzyme IDO in DCs in a CTLA-4 dependent manner [107]. Moreover, certain suppressive functions mediated by Tregs are dependent on intact IDO function in DCs. The investigators concluded that CTLA-4-mediated signals delivered to DCs via CTLA-4:B7 reverse signaling induce expression of IDO and result in local depletion of the essential amino acid tryptophan, thereby inhibiting conventional T cell priming and clonal expansion. The relative importance of this mechanism of CTLA-4 inhibitory function requires further clarification.
Preclinical models of cancer immunotherapy
Shortly after the inhibitory functions of CTLA-4 were discovered, antibody-mediated blockade of CTLA-4 function was found to enhance the rejection of transplanted mouse colon carcinoma and fibrosarcoma tumors and additionally, delay growth of established tumors [108]. Moreover, anti-CTLA-4 treatment resulted in immune memory, such that previously challenged mice could reject subsequently implanted tumors without additional CTLA-4 blockade. The anti-tumor effects of CTLA-4 blockade have been extended to numerous other murine tumor models, including prostate carcinoma, renal cell carcinoma, and lymphoma [109]. Importantly, the antibody used in these studies was a nonstimulatory, bivalent antibody that did not induce optimal cross-linking of CTLA-4 but rather, blocked the interaction of CTLA-4 with B7 ligands without affecting the activity of CD28.
In less immunogenic murine tumor models, such as B16 melanoma or SM1 mammary carcinoma, CTLA-4 blockade did not demonstrate efficacy as a single agent [110, 111]. However, a combination of CTLA blockade with a GM-CSF-based tumor vaccine was highly effective in controlling these tumors. Likewise, in a murine model of primary prostate cancer, a combination of CTLA-4 blockade with an irradiated GM-CSF-expressing tumor vaccine was capable of controlling disease, whereas neither component was effective on its own [112]. Numerous other therapeutic strategies were found to be effective in combination with CTLA-4 blockade, including radiation, chemotherapy, and a variety of vaccination strategies with tumor antigens. The common theme that emerged was that any method of increasing tumor antigen availability could potentially synergize with CTLA-4 blockade. Thus, CTLA-4 blockade in the setting of enhanced tumor antigen presentation likely functions to break T cell tolerance to tumor antigens and/or self-antigens expressed by tumors, thereby promoting effective anti-tumor immunity.
Clinical Question: What are the optimal strategies for enhancing tumor antigen delivery to synergize with CTLA-4 blockade as cancer immunotherapy, e.g., vaccination, cytokine delivery, radiation, cryoablation, or chemotherapy?
Significant efforts were devoted to elucidating the cellular and molecular correlates for anti-tumor activity in the mouse models of CTLA-4 blockade. In the B16 melanoma model, measurements of total Tregs or tumor-specific cytotoxic T cells in the blood did not correlate with anti-tumor activity [113]. Surprisingly, CTLA-4 blockade increased the numbers of Tregs in tumor-draining LNs. The most powerful correlate of anti-tumor activity proved to be an increased ratio of effector T cells to Tregs within the tumor [113]. These findings raised the question of whether the anti-tumor activity was primarily a result of CTLA-4 blockade on conventional T cells or Tregs. An elegant reconstitution system demonstrated that isolated blockade of CTLA-4 on Tregs conferred no anti-tumor activity toward established B16 melanoma, whereas isolated blockade of CTLA-4 on conventional T cells conferred modest anti-tumor activity [114]. Importantly, however, combined blockade of CTLA-4 on conventional T cells and Tregs provided robust and synergistic anti-tumor activity.
These preclinical mouse models also provided the first glimpses of immune-related adverse events that might result from CTLA-4 blockade as an anti-cancer therapeutic strategy. In contrast to CTLA-4-deficient mice, transient CTLA-4 blockade did not result in widespread systemic autoimmunity. Instead, self-directed immune pathology was observed primarily in the setting of vaccination strategies using irradiated tumor cells combined with CTLA-4 blockade, specifically in the tissues from which the tumors were derived. In the B16 melanoma model, mice from the experimental group developed vitiligo [110], whereas in the transgenic adenocarcinoma of the mouse prostate cancer model, the experimental treatment resulted in prostatitis, even in mice that did not have tumors [112].
Summary of CTLA-4 function
Several models, which help to clarify the potential mechanisms of anti-tumor activity mediated by CTLA-4 blockade, have been proposed to explain the inhibitory function of CTLA-4 [109, 115]. The threshold model suggests that CTLA-4 enforces an activation threshold that limits productive TCR engagement by low-affinity, self-reactive T cells. This could be achieved by cell-extrinsic and cell-intrinsic mechanisms. Competition between CD28 and CTLA-4 for B7 ligands prevents activation of T cells by weak TCR signals in situations where B7 ligands are limiting. Inhibition of access to B7 ligands might occur in cis on the T cell engaging the APC or in trans by Treg-mediated depletion of B7 ligands from the APC surface. Likewise, intracellular recruitment of phosphatases sets an intracellular signaling threshold that must be overcome for productive TCR/CD28 engagement.
The attenuation model suggests that in situations where B7 ligands are not limiting, CTLA-4 functions to attenuate T cell proliferation and clonal expansion after activation. This model fits well with the spatial and temporal pattern of CTLA-4:B7-1 interactions that occur relatively late in T cell activation. CTLA-4-mediated attenuation of T cell clonal expansion might function to maintain a broad repertoire of TCR specificities responding to a given antigenic challenge. In the absence of CTLA-4, T cells with high-affinity TCR specificities would selectively dominate the immune response. As CTLA-4 is induced in direct proportion to the strength of TCR signals, the degree of attenuation may be tailored to the affinity of the TCR for antigen. As a corollary, domination of CTLA-4-mediated inhibitory signals over TCR/CD28-mediated stimulatory signals might abort proliferation prematurely, leading to T cell anergy.
In the setting of tumor immunotherapy, CTLA-4 blockade might lower the threshold of activation for low-affinity, tumor-specific T cells, allowing effective priming and clonal expansion. The additional blockade of CTLA-4-mediated attenuation might then allow more robust proliferation and clonal expansion of tumor-specific T cells. The additional effects of CTLA-4 blockade on Treg function could further enhance tumor-specific T cell effector activity within the tumor itself.
PD-1
Discovery
PD-1 was identified originally as a gene induced by a T cell hybridoma undergoing apoptotic cell death [116]. The phenotype of PD-1-deficient mice provided insights into its function as an inhibitor of T cell activation. In contrast to the rapid-onset systemic autoimmunity observed in CTLA-4-deficient mice, PD-1 deficiency results in delayed-onset, organ-specific autoimmunity with incomplete penetrance that varies depending on the genetic background of the mouse. After 6 months of age, PD-1-deficient C57BL/6 mice begin to develop a lupus-like syndrome, characterized by glomerulonephritis and arthritis with Ig and complement deposition in the affected tissues [117]. PD-1-deficient BALB/c develop an autoimmune-dilated cardiomyopathy, mediated by anti-troponin I antibodies [118, 119]. Moreover, PD-1 deficiency accelerates tissue-specific autoimmune disease in a variety of contexts, including the lpr model of lupus and NOD model of diabetes [117, 120]. Thus, genetic evidence suggested that PD-1 might function as an inhibitor of lymphocyte responses at peripheral tissues.
PD-1: Inhibitory receptor expressed on the surface of activated T cells. Interactions between PD-1 and its ligands, PD-L1 or PD-L2, restrain T cell function in nonlymphoid organs and lymphoid organs, respectively.
Further insight into the role of PD-1 as a guardian against immune-mediated tissue destruction came from studies of PD-1 function in models of chronic infectious diseases. Considerable interest was generated by the observation that PD-1 is highly expressed by exhausted, dysfunctional T cells in the context of chronic infections [121, 122]. Antibody-mediated blockade of PD-L1, the major PD-L1, results in a restoration of T cell function and enhances control of viral replication. Importantly, however, chronic viral infection in PD-L1-deficient mice results in fatal autoimmune pathology. Thus, the PD-1 pathway appears to protect the host from immune-mediated tissue destruction in the setting of chronic antigen stimulation.
Molecular structure
Like CTLA-4, PD-1 is a type 1 transmembrane protein of the Ig superfamily [116]. The protein structure of PD-1 includes an extracellular N-terminal IgV-like domain, a transmembrane domain, and a cytoplasmic tail [123]. In distinction to CTLA-4, the cytoplasmic tail of PD-1, contains an ITIM and an ITSM, which are involved in the transmission of inhibitory signals [124–126]. Unlike CTLA-4, PD-1 is monomeric on the cell surface, as it lacks the extracellular cysteine residue required for covalent dimerization [123]. Similar to CTLA-4, numerous splice variants of PD-1 have been identified in activated human T cells [127]. These splice variants have not been thoroughly studied, although a splice variant lacking the transmembrane domain has been proposed to represent a soluble factor involved in the pathogenesis of rheumatoid arthritis [128].
