

PKC-ε−/− macrophages do not mobilize membrane in response to FcγR ligation; PKC-ε catalytic activity is required and cannot be replaced by a related kinase domain.
Keywords: patch-clamping, membrane fusion, macrophages, FcγR
Abstract
In RAW 264.7 cells [1], PKC-ε regulates FcγR-mediated phagocytosis. BMDM behave similarly; PKC-ε concentrates at phagosomes and internalization are reduced in PKC-ε−/− cells. Two questions were asked: what is the role of PKC-ε? and what domains are necessary for PKC-ε concentration? Function was studied using BMDM and frustrated phagocytosis. On IgG surfaces, PKC-ε−/− macrophages spread less than WT. Patch-clamping revealed that the spreading defect is a result of the failure of PKC-ε−/− macrophages to add membrane. The defect is specific for FcγR ligation and can be reversed by expression of full-length (but not the isolated RD) PKC-ε in PKC-ε−/− BMDM. Thus, PKC-ε function in phagocytosis requires translocation to phagosomes and the catalytic domain. The expression of chimeric PKC molecules in RAW cells identified the εPS as necessary for PKC-ε targeting. When placed into (nonlocalizing) PKC-δ, εPS was sufficient for concentration, albeit to a lesser degree than intact PKC-ε. In contrast, translocation of δ(εPSC1B) resembled that of WT PKC-ε. Thus, εPS and εC1B cooperate for optimal phagosome targeting. Finally, cells expressing εK437W were significantly less phagocytic than their PKC-ε-expressing counterparts, blocked at the pseudopod-extension phase. In summary, we have shown that εPS and εC1B are necessary and sufficient for targeting PKC-ε to phagosomes, where its catalytic activity is required for membrane delivery and pseudopod extension.
Introduction
The ability to recognize, internalize, and kill pathogens is fundamental to innate immunity. Ligation of FcγRs initiates a signaling cascade that culminates in target internalization, activation of the respiratory burst, and induction of inflammatory genes [2]. We have demonstrated that PKC is necessary for these responses and that different PKC isoforms mediate the different responses [3–6]. The localized nature of the phagocytic event makes it amenable to molecular study and has allowed the identification of PKC-ε as a major player influencing phagocytic rate [4]. The molecular mechanism of PKC-ε concentration and its role in phagocytosis remain to be elucidated.
PKC-ε belongs to a family of serine/threonine kinases having unique RDs linked to a homologous catalytic domain [7]. The homology among the catalytic domains (∼75%) has suggested that isoform specificity may reside within the RD. Indeed, PKC isoforms are grouped according to the structure of their RD: an autoinhibitory PS and two constant regions, C1 and C2, interspersed with V1s. C1 contains two cysteine-rich motifs, C1A and C1B, that bind DAG [8]; C2 binds Ca2+ and acidic phospholipids. Conventional PKCs (α, βI, βII, and γ) contain functional C1 and C2 domains; activation requires DAG, Ca2+, and phosphatidylserine. The novel PKCs (δ, ε, η, and θ) contain a C1 domain and a V1 with C2 homology but no Ca2+-binding activity; DAG and phosphatidylserine activate. The atypical PKCs (ζ and ι/λ) contain a single cysteine-rich motif and require phosphatidylserine for activity [9]. The binding of PKCs to receptors for activated C kinase is mediated by the C2 regions in PKC-β, -δ, and -ε [10].
The general consensus is that the regulatory and catalytic domains of PKCs serve different functions—that of localization and phosphorylation, respectively. However, there are notable exceptions. For example, although the catalytic domain of PKC-α is necessary for PLD activation, enzymatic activity is not [11]. Particularly relevant to our studies is the work of Zeidman et al. [12], who demonstrated that neurite extension (an actin-dependent process that may resemble pseudopod extension) requires the regulatory but not catalytic domain of PKC-ε. Thus, identification of a particular PKC isoform in a process does not a priori implicate the catalytic domain or catalytic activity in function.
Generation of PKC chimeras, in which the catalytic and RDs are derived from different isotypes, has provided insight into the relative roles of the domains in isotype-specific localization and function. For example, in K-562 erytholeukemia cells, nuclear translocation of β/α chimeras requires the β catalytic domain, whereas the α catalytic domain dictates proliferation [13]. Others have shown that the catalytic domain can modulate the membrane interactions of the RD [14]. Herein, we have used a chimeric strategy to dissect the role of the regulatory and catalytic domains of PKC-ε in phagocytosis and demonstrate that εPS is essential for localization and that PKC-ε catalytic activity is specifically required for efficient phagocytosis.
With respect to PKC-ε function, it has long been known that internal membranes are needed for phagocytosis [15], as the surface area of macrophages increases during phagocytosis [16], and retention of membrane in sucrose-laden vacuoles inhibits internalization [17]. In 2000, Bajno et al. [18] demonstrated focal exocytosis at the base of the developing phagosome. They constructed a GFP-VAMP3 plasmid, such that the GFP was luminal. Antibody to GFP, applied to the outside of the cell, was used as a read-out of vesicle fusion. As vesicles move on microtubules, it is not surprising that microtubule depolymerization with nocodazole inhibits phagocytosis, albeit to a modest extent [17].
PKC-ε is unique in that it contains an actin-binding motif in its RD. This implicates it in cytoskeletally associated functions. Indeed, it is associated with actin filaments and keratin and is involved in integrin-dependent cell motility and spreading [19]. Interestingly, its overexpression in NIH3T3 fibroblasts elicits a transformed phenotype similar to that of ras-overexpressing cells [20], and its down-regulation inhibits metastasis in a mouse prostate cancer model [21]. As metastatic cells are very motile, it is likely involved in cell motility in cancer models. Finally, PKC-ε is involved in various exocytic processes, including release of dense granules from chromaffin cells [22], secretion by lacrimal cells [23], and insulin exocytosis [24]. We have shown that PKC-ε regulates the rate of FcγR-mediated target internalization [4]. Based on its involvement in membrane mobilization in other models, we tested the hypothesis that PKC-ε is necessary for membrane delivery during phagocytosis.
MATERIALS AND METHODS
Buffers and reagents
ACK lysis buffer contains 0.15 M NH4Cl, 1 mM KHCO3, and 0.1 mM EDTA, pH 7.4; HBSS++ is HBSS containing 4 mM sodium bicarbonate, 10 mM HEPES, and 1.5 mM each CaCl2 and MgCl2; Mg/EGTA is HBSS containing 1.0 mM EGTA and 2.0 mM MgCl2; BM media is DMEM containing 10% FBS, 20% L-cell conditioned media, and 0.03% NaHCO3 and pen/strep; and PFA is 4% PFA in PBS. PLL was from Sigma-Aldrich (St. Louis, MO, USA).
Antibodies
Alexa 488 and Alexa 568 goat anti-rabbit IgG were purchased from Life Technologies (Carlsbad, CA, USA). For flow cytometry, 2.4G2-PE antibody was purchased from BD Biosciences PharMingen (Franklin Lakes, NJ, USA). F4/80-APC antibody and (unlabeled) 2.4G2 control antibody were from eBioscience (San Diego, CA, USA). CD64-PE antibody and IgG isotype control-PE antibody were obtained from R&D Systems (Minneapolis, MN, USA).
Mice
PKC-ε−/+ mice on the C57/Bl6 background were purchased from The Jackson Laboratory (Bar Harbor, ME, USA; Stock #004189) and bred in the Albany Medical Center Animal Resources Facility. Heterozygotes were crossed to produce the WT and PKC-ε−/− mice. All animal procedures were approved by the Albany Medical Center Institutional Animal Care and Use Committee. Cells obtained from one mouse represent one independent experiment.
Cells
BMDM.
PKC-ε+/+ and PKC-ε−/− mice were killed and their femurs removed. The BM was removed and the stem cells mixed with ACK lysis buffer to lyse red blood cells. BM stem cells were differentiated into BMDM by incubation in DMEM containing 10% FBS, 20% L cell conditioned media, and sodium bicarbonate. BMDM were used 7–10 days after harvesting.
RAW cells.
RAW cells [1] were a kind gift from Dr. Steven Greenberg (Presbyterian Hospital, New York, New York). Cells were cultured as described [4, 6]. This is a subclone of the ATTC line RAW 264.7 that expresses the human FMLPR. It has been used in all of our molecular studies, as it is more readily transfectable than the parental line.
Cloning and plasmid construction
All constructs used in these studies are listed in Fig. 1. Published plasmids are referenced. Primer sequences for constructions are given in Table 1.
Figure 1. Constructs used in this study.

Published plasmids are referenced.
