

Abstract
We report here the self-assembly of a rationally designed paclitaxel drug amphiphile into well-defined supramolecular filaments that possess a fixed 41% paclitaxel loading. These filaments can exert effective cytotoxicity against a number of cell lines comparable to that of free paclitaxel.
The design versatility and potential bioactive features, coupled with routine synthetic methods, have enabled peptides to become one of the most studied molecular building units for the creation of supramolecular biomaterials over the past two decades.1–8 Given their inherent biodegradable properties, peptide-based nanostructures and materials (e.g. hydrogels) have served as delivery vectors for cells,9 proteins,10 oligonucleotides,11 and anticancer drugs.12 Unlike cells, proteins and other water soluble molecules that can be easily incorporated into peptidic hydrogels, hydrophobic anticancer drugs must be loaded into the supramolecular nanostructure. Physical encapsulation into the hydrophobic compartments often results in very low drug loading contents, typically on the order of 2–5% by weight. The concept of covalently linking anticancer drugs to peptides has been explored for more than a decade now,13–15 with a majority of the work focused on the use of water soluble peptides, such as Tat or penetratin, to circumvent multidrug resistance. The resulting conjugates are often water soluble and do not present any self-assembling features. Recently, Xu and co-workers pioneered the rational design of a paclitaxel-peptide conjugate that can be triggered by enzymatic reactions to form hydrogels.16,17 This concept was later utilized by Yang and others to design a range of paclitaxel-based peptide hydrogelators.18,19 These hydrogelators show remarkable potential to improve cancer treatments through local delivery pathways. For systemic delivery, however, it is desirable to construct discrete and stable nanostructures of well-defined size and shape. We recently reported the concept of constructing self-assembling drug amphiphiles by conjugating a short peptide to varying numbers of the anticancer drug camptothecin, achieving control over both the self-assembled nanostructures and the quantitative drug loading per nanostructure.20 In this communication, we report the synthesis and assembly of a drug amphiphile (DA) containing a very bulky anticancer drug, paclitaxel (PTX). Notably, we found that conjugation of the bulky PTX to a short peptide does not prevent the peptide from assembling into one-dimensional nanostructures, and the conjugated peptide does not compromise the drug’s potency against a number of cancer cell lines.
Scheme 1A shows the chemical structure of the reported PTX DA containing three essential elements: the anticancer drug PTX, the β-sheet forming peptide, and the biodegradable linker. First, PTX serves as the hydrophobic segment in the design because of its very low water solubility. The use of nanoscale carriers to modify PTX’s pharmacokinetic profiles presents an effective strategy to improve PTX’s efficacy while minimizing possible side effects.21–23 However, the PTX loading capacity is very limited in these nanocarriers (typically below 5%),21–23 and varies from carriers to carriers. Nanoscale vehicles capable of delivering PTX with a high content and in a quantitative manner have yet to be developed. Second, the peptide VQIVYK is derived from the microtubule stabilizing Tau protein, and has been shown capable of forming filamentous nanostructures rich in β-sheets.24 We postulated that conjugation of the Tau peptide to PTX would lead the conjugated PTX to assemble into filamentous nanostructures (Scheme 1B). One extra lysine was added to the C-terminal of the Tau peptide to enhance the conjugate’s water solubility for the purposes of both controlled assembly and purification. Third, 4-(pyridin-2-yl-disulfanyl) butyrate (buSS) was chosen as the biodegradable linker to bridge the peptide and the drug. This disulfide linker breaks down in the presence of glutathione (GSH), a reducing agent that resides in the cytosol and is known to have an intracellular concentration 1000-fold higher than that in plasma.15 This linker was attached to the 2′-hydroxyl as studies have shown that esterification at this position results in considerable loss of activity, whereas reaction at the 7-hydroxyl has little effect.25 By attaching at the 2′-hydroxyl, the conjugate will have limited bioactivity until the payload is released.
Scheme 1.
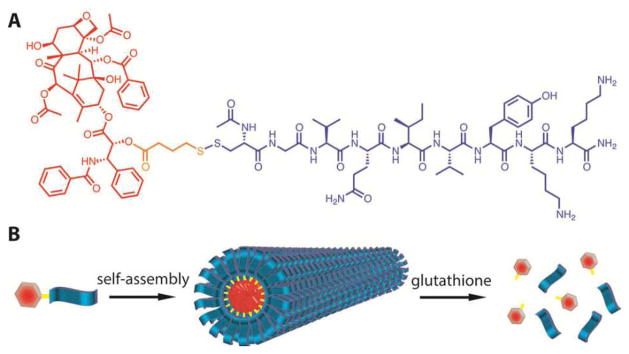
(A) Chemical structure of the designed PTX-Tau DA, and (B) schematic illustration of its assembly into supramolecular filaments. The drug content in the assembled filaments is the same as the molecular fraction of PTX in the DA (41%) which is defined by the mass of PTX divided by the total molecular weight of the conjugate. This drug loading can be tuned in a quantitative way by varying the peptide length.
