

Abstract
A series of new linear iron chelators containing hydroxypyridinone and terephthalamide moieties has been prepared. All are hexadentate ligands composed of a systematically varied combination of me-3,2-hydroxypyridinone and 2,3-dihydroxyterephthalamide binding units; most are based on a spermidine scaffold but one incorporates the bifunctional 2,3-dihydroxyterephthalamide unit as an integral part of the backbone. Protonation and ferric iron complex formation constants have been determined from solution thermodynamic studies giving log ε110 values of 25.7, 30.7, 36.3, 43.8, and 45.0 respectively. The ferric complexes display reversible reduction potentials from −276 mV to −1032 mV (measured relative to the normal hydrogen electrode NHE) in alkaline solution. The incremental replacement of hydroxypyridinone units by terephthalamide binding groups progressively reduces the ligand acidity, markedly increases the iron-chelate stability, and improves the selectivity for ferric ion over ferrous ion. While the majority of iron chelators forming very stable ferric complexes are based on a tripodal backbone such as TREN, the ferric 5-LIO(TAMmeg)2(TAM) complex, despite its non-tripodal scaffold, is one of the most stable iron complexes yet reported. Moreover, the high affinity for ferric ion of the discussed linear ligands strongly correlates with their ability to remove iron in vivo.
Keywords: Iron(III) Sequestering Agents, Iron(III)-chelating Properties, Equilibrium Constants, Thermodynamics, Siderophore Analogs
Introduction
While iron is essential for virtually all life,2 it can also mediate cell injury when present in excess.3 In humans the serious clinical condition of iron overload can be ameliorated by administration of iron-specific chelating agents which bind iron in vivo and promote its excretion as an iron complex.4 While desferrioxamine B (Desferal®) has been the drug of choice for chelation therapy for the past forty years, the use of this natural siderophore still presents disadvantages,5 and considerable effort has been invested in developing new iron chelating agents.6–12
While the hexadentate ligand TREN-Me-3,2-HOPO is composed of hydroxypyridinone units linked to a central tripodal amine backbone and has shown potential as a therapeutic iron chelator,13 the three hydroxamate binding units of desferrioxamine B are arranged in a linear fashion along a single chain, which may be part of the ligand’s therapeutic advantage (Figure 1). In this research project, a series of hexadentate ligands has been designed to investigate the effect on iron complex stability of the scaffold geometry and of the incorporation of specific binding units such as hydroxypyridinone (Me-3,2-HOPO) and terephthalamide (TAMmeg) units.7–9,11 Previous studies showed that the iron complex stability increases markedly upon progressive replacement of Me-3,2-HOPO units by TAMmeg units on a TREN scaffold.8 Moreover, incorporation of a bifunctional terephthalamide unit in the ligand backbone through alkylamine linkers provides linear ligands with high affinity for iron.9 However, thermodynamic and biological evaluation of such linear ligands is limited because of their poor solubility in aqueous solution. Only one linear ligand 5-LIO(Me-3,2-HOPO)2(TAM)9 (Figure 1) exhibited adequate solubility for utility.
Figure 1.

Desferrioxamine B (a.), previously investigated iron chelators TREN-Me-3,2-HOPO (b.), 5-LIO(Me-3,2-HOPO)2(TAM) (c.) and linear ligands presented in this study 5-LIO(TAMmeg)2(TAM) (d.), 3,4-LI(Me-3,2-HOPO) (e.), 3,4-LI(Me-3,2-HOPO)2(TAMmeg) (f.), 3,4-LI(Me-3,2-HOPO)(TAMmeg)2 (g.) and 3,4-LI(TAMmeg) (h.).