Table 1. Primers Used to Construct PKC Plasmids.
| Construct | Plasmid designation | Primer | Sequence |
|---|---|---|---|
| Δ(εPS)-GFP | BS1627 | Δ463F | TACATCAAGAACCACGAGTTCATCGCCACC |
| ε495R | GTGGACCCTGCGCCTGACAGCCCCTTGGCG | ||
| Δ(εPSC1B)-GFP | BS1626 | Δ463F | TACATCAAGAACCACGAGTTCATCGCCACC |
| ε495R | GTGGACCCTGCGCCTGACAGCCCCTTGGCG | ||
| ε439F (NruI)a | TTTCGCGAGCGGATGCGGCCAAGGAAG | ||
| Δ402R (PvuI)a | CTCCGATCGCATGGACTGTTTGCAATCCCC | ||
| εPSΔC1AεC1B-GFP | AMC068 | CP126F | AAGCTTACCATGTTTAGGGAGCGGATGCGGCCA |
| CP127R | CTCGAGACAATTGGGAGCCACGTTGGTCT | ||
| εRDδCAT-GFP | AMC109 | CP107F | GTCGACGTTCGAGAAGAAGACCGGAG |
| CP108R | GGATCCCCATCTTCCAGGAGGTGCTC | ||
| ε154AAA156-GFP | AMC125 | CP004F (PvuII)b | CGGATGCGGCCAGCAGCTGCCCAAGGGGCTGTC |
| CP005R (PvuII)b | GACAGCCCCTTGGGCAGCTGCTGGCCGCATCCG | ||
| ε161AAA163-GFP | AMC126 | CP006F (BbeI, PvuII)a,b | GGAAGCGCCAAGGCGCCGTCGCAGCTGCGGTCCACCAGGTC |
| CP007R (PvuII, BbeI)a,b | GACCTGGTGGACCGCAGCTGCGACGGCGCCTTGGCGCTTCC | ||
| εK437W-GFP | AMC148 | CP132F | GGATGAAGTCTATGCTGTGTGGGTCTTAAAGAAGG3c |
| CP133R | TTACCCTTGAGCTCGGCCAGCATGACCTT |
Identifies the restriction sites with silent mutations to distinguish the PCR product and templates.
PvuII sites are where the mutation within the PS region is located.
Boldface letters identify the mutated residues.
δ(εPS)-GFP (BS1627).
The PCR product [δ(εPS); BS1618] was produced using BS751 and BS1614 as templates and ε495R/δ463F as primers and subcloned into BS340.
δ(εPSC1B)-GFP (BS1626).
The chimera was produced by two-step PCR reaction, using two templates in one reaction. First, δ(εV1PSC1B) was produced using rat PKC-ε in pCR 2.1 (BS495) [25] and δ(εC1B) in pCR 2.1 (BS789) [27] as templates and ε495R and δ463F as primers (Fig. 1). The amplified PCR product [δ(εV1PSC1B); BS1448] and PKC-δ in pCR 2.1 (BS751) [27] were used as templates and δ402R/ε439F as primers for the second PCR reaction. The final PCR product [δ(εPSC1B); BS1614] was subcloned into BglII sites of BS340 (pTB701 containing eGFP) [26].
εPSδC1AεC1B-GFP (AMC068).
The PCR product (εPSδC1AεC1B; TOPO17) was produced using BS1626 as a template and CR126 and CR127 as primers and cloned into pCR4-TOPO (Invitrogen, Carlsbad, CA, USA) for sequence verification. The insert was excised with HindIII and XhoI and subsequently subcloned into HindIII and SalI sites of pEGFP-N3 (Clontech Laboratories, Mountain View, CA, USA).
εRD-δCAT (AMC109).
Construction of the PKC-ε RD plasmid (εRD-GFP; AMC006) and PKC-δ (AMC038) has been published [12]. The PCR product (δCAT; TOPO14) was produced using AMC038 as a template and CR107 and CR108 as primers and cloned into pCR4-TOPO (Invitrogen) for sequence verification. The insert was isolated with SalI and BamHI and subsequently subcloned into SalI and BamHI sites of AMC006.
ε154AAA156 (AMC125) and ε161AAA163 (AMC126).
PKC-ε (AMC004) was used as a template in the production of both εPS mutant constructs using QuikChange II XL site-directed mutagenesis kit, per the manufacturer's instructions (Stratagene, La Jolla, CA, USA).
εK437W (AMC148).
Construction of the PKC-ε (AMC004) has been published [4]. With the use of AMC004 as a template, site-directed mutagenesis using Phusion PCR (Thermo Fisher Scientific, Waltham, MA, USA) was performed using phosphorylated CP132F (introducing the mutation) and CP133R. The resulting PCR product was ligated and sequenced to screen for the introduction of the mutation.
Expression of exogenous proteins
Nucleofection of BMDM.
Plasmids εRD-GFP and GFP were delivered to BMDM by nucleofection, according to the manufacturers' instructions. Briefly, 3 × 106 BMDM were suspended in 82 μl macrophage solution and 18 μl supplement. DNA (5 μg) was then added and the cells transferred to a sterile electroporation cuvette. The cuvette was placed into the nucleofector set to the Y-001 program. Once transfected, the cells were transferred gently to wells in a six-well plate and maintained in 2 ml BMDM media.
Viral transduction
Viral plasmids.
The pBM-IRES-Puro vector was a kind gift from Dr. Elaine Raines (University at Washington, Seattle, WA, USA). pBM-eGFP-IRES-Puro was created by excising the BglII/NotI fragment of peGFPN3 (Clontech Laboratories) and ligating it to the NotI/BamHI fragment of pBM-IRES-Puro. The resulting vector was cut with EcoRI, blunted, and ligated with a gateway conversion cassette (Life Technologies, Carlsbad, CA, USA). The resulting gateway-compatible vector (pBM-GW-eGFP-IRES-Puro) was recombined with human PKC-ε cDNA, human PKC-δ cDNA, or rat VAMP3, encoded in pENTR/D-TOPO. The pBM-eGFP-IRES-Puro, human PKC-ε in pBM-GW-eGFP-IRES-Puro, human PKC-δ in pBM-GW-eGFP-IRES-Puro, and rat VAMP3 in pBM-GW-eGFP-IRES-Puro were used for retroviral production.
Virus production.
Retrovirus was produced by calcium phosphate-mediated transfection of the retroviral plasmids into Phoenix ecotropic packaging cells (National Gene Vector Biorepository, Indianapolis, IN, USA) as described [28]. Virus-containing supernatants were harvested at 72 h post-transfection and frozen, stored at −80°C.
Viral transduction of primary macrophages.
Viral transduction was done essentially as described [29]. Briefly, freshly isolated BM cells were transduced in OptiMEM containing 10% heat-inactivated FBS, 50% retroviral containing supernatant, 8 μg/ml polybrene, and 20% L cell conditioned media by two consecutive spinfections (900 g, 2 h, 22 h apart). Twenty-two hours after the second spinfection, the media were changed to DMEM with 10% heat-inactivated FBS, 20% L cell conditioned media, and 2 μg/μl puromycin. Differentiated BMDM were used 8–10 days after isolation, at which time, >90% of the cells were GFP-positive.
Transfection.
RAW 264.7 cells were cultured as described. Cells (2.0×105), seeded onto 12-mm coverslips (Thermo Fisher Scientific, Waltham, MA, USA), were transiently transfected with 0.5–1 μg DNA, complexed to TransIT-Neural (Mirus Bio, Madison, WI, USA). After 18–24 h, transfectants were incubated with BIgG and imaged by spinning disk confocal microscopy. Flow cytometry established that expression levels (as determined by MFI) of the different constructs were similar. Transfection efficiencies ranged from 12% to 20%. Only transfected cells were scored. Concentration at the phagosome was compared with PKC-ε-GFP (positive control) and PKC-δ-GFP, and PKC-δ-GFP was used as the baseline, as it is expressed in macrophages and neither concentrates during phagocytosis nor alters the phagocytic rate [4].
IgG-opsonized targets and surfaces
BIgG.
BIgG were prepared as described previously [4]. Briefly, 2 μm borosilicate microspheres (Duke Standards, Thermo Scientific USA) were coated sequentially with PLL, activated with dimethylpimelimidate · 2 HCl, washed, and incubated with Alexa 568-labeled BSA (IgG-free) overnight. BSA beads were blocked, washed, and opsonized with rabbit anti-BSA IgG.
IgG-coated surfaces.
Frustrated phagocytosis uses IgG-coated surfaces produced by sequentially coating surfaces with an antigen and its cognate antibody [17, 30]. In initial studies, we observed that macrophages strip the IgG from the surface, thus changing the surface with time. To prevent this, our glass surfaces were silane-treated. Briefly, glass coverslips were rinsed with ethanol and distilled H2O and dried with compressed air to prepare for coating. The coverslips were aldehyde silane-coated by sequential incubation with an oxygen plasma cleaner (5 min) and vaporized silane [11-(triethoxysilyl) undecanal, 70°C, 1 h, sealed in curing oven]. To covalently link proteins to the silane-coated coverslips, the coverslips were washed with PBS and incubated with rabbit IgG or PLL (25 μg/ml, 60 min, room temperature). After protein attachment, the remaining active sites were blocked (0.5 M Tris, pH 8, overnight). Before use, the coverslips were washed twice in HBSS++. With the use of fluorescent IgG, we determined that cells cannot endocytose IgG attached via silane.
Phagocytosis
Frustrated phagocytosis.