Expression pattern
PD-1 exhibits minimal expression on resting cells of the immune system; however, upon activation, PD-1 expression is broadly induced on T cells, B cells, NK cells, NKT cells, DCs, and macrophages [129] (Table 1 and Fig. 1). Induction of PD-1 in peripheral T cells occurs downstream of TCR signaling [130]. As mentioned above, PD-1 is highly expressed by dysfunctional T cells in the setting of chronic infections but is not expressed on resting memory T cells that arise after an acute infection [121, 122].
Ligand distribution
PD-1 binds two distinct ligands, known as PD-L1 (B7-H1) and PD-L2 (B7-DC), which are members of the B7 family of proteins (Table 1 and Fig. 1) [129]. PD-L1 is expressed constitutively on hematopoietic cells, including DCs, macrophages, mast cells, B cells, and T cells [131]. In contrast to B7 ligands, PD-L1 is also expressed on a wide range of nonhematopoietic cells, including endothelial cells and numerous types of epithelial cells [129, 132]. Inflammatory signals, including type I and type II IFNs and TNF-α, result in further induction of PD-L1 on hematopoietic and nonhematopoietic cells [132, 133]. Thus, the broad distribution of PD-L1 expression supports the importance of PD-1:PD-L1 interactions in regulating effector T cell responses in peripheral tissues, particularly at sites of inflammation.
PD-L1 (B7-H1): PD-L1 broadly expressed by hematopoietic and nonhematopoietic cells. Numerous tumors express PD-L1, thereby inhibiting effective anti-tumor immune responses by PD-1-expressing T cells.
In addition to its function as a ligand for PD-1, PD-L1 is reported to bind to B7-1 [134]. Experimental evidence suggests that B7-1:PD-L1 interactions inhibit T cell function independently of PD-1. In the NOD mouse model of diabetes, selective blockade of B7-1:PD-L1 interactions accelerates autoimmune destruction of the pancreas in already established diabetes but fails to precipitate disease in young mice [135]. Thus, the B7-1:PD-L1 interaction appears to function primarily in restraining the activity of effector T cells at sites of ongoing autoimmune pathology. This additional layer of complexity in the coinhibitory receptor/ligand system predicts possible differences in the therapeutic activities of antibodies that target PD-L1 compared with antibodies that target PD-1.
Clinical Question: PD-L1 interacts with B7-1 and PD-1. Does this predict distinct clinical responses to PD-1 versus PD-L1 blockade as cancer immunotherapy?
In contrast to PD-L1, PD-L2 exhibits a much more restricted expression pattern, with expression restricted to cells of the immune system, including DCs, macrophages, and mast cells [136, 137]. Like PD-L1, PD-L2 expression is enhanced by inflammatory signals, including IFN-γ, GM-CSF, and IL-4 [137]. The relative contributions of PD-L1 and PD-L2 to the inhibitory functions of PD-1 have been investigated through a variety of genetic approaches. Studies of PD-L1-deficient mice demonstrated that PD-L1 expression on hematopoietic cells inhibits T cell cytokine production in lymphoid tissues, whereas PD-L1 expression on nonhematopoietic cells plays a unique role in limiting pathologic immune responses in peripheral tissues [138]. The specific functions of PD-L2 are less clear, as PD-L2-deficient mice have variably been reported to have enhanced [138] or impaired T cell responses [139].
PD-L2 (B7-DC): PD-L1 with expression restricted to APCs and certain hematopoietic malignancies. PD-1:PD-L2 interactions appear to restrain effector T cell function within lymphoid organs.
Intracellular signaling
PD-1-mediated signals inhibit T lymphocyte glucose consumption, cytokine production, proliferation, and survival [129] (Fig. 2). Upon TCR stimulation, PD-1 undergoes phosphorylation of the tyrosine residues in the ITIM and ITSM motifs of the cytoplasmic tail, allowing recruitment of the phosphatases SHP-1 and SHP-2, which in turn, dephosphorylate proximal signaling molecules downstream of the TCR and CD28 [94, 132, 137]. Positional mutagenesis studies demonstrated that the ITSM motif is necessary for the inhibitory function of PD-1 [125, 126]. Moreover, PD-1 ligation and recruitment to the immune synapse appear to be necessary to mediate the inhibitory effects on proximal TCR signaling [126, 140].
CTLA-4 and PD-1 inhibit T cell activation of Akt and thereby, block CD28-mediated induction of glucose uptake; however, the level of the blockade is distinct [94]. PD-1-mediated recruitment of SHP-2 blocks proximal activation of PI3K, which prevents activation of Akt. In contrast, CTLA-4 ligation does not affect PI3K activation but instead, blocks Akt induction downstream of PI3K, in part, through the activity of PP2A. PD-1-mediated inhibition of PI3K prevents induction of Bcl-xL, thus potentially explaining the ability of PD-1 ligation to lead to apoptosis in some settings [94, 141]. In addition, suppression of IL-2 production may sensitize PD-1-expressing lymphocytes to cell death. PD-1-mediated inhibition of PKCθ and ERK pathway activation appear to be particularly important for suppression of IL-2 production [142, 143]. Of note, exogenous IL-2 can over-ride PD-1 inhibitory signals [144, 145].
SHP-2: Intracellular tyrosine phosphatase that is recruited to the cytoplasmic tail of PD-1, inhibiting T cell function by preventing activation of PI3K.
Thus, partially overlapping but distinct intracellular signaling pathways are activated by ligation of CTLA-4 and PD-1. The more proximal blockade of TCR/CD28 by PD-1 is consistent with the more restrictive gene expression profile exhibited by activated T cells following ligation of PD-1 compared with ligation of CTLA-4 [94, 98]. The distinct mechanisms of inhibitory signaling by CTLA-4 and PD-1 suggest that combined inhibition of these pathways may result in therapeutic synergy.
Role on Tregs
In addition to its well-established role as a cell-intrinsic inhibitor of conventional T cell activation and function, PD-1 has been demonstrated to regulate the development, maintenance, and function of Tregs. Specifically, it was shown that PD-L1 expression by APCs plays a critical role in inducing differentiation of Tregs and maintaining their suppressive function [146]. Given the prominent expression of PD-L1 by tumors and the established role of Tregs as inhibitors of tumor-specific immune responses, PD-1:PD-L1-mediated generation of Tregs within the tumor microenvironment may represent an additional mechanism of immune evasion (Fig. 1). Thus, therapeutic blockade of PD-1 might mediate its effects via simultaneous disinhibition of effector T cells and inhibition of Treg development and function.
Clinical Question: How does antagonism of PD-1 function on Tregs contribute to the anti-tumor activities of PD-1 blocking agents in humans?
PD-1 blockade for cancer immunotherapy
The expression of PD-1 by tumor-infiltrating lymphocytes [147, 148] along with the expression of PD-L1 by numerous tumor types [149] suggest that the pathway might be involved in immune evasion by human cancers (Fig. 1). In particular, the broad expression pattern of PD-L1 on normal, nonhematopoietic cells is mirrored by PD-L1 expression on carcinomas of the lung, breast, colon, kidney, bladder, ovary, and cervix, as well as nonepithelial tumors, including melanoma, glioblastoma, multiple myeloma, T cell lymphoma, and various leukemias [150–155]. In addition, PD-L1 expression by myeloid cells within certain tumors correlates with impaired anti-tumor immunity [156, 157]. The prognostic significance of PD-L1 expression by tumors has been variable, correlating with inferior outcomes in some cases [151, 158–160] and superior outcomes in others [161]. In contrast to PD-L1, which exhibits aberrant expression in many tumors of epithelial origin, PD-L2 is highly expressed by a number of lymphoid malignancies [162], likely as a result of amplification of the chromosomal region encoding PD-L2 [163].
Clinical Question: Does PD-L1 or PD-L2 expression on tumor cells predict efficacy of agents targeting the PD-1 pathway in cancer patients? This remains controversial.
Various signaling mechanisms induce PD-L1 expression on malignant cells. Human glioblastoma specimens demonstrate increased expression of PD-L1 that is dependent on loss of phosphatase and tensin homolog and activation of the PI3K pathway [164]. Likewise, in ALK-positive T cell lymphoma, an ALK/STAT3 signaling pathway results in induction of high-level PD-L1 expression [165]. Expression of PD-L1 by multiple myeloma cells is induced by signaling via MEK/ERK/STAT1 and MYD88/TRAF6 pathways [153]. In numerous carcinomas, PD-L1 expression is induced and sustained by signaling through the IFN-γ/JAK2/IFN regulatory factor 1 pathway [150, 166]. Recent evidence demonstrates that tumor-infiltrating lymphocytes can directly induce PD-L1 expression on tumor cells via production of IFN-γ [161]. This feedback-inhibitory loop is analogous to that observed in chronic viral infection, whereby inflammatory cytokines produced by infiltrating T cells may induce tissue expression of PD-L1 to inhibit T cell function and limit tissue damage [121]. Thus, PD-L1 expression by tumors can result from activation of common oncogenic pathways or exposure to inflammatory cytokine produced by infiltrating immune cells.