The work presented here extends those studies. The analog of 5-LIO(Me-3,2-HOPO)2(TAM) incorporating three terephthalamide units has been synthesized, as well as a new series of hexadentate ligands based on a spermidine backbone and incorporating Me-3,2-HOPO and TAMmeg units (Figure 1). Linear polyamines such as spermine have been used before in ligands designed for actinide decorporation with appreciable success,14 hence the new ligands are envisaged to exhibit good in vivo iron chelation. The spermidine backbone 3,4-LI (the abbreviation stands for a linear triamine backbone, shortened to LI, in which the three amine nitrogen atoms are separated by 3 and 4 carbon atoms, respectively) represents an intermediate between the branched TREN and the linear 5-LIO(TAM) scaffolds, and improves the solubility of the chelating agents. We present here the synthesis, characterization, and solution thermodynamics of these new chelating agents, which confirms the greater iron affinity of terephthalamide as compared to hydroxypyridinone, independent of the backbone. Furthermore, all the ligands form very strong iron complexes, which validates the linear design concept for therapeutic purposes. The high affinity for ferric ion of these linear ligands is strongly reflected in their efficacy at removing iron in vivo, as reported elsewhere.15
Experimental Section
Synthesis. General
All chemicals were obtained from commercial suppliers and were used as received. The starting materials methyl-2,3-dibenzyloxyterephthalic acid (Bn(MeTAM) acid ),16 1-methyl-3-benzyloxy-2-pyridinone acid chloride (Me-3,2-HOPOBn acid chloride),14 3-benzyloxy-4-(2-thioxothazolidin-1-yl)carbonyl-2-pyridinone (Me-3,2-HOPOBn thiazolide),8 2,3-dibenzyloxy-1-(methoxyethylcarbamoyl)-4-(2-thioxothazolidin-1-yl)terephthalamide (BnTAMmeg thiazolide)8 and 5-LIO(TAM) diamine9 were prepared according to procedures described in the designated references. Flash silica gel chromatography was performed using Merck 40–70 mesh silica gel. Melting points were taken on a Büchi melting apparatus and are uncorrected. All NMR spectra were recorded at ambient temperature on Bruker FT-NMR spectrometers at the NMR Laboratory, University of California, Berkeley. Microanalyses were performed by the Microanalytical Services Laboratory, College of Chemistry, University of California, Berkeley. Mass spectra were recorded at the Mass Spectrometry Laboratory, College of Chemistry, University of California, Berkeley. Infrared spectra were measured using a Thermo Nicolet IR Avatar 370 Fourier transform spectrometer.
Bn(MeTAMmeg) (1)
Methyl-2,3-dibenzyloxyterephthalic acid ( 2.0 g, 5.1 mmol) was dissolved in 10 mL of oxalyl chloride with stirring at 0 °C. After 2 h, the excess oxalyl chloride was removed under vacuum to give a light yellow solid. The solid was added to a methoxyethylamine (0.28 mL, 5.2 mmol) solution in 150 mL of CH2Cl2 and stirred at room temperature overnight. After filtration of the solution, the filtrate was condensed and loaded onto a silica gel column. Elution with 5% MeOH in CH2Cl2 afforded the product as a beige foam. Yield: 1.84 g (81%). 1H NMR (400 MHz, CDCl3): ε 3.25 (s, 3H), 3.43 (t, J = 5.2 Hz, 2H), 3.54 (dt, J = 5.6, 5.2 Hz, 2H), 3.87 (s, 3H), 5.12 (s, 2H), 5.14 (s, 2H), 7.35–7.45 (m, 10H), 7.62 (d, J = 8.4 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H), 8.19 (br, 1H) ppm. (+)-FABMS: m/z 450 (MH+).
Bn(TAMmeg acid) (2)
An aqueous NaOH solution (4 M, 10 mL) was added to Bn(MeTAMmeg) (1.84 g, 4.1 mmol) in 50 mL of MeOH. The mixture was stirred at room temperature for 4 h. After removal of the solvents, the residue was dissolved in 30 mL of water. The resulting solution was acidified to pH 3 with 6 M HCl to give an off-white precipitate. It was recrystallized from hot water to give 1.57 g (88%) of white crystals. 1H NMR (400 MHz, CDCl3): ε 3.28 (s, 3H), 3.47 (t, J = 4.8 Hz, 2H), 3.57 (dt, J = 5.2, 4.8 Hz, 2H), 5.15 (s, 2H), 5.25 (s, 2H), 7.36–7.39 (m, 10H), 7.89 (d, J = 8.4 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 8.05 (t, br, 1H) ppm. (+)-FABMS: m/z 436 (MH+).