WT and εKO macrophages were calcium-depleted (Mg/EGTA, 45 min, 37°C) or kept in HBSS++. The cells were cooled (ice, 30 min), added to IgG-coated coverslips (3.2×104/well), and centrifuged (1000 g, 5 min, 4°C) to facilitate attachment. The cells were transferred to 37°C and allowed to spread for 0–15 min, after which time, they were fixed (4% PFA). To visualize the bound cells, the exposed IgG surface stained with Alexa 488-conjugated anti-rabbit antibody. Stained coverslips were washed with PBS, mounted, and imaged by epifluorescence. ImageJ was used to trace the outline of the cells (which appeared as black holes against the fluorescent surface; see Fig. 3), and the area of the black holes was calculated. A minimum of 100 cells/time-point was measured from three animals of each genotype. For calculation of spreading rates, the slope of the lines between 0 and 15 min was determined. Statistics were calculated using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA) and the results analyzed using a two-way ANOVA with Tukey's post-test (to compare the rate of spreading of WT with εKO macrophages) or a one-way ANOVA to compare the spread area of WT with εKO reconstituted with PKC-ε or εRD-GFP.
Figure 3. IgG-mediated spreading in BMDM is Ca2+-independent; εKO spreading is significantly slower.

WT and εKO BMDM were calcium-depleted in Mg/EGTA (EGTA) or maintained in HBSS++ (Ca2+). Cells were seeded onto IgG-coated coverslips and allowed to undergo frustrated phagocytosis. At the indicated time, cells were fixed and the exposed IgG visualized with Alexa 488 anti-IgG. Area measurements of black holes (inset) were made from three independent experiments; results are reported as average spread area (μm2) ± sem (>150 total cells/time-point). Frustrated phagocytosis was Ca2+-independent. The spread area of εKO was significantly smaller than WT, from 7.5 to 15 min. Statistical differences were calculated by two-way ANOVA with Tukey's post-test. *P < 0.50; ***P < 0.001.
To ensure that the black holes were an accurate measure of the cell periphery, we compared the area measurements obtained from black-hole measurements with those obtained from the same cells, followed by surface staining with wheat-germ agglutinin-Alexa 488. The area measurements were not different. As black holes were more amenable to computer-based analysis, we chose the black-hole method for cell-area quantitation.
Synchronized phagocytosis.
Synchronized time courses with 2-μm BIgG were performed as reported [4, 6]. Briefly, the media were removed from cells (2×105) and replaced with HBSS2+ (HBSS containing 10 mM HEPES, supplemented with 4 mM sodium bicarbonate and 1.5 mM each CaCl2 and MgCl2). Cells were cooled (30 min on ice), and BIgG were added (4:1; BIgG:macrophage, 15 min on ice). Phagocytosis was initiated by transferring the cells to a 37°C waterbath. At varying times (0–15 min), cells were fixed (4% PFA), mounted, and imaged by confocal microscopy. For calculation of PI, a synchronized round of phagocytosis was performed with a 10:1-unlabeled BIgG:macrophage ratio and fixed after 7.5 min at 37°C. Incomplete phagosomes were detected by the addition of Alexa 568-conjugated goat anti-rabbit IgG (Invitrogen) to label the IgG on the exposed targets. For each experiment, the number of internal beads associated with 100 transfected cells was determined. PI = (# internal beads/# cells counted) × 100. Real-time confocal imaging was conducted as described previously [4, 6]. Briefly, BIgG were added to live cells and images collected every 5 s for 12 min.
Patch-clamping
WT and εKO were prepared as for frustrated phagocytosis. Cells were seeded on IgG or PLL-coated coverslips in six-well plates, precooled on ice; PLL surfaces served as the attachment control. Cells were spun down onto the coverslips (1000 g, 5 min, 4°C). Plates were then transferred to a 37°C water bath. After transfer (7.5 min) to 37°C, the coverslips were placed into a coverslip holder and the cells bathed in a balanced salt solution (10 mM HEPES, pH 7.4, containing 145 mM NaCl, 5 mM KCl, 1 mM CaCl2, 2 mM MgCl2, and 5 mM glucose). Coverslips were placed into the patch-clamping apparatus, and the membrane capacitance of individual cells was measured using patch-clamping equipment, Axopatch 200B, and Digidata 1440A (Axon Instruments, New York, NY, USA). Pipettes were pulled from borosilicate glass capillaries (World Precision Instruments, Sarasota, FL, USA) with a P-97 flaming/brown micropipette puller (Sutter Instrument, Novato, CA, USA) and polished with DMF1000 (World Precision Instruments). The pipettes had a resistance of 2–4 MΩ when filled with a pipette solution, pH 7.2, consisting of 140 μM K-methanesulfonate, 2 mM KCl, 5 mM EGTA, 0.5 mM CaCl2, 4 mM MgCl2, 10 mM HEPES, and 3 mM ATP-Mg2. When a tight seal (>16 GΩ) was formed between the pipette tip and the cell membrane, pipette capacitance was compensated and the cell membrane broken to achieve whole-cell configuration. The whole-cell capacitance button was then turned on to compensate for the whole-cell capacitance. Cell capacitance was then read. Cells were maintained at a 0-mV holding potential during experiments. The experiment was done with macrophages from six mice (three WT; three εKO); ≥10 cells/condition/experiment were collected. Data are reported as mean capacitance ± sem. Statistical significance was determined by ANOVA with Tukey's post-test.
Flow cytometry
PKC-ε+/+ and PKC-ε−/− BMDM (2×106 cells) were incubated with the following: (1) IgG isotype control-PE, (2) CD64-PE and F4/80-APC, (3) 2.4G2-PE and F4/80-APC, or (4) left unstained. For CD64 (FcγR I) staining, endogenous FcγRs were blocked with (unlabeled) 2.4G2 (10 min). The cells were then stained with primary antibodies and incubated (4°C, dark, 45 min). Cells that were stained with 2.4G2-PE were not blocked. After staining, cells were washed three times and fixed with PFA (10 min, 4°C). The cells were read using a FACSCalibur flow cytometer and the results analyzed by FlowJo. Cells (>95%) were macrophages, as determined by F4/80 positivity.
Live imaging, calculation of phagocytic rates, and LI
Transfected/transduced BMDM were removed from plates with PBS/5 mM EDTA (30 min, 37°C), washed in HBSS++, and replated in Mat-Tek glass-bottom dishes (2×105 cells/dish). Fluorescent cells were visualized by spinning disk confocal microscopy (Olympus IX81-DSU) with a 100×/1.4 numerical aperture oil objective. GFP was detected with a dichroic Q495lp filter (excitation 470/40; Chroma Technology, Bellows Falls, VT, USA) and Alexa 568 with a dichroic Q595lp filter (excitation 560/55; Chroma Technology). Images were captured using a Hamamatsu electron-multiplying charge-coupled device camera, driven by MetaMorph software (Molecular Devices, Sunnyvale, CA, USA). With cells in the field of view, Alexa 568-labeled BIgG were added. Cells were imaged for 10 min, with data collected at 5-s intervals. The phagocytic rate was determined from the first frame with membrane-bead contact to the frame, in which the GFP surrounded the bead completely [4]. Phagocytic rate (s/bead) = (time of completion−first frame of contact) × 5. Images are presented as the red/green merge and as gray-scale images of the GFP intensity.
Synchronized phagocytosis was also followed using spinning disk confocal microscopy. GFP at the phagosome was quantified, as detailed previously [6], and reported as the LI. Briefly, the cell images are enlarged to visualize individual pixels. NIH ImageJ was used to determine the intensity of the 10 brightest pixels in the phagosome and in an uninvolved region of the membrane. The average pixel intensity in the phagosome was then normalized to the average on an uninvolved membrane. This strategy is rigorous, as pixel intensity in the phagosome must be higher than that of the brightest region of the plasma membrane to be considered concentrated. It should also be noted that the PKC-ε concentration in some phagosomes (e.g., PKC-ε control, see Fig. 6; ε(δPSC1B), see Fig. 9; and εK437W, see Fig. 10) is punctate, and LI is based on those bright pixels. Thus, whereas the overall concentration of a phagosome may not be dramatic, the LI (bar graphs) may be significant. Images were prepared in Adobe Photoshop 7.0 (Adobe Systems, Mountain View, CA, USA).
Figure 6. εC1B is not sufficient for PKC translocation to IgG-containing phagosomes.
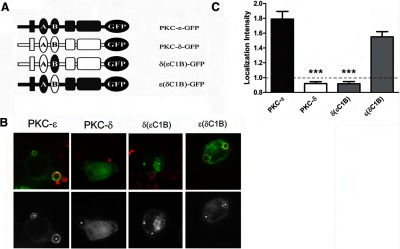
RAW 264.7 cells were transfected with plasmids encoding the indicated GFP-conjugated proteins (A) and subjected to synchronized phagocytosis. (B) Representative images showing red BIgG (upper); the same image represented in gray scale (lower). Asterisks designate phagosomes. (C) LI was calculated as detailed in Materials and Methods. Data are presented as mean ± sem (80 events for each construct; three to four experiments). Significance was determined by ANOVA with Tukey's post-test; ***P < 0.001 compared with PKC-ε-expressing cells. Dotted line indicates no concentration (i.e., GFP intensity at phagosomes equivalent to that of uninvolved membrane).