The synthesis of PTX-Tau DA consists of three steps (details in the ESI†). In brief, the peptide segment was first synthesized on an automatic peptide synthesizer using standard Fmoc solid phase synthesis protocols. Second, the activated disulfide paclitaxel ester was synthesized by reaction of paclitaxel with 4-(pyridin-2-yl-disulfanyl) butyric acid using a previously reported method.6 The last step involves the coupling of the two components using a directed disulfide bond formation reaction. The final product was purified using preparative HPLC, and its expected mass and purity were confirmed using mass spectrometry and analytical HPLC, respectively.
The self-assembly of the PTX-Tau conjugate can be promoted by directly dissolving a predetermined amount of the molecule into deionized water or phosphate buffered saline. Figures 1A and 1B show two representative transmission electron microscopy (TEM) images taken from a 200 μM aqueous solution after aging for 48 h, clearly revealing dominant filamentous nanostructures. This morphology was further confirmed by atomic force microscopy (Fig. S6 in ESI†) and cryogenic TEM imaging (Fig. S7 in ESI†). These observed nanofibers are typically a few micrometers in length and have a diameter of 11.8 ± 1.3 nm. Given the amphiphilic nature of the designed molecule with its fully extended length of ~5 nm, it is reasonable to assume that the molecules within the observed nanostructures are packed with a cylindrical geometry, similar to those observed in other peptide-based amphiphile systems.8 With respect to their bulky three-dimensional nature, it is remarkable that the PTX molecules can pack together inside these nanostructures.
Fig. 1.

(A) and (B) Representative TEM images of PTX-Tau (200 μM in H2O) showing filamentous nanostructures; (C) Fluorescence measurment to determine the CMC value of PTX-Tau based on Nile Red encapsulation within the assembled nanostructures; (D) Circular dichroism spectrum of 5μM PTX-Tau in water.
The critical micellization concentration (CMC) was evaluated using the solvatochromic fluorescent dye, Nile Red.26 Due to its low water solubility, Nile Red can partition into the hydrophobic compartment of assembled nanostructures, leading to changes in both fluorescence intensity and emission maximum. Figure 1C shows a plot of PTX DA concentration versus Nile Red fluorescence intensity and maximum emission wavelength, suggesting a CMC value around 10 μM. However, circular dichroism (CD) measurement (Figure 1D) can still reveal the characteristic absorption of β-sheets at 216 nm even below the measured CMC value, suggesting that some of the PTX DA are present in the β-sheet form below the detected CMC. It is likely that these non-micellar β-sheets do not have the ability to encapsulate Nile Red. The negative absorption at 291 nm with a shoulder around 268 nm, and the positive absorption at 233 nm are all attributed to the PTX chromophores associated with different asymmetric centres. The CD absorption of the PTX-Tau DA observed here is remarkably similar to that of free PTX in non-polar solvents.27 However, it is unclear if intermolecular hydrogen bonding exists among the PTX molecules within the nanofiber cores and how the β-sheet formation among the Tau peptides contributes to the PTX absorption peaks in CD.
Since release of the active compound represents a critical step for prodrugs to exert the desired function, experiments to determine PTX release were carried out in PBS buffer at 37 °C in the presence or absence of 10 mM GSH (Fig. 2). As expected, release of PTX-Tau at 5 μM (below its CMC) showed faster release rates than those above (100 μM). For example, after 2 h incubation, a 5 μM solution of PTX-Tau released 70% of the available PTX, while only 40% was released from a 100 μM solution. Similarly, a faster release rate under GSH-free conditions, in which hydrolysis of the PTX ester bond is solely responsible for release, was observed at 5 μM compared with 100 μM. Since the PTX DA is expected to exist in the monomeric form at 5 μM (with some non-micellar β-sheets) but in the assembled form at 10 μM, the release data in Fig. 2 clearly suggest that formation of nanostructure provides a mechanism to regulate the drug release kinetics. It is very likely that in the assembled form the degradable linker is deeply embedded inside the filaments inaccessible to GSH reduction, thus affording some protection from both hydrolysis and reduction.