3,4-LI(Me-3,2-HOPO)Bn (3)
A solution of crude Me-3,2-HOPOBn acid chloride (1.32 g, 4.75 mmol) in THF (40 mL) was added dropwise to a solution of spermidine (203 mg, 1.39 mmol) and triethylamine (5.1 mg, 0.050 mmol) in THF (60 mL). The mixture was heated to 60 °C and stirred overnight under nitrogen. The solvent was removed with a rotary evaporator, and the residue was partitioned into a mixture of water (50 mL) and CH2Cl2 (50 mL). The organic phase was then washed successively with 1 M NaOH (100 mL), 1 M HCl (100 mL), brine (100 mL), and dried over Na2SO4. After removal of the solvent, the product was loaded onto a flash silica gel column. Elution with a gradient of MeOH (3–7%) in CH2Cl2 allowed separation of the benzyl protected precursor as a white foam. Yield: 0.91 g (75%). 1H NMR (500 MHz, CDCl3): ε 1.02–1.63 (m, 6H), 2.74–3.24 (m, 8H), 3.53 (s, 3H), 3.56 (s, 3H), 3.59 (s, 3H), 4.90 (d, J = 11 Hz, 1H), 5.23 (d, J = 11 Hz, 1H), 5.29–5.40 (m, 3H), 5.48–5.50 (m, 1H), 5.90 (dd, J = 15, 7.0 Hz, 1H), 6.70–6.78 (m, 2H), 6.99 (d, J = 7.0 Hz, 1H), 7.09–7.22 (m, 2H), 7.26–7.44 (m, 15H), 7.75 (br, 1H), 7.87 (br, 1H) ppm. (+)-FABMS: m/z 869 (MH+).
3,4-LI(Me-3,2-HOPO) (4)
The precursor 3,4-LI(Me-3,2-HOPO)Bn (3) (0.87 g, 1.0 mmol) was deprotected at rt with 10 mL of 1:1 HCl (37%):glacial HOAc for 4 days. All of the volatiles were removed in vacuo. The residue was dissolved in a minimum amount of methanol and slowly dropped into diethyl ether (50 mL). The product precipitated as a white powder. Yield: 0.48 g (77%). 1H NMR (500 MHz, DMSO-d6): ε 1.32–1.77 (m, 6H), 3.11–3.42 (m, 11H), 3.45 (s, 6H), 5.94 (d, J = 7.0 Hz, 1H), 6.38–6.52 (m, 2H), 7.05 (d, J = 7.0 Hz, 1H), 7.14–7.19 (m, 2H), 8.29–8.37 (m, 1H), 8.49–8.54 (m, 1H), 9.30 (br, 1H), 11.70 (br, 2H) ppm. Mp: 93–95 °C. (+)-FABMS: m/z 599 (MH+). Anal. Calcd (Found) for C28H34N6O9·1.5H2O: C, 53.75 (53.99); H, 5.96 (6.06); N, 13.43 (13.07).
3,4-LI(TAMmeg)Bn (5)
Bn(TAMmeg acid) (2) (1.31 g, 3.0 mmol) was dissolved in 15 mL of oxalyl chloride with stirring at 0 °C. After 2 h, the excess oxalyl chloride was removed under vacuum to give a light yellow solid. A solution of the crude BnTAMmeg acid chloride in THF (50 mL) was added dropwise to a solution of spermidine (131 mg, 0.90 mmol) and triethylamine (3.60 mg, 0.036 mmol) in THF (50 mL), under nitrogen. The mixture was heated to 60 °C and stirred overnight. The solvent was then removed with a rotary evaporator and the residue was partitioned into a mixture of water (30 mL) and CH2Cl2 (30 mL). The organic phase was then washed successively with 1 M NaOH (60 mL), 1 M HCl (60 mL), brine (60 mL), and dried over Na2SO4. After removal of the solvent, the product was loaded onto a flash silica gel column. Elution with a gradient of MeOH (3–7%) in CH2Cl2 allowed separation of the benzyl protected precursor as a white foam. Yield: 0.90 g (66%). 1H NMR (500 MHz, CDCl3): ε 1.02–1.69 (m, 8H), 2.92–3.53 (m, 6H), 3.24 (s, 9H), 3.54 (t, J = 9.0 Hz, 6H), 3.58 (q, J = 4.5, 7.0 Hz, 6H), 5.30–5.39 (m, 12H), 6.65–6.79 (m, 6H), 6.97–7.12 (m, 9H), 7.26–7.42 (m, 21H), 7.71–8.16 (m, 5H) ppm. (+)-FABMS: m/z 1404 (MLi+). Anal. Calcd (Found) for C82H88N6O15·7H2O: C, 64.64 (64.75); H, 6.75 (6.56); N, 5.52 (5.53).