Figure 9. εPS and εC1B synergize to recapitulate WT PKC-ε concentration.
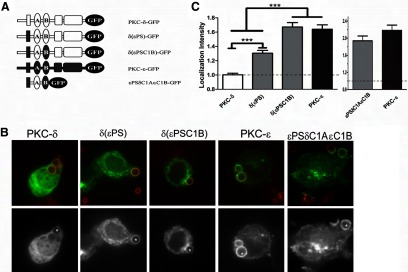
(A) Cells were transfected with the indicated constructs and assayed as in Fig. 6. (B) Representative images showing red BIgG (upper); the same image represented in gray scale (lower). Asterisks designate phagosomes. (C) LI was calculated as in Materials and Methods (mean±sem of 80 total events/construct from three to four experiments). Significance was determined by ANOVA with Tukey's post-test; ***P < 0.001. Dotted line indicates no concentration. (C) Localization of a chimeric fragment consisting of εPSδC1AεC1B-GFP is equivalent to PKC-ε with respect to distribution (B) and intensity (C).
Figure 10. PKC-ε catalytic activity is necessary for efficient phagocytosis.
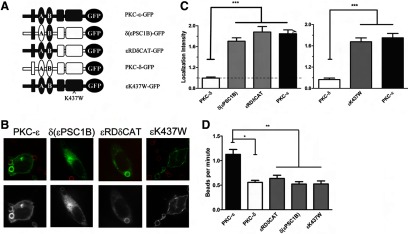
(A) Cells were transfected with the indicated constructs and assayed as in Fig. 6. (B) Representative images showing red BIgG (upper) and the same image represented in gray scale (lower). (C) LI is presented as mean ± sem (80 total events/construct from three to four experiments). Dotted line indicates no concentration. (D) Transfected cells were subjected to live imaging and internalization rates determined from the first frame of indentation until the particle was completely surrounded. Significance was determined by ANOVA with Tukey's post-test; *P < 0.05; **P < 0.01;***P < 0.001. Although the LI was similar to WT PKC-ε, expression of the chimeras did not increase phagocytosis, suggesting that the PKC-δ catalytic domain cannot functionally replace that of PKC-ε.
Statistical analysis
All data are expressed as mean ± sem, and significance was calculated by ANOVA with a Tukey's post-test. P ≤ 0.05 was considered significant.
RESULTS
Primary mouse macrophages require PKC-ε for efficient phagocytosis
With the use of the RAW 264.7 cell line, we reported that PKC-ε concentrates at phagocytic cups and formed phagosomes and that it is necessary for efficient phagocytosis [4, 6]. The availability of PKC-ε−/− mice enabled us to validate the RAW cell results in primary BMDM. Like RAW cells, PKC-ε-GFP concentrated at phagocytic cups and internalized phagosomes in WT BMDM (Fig. 2 and Supplemental Video 1). Calculated from live imaging, PKC-ε−/− BMDM (εKO) internalized 2 μm IgG-opsonized beads at a significantly slower rate than their WT counterparts (εKO: 78.1±4.9 s/target; WT: 53.7±3.9 s/target; P<0.001, n=40 from three independent experiments). The phagocytosis rate was restored upon expression of PKC-ε-GFP in εKO; the average time for internalization of a single bead was 54.3 ± 3.3 s for reconstituted εKO cells and 87.1 ± 6.5 s for εKO-expressing GFP (n=77–79, three experiments; P<0.001). Flow cytometry established that FcγR expression is equivalent in WT and PKC-ε−/− BMDM (Supplemental Fig. 1), eliminating this trivial explanation for the observed decrease in phagocytosis in εKO cells. Thus, RAW cells and BMDM require PKC-ε for efficient phagocytosis.
Figure 2. PKC-ε concentrates at phagosomes during IgG-mediated phagocytosis in primary mouse macrophages.
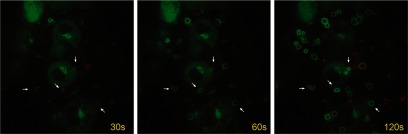
C57/Bl6 BMDM were transduced with a retrovirus encoding human PKC-ε-GFP. Alexa 568-labeled BIgG were added to the cells (15 targets:BMDM) and internalization followed with time. PKC-ε concentration is apparent in phagocytic cups, as well as on fully formed phagosomes (examples indicated by arrows). The three panels show the same field of cells taken over 90 s. Taken from Supplemental Video 1.
PKC-ε is necessary for membrane mobilization in response to FcγR ligation
As phagocytosis involves pseudopod extension, and PKC-ε is involved in cell spreading [31] and neurite extension [12], we asked whether PKC-ε contributes to membrane mobilization in response to FcγR ligation. To maximize membrane recruitment, BMDM were subjected to frustrated phagocytosis on IgG surfaces [17]. As a result of the tight binding of cells to the IgG surface, the area of attached cells can be calculated from the “black holes” produced by labeling the exposed IgG with Alexa 488 secondary antibodies (Fig. 3, inset). When measured with time, WT were significantly larger than their εKO counterparts after 5 min (Fig. 3). The slopes of the time-course plots provided a measure of spreading rate. A comparison of the rates confirmed that WT cells spread significantly faster than εKO (60±3 μm2/min vs. 33±2 μm2/min; P<0.05). Finally, the difference between WT and εKO cells was Ca2+-independent and was seen for primary BMDM (Fig. 3) and elicited peritoneal macrophages (not shown), suggesting that the spreading defect is intrinsic and not a function of the in vitro differentiation of BM precursors.
Flow cytometry indicates that WT and εKO cells are similar in size and granularity (forward- and side-scatter, respectively; Supplemental Fig. 1). Thus, the smaller spread area in εKO cells could be a result of less overall plasma membrane or an inability of PKC-ε−/− cells to access their plasma membrane (as a result of defects in cortical actin structure, etc.). Whole-cell patch-clamping, the gold standard for quantitation of membrane area, was used to measure the capacitance of the plasma membrane. WT and εKO macrophages were patched 7.5–10 min into frustrated phagocytosis. On IgG surfaces, WT had a significantly higher capacitance than εKO (25.5±0.76 pF vs. 21.9±0.53 pF; P<0.001), consistent with having more plasma membrane (Fig. 4). To determine whether membrane delivery was specific to receptor ligation or a response to cell attachment, we repeated the experiment on PLL surfaces, to which cells attach via electrostatic interactions [32]. On PLL, the capacitance of WT and εKO cells was equivalent (WT: 19.7±0.37 pF vs. εKO: 20.3±0.49 pF) and not different from εKO cells on IgG (Fig. 4). This validates the flow data and demonstrates that WT, but not εKO, cells add membrane to their surface upon FcγR ligation. It is notable that the 5-pF capacitance increase of WT on IgG translates to a 30% increase in surface membrane. To probe the requirement for PKC-ε in spreading, PKC-ε-GFP was expressed in εKO cells. Reconstituted cells were significantly more spread than their εKO counterparts (Fig. 5A). The defects in frustrated (Fig. 5A) phagocytosis and membrane mobilization seen in εKO cells can be attributed to the absence of PKC-ε and lead to the conclusion that PKC-ε is necessary for membrane fusion upon FcγR ligation.
Figure 4. Plasma membrane of WT, but not εKO, cells increases during frustrated phagocytosis.
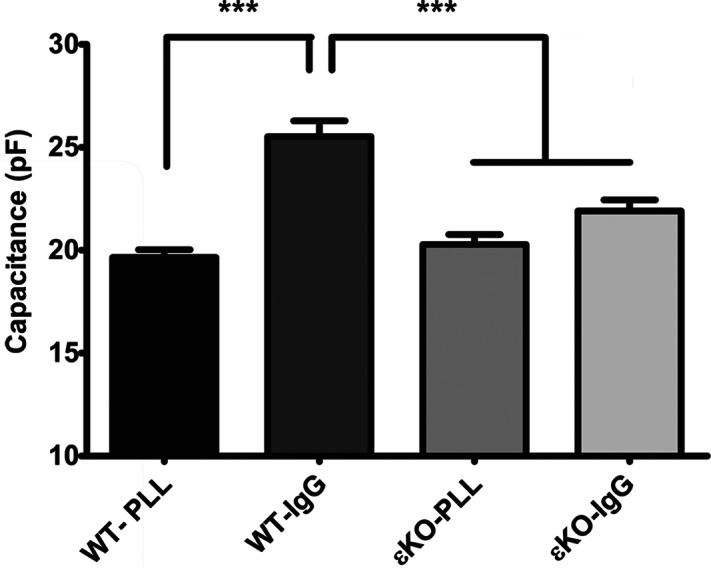
WT and εKO BMDM were seeded onto PLL or IgG surfaces at 4°C. Plates were transferred to 37°C and membrane capacitance determined by whole-cell patch-clamping at 7.5– 10 min. The average capacitance ± sem is reported (n=31–36 cells from three independent experiments). Statistical differences were calculated by two-way ANOVA with Tukey's post-test. WT on IgG has significantly more membranes than WT on PLL or εKO on either surface, which are not significantly different. ***P < 0.001.
Figure 5. PKC-ε re-expression rescues the spreading defect in εKO BMDM.