Experimental manipulation of the PD-1:PD-L1 pathway confirms its importance in immune evasion by tumors. Overexpression of PD-L1 on a mouse mastocytoma tumor inhibits tumor-directed T cell cytotoxicity in vitro and promotes immune evasion leading to enhanced growth of the tumor in vivo [150, 154]. These effects are abrogated by antibody-mediated blockade of PD-L1, which promotes immune-mediated tumor rejection. Likewise, blockade of PD-L1 promotes immune-mediated destruction of tumors that naturally express PD-L1, including myeloma, melanoma, and mammary carcinoma [150, 167, 168]. In some settings, PD-L1 expression by the tumor promotes apoptosis of tumor-specific T cells [150], whereas in other systems, tumor expression of PD-L1 inhibits tumor cell lysis by cytotoxic T cells in the absence of apoptosis or anergy [167].
Of note, the effects of PD-1 blockade as an anti-cancer therapeutic may be mediated partially by immune cells other than T cells, including NK cells or B cells. Tumor-mediated production of IL-18 enhances PD-1 expression and inhibits the function of NK cells, another type of immune cell important for anti-tumor immunity [169]. Early studies demonstrated that BCR signaling induces PD-1 expression, which negatively regulates B cell proliferation and function [125, 170]. In fact, the lupus-like disease and dilated cardiomyopathy observed in PD-1-deficient mice may be, at least in part, a result of aberrant B cell function, given the predominant, antibody-mediated pathology [117, 118]. Blockade of PD-1 in chronic SIV infection enhanced proliferation of memory B cells and boosted anti-SIV antibody titers [171]. Thus, restoration of NK cell and B cell function might also play a role in the anti-tumor activity of PD-1 blockade. Although, the effects of PD-1 blockade on immune cells other than T cells are less well-studied, they may be relevant to manipulating the PD-1 pathway therapeutically.
Clinical Question: What are the relative contributions of T cells, B cells, NK cells, NKT cells, DCs, and macrophages to the anti-cancer activity of PD-1 blockade, as PD-1 is expressed on all of these types of immune cells?
Importantly, there were no significant adverse, immune-related events reported in preclinical models of PD-1 or PD-L1 blockade. The lack of immune-mediated pathology is consistent with the lack of systemic autoimmunity observed in PD-1-deficient mice compared with CTLA-4-deficient mice. Nonetheless, the preclinical models predict that PD-1 or PD-L1 blockade could exacerbate pre-existing autoimmune disease or lead to organ damage in the setting of chronic infections.
Summary of PD-1 function
PD-1 can be conceived of as a dampener of effector T lymphocyte function in nonlymphoid organs. Genetic evidence indicates that in a healthy host, PD-1 does not play a critical role in regulating immune homeostasis or preventing widespread autoimmune pathology. However, there may be specific organs where PD-1 exerts inhibitory effects on self-reactive lymphocytes, as evidenced by the strain-specific autoimmunity observed in PD-1-deficient mice. In contrast to the healthy host, PD-1 plays a vital role in limiting immune-mediated tissue destruction at sites of ongoing infection or inflammation. The inflammatory environment induces expression of PD-L1 on involved tissues, which provides feedback inhibition via the PD-1 receptor and reduced responsiveness to antigen stimulation by lymphocytes at the site of inflammation. PD-L1 expression by inflamed tissues also promotes the differentiation of Tregs and maintenance of their suppressive function, thus providing an additional mechanism of cell-extrinsic inhibition of effector T cell function.
Tumors appear to use the PD-1 axis in a similar fashion to inflamed tissues, thereby shielding themselves from immune-mediated destruction. Tumor cells can activate expression of PD-L1 via common oncogenic signaling pathways. Alternatively, tumor-infiltrating lymphocytes can thwart their own efforts by producing inflammatory cytokines that induce PD-L1 on tumor cells and myeloid cells within the tumor. PD-1:PD-L1 interactions can transmit cell-intrinsic inhibitory signals that block tumor-directed cytolysis and promote T cell apoptosis. Moreover, PD-L1 expression by tumor cells promotes the development of suppressive function of Tregs, which offer an additional layer of protection to the tumor. Therapeutic blockade of PD-1 or PD-L1 might break these multiple layers of immune inhibition, allowing effective anti-tumor T cell responses that could additionally be assisted by enhanced B cell or NK cell function.
Clinical Question: Will combined blockade of CTLA-4 and PD-1 yield synergistic anti-tumor immunity, or will this strategy be limited by synergistic toxicity?
CONCLUDING REMARKS
The mechanisms whereby CTLA-4 and PD-1 exert their inhibitory effects on T cell activation are multifaceted, including cell-intrinsic and cell-extrinsic mechanisms. CTLA-4 functions primarily to limit T cell activation and clonal expansion, whereas PD-1 functions primarily to limit effector T cell function in the peripheral tissues. Their distinct molecular structures, spatiotemporal regulation, intracellular signaling pathways, ligand distribution, and function on Tregs and other immune cells suggest that combined therapeutic blockade of CTLA-4 and PD-1 could synergize to mediate robust anti-tumor immunity. On the other hand, combined therapeutic blockade of CTLA-4 and PD-1 might also result in synergistic immune-mediated toxicity.
Therapeutic synergy could potentially result from blockade of CTLA-4 and PD-1 on the same cell or different cells. CTLA-4 blockade might lower the threshold of activation, thereby allowing recruitment and activation of a low-affinity, tumor-specific T cell. The tumor-specific T cell would undergo enhanced clonal expansion as a result of blockade of CTLA-4-mediated attenuation of the proliferative response. Upon activation and clonal expansion, the tumor-specific T cell and its progeny would induce expression of PD-1. The tumor-specific T cell would then migrate to the site of the tumor, where PD-L1 might be highly expressed. Blockade of PD-1 interactions would allow the tumor-specific T cell to unleash its full armamentarium of effector function on the tumor cells. Alternatively, PD-1 blockade might function predominantly to restore the effector functions of pre-existing tumor-infiltrating lymphocytes within the tumor microenvironment that have been rendered impotent by PD-L1 expression on the tumor cells. In addition, CTLA-4 and PD-1 blockade would be expected to synergize in removing the cell-extrinsic, inhibitory effects of Tregs within the tumor microenvironment.
Initial preclinical models of combined CTLA-4 and PD-1 blockade have demonstrated promising results [172–174]. In the B16 model of melanoma, a Fms-like tyrosine kinase 3-based tumor cell vaccine was combined with blocking antibodies directed against CTLA-4, PD-1, and PD-L1 [172]. CTLA-4 blockade alone resulted in rejection of 10% of tumors, whereas PD-1 blockade alone resulted in rejection of 25% of tumors. However, mice treated with a combination of CTLA-4 plus PD-1 blockade rejected 50% of tumors, with a further increase to 65% tumor rejection when CTLA-4, PD-1, and PD-L1 were targeted simultaneously. Combined blockade induced highly favorable effector T cell:Treg ratios within the tumors and resulted in a significantly reduced proportion of suppressive myeloid cells. Interestingly, the tumor-infiltrating T lymphocytes, present in the setting of combined CTLA-4/ PD-1/PD-L1 blockade, exhibited significant coexpression of CTLA-4 and PD-1, thus implying that combined blockade of these pathways was unleashing the activity of tumor-specific T cells that would have otherwise been suppressed. Moreover, this observation suggests that therapeutic synergy results from combined blockade of CTLA-4 and PD-1 on the same tumor-specific T cell. A significant increase in toxicity from combined blockade was not reported.
It will be exciting to follow up these promising preclinical observations with combined blockade of CTLA-4 and PD-1/PD-L1 in human subjects, particularly in the setting of advanced melanoma. However, the safety and feasibility of combined blockade will need to be investigated rigorously, given the poor correlation between adverse immune-related events in preclinical models and those observed in patients. In addition to CTLA-4 and PD-1, numerous other immune inhibitory receptors and ligands are being investigated as potential targets for cancer immunotherapy, including B and T lymphocyte attenuator, lymphocyte activation gene 3, and T cell membrane protein 3 [175]. The potential for combined blockade of immune checkpoints offers new hope for cancer patients. A sound understanding of the cellular and molecular mechanisms whereby these immune modulators function will be essential to guide their optimal use in the clinic (Table 2).