3,4-LI(TAMmeg) (6)
The precursor 3,4-LI(TAMmeg)Bn (5) (250 mg, 0.16 mmol) was deprotected at rt with 10 mL of 1:1 HCl (37%):glacial HOAc for 3 days. All of the volatiles were removed in vacuo. The residue was dissolved in a minimum amount of methanol and slowly dropped in diethyl ether (50 mL). The product precipitated as a beige powder and was isolated in quantitative yield. 1H NMR (400 MHz, DMSO-d6): ε 1.32–1.86 (m, 10H), 2.97–3.46 (m, 25H), 7.26–7.33 (m, 6H), 8.66 (br, 1H), 8.78 (br, 1H), 8.92 (br, 3H), 12.46–12.89 (br, 6H) ppm. Mp: 115–116 °C. (+)-FABMS: m/z 857 (M+). Anal. Calcd (Found) for C40H52N6O15·2.5H2O: C, 53.27 (53.21); H, 6.37 (6.49); N, 9.32 (9.52).
3,4-LI-bis(Me-3,2-HOPO)Bn (7)
Me-3,2-HOPOBn thiazolide (721 mg, 2.00 mmol) was added while stirring to a solution of spermidine (145 mg, 0.998 mmol) in CH2Cl2 (40 mL). The mixture was stirred at rt over night and washed with 1 M KOH solution (3 × 30 mL). The organic phase was dried in vacuo to afford a yellow-brown foam as raw product. Yield: 0.55 g (88%). 1H NMR (400 MHz, CDCl3): ε 1.34 (m, br, 4H), 1.55–1.59 (m, 4H), 2.50 (t, J = 6.4 Hz, 2H), 3.18–3.29 (m, 4H), 3.60 (s, 6H), 5.30 (s, 2H), 5.37 (s, 2H), 6.73–6.77 (m, 2H), 7.11 (d, J = 6 Hz, 2H), 7.35–7.41 (m, 10H) ppm. Mp: 70–71 °C. (+)-FABMS: m/z 628 (MH+).
3,4-LI[(Me-3,2-HOPO)2(TAMmeg)]Bn (8)
3,4-LI-bis(Me-3,2-HOPO)Bn (7) (377 mg, 0.600 mmol) was dissolved in 20 mL of dry THF containing triethylamine (0.40 mL) and the solution was slowly added to a solution of crude BnTAMmeg acid chloride (synthesized as described in the preparation of compound 5) (295 mg, 0.650 mmol) in 50 mL of dry THF while stirring. The reaction mixture was heated at 50 °C overnight. After the solvents were removed, the residue was partitioned into a mixture of water (20 mL) and dichloromethane (20 mL). The organic phase was washed with 1 M NaOH solution (40 mL), 1 M HCl solution (40 mL), brine (40 mL) and loaded on a flash silica plug. Elution of the mixture with a gradient of MeOH (1–7%) in CH2Cl2 provided the product as a beige foam. Yield: 451 mg (72%). 1H NMR (300 MHz, CDCl3): ε 1.09–1.57 (m, 8H), 3.03–3.37 (m, 19H), 5.11 (s, 2H), 5.14 (s, 2H), 5.24 (s, 2H), 5.27 (s, 2H), 6.99–7.08 (m, 4H), 7.18–7.23 (m, 6H), 7.28–7.45 (m, 16H), 7.62 (br, 1H), 7.93 (br, 1H) ppm. (+)-FABMS: m/z 1045 (MH+).