WT and εKO BMDM were (A) virally transduced with plasmids encoding GFP and PKC-ε-GFP or (B) nucleofected with plasmids encoding εRD-GFP and GFP and subjected to frustrated phagocytosis for 10 or 15 min (A and B). Area measurements were made from three experiments (100 total cells/time-point); results are reported as average spread area (μm2) ± sem. Statistical differences were calculated by two-way ANOVA with Tukey's post-test. *P < 0.1; **P < 0.01; ***P < 0.001.
PKC-ε mediates neurite extension in neuroblastoma cells, but the effect is independent of the catalytic domain [12, 33]. As phagocytosis and neurite extension are actin-based membrane-extension processes, we asked whether the catalytic domain of PKC-ε was necessary for frustrated phagocytosis in BMDM. Expression of the RD (εRD) alone did not restore spreading in εKO cells and blocked spreading in WT cells (Fig. 5B). This result, which is consistent with the observation that εRD blocks pseudopod extension in RAW cells [4], demonstrates that FcγR-dependent membrane mobilization requires full-length PKC-ε.
In conclusion, we have established that PKC-ε is necessary for membrane delivery in response to FcγR ligation. Additionally, we have confirmed in BMDM the results obtained using the RAW 264.7 cell line (i.e., that IgG-mediated phagocytosis is Ca2+-independent, that PKC-ε accumulates at targets during internalization and is necessary for efficient phagocytosis, that pseudopod-extension/membrane delivery is defective in the absence of PKC-ε, and that the catalytic domain of PKC-ε is involved). Given their relative ease of transfection, RAW 264.7 cells were used to probe the mechanism of PKC-ε recruitment to the phagosome.
εPS is required for PKC-ε translocation to the phagosome
PKC-ε translocation to phagosomes requires εC1B-DAG binding [6]. To determine whether εC1B is sufficient for translocation, we constructed reciprocal chimeras in which the C1B region of PKC-δ was replaced with that of PKC-ε and vice versa (Fig. 6A). The constructs were expressed and the cells subjected to synchronized phagocytosis. If sufficient, the presence of εC1B in (nonlocalizing) PKC-δ would produce a chimera that accumulated at phagosomes; the reciprocal chimera would lack the critical targeting domain and fail to concentrate. Substitution of εC1B for δC1B produced a chimera [δ(εC1B)] that did not accumulate (Fig. 6B) by visual inspection and by quantitation of phagosomally associated fluorescence (Fig. 6C). In contrast, the reverse chimera [ε(δC1B)] resembled PKC-ε in both localization pattern (Fig. 6B) and intensity (Fig. 6C). These results suggest that εC1B, although necessary [6], is not sufficient to bring PKC-δ to the phagosome.
As it has been shown that the V1 and PS regions of PKCs encode membrane-targeting information in other systems [34–36], we tested their involvement in FcγR-dependent PKC-ε translocation. Removal of εV1 did not significantly alter the intensity or pattern of concentration (Fig. 7B and C). In contrast, translocation was reduced significantly upon loss of εPS, by truncation (εΔV1PS) or deletion (εΔPS) (Fig. 7B and C). When compared, the LIs (based on the brightest pixel intensity in the phagosomes) of εΔV1PS and εΔPS were equivalent, both significantly lower than PKC-ε but higher than PKC-δ (Fig. 7C), suggesting that εPS is important in targeting PKC-ε to phagosomes.
Figure 7. The PS is required for PKC-ε translocation.
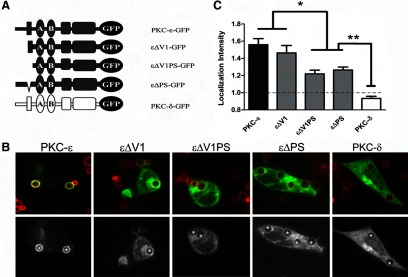
Cells were transfected with the indicated constructs (A) and treated as in Fig. 6. (B) Representative images showing red BIgG (upper); the same image represented in gray scale (lower). Asterisks designate phagosomes. (C) LI was calculated as detailed in Materials and Methods. Data are presented as mean ± sem (80 total events/construct; three to four experiments). Significance was determined by ANOVA with Tukey's post-test, *P < 0.05; **P < 0.01. Dotted line indicates no concentration.
A mutational approach was taken to further define the localization determinants in εPS. Scanning of the εPS sequence (aa 147–165) revealed that >50% of the constituent amino acids (10/19) are positively charged with six residues present in polybasic triplets. In contrast, the homologous region of PKC-δ (aa 136–154) has five basic residues with a single tandem RR sequence (Fig. 8A). Focusing on the polybasic triplets, we mutated ε154RKR156 and ε161RRR163 to AAA and quantified the concentration of the mutants during phagocytosis. Mutation of 161RRR163 had little effect on localization, suggesting that εPS mutation per se does not change the molecule dramatically (Fig. 8B and C). However, the mutant containing the 154AAA156 sequence failed to accumulate, confirming the εΔPS results and flagging this region as critical for PKC-ε targeting (Fig. 8B and C).
Figure 8. ε154RKR156 plays a role in PKC-ε phagosomal localization.
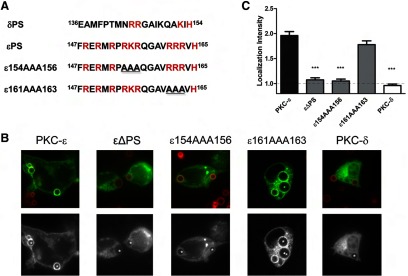
Macrophages were transfected with the indicated constructs (A) and subjected to synchronized phagocytosis. (B) ε161AAA163, but not ε154AAA156, concentrated at phagosomes; representative images showing red BIgG (upper) and the same image represented in gray scale (lower). Asterisks designate phagosomes. (C) LI was calculated as detailed in Materials and Methods. Data are presented as mean ± sem (60 total events from two experiments). Statistical significance was determined by ANOVA with Tukey's post-test. ***P < 0.001.
εPS confers phagosomal accumulation but cooperates with εC1B for optimal localization
Based on our findings that εPS and εC1B contain targeting information and that δ(εC1B) did not localize (Fig. 6) and knowing that εC1B and δC1B are ∼59% identical, we asked whether εPS plus any C1B was sufficient for localization. To answer this question, εPS was expressed in the context of PKC-δ [δ(εPS)] (Fig. 9A). In this chimera, εPS conferred a modest level of accumulation (Fig. 9B); localization was lower than WT PKC-ε but significantly higher than PKC-δ (Fig. 9Β). In contrast, the δ(εPSC1B) exhibited a LI similar to WT PKC-ε (Fig. 9B and C). That the εPSδC1AεC1B resembled full-length PKC-ε with respect to localization pattern and intensity (Fig. 9B and C) provides convincing evidence that εPS and εC1B are sufficient for phagosomal targeting.
Results with the δ(εPS) chimera suggest that εPS can cooperate with δC1B to facilitate phagosomal accumulation. However, the fact that δ(εPSC1B) localizes to a significantly greater extent than δ(εPS), which lacks εC1B, argues that εPS and εC1B synergize for optimal PKC targeting (Fig. 9B). In conclusion, the information contained within εPS is sufficient to target PKC-δ to the phagosome, but εPS and εC1B cooperate to convert the PKC-δ into a “PKC-ε-like” molecule. Thus, although a C1B domain is necessary for localization, δC1B is not functionally equivalent to εC1B.
The catalytic domain of PKC-ε is unique in its ability to facilitate phagocytosis
The RD of PKC-ε suppresses phagocytosis in RAW cells [4] and frustrated phagocytosis in BMDM (Fig. 5), implicating the catalytic domain in PKC-ε function. The high homology among the PKC catalytic regions raises the following questions. Is PKC-ε specifically required, or can any catalytic domain suffice, as long as it is properly targeted? If the PKC-ε catalytic domain is required, does it act enzymatically or structurally? Knowing that δ(εPSC1B) mimics PKC-ε with respect to localization, we asked whether expression of this chimera enhanced phagocytosis. If so, the critical domains for PKC-ε function are εPS and εC1B, and they act to deliver the catalytic domain to the phagosome. Alternatively, the PKC-ε catalytic domain may provide a signaling scaffold necessary for phagocytosis, with the requirement for structural integrity rather than catalytic activity. Finally, PKC-ε may phosphorylate isoform-specific targets required for internalization, and thus, its catalytic activity is required. To distinguish among these possibilities, we calculated internalization rates for two chimeras: δ(εPSC1B) and a chimera in which the RD of PKC-ε was fused to the catalytic domain of PKC-δ (εRDδCAT) (Fig. 10A). Despite the fact that the chimeras had localization patterns and intensities similar to PKC-ε (Figs. 9B and C and 10B and C) and higher overall expression levels (based on MFI by flow cytometry; Supplemental Table 1), the rates of internalization were significantly slower than PKC-ε (Fig. 10D). These data suggest that although εPS and εC1B are targeting domains, the PKC-ε catalytic domain is specifically required for efficient phagocytosis. To address the requirement for catalytic activity, RAW cells were transfected with a kinase dead PKC-ε (εK437W) [10]. Similar to the chimeras, εK437W-expressing cells internalized targets at a significantly reduced rate (Fig. 10D). Together, these data provide evidence that the catalytic activity of PKC-ε is critical for target internalization.