Table 2. Summary of questions for future clinical-translational research.
|
ACKNOWLEDGMENTS
The Thompson laboratory is supported by grants from the U.S. National Institutes of Health and National Cancer Institute.
The authors thank Jedd Wolchok, Maggie Callahan, and members of the Thompson laboratory for helpful discussions.
SEE CORRESPONDING ARTICLE ON PAGE 41
- ALK
- anaplastic lymphoma kinase
- CTLA-4
- cytotoxic T lymphocyte activation antigen 4
- Foxp3
- forkhead box p3
- ITSM
- immunoreceptor tyrosine-based switch motif
- PD-1
- programmed death 1
- PD-L1/2
- programmed death ligand 1/2
- PP2A
- protein phosphatase 2A
- SHP-1/2
- Src homology 2-containing tyrosine phosphatase 1/2
- Treg
- regulatory T cell
- Y165/182
- tyrosine 165/182
AUTHORSHIP
A.M.I. and C.B.T. wrote the review.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Rosenberg S. A., Spiess P., Lafreniere R. (1986) A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 233, 1318–1321 [DOI] [PubMed] [Google Scholar]
- 2. Matsushita H., Vesely M. D., Koboldt D. C., Rickert C. G., Uppaluri R., Magrini V. J., Arthur C. D., White J. M., Chen Y. S., Shea L. K.., et al. (2012) Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shankaran V., Ikeda H., Bruce A. T., White J. M., Swanson P. E., Old L. J., Schreiber R. D. (2001) IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410, 1107–1111 [DOI] [PubMed] [Google Scholar]
- 4. Koebel C. M., Vermi W., Swann J. B., Zerafa N., Rodig S. J., Old L. J., Smyth M. J., Schreiber R. D. (2007) Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907 [DOI] [PubMed] [Google Scholar]
- 5. Topalian S. L., Hodi F. S., Brahmer J. R., Gettinger S. N., Smith D. C., McDermott D. F., Powderly J. D., Carvajal R. D., Sosman J. A., Atkins M. B.., et al. (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brahmer J. R., Tykodi S. S., Chow L. Q., Hwu W. J., Topalian S. L., Hwu P., Drake C. G., Camacho L. H., Kauh J., Odunsi K.., et al. (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hodi F. S., O'Day S. J., McDermott D. F., Weber R. W., Sosman J. A., Haanen J. B., Gonzalez R., Robert C., Schadendorf D., Hassel J. C.., et al. (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Robert C., Thomas L., Bondarenko I., O'Day S., Weber J., Garbe C., Lebbe C., Baurain J. F., Testori A., Grob J. J.., et al. (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, 2517–2526 [DOI] [PubMed] [Google Scholar]
- 9. Schwartz R. H. (1990) A cell culture model for T lymphocyte clonal anergy. Science 248, 1349–1356 [DOI] [PubMed] [Google Scholar]
- 10. Webb S., Morris C., Sprent J. (1990) Extrathymic tolerance of mature T-cells—clonal elimination as a consequence of immunity. Cell 63, 1249–1256 [DOI] [PubMed] [Google Scholar]
- 11. Bretscher P., Cohn M. (1970) A theory of self-nonself discrimination. Science 169, 1042–1049 [DOI] [PubMed] [Google Scholar]
- 12. Lafferty K. J., Cunningham A. J. (1975) A new analysis of allogeneic interactions. Aust. J. Exp. Biol. Med. Sci. 53, 27–42 [DOI] [PubMed] [Google Scholar]
- 13. Hara T., Fu S. M., Hansen J. A. (1985) Human T cell activation. II. A new activation pathway used by a major T cell population via a disulfide-bonded dimer of a 44 kilodalton polypeptide (9.3 antigen). J. Exp. Med. 161, 1513–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. June C. H., Ledbetter J. A., Gillespie M. M., Lindsten T., Thompson C. B. (1987) T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol. Cell. Biol. 7, 4472–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thompson C. B., Lindsten T., Ledbetter J. A., Kunkel S. L., Young H. A., Emerson S. G., Leiden J. M., June C. H. (1989) CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc. Natl. Acad. Sci. USA 86, 1333–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Green J. M., Noel P. J., Sperling A. I., Walunas T. L., Gray G. S., Bluestone J. A., Thompson C. B. (1994) Absence of B7-dependent responses in CD28-deficient mice. Immunity 1, 501–508 [DOI] [PubMed] [Google Scholar]
- 17. Shahinian A., Pfeffer K., Lee K. P., Kundig T. M., Kishihara K., Wakeham A., Kawai K., Ohashi P. S., Thompson C. B., Mak T. W. (1993) Differential T cell costimulatory requirements in CD28-deficient mice. Science 261, 609–612 [DOI] [PubMed] [Google Scholar]
- 18. Boise L. H., Minn A. J., Noel P. J., June C. H., Accavitti M. A., Lindsten T., Thompson C. B. (1995) CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 3, 87–98 [DOI] [PubMed] [Google Scholar]
- 19. Frauwirth K. A., Riley J. L., Harris M. H., Parry R. V., Rathmell J. C., Plas D. R., Elstrom R. L., June C. H., Thompson C. B. (2002) The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777 [DOI] [PubMed] [Google Scholar]
- 20. Viola A., Lanzavecchia A. (1996) T cell activation determined by T cell receptor number and tunable thresholds. Science 273, 104–106 [DOI] [PubMed] [Google Scholar]
- 21. Lindsten T., June C. H., Ledbetter J. A., Stella G., Thompson C. B. (1989) Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science 244, 339–343 [DOI] [PubMed] [Google Scholar]
- 22. Brunet J. F., Denizot F., Luciani M. F., Roux-Dosseto M., Suzan M., Mattei M. G., Golstein P. (1987) A new member of the immunoglobulin superfamily—CTLA-4. Nature 328, 267–270 [DOI] [PubMed] [Google Scholar]
- 23. Harper K., Balzano C., Rouvier E., Mattei M. G., Luciani M. F., Golstein P. (1991) CTLA-4 and CD28 activated lymphocyte molecules are closely related in both mouse and human as to sequence, message expression, gene structure, and chromosomal location. J. Immunol. 147, 1037–1044 [PubMed] [Google Scholar]
- 24. Linsley P. S., Brady W., Urnes M., Grosmaire L. S., Damle N. K., Ledbetter J. A. (1991) CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 174, 561–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Linsley P. S., Greene J. L., Tan P., Bradshaw J., Ledbetter J. A., Anasetti C., Damle N. K. (1992) Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J. Exp. Med. 176, 1595–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walunas T. L., Lenschow D. J., Bakker C. Y., Linsley P. S., Freeman G. J., Green J. M., Thompson C. B., Bluestone J. A. (1994) CTLA-4 can function as a negative regulator of T cell activation. Immunity 1, 405–413 [DOI] [PubMed] [Google Scholar]
- 27. Krummel M. F., Allison J. P. (1995) CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 182, 459–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krummel M. F., Allison J. P. (1996) CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 183, 2533–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Walunas T. L., Bakker C. Y., Bluestone J. A. (1996) CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 183, 2541–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Greenwald R. J., Boussiotis V. A., Lorsbach R. B., Abbas A. K., Sharpe A. H. (2001) CTLA-4 regulates induction of anergy in vivo. Immunity 14, 145–155 [DOI] [PubMed] [Google Scholar]
- 31. Waterhouse P., Penninger J. M., Timms E., Wakeham A., Shahinian A., Lee K. P., Thompson C. B., Griesser H., Mak T. W. (1995) Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988 [DOI] [PubMed] [Google Scholar]
- 32. Tivol E. A., Borriello F., Schweitzer A. N., Lynch W. P., Bluestone J. A., Sharpe A. H. (1995) Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547 [DOI] [PubMed] [Google Scholar]
- 33. Chambers C. A., Cado D., Truong T., Allison J. P. (1997) Thymocyte development is normal in CTLA-4-deficient mice. Proc. Natl. Acad. Sci. USA 94, 9296–9301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Waterhouse P., Bachmann M. F., Penninger J. M., Ohashi P. S., Mak T. W. (1997) Normal thymic selection, normal viability and decreased lymphoproliferation in T cell receptor-transgenic CTLA-4-deficient mice. Eur. J. Immunol. 27, 1887–1892 [DOI] [PubMed] [Google Scholar]
- 35. Chambers C. A., Sullivan T. J., Allison J. P. (1997) Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity 7, 885–895 [DOI] [PubMed] [Google Scholar]