Fig. 2.

Paclitaxel release study of PTX-Tau in PBS buffer at 37 °C at two concentrations (5 and 100 μM). Solutions were incubated in the prsence or absence of 10 mM GSH. Data are given as mean ± s.d. (n = 3). For samples treated with 10 mM GSH, the remaining conjugates were no more detectable by analytical HPLC after 4 h for the 5 uM solution, and 48 h for the 100 uM solution.
The in vitro cytotoxicity of PTX-Tau was assessed and compared to free PTX against three cancer cell lines, including MCF-7 human breast cancer, A549 non-small cell lung cancer, and PC3-flu human prostate cancer cell lines (Figs. 3 and S8 in ESI†). Cells were incubated for 48 h with varying PTX and PTX-Tau concentrations and cell viability was determined by sulforhodamine B (SRB) assay. For all three cell lines tested, PTX-Tau showed near-identical toxicity to free PTX, indicating that the conjugation of PTX to Tau peptide does not compromise PTX’s potency. This observed comparable in vitro efficacy is impressive given the fact that the PTX DA need to be first cleaved by intracellular GSH and then go through a hydrolysis process to release the bioactive PTX for the desired toxicity. This delay in release from prodrugs often increases the drug’s IC50 value and reduces the drug’s in vitro efficacy compared to the free drug. We speculate that in our system the Tau peptide may facilitate the intracellular accumulation of the PTX DA, and the internalized PTX DA can be quickly converted to the active PTX. Our release study (Fig. 2) has shown that PTX can be effectively liberated from the unassembled DA in the presence of GSH.
Fig. 3.

Cytotoxicity evaluation of PTX-Tau against MCF-7 breast cancer cells, A549 non-small cell lung cancer cells, and PC-3 FLU prostate cancer cells. Dose–response study of PTX-Tau and free PTX against MCF-7 (A), A549 (D), and PC-3 FLU (G). Cells were incubated with the PTX or PTX-Tau for 48 h and cell viability was determined by SRB assay. Data are given as mean ± s.d. (n = 3). Live/dead assay cell images of MCF-7 (B, C), A549 (E, F), and PC-3 FLU (H, I) cancer cells after 48 h incubation with DMSO (B, E, H), PTX (see Fig. S8 in ESI†) or PTX-Tau (C, F, I).
To further demonstrate the efficacy of the synthesized DA, live/dead assays were performed. After 48 h exposure to PTX or PTX-Tau (both 50 nM), MCF-7 tumour cells were treated with calcein-AM and EthD-1 to give green or red fluorescence indicating live or dead cells, respectively. DMSO was utilized as a negative control (Fig. 3B), showing no significant inhibition in cell growth. After treatment with PTX or PTX-Tau (Fig. 3C), the cell populations were significantly reduced with individual cells appearing larger in size, a morphological change that is characteristic of cells in G2-M arrest caused by PTX inhibition of mitosis. Similar behaviour was observed for live/dead assays performed using the A549 and PC3-flu cell lines (Figs. 3D, 3E, 3H and 3I, and Fig. S8 in ESI†). These observations provide further evidence that the active agent released from PTX-Tau shows no difference in behaviour to that of free PTX.
In summary, we have synthesized a new amphiphilic PTX-peptide conjugate with a high and fixed drug content that has significantly improved solubility. Self-assembly into filaments imparts improved stability, and our in vitro cell experiments reveal that the bioactive payload can be effectively released with no detrimental effect on its ability to inhibit cell proliferation.
We thank National Science Foundation (DMR 1255281) for support of the project, The Johns Hopkins University (JHU) for the Startup Fund, and NIH for funding A.C. (T-32CA130840) and Y.L. (R25CA153952). We thank the JHU Integrated Imaging Center (IIC) for the use of the TEM facility, and the JHU Department of Chemistry Mass Spectrometry facility for MALDI-ToF (NSF CHE-0840463) and ESI analysis. We also thank Prof. Kalina Hristova (MSE, JHU) for the use of CD and fluorescence instrumentation.