In summary, we have defined a role for PKC-ε in IgG-mediated phagocytosis—that of membrane mobilization in response to FcγR ligation. Concentration at the phagosome is critical for this function, and εPS and εC1B cooperate as minimal targeting elements. Finally, we have shown that the PKC-ε catalytic activity is unique in its ability to support efficient phagocytosis. Thus, with the use of data generated in WT and PKC-ε−/− BMDM and RAW 264.7 cells, we conclude that the role of PKC-ε in IgG-mediated phagocytosis is to phosphorylate substrates necessary for the delivery and/or fusion of vesicles into the nascent phagosome, providing the membrane necessary for pseudopod extension and target ingestion.
DISCUSSION
RAW cells and primary macrophages require PKC-ε for efficient phagocytosis
Much of the information that we have on the molecular mechanisms of phagocytosis derives from macrophage cell lines, predominantly the mouse macrophage-like line RAW 264.7. This is a result of the difficulty in molecular manipulation of primary macrophages. However, recent developments in viral transduction have made studies in primary cells more approachable. With the use of BMDM from WT and PKC-ε−/− mice, we confirmed the involvement of PKC-ε in IgG-mediated phagocytosis and identified its role in membrane mobilization (Fig. 4). Reconstitution experiments established that the membrane mobilization defect in PKC-ε−/− is a result of PKC-ε and that catalytic activity is important (Fig. 5).
PKC-ε and PKC-δ are homologous but not interchangeable
PKC is a component of virtually every known signaling pathway. It is a superfamily of eight closely related isoforms (α, βΙ, βΙΙ, γ, δ, ε, η, θ) and two more distant, atypical family members (ζ, ι/λ). PKCs are modular proteins composed of a RD and highly homologous catalytic domains. The efficacy of commercial PKC activity assays underscores the similarity in PKC substrate specificity. Why then would cells express multiple isoforms? Our results argue that there are isoform-specific differences in targeting, as well as substrate specificity, that are revealed in intact cell studies but not apparent by in vitro enzyme assays. Specifically, PKC-ε and PKC-δ are both members of the novel PKC subfamily, with an overall 42% identity and 61% homology. Despite their similarities, only PKC-ε accumulates at phagosomes. This targeting requires εPS and εC1B. As the C1Bs share 60% identity (78% homology) and bind DAG, it is not surprising that a PKC-ε chimera containing δC1B translocates (Fig. 6). However, the fact that εC1B could not induce the translocation of PKC-δ [the δ(εC1B) chimera; Fig. 6] implicated the involvement of (an)other domain(s). The logical domain was εV1, which binds β′-coatomer protein, a resident Golgi protein [34]; in resting cells, PKC-ε is often expressed at the Golgi (Figs. 9 and 10) [3, 4, 6]. Surprisingly, deletion of this domain had little effect on PKC-ε translocation; although localization of εΔV1 was always lower than PKC-ε, the differences did not reach statistical significance (Fig. 7C). This suggests that the docking partners for PKC-ε at the phagosome are different from those in the Golgi.
One criticism of transfection studies is the concern that effects may be artifacts of differential expression of plasmids. In our studies, differential expression might lead to localization (or not). Transfection efficiencies are variable; smaller plasmids tend to transfect better. To determine the relative expression of the constructs, we analyzed transfected cell populations by flow cytometry, quantifying GFP on a cell-by-cell basis (reported as MFI; Supplemental Table 1). Notably, percent GFP positivity did not correlate with localization. For example, unconjugated GFP had a MFI of 1267, PKC-ε was 711, and PKC-δ was 835. PKC-ε concentrated, but PKC-δ and GFP did not. Of particular interest are the εRDδCAT, δ(εPSC1B), and εK437W constructs. Their MFIs vary considerably (1878, 2187, and 551, respectively) with PKC-ε expression intermediate (711), but they concentrate with similar localization intensities (Fig. 10C), and all depressed phagocytic rate (Fig. 10D). It should be noted that LI is a measure of pixel intensity in the phagosomes normalized to that in an uninvolved region of the membrane. This strategy normalizes the intensity values, taking into account cell-to-cell variability in GFP expression.
The signals generated at the phagosome recruit PKC-ε but not the closely related PKC-δ. Truncation and deletion analyses (Fig. 7) implicate εPS, which bears no homology to δPS. However, there may be concern that domain deletion causes nonspecific changes in the molecule, resulting in failure to translocate. Thus, we followed up the deletion studies with εPS mutations. When scanning εPS, the obvious first approach for mutation targeted the polybasic triplets, which were mutated to AAA. Although these mutations equivalently altered the change of the domain, only 154AAA156 altered localization, suggesting that the charge per se did not impact localization. Thus, we have pinpointed a triplet of amino acids, some or all of which are involved in PKC-ε targeting.
When εPS is expressed in PKC-δ, the chimera did localize, significantly more than PKC-δ but much less than PKC-δ containing both εPS and εC1B) (Fig. 9C). From these data, we conclude that εPS and εC1B synergize for optimal targeting. However, εPS can interact with δC1B, as shown by the translocation of ε(δC1B) (Fig. 6C) and δ(εPS) (Fig. 9). In contrast, εC1B cannot act without εPS (Fig. 5) as its expression in PKC-δ produced a chimera (δ(εC1B)) that did not translocate (Fig. 6C). Finally, when combined, the fragment εPSδC1AεC1B concentrated with a pattern indistinguishable from that of full-length PKC-ε (Fig. 9B). δC1A was included as a physiological spacer in this fragment; C1A domains have very low affinity for lipids (ten- to 100-fold lower than εC1B) [37] and do not contribute to translocation of PKC-ε or PKC-δ [6]. The fact that the isolated εPSδC1AεC1B fragment translocated to phagosomes (Fig. 9C) confirms that εPS and εC1B are critical targeting domains and validates the results obtained with the deletion constructs and chimeras.
The catalytic domain of PKC-ε uniquely supports phagocytosis
Expression of εK437W did not facilitate phagocytosis (Fig. 10D), suggesting that catalytic activity is necessary for its function in phagocytosis. However, based on the sequence homology of PKC catalytic domains, we hypothesized that inducing translocation of PKC-δ by inserting εPS and εC1B would result in the efficient phagocytosis seen with full-length PKC-ε. Surprisingly, this was not the case (Fig. 10D). As localization of δ(εPSC1B) was similar to PKC-ε (Fig. 10C), and expression was higher than PKC-ε (Supplemental Table 1), the defect was not in translocation. It is possible that the chimeric RD might have an altered conformation (although this is unlikely to decrease activity, as the active site would be exposed as a result of the absence of δPS). To address this, we made a chimera with the PKC-ε regulatory and PKC-δ catalytic domains and established that this fusion protein was also unable to enhance phagocytosis (Fig. 10D). As the PKC-δ catalytic domain is constitutively active when freed of its RD [10], it is not that PKC-δ is catalytically inactive but more likely, that the PKC-ε catalytic domain is unique in its ability to facilitate phagocytosis. This finding is consistent with an earlier report that unlike the other PKCs, PKC-ε is a poor substrate for histone-IIIs in in vitro assays and thus, is likely to have unique substrate specificity [38].
Role for PKC-ε in membrane mobilization
Scanning electron microscopy has shown that macrophages have ruffled membranes [39]. Thus, it could be argued that the defect in εKO cells is a result of their failure to flatten out ruffles. Although Fig. 3 shows that the size of the black holes made by εKO cells increases with time, capacitance measurements (Fig. 4) reveal that the amount of plasma membrane does not. The fact that the εKO “cellprint” increases without a corresponding increase in cell membrane is consistent with cell flattening. Thus, εKOs have some plasma membrane that they can recruit for limited phagocytosis, consistent with published data that inhibition/deletion of PKC-ε decreases, but does not abolish, phagocytosis [4]. However, at some point during phagocytosis (presumably when membrane ruffles have been exhausted), cells must access internal membranes. This is where PKC-ε comes into play.
On IgG surfaces, WT cells are significantly larger (Fig. 3) and have a higher capacitance (Fig. 4) than those attached to PLL. That εKO cells have the same capacitance on both surfaces, and this was the same as WT on PLL (Fig. 4) suggests that FcγR ligation transduces a membrane delivery signal. The internal membranes are most likely the VAMP3+ vesicles shown to insert at the base of phagocytic cups [18]. Although VAMP3 is not required for phagocytosis [40], it does facilitate the release of TNF-α from LPS-stimulated macrophages [41]. TNF-α-loaded vesicles originate in the golgi, acquireVAMP3 by fusion with recycling endosomes, and then fuse into the phagocytic cup [41]. As PKC-ε is present in the golgi (Figs. 6–10), it is plausible that it travels on the TNF-α vesicles and mediates their fusion into the base of the phagocytic cup [18]. Based on the fact that PKC-ε catalytic activity is required (Fig. 5), a likely scenario is that PKC-ε phosphorylates substrate(s) involved in fusion of VAMP3+ vesicles. Whereas specific substrates remain to be identified, the iPLA2 is a likely candidate. iPLA2 is activated by a novel PKC [42], releases arachidonic acid—a known fusogen [43]—and is downstream of PKC in the FcγR signaling cascade [44, 45], and its inhibition results in the accumulation of vesicles beneath bound targets [43].