- 36. Tivol E. A., Boyd S. D., McKeon S., Borriello F., Nickerson P., Strom T. B., Sharpe A. H. (1997) CTLA4Ig prevents lymphoproliferation and fatal multiorgan tissue destruction in CTLA-4-deficient mice. J. Immunol. 158, 5091–5094 [PubMed] [Google Scholar]
- 37. Tai X., Van Laethem F., Sharpe A. H., Singer A. (2007) Induction of autoimmune disease in CTLA-4−/− mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc. Natl. Acad. Sci. USA 104, 13756–13761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ostrov D. A., Shi W., Schwartz J. C., Almo S. C., Nathenson S. G. (2000) Structure of murine CTLA-4 and its role in modulating T cell responsiveness. Science 290, 816–819 [DOI] [PubMed] [Google Scholar]
- 39. Schwartz J. C., Zhang X., Fedorov A. A., Nathenson S. G., Almo S. C. (2001) Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 410, 604–608 [DOI] [PubMed] [Google Scholar]
- 40. Stamper C. C., Zhang Y., Tobin J. F., Erbe D. V., Ikemizu S., Davis S. J., Stahl M. L., Seehra J., Somers W. S., Mosyak L. (2001) Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature 410, 608–611 [DOI] [PubMed] [Google Scholar]
- 41. Linsley P. S., Nadler S. G., Bajorath J., Peach R., Leung H. T., Rogers J., Bradshaw J., Stebbins M., Leytze G., Brady W.., et al. (1995) Binding stoichiometry of the cytotoxic T lymphocyte-associated molecule-4 (CTLA-4). A disulfide-linked homodimer binds two CD86 molecules. J. Biol. Chem. 270, 15417–15424 [DOI] [PubMed] [Google Scholar]
- 42. Greene J. L., Leytze G. M., Emswiler J., Peach R., Bajorath J., Cosand W., Linsley P. S. (1996) Covalent dimerization of CD28/CTLA-4 and oligomerization of CD80/CD86 regulate T cell costimulatory interactions. J. Biol. Chem. 271, 26762–26771 [DOI] [PubMed] [Google Scholar]
- 43. Lindsten T., Lee K. P., Harris E. S., Petryniak B., Craighead N., Reynolds P. J., Lombard D. B., Freeman G. J., Nadler L. M., Gray G. S.., et al. (1993) Characterization of CTLA-4 structure and expression on human T cells. J. Immunol. 151, 3489–3499 [PubMed] [Google Scholar]
- 44. Freeman G. J., Borriello F., Hodes R. J., Reiser H., Hathcock K. S., Laszlo G., McKnight A. J., Kim J., Du L., Lombard D. B.., et al. (1993) Uncovering of functional alternative CTLA-4 counter-receptor in B7-deficient mice. Science 262, 907–909 [DOI] [PubMed] [Google Scholar]
- 45. Freeman G. J., Gribben J. G., Boussiotis V. A., Ng J. W., Restivo V. A., Jr., Lombard L. A., Gray G. S., Nadler L. M. (1993) Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science 262, 909–911 [DOI] [PubMed] [Google Scholar]
- 46. Ueda H., Howson J. M., Esposito L., Heward J., Snook H., Chamberlain G., Rainbow D. B., Hunter K. M., Smith A. N., Di Genova G.., et al. (2003) Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423, 506–511 [DOI] [PubMed] [Google Scholar]
- 47. Vijayakrishnan L., Slavik J. M., Illes Z., Greenwald R. J., Rainbow D., Greve B., Peterson L. B., Hafler D. A., Freeman G. J., Sharpe A. H., Wicker L. S., Kuchroo V. K. (2004) An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity 20, 563–575 [DOI] [PubMed] [Google Scholar]
- 48. Pioli C., Gatta L., Ubaldi V., Doria G. (2000) Inhibition of IgG1 and IgE production by stimulation of the B cell CTLA-4 receptor. J. Immunol. 165, 5530–5536 [DOI] [PubMed] [Google Scholar]
- 49. Alegre M. L., Noel P. J., Eisfelder B. J., Chuang E., Clark M. R., Reiner S. L., Thompson C. B. (1996) Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J. Immunol. 157, 4762–4770 [PubMed] [Google Scholar]
- 50. Perkins D., Wang Z., Donovan C., He H., Mark D., Guan G., Wang Y., Walunas T., Bluestone J., Listman J., Finn P. W. (1996) Regulation of CTLA-4 expression during T cell activation. J. Immunol. 156, 4154–4159 [PubMed] [Google Scholar]
- 51. Luhder F., Chambers C., Allison J. P., Benoist C., Mathis D. (2000) Pinpointing when T cell costimulatory receptor CTLA-4 must be engaged to dampen diabetogenic T cells. Proc. Natl. Acad. Sci. USA 97, 12204–12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chambers C. A., Kuhns M. S., Allison J. P. (1999) Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates primary and secondary peptide-specific CD4(+) T cell responses. Proc. Natl. Acad. Sci. USA 96, 8603–8608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Takahashi T., Tagami T., Yamazaki S., Uede T., Shimizu J., Sakaguchi N., Mak T. W., Sakaguchi S. (2000) Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192, 303–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Read S., Malmstrom V., Powrie F. (2000) Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 192, 295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gattinoni L., Ranganathan A., Surman D. R., Palmer D. C., Antony P. A., Theoret M. R., Heimann D. M., Rosenberg S. A., Restifo N. P. (2006) CTLA-4 dysregulation of self/tumor-reactive CD8+ T-cell function is CD4+ T-cell dependent. Blood 108, 3818–3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wing K., Onishi Y., Prieto-Martin P., Yamaguchi T., Miyara M., Fehervari Z., Nomura T., Sakaguchi S. (2008) CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 [DOI] [PubMed] [Google Scholar]
- 57. Chambers C. A., Sullivan T. J., Truong T., Allison J. P. (1998) Secondary but not primary T cell responses are enhanced in CTLA-4-deficient CD8+ T cells. Eur. J. Immunol. 28, 3137–3143 [DOI] [PubMed] [Google Scholar]
- 58. Linsley P. S., Bradshaw J., Greene J., Peach R., Bennett K. L., Mittler R. S. (1996) Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 4, 535–543 [DOI] [PubMed] [Google Scholar]
- 59. Leung H. T., Bradshaw J., Cleaveland J. S., Linsley P. S. (1995) Cytotoxic T lymphocyte-associated molecule-4, a high-avidity receptor for CD80 and CD86, contains an intracellular localization motif in its cytoplasmic tail. J. Biol. Chem. 270, 25107–25114 [DOI] [PubMed] [Google Scholar]
- 60. Zhang Y., Allison J. P. (1997) Interaction of CTLA-4 with AP50, a clathrin-coated pit adaptor protein. Proc. Natl. Acad. Sci. USA 94, 9273–9278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chuang E., Alegre M. L., Duckett C. S., Noel P. J., Vander Heiden M. G., Thompson C. B. (1997) Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. J. Immunol. 159, 144–151 [PubMed] [Google Scholar]
- 62. Bradshaw J. D., Lu P., Leytze G., Rodgers J., Schieven G. L., Bennett K. L., Linsley P. S., Kurtz S. E. (1997) Interaction of the cytoplasmic tail of CTLA-4 (CD152) with a clathrin-associated protein is negatively regulated by tyrosine phosphorylation. Biochemistry 36, 15975–15982 [DOI] [PubMed] [Google Scholar]
- 63. Schneider H., Martin M., Agarraberes F. A., Yin L., Rapoport I., Kirchhausen T., Rudd C. E. (1999) Cytolytic T lymphocyte-associated antigen-4 and the TCR ζ/CD3 complex, but not CD28, interact with clathrin adaptor complexes AP-1 and AP-2. J. Immunol. 163, 1868–1879 [PubMed] [Google Scholar]
- 64. Shiratori T., Miyatake S., Ohno H., Nakaseko C., Isono K., Bonifacino J. S., Saito T. (1997) Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity 6, 583–589 [DOI] [PubMed] [Google Scholar]
- 65. Valk E., Leung R., Kang H., Kaneko K., Rudd C. E., Schneider H. (2006) T cell receptor-interacting molecule acts as a chaperone to modulate surface expression of the CTLA-4 coreceptor. Immunity 25, 807–821 [DOI] [PubMed] [Google Scholar]
- 66. Baroja M. L., Luxenberg D., Chau T., Ling V., Strathdee C. A., Carreno B. M., Madrenas J. (2000) The inhibitory function of CTLA-4 does not require its tyrosine phosphorylation. J. Immunol. 164, 49–55 [DOI] [PubMed] [Google Scholar]
- 67. Egen J. G., Allison J. P. (2002) Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 16, 23–35 [DOI] [PubMed] [Google Scholar]
- 68. Linsley P. S., Greene J. L., Brady W., Bajorath J., Ledbetter J. A., Peach R. (1994) Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1, 793–801 [DOI] [PubMed] [Google Scholar]