3,4-LI(Me-3,2-HOPO)2(TAMmeg) (9)
The precursor 3,4-LI[(Me-3,2-HOPO)2(TAMmeg)]Bn (8) (451 mg, 0.432 mmol) was deprotected at rt with 10 mL of 1:1 HCl (37%):glacial HOAc for 4 days. All of the volatiles were removed in vacuo. The residue was dissolved in a minimum amount of methanol and slowly dropped into diethyl ether (50 mL). The product precipitated as a white powder and was isolated in quantitative yield. 1H NMR (500 MHz, DMSO-d6): ε 1.33–1.99 (m, 8H), 3.11–3.56 (m, 19H), 6.45–6.50 (m, 4H), 7.15–7.18 (m, 2H), 8.32 (br, 1H), 8.49 (br, 1H), 11.70 (br, 2H) ppm. Mp: 137–138 °C. (+)-FABMS: m/z 685 (MH+). Anal. Calcd (Found) for C32H40N6O11·1H2O: C, 54.70 (54.81); H, 6.02 (5.85); N, 11.96 (12.22).
3,4-LI-bis(TAMmeg)Bn (10)
BnTAMmeg thiazolide (1.13 g, 2.11 mmol) was added while stirring to a solution of spermidine (145 mg, 0.998 mmol) in CH2Cl2 (40 mL). The mixture was stirred at rt over night and was then washed with 1 M KOH solution (3 × 50 mL). The organic phase was dried in vacuo to afford a yellow-brown foam as raw product. Yield: 0.95 g (97%). 1H NMR (300 MHz, CDCl3): ε 1.37–1.60 (m, 6H), 2.51–2.55 (m, 4H), 3.24 (s, 6H), 3.29–3.35 (m, 4H), 3.43 (t, J = 5.1 Hz, 4H), 3.55 (q, J = 5.1, 5.1 Hz, 4H), 5.12 (s, 4H), 5.14 (s, 4H), 7.32–7.43 (m, 20H), 7.80 (t, br, 1H), 7.91–7.97 (m, 4H), 8.08 (t, br, 2H) ppm. (+)-FABMS: m/z 981 (MH+). Anal. Calcd (Found) for C57H65N5O10: C, 69.85 (69.65); H, 6.68 (6.99); N, 7.15 (7.20).
3,4-LI[(Me-3,2-HOPO)(TAMmeg)2]Bn (11)
3,4-LI-bis(TAMmeg)Bn (10) (952 mg, 0.971 mmol) was dissolved in 50 mL of dry THF containing triethylamine (1.00 mL) and slowly added to a solution of Me-3,2-HOPOBn acid chloride (694 mg, 2.50 mmol) in 50 mL of dry THF while stirring. The reaction mixture was heated at 50 °C over night. After the solvents were removed, the residue was partitioned into a mixture of water (25 mL) and dichloromethane (25 mL). The organic phase was washed with 1 M NaOH solution (50 mL), 1 M HCl solution (50 mL), brine (50 mL) and loaded on a flash silica plug. Elution of the mixture with a gradient of MeOH (2–8%) in CH2Cl2 provided the product as a slightly beige foam. Yield: 692 mg (58%). 1H NMR (300 MHz, CDCl3): ε 1.09–1.62 (m, 8H), 2.52–3.30 (m, 6H), 3.24 (s, 6H), 3.38–3.44 (m, 4H), 3.55–3.58 (m, 7H), 4.85–5.23 (m, 10H), 6.99 (d, J = 4.2 Hz, 1H), 7.08 (d, J = 4.2 Hz, 1H), 7.18–7.43 (m, 25H), 7.62–7.94 (m, 4H), 8.09 (m, br, 2H) ppm. (+)-FABMS: m/z 1221 (MH+).