Model for PKC-ε translocation and membrane mobilization
Based on the εPS sequence, polybasic with intervening hydrophobic amino acids (Fig. 4), the phagosomal docking partner is likely a phosphoinositide [46]. Indeed, others have reported that peptides resembling the PKC-β PS bind acidic lipids [47] and that the PKC-ε RD binds phosphoinositides [48]. The finding that PKC-ε is necessary for membrane delivery (Fig. 4) parallels results showing that the spread area of macrophages and pseudopod extension is decreased by PI3K inhibitors [39, 49]; the requirement for εPS provides the link between these findings.
A model that fits the data is that a PI3K product [e.g., PI(3,4)P2, PI(3,4,5)P3] [50] is produced at the phagosome and binds εPS. Together with the DAG that is generated by PLCγ1 and binds εC1B [6], εPS/PIP binding conforms to a “tether and fix” mechanism for PKC-ε activation (note that this term was used originally by Rhee and Bae [51] to describe PLCδ activation). Briefly, DAG/εC1B tethers PKC-ε to target membranes. PI3K phosphoinositide phosphorylation produces binding site(s) for εPS. The binding of εPS to this PIP exposes the active site for substrate phosphorylation and signal propagation (see the “foot in mouth” model of Mosior and McLaughlin [47] for the Ca-dependent PKCs). Active PKC-ε then phosphorylates iPLA2, which releases the arachidonic acid that facilitates the fusion of the VAMP3+ vesicles for pseudopod extension [18].
The caveat to this model is that it has been reported that phagocytosis of small particles is insensitive to PI3K activity [49]. We used 2 μm IgG beads, which are similar in size to the 3-μm latex beads used for the VAMP3 studies [18]. Several possibilities present themselves: the εPS docking lipid is not a product of PI3K, and/or the εPS docking lipid is produced by an enzyme insensitive to classical PI3K inhibitors. Alternatively, the differences may arise from the approach. The published studies were endpoint assays, quantifying total internalization after 30 min. As PKC-ε affects the rate of internalization, it is possible that by 30 min, the cells have reached a plateau, and despite a slower rate, the KO cells have reached the same point. Only when target size is increased (or during frustrated phagocytosis) is the demand for internal membrane too great for the PKC-ε−/− cells to overcome. The answers to these questions await identification of the εPS binding partner and the PKC-ε substrate(s), the next phase of these studies.
Supplementary Material
ACKNOWLEDGMENTS
The study was supported by U.S. National Institutes of Health grants AI50821 and GM090325 (to M.R.L.).
The authors acknowledge Alex Granick for preliminary BMDM results, Dr. Keylon Cheeseman for performing preliminary RAW cells studies, William Frederick Hynes for silane-activated surfaces, Elaine Raines and lab for guidance on viral constructs and production of pseudovirions, Dr. Paul Feustel for assistance with statistical analyses, and Drs. Daniel J. Loegering and Erin Harmon for helpful discussions. Finally, many thanks go to Rosemary Naftalis for all of her work on the images and help with formatting the manuscript. This paper is dedicated to the memory of William L. Sheets.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- δ(εPS)
- PKC-δ containing εPS
- δ(εPSC1B)
- PKC-δ chimera containing εPS and εC1B
- δ(εV1PSC1B)
- PKC-δ containing εV1, εPS, and εC1B
- δCAT
- catalytic domain of PKC-δ
- εK437W
- catalytically inactive PKC-ε
- εKO
- PKC-ε knockout mice
- εPSδC1AεC1B
- PKC fragment containing εPS and εC1B linked by δC1A
- ACK
- ammonium-chloride-potassium
- APC
- allophycocyanin
- BIgG
- IgG-opsonized glass beads
- BM
- bone marrow
- BMDM
- bone marrow-derived macrophages
- iPLA2
- calcium-independent PLA2
- KO
- knockout
- LI
- localization intensity
- MFI
- mean fluorescence intensity
- PI
- phagocytic index
- PIP
- phosphatidylinositol phosphate
- PKC-ε−/−
- PKC-ε-deficient (knockout)
- PLL
- poly-l-lysine
- PS
- pseudosubstrate
- RAW cells
- RAW LacR/FMLPR.2 mouse macrophage-like cell line
- RD
- regulatory domain
- V1
- variable region
- VAMP
- vesicle-associated membrane protein
AUTHORSHIP
T.R.W., R.Y.C., and C.M.H. performed and analyzed experiments, prepared figures, and performed statistical analyses. C.M.H., K.K., Y.S., and N.S. constructed plasmids and contributed to the design of experiments. X.Z. and M.T. performed patch-clamping and assisted with its analysis. D.J.L. assisted with design of experiments, discussion of work in progress, and writing the manuscript. M.R.L. conceived of the project, was responsible for the design of the experiments, assisted in analysis of results, and wrote the manuscript.
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Cox D., Chang P., Zhang Q., Reddy P. G., Bokoch G. M., Greenberg S. (1997) Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 186, 1287–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garcia-Garcia E., Rosales C. (2002) Signal transduction during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 72, 1092–1108 [PubMed] [Google Scholar]
- 3. Larsen E. C., DiGennaro J. A., Saito N., Matha S., Loegering D. J., Mazurkiewicz J. M., Lennartz M. R. (2000) Differential requirement for classic and novel PKC isoforms in respiratory burst and phagocytosis in RAW 264.7 cells. J. Immunol. 165, 2809–2817 [DOI] [PubMed] [Google Scholar]
- 4. Larsen E. C., Ueyama T., Brannock P. M., Shirai Y., Saito N., Larsson C., Loegering D., Weber P. B., Lennartz M. R. (2002) A role for PKC-varε in FcγR-mediated phagocytosis by RAW 264.7 cells. J. Cell Biol. 159, 939–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fronhofer V., Lennartz M. R., Loegering D. J. (2006) Role of PKC isoforms in the Fc(γ)R-mediated inhibition of LPS-stimulated IL-12 secretion by macrophages. J. Leukoc. Biol. 79, 408–415 [DOI] [PubMed] [Google Scholar]
- 6. Cheeseman K. L., Ueyama T., Michaud T. M., Kashiwagi K., Wang D., Flax L. A., Shirai Y., Loegering D. J., Saito N., Lennartz M. R. (2006) Targeting of PKC-ε during FcγR-dependent phagocytosis requires the εC1B domain and phospholipase C-γ1. Mol. Biol. Cell 17, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corbalan-Garcia S., Gomez-Fernandez J. C. (2006) Protein kinase C regulatory domains: the art of decoding many different signals in membranes. Biochim. Biophys. Acta 1761, 633–654 [DOI] [PubMed] [Google Scholar]
- 8. Colon-Gonzalez F., Kazanietz M. G. (2006) C1 domains exposed: from diacylglycerol binding to protein-protein interactions. Biochim. Biophys. Acta 1761, 827–837 [DOI] [PubMed] [Google Scholar]
- 9. Newton A. C. (1997) Regulation of protein kinase C. Curr. Opin. Cell Biol. 9, 161–167 [DOI] [PubMed] [Google Scholar]
- 10. Steinberg S. F. (2008) Structural basis of protein kinase C isoform function. Physiol. Rev. 88, 1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oka M., Hitomi T., Okada T., Nakamura Si S., Nagai H., Ohba M., Kuroki T., Kikkawa U., Ichihashi M. (2002) Dual regulation of phospholipase D1 by protein kinase C α in vivo. Biochem. Biophys. Res. Commun. 294, 1109–1113 [DOI] [PubMed] [Google Scholar]
- 12. Zeidman R., Lofgren B., Pahlman S., Larsson C. (1999) PKCε, via its regulatory domain and independently of its catalytic domain, induces neurite-like processes in neuroblastoma cells. J. Cell Biol. 145, 713–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walker S. D., Murray N. R., Burns D. J., Fields A. P. (1995) Protein kinase C chimeras: catalytic domains of α and β II protein kinase C contain determinants for isotype-specific function. Proc. Natl. Acad. Sci. USA 92, 9156–9160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Acs P., Wang Q. J., Bogi K., Marquez A. M., Lorenzo P. S., Biro T., Szallasi Z., Mushinski J. F., Blumberg P. M. (1997) Both the catalytic and regulatory domains of protein kinase C chimeras modulate the proliferative properties of NIH 3T3 cells. J. Biol. Chem. 272, 28793–28799 [DOI] [PubMed] [Google Scholar]
- 15. Lee W. L., Mason D., Schreiber A. D., Grinstein S. (2007) Quantitative analysis of membrane remodeling at the phagocytic cup. Mol. Biol. Cell 18, 2883–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Steinman R. M., Bordie S. E., Cohn Z. A. (1976) Membrane flow during pinocytosis: a stereologic analysis. J. Cell Biol. 68, 665–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cannon G. J., Swanson J. A. (1992) The macrophage capacity for phagocytosis. J. Cell Sci. 101, 907–913 [DOI] [PubMed] [Google Scholar]