- 69. Hathcock K. S., Laszlo G., Pucillo C., Linsley P., Hodes R. J. (1994) Comparative analysis of B7-1 and B7-2 costimulatory ligands: expression and function. J. Exp. Med. 180, 631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lenschow D. J., Su G. H., Zuckerman L. A., Nabavi N., Jellis C. L., Gray G. S., Miller J., Bluestone J. A. (1993) Expression and functional significance of an additional ligand for CTLA-4. Proc. Natl. Acad. Sci. USA 90, 11054–11058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Collins A. V., Brodie D. W., Gilbert R. J., Iaboni A., Manso-Sancho R., Walse B., Stuart D. I., van der Merwe P. A., Davis S. J. (2002) The interaction properties of costimulatory molecules revisited. Immunity 17, 201–210 [DOI] [PubMed] [Google Scholar]
- 72. Pentcheva-Hoang T., Egen J. G., Wojnoonski K., Allison J. P. (2004) B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity 21, 401–413 [DOI] [PubMed] [Google Scholar]
- 73. Qureshi O. S., Zheng Y., Nakamura K., Attridge K., Manzotti C., Schmidt E. M., Baker J., Jeffery L. E., Kaur S., Briggs Z., Hou T. Z., Futter C. E., Anderson G., Walker L. S., Sansom D. M. (2011) Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Onishi Y., Fehervari Z., Yamaguchi T., Sakaguchi S. (2008) Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. USA 105, 10113–10118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Oderup C., Cederbom L., Makowska A., Cilio C. M., Ivars F. (2006) Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology 118, 240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Carreno B. M., Bennett F., Chau T. A., Ling V., Luxenberg D., Jussif J., Baroja M. L., Madrenas J. (2000) CTLA-4 (CD152) can inhibit T cell activation by two different mechanisms depending on its level of cell surface expression. J. Immunol. 165, 1352–1356 [DOI] [PubMed] [Google Scholar]
- 77. Masteller E. L., Chuang E., Mullen A. C., Reiner S. L., Thompson C. B. (2000) Structural analysis of CTLA-4 function in vivo. J. Immunol. 164, 5319–5327 [DOI] [PubMed] [Google Scholar]
- 78. Chikuma S., Abbas A. K., Bluestone J. A. (2005) B7-independent inhibition of T cells by CTLA-4. J. Immunol. 175, 177–181 [DOI] [PubMed] [Google Scholar]
- 79. Miyatake S., Nakaseko C., Umemori H., Yamamoto T., Saito T. (1998) Src family tyrosine kinases associate with and phosphorylate CTLA-4 (CD152). Biochem. Biophys. Res. Commun. 249, 444–448 [DOI] [PubMed] [Google Scholar]
- 80. Schneider H., Schwartzberg P. L., Rudd C. E. (1998) Resting lymphocyte kinase (Rlk/Txk) phosphorylates the YVKM motif and regulates PI 3-kinase binding to T-cell antigen CTLA-4. Biochem. Biophys. Res. Commun. 252, 14–19 [DOI] [PubMed] [Google Scholar]
- 81. Chuang E., Lee K. M., Robbins M. D., Duerr J. M., Alegre M. L., Hambor J. E., Neveu M. J., Bluestone J. A., Thompson C. B. (1999) Regulation of cytotoxic T lymphocyte-associated molecule-4 by Src kinases. J. Immunol. 162, 1270–1277 [PubMed] [Google Scholar]
- 82. Chikuma S., Murakami M., Tanaka K., Uede T. (2000) Janus kinase 2 is associated with a box 1-like motif and phosphorylates a critical tyrosine residue in the cytoplasmic region of cytotoxic T lymphocyte associated molecule-4. J. Cell. Biochem. 78, 241–250 [PubMed] [Google Scholar]
- 83. Marengere L. E., Waterhouse P., Duncan G. S., Mittrucker H. W., Feng G. S., Mak T. W. (1996) Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science 272, 1170–1173 [DOI] [PubMed] [Google Scholar]
- 84. Lee K. M., Chuang E., Griffin M., Khattri R., Hong D. K., Zhang W., Straus D., Samelson L. E., Thompson C. B., Bluestone J. A. (1998) Molecular basis of T cell inactivation by CTLA-4. Science 282, 2263–2266 [DOI] [PubMed] [Google Scholar]
- 85. Yokosuka T., Kobayashi W., Takamatsu M., Sakata-Sogawa K., Zeng H., Hashimoto-Tane A., Yagita H., Tokunaga M., Saito T. (2010) Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity 33, 326–339 [DOI] [PubMed] [Google Scholar]
- 86. Chikuma S., Imboden J. B., Bluestone J. A. (2003) Negative regulation of T cell receptor-lipid raft interaction by cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 197, 129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schneider H., Smith X., Liu H., Bismuth G., Rudd C. E. (2008) CTLA-4 disrupts ZAP70 microcluster formation with reduced T cell/APC dwell times and calcium mobilization. Eur. J. Immunol. 38, 40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schneider H., Downey J., Smith A., Zinselmeyer B. H., Rush C., Brewer J. M., Wei B., Hogg N., Garside P., Rudd C. E. (2006) Reversal of the TCR stop signal by CTLA-4. Science 313, 1972–1975 [DOI] [PubMed] [Google Scholar]
- 89. Li D., Gal I., Vermes C., Alegre M. L., Chong A. S., Chen L., Shao Q., Adarichev V., Xu X., Koreny T., Mikecz K., Finnegan A., Glant T. T., Zhang J. (2004) Cutting edge: Cbl-b: one of the key molecules tuning CD28- and CTLA-4-mediated T cell costimulation. J. Immunol. 173, 7135–7139 [DOI] [PubMed] [Google Scholar]
- 90. Chuang E., Fisher T. S., Morgan R. W., Robbins M. D., Duerr J. M., Vander Heiden M. G., Gardner J. P., Hambor J. E., Neveu M. J., Thompson C. B. (2000) The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 13, 313–322 [DOI] [PubMed] [Google Scholar]
- 91. Baroja M. L., Vijayakrishnan L., Bettelli E., Darlington P. J., Chau T. A., Ling V., Collins M., Carreno B. M., Madrenas J., Kuchroo V. K. (2002) Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J. Immunol. 168, 5070–5078 [DOI] [PubMed] [Google Scholar]
- 92. Schneider H., Prasad K. V., Shoelson S. E., Rudd C. E. (1995) CTLA-4 binding to the lipid kinase phosphatidylinositol 3-kinase in T cells. J. Exp. Med. 181, 351–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schneider H., Valk E., Leung R., Rudd C. E. (2008) CTLA-4 activation of phosphatidylinositol 3-kinase (PI 3-K) and protein kinase B (PKB/AKT) sustains T-cell anergy without cell death. PLoS One 3, e3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Parry R. V., Chemnitz J. M., Frauwirth K. A., Lanfranco A. R., Braunstein I., Kobayashi S. V., Linsley P. S., Thompson C. B., Riley J. L. (2005) CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 25, 9543–9553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fraser J. H., Rincon M., McCoy K. D., Le Gros G. (1999) CTLA4 ligation attenuates AP-1, NFAT and NF-κB activity in activated T cells. Eur. J. Immunol. 29, 838–844 [DOI] [PubMed] [Google Scholar]
- 96. Olsson C., Riesbeck K., Dohlsten M., Michaelsson E. (1999) CTLA-4 ligation suppresses CD28-induced NF-κB and AP-1 activity in mouse T cell blasts. J. Biol. Chem. 274, 14400–14405 [DOI] [PubMed] [Google Scholar]
- 97. Brunner M. C., Chambers C. A., Chan F. K., Hanke J., Winoto A., Allison J. P. (1999) CTLA-4-mediated inhibition of early events of T cell proliferation. J. Immunol. 162, 5813–5820 [PubMed] [Google Scholar]
- 98. Riley J. L., Mao M., Kobayashi S., Biery M., Burchard J., Cavet G., Gregson B. P., June C. H., Linsley P. S. (2002) Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc. Natl. Acad. Sci. USA 99, 11790–11795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bachmann M. F., Kohler G., Ecabert B., Mak T. W., Kopf M. (1999) Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J. Immunol. 163, 1128–1131 [PubMed] [Google Scholar]
- 100. Tivol E. A., Gorski J. (2002) Re-establishing peripheral tolerance in the absence of CTLA-4: complementation by wild-type T cells points to an indirect role for CTLA-4. J. Immunol. 169, 1852–1858 [DOI] [PubMed] [Google Scholar]
- 101. Bachmann M. F., Gallimore A., Jones E., Ecabert B., Acha-Orbea H., Kopf M. (2001) Normal pathogen-specific immune responses mounted by CTLA-4-deficient T cells: a paradigm reconsidered. Eur. J. Immunol. 31, 450–458 [DOI] [PubMed] [Google Scholar]
- 102. Marson A., Kretschmer K., Frampton G. M., Jacobsen E. S., Polansky J. K., MacIsaac K. D., Levine S. S., Fraenkel E., von Boehmer H., Young R. A. (2007) Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 445, 931–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zheng Y., Josefowicz S. Z., Kas A., Chu T. T., Gavin M. A., Rudensky A. Y. (2007) Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445, 936–940 [DOI] [PubMed] [Google Scholar]
- 104. Ono M., Yaguchi H., Ohkura N., Kitabayashi I., Nagamura Y., Nomura T., Miyachi Y., Tsukada T., Sakaguchi S. (2007) Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446, 685–689 [DOI] [PubMed] [Google Scholar]