Supplementary Material
Footnotes
Electronic Supplementary Information (ESI) available: experimental methods, characterization data, cell studies See DOI: 10.1039/b000000x/
Notes and references
- 1.Hartgerink JD, Beniash E, Stupp SI. Proc Natl Acad Sci USA. 2002;99:5133. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Branco MC, Sigano DM, Schneider JP. Curr Opin Chem Biol. 2011;15:427. doi: 10.1016/j.cbpa.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jonker AM, Lowik D, van Hest JCM. Chem Mater. 2012;24:759. [Google Scholar]
- 4.Trent A, Marullo R, Lin B, Black M, Tirrell M. Soft Matter. 2011;7:9572. [Google Scholar]
- 5.Zelzer M, Ulijn RV. Chem Soc Rev. 2010;39:3351. doi: 10.1039/c0cs00035c. [DOI] [PubMed] [Google Scholar]
- 6.Zhang SG. Accounts Chem Res. 2012;45:2142. doi: 10.1021/ar300034v. [DOI] [PubMed] [Google Scholar]
- 7.Fallas JA, O’Leary LER, Hartgerink JD. Chemical Society Reviews. 2010;39:3510. doi: 10.1039/b919455j. [DOI] [PubMed] [Google Scholar]
- 8.Cui HG, Webber MJ, Stupp SI. Biopolymers. 2010;94:1. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, Lamm MS, Pochan DJ, Schneider JP. Proc Natl Acad Sci USA. 2007;104:7791. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thornton PD, Mart RJ, Webb SJ, Ulijn RV. Soft Matter. 2008;4:821. doi: 10.1039/b714750c. [DOI] [PubMed] [Google Scholar]
- 11.Bulut S, Erkal TS, Toksoz S, Tekinay AB, Tekinay T, Guler MO. Biomacromolecules. 2011;12:3007. doi: 10.1021/bm200641e. [DOI] [PubMed] [Google Scholar]
- 12.Soukasene S, Toft DJ, Moyer TJ, Lu HM, Lee HK, Standley SM, Cryns VL, Stupp SI. ACS Nano. 2011;5:9113. doi: 10.1021/nn203343z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagy A, Schally AV, Halmos G, Armatis P, Cai RZ, Csernus V, Kovacs M, Koppan M, Szepeshazi K, Kahan Z. Proc Natl Acad Sci USA. 1998;95:1794. doi: 10.1073/pnas.95.4.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazel M, Clair P, Rousselle C, Vidal P, Scherrmann JM, Mathieu D, Temsamani J. Anti-Cancer Drug. 2001;12:107. doi: 10.1097/00001813-200102000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Dubikovskaya EA, Thorne SH, Pillow TH, Contag CH, Wender PA. Proc Natl Acad Sci USA. 2008;105:12128. doi: 10.1073/pnas.0805374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao Y, Kuang Y, Guo ZF, Guo ZH, Krauss IJ, Xu B. J Am Chem Soc. 2009;131:13576. doi: 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]
- 17.Zhao F, Ma ML, Xu B. Chem Soc Rev. 2009;38:883. doi: 10.1039/b806410p. [DOI] [PubMed] [Google Scholar]
- 18.Mao LN, Wang HM, Tan M, Ou LL, Kong DL, Yang ZM. Chem Commun. 2012;48:395. doi: 10.1039/c1cc16250k. [DOI] [PubMed] [Google Scholar]
- 19.Wang HM, Wei J, Yang CB, Zhao HY, Li DX, Yin ZN, Yang ZM. Biomaterials. 2012;33:5848. doi: 10.1016/j.biomaterials.2012.04.047. [DOI] [PubMed] [Google Scholar]
- 20.Cheetham AG, Zhang P, Lin Y-a, Lock LL, Cui H. J Am Chem Soc. 2013;135:2907. doi: 10.1021/ja3115983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aryal S, Hu CMJ, Fu V, Zhang LF. J Mater Chem. 2012;22:994. [Google Scholar]
- 22.Deepa G, Ashwanikumar N, Pillai JJ, Kumar GSV. Curr Med Chem. 2012;19:6207. [PubMed] [Google Scholar]
- 23.Singla AK, Garg A, Aggarwal D. Int J Pharm. 2002;235:179. doi: 10.1016/s0378-5173(01)00986-3. [DOI] [PubMed] [Google Scholar]
- 24.von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Proc Natl Acad Sci USA. 2000;97:5129. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellado W, Magri NF, Kingston DGI, Garciaarenas R, Orr GA, Horwitz SB. Biochem Biophys Res Commun. 1984;124:329. doi: 10.1016/0006-291x(84)91557-2. [DOI] [PubMed] [Google Scholar]
- 26.Stuart MCA, van de Pas JC, Engberts J. J Phys Org Chem. 2005;18:929. [Google Scholar]
- 27.Balasubramanian SV, Alderfer JL, Straubinger RM. J Pharm Sci. 1994;83:1470. doi: 10.1002/jps.2600831021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.