- 18. Bajno L., Peng X. R., Schreiber A. D., Moore H. P., Trimble W. S., Grinstein S. (2000) Focal exocytosis of VAMP3-containing vesicles at sites of phagosome formation. J. Cell Biol. 149, 697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Akita Y. (2008) Protein kinase Cε: multiple roles in the function of, and signaling mediated by, the cytoskeleton. FEBS J. 275, 3995–4004 [DOI] [PubMed] [Google Scholar]
- 20. Tachado S. D., Mayhew M. W., Wescott G. G., Foreman T. L., Goodwin C. D., McJilton M. A., Terrian D. M. (2002) Regulation of tumor invasion and metastasis in protein kinase C ε-transformed NIH3T3 fibroblasts. J. Cell. Biochem. 85, 785–797 [DOI] [PubMed] [Google Scholar]
- 21. Hafeez B. B., Zhong W., Weichert J., Dreckschmidt N. E., Jamal M. S., Verma A. K. (2011) Genetic ablation of PKC ε inhibits prostate cancer development and metastasis in transgenic mouse model of prostate adenocarcinoma. Cancer Res. 71, 2318–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park Y. S., Hur E. M., Choi B. H., Kwak E., Jun D. J., Park S. J., Kim K. T. (2006) Involvement of protein kinase C-ε in activity-dependent potentiation of large dense-core vesicle exocytosis in chromaffin cells. J. Neurosci. 26, 8999–9005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jerdeva G. V., Yarber F. A., Trousdale M. D., Rhodes C. J., Okamoto C. T., Dartt D. A., Hamm-Alvarez S. F. (2005) Dominant-negative PKC-epsilon impairs apical actin remodeling in parallel with inhibition of carbachol-stimulated secretion in rabbit lacrimal acini. Am. J. Physiol. Cell. Physiol. 289, C1052–C1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mendez C. F., Leibiger I. B., Leibiger B., Hoy M., Gromada J., Berggren P. O., Bertorello A. M. (2003) Rapid association of protein kinase C-ε with insulin granules is essential for insulin exocytosis. J. Biol. Chem. 278, 44753–44757 [DOI] [PubMed] [Google Scholar]
- 25. Shirai Y., Kashiwagi K., Yagi K., Sakai N., Saito N. (1998) Distinct effects of fatty acids on translocation of γ- and ε-subspecies of protein kinase C. J. Cell Biol. 143, 511–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ohmori S., Shirai Y., Sakai N., Fujii M., Konishi H., Kikkawa U., Saito N. (1998) Three distinct mechanisms for translocation and activation of the δ subspecies of protein kinase C. Mol. Cell. Biol. 18, 5263–5271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kashiwagi K., Shirai Y., Kuriyama M., Sakai N., Saito N. (2002) Importance of C1B domain for lipid messenger-induced targeting of protein kinase C. J. Biol. Chem. 277, 18037–18045 [DOI] [PubMed] [Google Scholar]
- 28. Hitoshi Y., Lorens J., Kitada S. I., Fisher J., LaBarge M., Ring H. Z., Francke U., Reed J. C., Kinoshita S., Nolan G. P. (1998) Toso, a cell surface, specific regulator of Fas-induced apoptosis in T cells. Immunity 8, 461–471 [DOI] [PubMed] [Google Scholar]
- 29. Gough P. J., Raines E. W. (2003) Gene therapy of apolipoprotein E-deficient mice using a novel macrophage-specific retroviral vector. Blood 101, 485–491 [DOI] [PubMed] [Google Scholar]
- 30. McNeil P. L., Swanson J. A., Wright S. D., Silverstein S. C., Taylor D. L. (1986) Fc-receptor-mediated phagocytosis occurs in macrophages without an increase in average [Ca++]i. J. Cell Biol. 102, 1586–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berrier A. L., Mastrangelo A. M., Downward J., Ginsberg M., LaFlamme S. E. (2000) Activated R-ras, Rac1, PI 3-kinase and PKCε can each restore cell spreading inhibited by isolated integrin β1 cytoplasmic domains. J. Cell Biol. 151, 1549–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heuser J. (2000) The production of ‘cell cortices’ for light and electron microscopy. Traffic 1, 545–552 [DOI] [PubMed] [Google Scholar]
- 33. Zeidman R., Troller U., Raghunath A., Pahlman S., Larsson C. (2002) Protein kinase C-ε actin-binding site is important for neurite outgrowth during neuronal differentiation. Mol. Biol. Cell 13, 12–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Csukai M., Chen C. H., De Matteis M. A., Mochly-Rosen D. (1997) The coatomer protein β′-COP, a selective binding protein (RACK) for protein kinase Cε. J. Biol. Chem. 272, 29200–29206 [DOI] [PubMed] [Google Scholar]
- 35. Jose Lopez-Andreo M., Gomez-Fernandez J. C., Corbalan-Garcia S. (2003) The simultaneous production of phosphatidic acid and diacylglycerol is essential for the translocation of protein kinase Cε to the plasma membrane in RBL-2H3 cells. Mol. Biol. Cell 14, 4885–4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maissel A., Marom M., Shtutman M., Shahaf G., Livneh E. (2006) PKCη is localized in the Golgi, ER and nuclear envelope and translocates to the nuclear envelope upon PMA activation and serum-starvation: C1b domain and the pseudosubstrate containing fragment target PKCη to the Golgi and the nuclear envelope. Cell. Signal. 18, 1127–1139 [DOI] [PubMed] [Google Scholar]
- 37. Irie K., Nakahara A., Nakagawa Y., Ohigashi H., Shindo M., Fukuda H., Konishi H., Kikkawa U., Kashiwagi K., Saito N. (2002) Establishment of a binding assay for protein kinase C isozymes using synthetic C1 peptides and development of new medicinal leads with protein kinase C isozyme and C1 domain selectivity. Pharmacol. Ther. 93, 271–281 [DOI] [PubMed] [Google Scholar]
- 38. Schaap D., Parker P. J., Bristol A., Kriz R., Knopf J. (1989) Unique substrate specificity and regulatory properties of PKC-ε: a rationale for diversity. FEBS Lett. 243, 351–357 [DOI] [PubMed] [Google Scholar]
- 39. Araki N., Johnson M., Swanson J. (1996) A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 135, 1249–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Allen L. A., Yang C., Pessin J. E. (2002) Rate and extent of phagocytosis in macrophages lacking VAMP3. J. Leukoc. Biol. 72, 217–221 [PMC free article] [PubMed] [Google Scholar]
- 41. Murray R. Z., Kay J. G., Sangermani D. G., Stow J. L. (2005) A role for the phagosome in cytokine secretion. Science 310, 1492–1495 [DOI] [PubMed] [Google Scholar]
- 42. Steer S. A., Wirsig K. C., Creer M. H., Ford D. A., McHowat J. (2002) Regulation of membrane-associated iPLA2 activity by a novel PKC isoform in ventricular myocytes. Am. J. Physiol. Cell. Physiol. 283, C1621–C1626 [DOI] [PubMed] [Google Scholar]
- 43. Lennartz M. R., Yuen A. F., Masi S. M., Russell D. G., Buttle K. F., Smith J. J. (1997) Phospholipase A2 inhibition results in sequestration of plasma membrane into electronlucent vesicles during IgG-mediated phagocytosis. J. Cell Sci. 110, 2041–2052 [DOI] [PubMed] [Google Scholar]
- 44. Karimi K., Gemmill T. R., Lennartz M. R. (1999) Protein kinase C and a calcium-independent phospholipase are required for IgG-mediated phagocytosis by Mono-Mac-6 cells. J. Leukoc. Biol. 65, 854–862 [DOI] [PubMed] [Google Scholar]
- 45. Karimi K., Lennartz M. R. (1995) Protein kinase C activation precedes arachidonic acid release during IgG-mediated phagocytosis. J. Immunol. 155, 5786–5794 [PubMed] [Google Scholar]
- 46. Heo W. D., Inoue T., Park W. S., Kim M. L., Park B. O., Wandless T. J., Meyer T. (2006) PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314, 1458–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mosior M., McLaughlin S. (1991) Peptides that mimic the pseudosubstrate region of protein kinase C bind to acidic lipids in membranes. Biophys. J. 60, 149–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shirai Y., Murakami T., Kuramasu M., Iijima L., Saito N. (2007) A novel PIP2 binding of εPKC and its contribution to the neurite induction ability. J. Neurochem. 102, 1635–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cox D., Tseng C. C., Bjekic G., Greenberg S. (1999) A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J. Biol. Chem. 274, 1240–1247 [DOI] [PubMed] [Google Scholar]
- 50. Kamen L. A., Levinsohn J., Swanson J. A. (2007) Differential association of phosphatidylinositol 3-kinase, SHIP-1, and PTEN with forming phagosomes. Mol. Biol. Cell 18, 2463–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rhee S. G., Bae Y. S. (1997) Regulation of phosphoinositide-specific phospholipase C isozymes. J. Biol. Chem. 272, 15045–15048 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.