- 105. Read S., Greenwald R., Izcue A., Robinson N., Mandelbrot D., Francisco L., Sharpe A. H., Powrie F. (2006) Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J. Immunol. 177, 4376–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Grohmann U., Orabona C., Fallarino F., Vacca C., Calcinaro F., Falorni A., Candeloro P., Belladonna M. L., Bianchi R., Fioretti M. C., Puccetti P. (2002) CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 3, 1097–1101 [DOI] [PubMed] [Google Scholar]
- 107. Fallarino F., Grohmann U., Hwang K. W., Orabona C., Vacca C., Bianchi R., Belladonna M. L., Fioretti M. C., Alegre M. L., Puccetti P. (2003) Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 4, 1206–1212 [DOI] [PubMed] [Google Scholar]
- 108. Leach D. R., Krummel M. F., Allison J. P. (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 [DOI] [PubMed] [Google Scholar]
- 109. Peggs K. S., Quezada S. A., Allison J. P. (2008) Cell intrinsic mechanisms of T-cell inhibition and application to cancer therapy. Immunol. Rev. 224, 141–165 [DOI] [PubMed] [Google Scholar]
- 110. Van Elsas A., Hurwitz A. A., Allison J. P. (1999) Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 190, 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hurwitz A. A., Yu T. F., Leach D. R., Allison J. P. (1998) CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc. Natl. Acad. Sci. USA 95, 10067–10071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hurwitz A. A., Foster B. A., Kwon E. D., Truong T., Choi E. M., Greenberg N. M., Burg M. B., Allison J. P. (2000) Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 60, 2444–2448 [PubMed] [Google Scholar]
- 113. Quezada S. A., Peggs K. S., Curran M. A., Allison J. P. (2006) CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Invest. 116, 1935–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Peggs K. S., Quezada S. A., Chambers C. A., Korman A. J., Allison J. P. (2009) Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 206, 1717–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chambers C. A., Kuhns M. S., Egen J. G., Allison J. P. (2001) CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 19, 565–594 [DOI] [PubMed] [Google Scholar]
- 116. Ishida Y., Agata Y., Shibahara K., Honjo T. (1992) Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nishimura H., Nose M., Hiai H., Minato N., Honjo T. (1999) Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151 [DOI] [PubMed] [Google Scholar]
- 118. Nishimura H., Okazaki T., Tanaka Y., Nakatani K., Hara M., Matsumori A., Sasayama S., Mizoguchi A., Hiai H., Minato N., Honjo T. (2001) Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291, 319–322 [DOI] [PubMed] [Google Scholar]
- 119. Okazaki T., Tanaka Y., Nishio R., Mitsuiye T., Mizoguchi A., Wang J., Ishida M., Hiai H., Matsumori A., Minato N., Honjo T. (2003) Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 9, 1477–1483 [DOI] [PubMed] [Google Scholar]
- 120. Wang J., Yoshida T., Nakaki F., Hiai H., Okazaki T., Honjo T. (2005) Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. USA 102, 11823–11828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Barber D. L., Wherry E. J., Masopust D., Zhu B., Allison J. P., Sharpe A. H., Freeman G. J., Ahmed R. (2006) Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 [DOI] [PubMed] [Google Scholar]
- 122. Day C. L., Kaufmann D. E., Kiepiela P., Brown J. A., Moodley E. S., Reddy S., Mackey E. W., Miller J. D., Leslie A. J., DePierres C., Mncube Z., Duraiswamy J., Zhu B., Eichbaum Q., Altfeld M., Wherry E. J., Coovadia H. M., Goulder P. J., Klenerman P., Ahmed R., Freeman G. J., Walker B. D. (2006) PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443, 350–354 [DOI] [PubMed] [Google Scholar]
- 123. Zhang X., Schwartz J. C., Guo X., Bhatia S., Cao E., Lorenz M., Cammer M., Chen L., Zhang Z. Y., Edidin M. A., Nathenson S. G., Almo S. C. (2004) Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 20, 337–347 [DOI] [PubMed] [Google Scholar]
- 124. Riley J. L. (2009) PD-1 signaling in primary T cells. Immunol. Rev. 229, 114–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Okazaki T., Maeda A., Nishimura H., Kurosaki T., Honjo T. (2001) PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 98, 13866–13871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Chemnitz J. M., Parry R. V., Nichols K. E., June C. H., Riley J. L. (2004) SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 173, 945–954 [DOI] [PubMed] [Google Scholar]
- 127. Nielsen C., Ohm-Laursen L., Barington T., Husby S., Lillevang S. T. (2005) Alternative splice variants of the human PD-1 gene. Cell. Immunol. 235, 109–116 [DOI] [PubMed] [Google Scholar]
- 128. Wan B., Nie H., Liu A., Feng G., He D., Xu R., Zhang Q., Dong C., Zhang J. Z. (2006) Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J. Immunol. 177, 8844–8850 [DOI] [PubMed] [Google Scholar]
- 129. Keir M. E., Butte M. J., Freeman G. J., Sharpe A. H. (2008) PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Agata Y., Kawasaki A., Nishimura H., Ishida Y., Tsubata T., Yagita H., Honjo T. (1996) Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 8, 765–772 [DOI] [PubMed] [Google Scholar]
- 131. Yamazaki T., Akiba H., Iwai H., Matsuda H., Aoki M., Tanno Y., Shin T., Tsuchiya H., Pardoll D. M., Okumura K., Azuma M., Yagita H. (2002) Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 169, 5538–5545 [DOI] [PubMed] [Google Scholar]
- 132. Freeman G. J., Long A. J., Iwai Y., Bourque K., Chernova T., Nishimura H., Fitz L. J., Malenkovich N., Okazaki T., Byrne M. C., Horton H. F., Fouser L., Carter L., Ling V., Bowman M. R., Carreno B. M., Collins M., Wood C. R., Honjo T. (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Eppihimer M. J., Gunn J., Freeman G. J., Greenfield E. A., Chernova T., Erickson J., Leonard J. P. (2002) Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation 9, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Butte M. J., Keir M. E., Phamduy T. B., Sharpe A. H., Freeman G. J. (2007) Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Paterson A. M., Brown K. E., Keir M. E., Vanguri V. K., Riella L. V., Chandraker A., Sayegh M. H., Blazar B. R., Freeman G. J., Sharpe A. H. (2011) The programmed death-1 ligand 1: B7-1 pathway restrains diabetogenic effector T cells in vivo. J. Immunol. 187, 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Liang S. C., Latchman Y. E., Buhlmann J. E., Tomczak M. F., Horwitz B. H., Freeman G. J., Sharpe A. H. (2003) Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur. J. Immunol. 33, 2706–2716 [DOI] [PubMed] [Google Scholar]
- 137. Latchman Y., Wood C. R., Chernova T., Chaudhary D., Borde M., Chernova I., Iwai Y., Long A. J., Brown J. A., Nunes R.., et al. (2001) PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268 [DOI] [PubMed] [Google Scholar]
- 138. Keir M. E., Liang S. C., Guleria I., Latchman Y. E., Qipo A., Albacker L. A., Koulmanda M., Freeman G. J., Sayegh M. H., Sharpe A. H. (2006) Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 203, 883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Shin T., Yoshimura K., Shin T., Crafton E. B., Tsuchiya H., Housseau F., Koseki H., Schulick R. D., Chen L., Pardoll D. M. (2005) In vivo costimulatory role of B7-DC in tuning T helper cell 1 and cytotoxic T lymphocyte responses. J. Exp. Med. 201, 1531–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Pentcheva-Hoang T., Chen L., Pardoll D. M., Allison J. P. (2007) Programmed death-1 concentration at the immunological synapse is determined by ligand affinity and availability. Proc. Natl. Acad. Sci. USA 104, 17765–17770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Blair P. J., Riley J. L., Levine B. L., Lee K. P., Craighead N., Francomano T., Perfetto S. J., Gray G. S., Carreno B. M., June C. H. (1998) CTLA-4 ligation delivers a unique signal to resting human CD4 T cells that inhibits interleukin-2 secretion but allows Bcl-X(L) induction. J. Immunol. 160, 12–15 [PubMed] [Google Scholar]
- 142. Sheppard K. A., Fitz L. J., Lee J. M., Benander C., George J. A., Wooters J., Qiu Y., Jussif J. M., Carter L. L., Wood C. R., Chaudhary D. (2004) PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 574, 37–41 [DOI] [PubMed] [Google Scholar]
- 143. Saunders P. A., Hendrycks V. R., Lidinsky W. A., Woods M. L. (2005) PD-L2: PD-1 involvement in T cell proliferation, cytokine production, and integrin-mediated adhesion. Eur. J. Immunol. 35, 3561–3569 [DOI] [PubMed] [Google Scholar]
- 144. Carter L., Fouser L. A., Jussif J., Fitz L., Deng B., Wood C. R., Collins M., Honjo T., Freeman G. J., Carreno B. M. (2002) PD-1: PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur. J. Immunol. 32, 634–643 [DOI] [PubMed] [Google Scholar]
- 145. Bennett F., Luxenberg D., Ling V., Wang I. M., Marquette K., Lowe D., Khan N., Veldman G., Jacobs K. A., Valge-Archer V. E., Collins M., Carreno B. M. (2003) Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J. Immunol. 170, 711–718 [DOI] [PubMed] [Google Scholar]
- 146. Francisco L. M., Salinas V. H., Brown K. E., Vanguri V. K., Freeman G. J., Kuchroo V. K., Sharpe A. H. (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Sfanos K. S., Bruno T. C., Meeker A. K., De Marzo A. M., Isaacs W. B., Drake C. G. (2009) Human prostate-infiltrating CD8+ T lymphocytes are oligoclonal and PD-1+. Prostate 69, 1694–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Ahmadzadeh M., Johnson L. A., Heemskerk B., Wunderlich J. R., Dudley M. E., White D. E., Rosenberg S. A. (2009) Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zou W., Chen L. (2008) Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 8, 467–477 [DOI] [PubMed] [Google Scholar]
- 150. Dong H., Strome S. E., Salomao D. R., Tamura H., Hirano F., Flies D. B., Roche P. C., Lu J., Zhu G., Tamada K., Lennon V. A., Celis E., Chen L. (2002) Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800 [DOI] [PubMed] [Google Scholar]
- 151. Thompson R. H., Gillett M. D., Cheville J. C., Lohse C. M., Dong H., Webster W. S., Krejci K. G., Lobo J. R., Sengupta S., Chen L., Zincke H., Blute M. L., Strome S. E., Leibovich B. C., Kwon E. D. (2004) Costimulatory B7-H1 in renal cell carcinoma patients: indicator of tumor aggressiveness and potential therapeutic target. Proc. Natl. Acad. Sci. USA 101, 17174–17179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Dorfman D. M., Brown J. A., Shahsafaei A., Freeman G. J. (2006) Programmed death-1 (PD-1) is a marker of germinal center-associated T cells and angioimmunoblastic T-cell lymphoma. Am. J. Surg. Pathol. 30, 802–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Liu J., Hamrouni A., Wolowiec D., Coiteux V., Kuliczkowski K., Hetuin D., Saudemont A., Quesnel B. (2007) Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 110, 296–304 [DOI] [PubMed] [Google Scholar]
- 154. Iwai Y., Ishida M., Tanaka Y., Okazaki T., Honjo T., Minato N. (2002) Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 99, 12293–12297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ishida M., Iwai Y., Tanaka Y., Okazaki T., Freeman G. J., Minato N., Honjo T. (2002) Differential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol. Lett. 84, 57–62 [DOI] [PubMed] [Google Scholar]
- 156. Kuang D. M., Zhao Q., Peng C., Xu J., Zhang J. P., Wu C., Zheng L. (2009) Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 206, 1327–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Curiel T. J., Wei S., Dong H., Alvarez X., Cheng P., Mottram P., Krzysiek R., Knutson K. L., Daniel B., Zimmermann M. C.., et al. (2003) Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 9, 562–567 [DOI] [PubMed] [Google Scholar]
- 158. Hamanishi J., Mandai M., Iwasaki M., Okazaki T., Tanaka Y., Yamaguchi K., Higuchi T., Yagi H., Takakura K., Minato N., Honjo T., Fujii S. (2007) Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 104, 3360–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Inman B. A., Sebo T. J., Frigola X., Dong H., Bergstralh E. J., Frank I., Fradet Y., Lacombe L., Kwon E. D. (2007) PD-L1 (B7-H1) expression by urothelial carcinoma of the bladder and BCG-induced granulomata: associations with localized stage progression. Cancer 109, 1499–1505 [DOI] [PubMed] [Google Scholar]
- 160. Ohigashi Y., Sho M., Yamada Y., Tsurui Y., Hamada K., Ikeda N., Mizuno T., Yoriki R., Kashizuka H., Yane K., Tsushima F., Otsuki N., Yagita H., Azuma M., Nakajima Y. (2005) Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin. Cancer Res. 11, 2947–2953 [DOI] [PubMed] [Google Scholar]
- 161. Taube J. M., Anders R. A., Young G. D., Xu H., Sharma R., McMiller T. L., Chen S., Klein A. P., Pardoll D. M., Topalian S. L., Chen L. (2012) Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 4, 127ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Rosenwald A., Wright G., Leroy K., Yu X., Gaulard P., Gascoyne R. D., Chan W. C., Zhao T., Haioun C., Greiner T. C.., et al. (2003) Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 198, 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Lenz G., Wright G. W., Emre N. C., Kohlhammer H., Dave S. S., Davis R. E., Carty S., Lam L. T., Shaffer A. L., Xiao W.., et al. (2008) Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 105, 13520–13525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Parsa A. T., Waldron J. S., Panner A., Crane C. A., Parney I. F., Barry J. J., Cachola K. E., Murray J. C., Tihan T., Jensen M. C., Mischel P. S., Stokoe D., Pieper R. O. (2007) Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 13, 84–88 [DOI] [PubMed] [Google Scholar]
- 165. Marzec M., Zhang Q., Goradia A., Raghunath P. N., Liu X., Paessler M., Wang H. Y., Wysocka M., Cheng M., Ruggeri B. A., Wasik M. A. (2008) Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 105, 20852–20857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Lee S. J., Jang B. C., Lee S. W., Yang Y. I., Suh S. I., Park Y. M., Oh S., Shin J. G., Yao S., Chen L., Choi I. H. (2006) Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-γ-induced upregulation of B7-H1 (CD274). FEBS Lett. 580, 755–762 [DOI] [PubMed] [Google Scholar]
- 167. Hirano F., Kaneko K., Tamura H., Dong H., Wang S., Ichikawa M., Rietz C., Flies D. B., Lau J. S., Zhu G., Tamada K., Chen L. (2005) Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 65, 1089–1096 [PubMed] [Google Scholar]
- 168. Blank C., Brown I., Peterson A. C., Spiotto M., Iwai Y., Honjo T., Gajewski T. F. (2004) PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 64, 1140–1145 [DOI] [PubMed] [Google Scholar]
- 169. Terme M., Ullrich E., Aymeric L., Meinhardt K., Desbois M., Delahaye N., Viaud S., Ryffel B., Yagita H., Kaplanski G., Prevost-Blondel A., Kato M., Schultze J. L., Tartour E., Kroemer G., Chaput N., Zitvogel L. (2011) IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 71, 5393–5399 [DOI] [PubMed] [Google Scholar]
- 170. Nishimura H., Minato N., Nakano T., Honjo T. (1998) Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 10, 1563–1572 [DOI] [PubMed] [Google Scholar]
- 171. Velu V., Titanji K., Zhu B., Husain S., Pladevega A., Lai L., Vanderford T. H., Chennareddi L., Silvestri G., Freeman G. J., Ahmed R., Amara R. R. (2009) Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 458, 206–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Curran M. A., Montalvo W., Yagita H., Allison J. P. (2010) PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 107, 4275–4280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Liu Y., Yu Y., Yang S., Zeng B., Zhang Z., Jiao G., Zhang Y., Cai L., Yang R. (2009) Regulation of arginase I activity and expression by both PD-1 and CTLA-4 on the myeloid-derived suppressor cells. Cancer Immunol. Immunother. 58, 687–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Mangsbo S. M., Sandin L. C., Anger K., Korman A. J., Loskog A., Totterman T. H. (2010) Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J. Immunother. 33, 225–235 [DOI] [PubMed] [Google Scholar]
- 175. Pardoll D. M. (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 [DOI] [PMC free article] [PubMed] [Google Scholar]