3,4-LI(Me-3,2-HOPO)(TAMmeg)2 (12)
The precursor 3,4-LI[(Me-3,2-HOPO)(TAMmeg)2]Bn (690 mg, 0.56 mmol) was deprotected at rt with 25 mL of 1:1 HCl (37%):glacial HOAc for 4 days. All of the volatiles were removed in vacuo. The residue was dissolved in a minimum amount of methanol and slowly dropped into diethyl ether (50 mL). The product precipitated as a white powder and was isolated in quantitative yield. 1H NMR (400 MHz, DMSO-d6): ε 1.37–1.82 (m, 10H), 3.16–3.47 (m, 21H), 6.97–7.34 (m, 6H), 8.73 (t, br, 1H), 8.81 (t, br, 1H), 8.92 (br, 5H), 9.30–9.50 (m, br, 2H) ppm. Mp: 122–124 °C. (+)-FABMS: m/z 771 (MH+). Anal. Calcd (Found) for C36H46N6O13·2H2O: C, 53.59 (53.84); H, 6.25 (6.23); N, 10.42 (10.25).
5-LIO[(TAMmeg)2(TAM)]Bn (13)
A solution of BnTAMmeg thiazolide (1.23 g, 2.29 mmol) in 20 mL of dichloromethane was added to a solution of 5-LIO(TAM) diamine (0.63 g, 1.14 mmol) in dichloromethane (20 mL). The reaction mixture was stirred overnight, washed with 2M KOH in brine (2 × 40 mL), dried and condensed. The residue was applied to a silica column and eluted with ethyl acetate to remove unreacted starting material; the product was then eluted with 10% MeOH in CH2Cl2. The product fractions were pooled and evaporated to dryness to afford the product as a white foam. Yield: 1.38 g (85%). 1H NMR (300 MHz, CDCl3): ε 3.23 (s, 6H), 3.33 (d, J = 5.1 Hz, 8H), 3.41 (t, J = 5.1 Hz, 12H), 3.55 (q, J = 5.1, 5.4 Hz, 4H), 5.04 (s, 4H), 5.07 (s, 4H), 5.08 (s, 4H), 7.29 (s, 20H), 7.36 (s, 10H), 7.83–8.03 (m, 12H) ppm. (+)-FABMS: m/z 1385.4 (MH+). Anal. Calcd (Found) for C80H84N6O16·2H2O: C, 67.59 (67.32); H, 6.24 (6.08); N, 5.91 (5.89).
5-LIO(TAMmeg)2(TAM) (14)
The precursor 5-LIO[(TAMmeg)2 TAM)]Bn (13) (831 mg, 0.585 mmol) was deprotected at rt with 20 mL of 1:1 HCl (37%):glacial HOAc for 3 days. All of the volatiles were removed in vacuo. The residue was dissolved in a minimum amount of methanol and slowly dropped in diethyl ether (50 mL). The product precipitated as a grey powder and was isolated in quantitative yield. 1H NMR (500 MHz, DMSO-d6): ε 3.26 (s, 6H), 3.37 (t, J = 6.8 Hz, 8H), 3.46–3.49 (m, 12H), 3.59 (t, J = 6.4 Hz, 4H), 7.32 (d, J = 9.5 Hz, 6H), 8.91 (t, br, 6H), 12.62 (br, 6H) ppm. Mp: 117 °C. (+)-FABMS: m/z 845 (MH+). Anal. Calcd (Found) for C38H48N6O16·4H2O: C, 49.78 (49.72); H, 6.16 (5.98); N, 9.17 (9.19).
Synthesis of Iron Complexes
In each case, a degassed solution of Fe(acac)3 (26.5 mg, 75.0 µmol) in MeOH (5 mL) was added to a stirred solution of the appropriate ligand (75.0 µmol) in degassed MeOH (20 mL). The mixture was stirred for 2 hours under a nitrogen atmosphere and one equivalent per terephthalamide unit of KOH (0.5 N in MeOH) was added. The solution was stirred for at least two more hours, condensed to 2–4 mL, and added dropwise to 50 mL of diethyl ether. The precipitated iron complex was isolated by centrifugation. Once the supernatant was decanted, the solid powder was dried under vacuum at 50 °C overnight.
Fe[3,4-LI(Me-3,2-HOPO)] (15)
A 47 mg portion of the ligand was used, yielding 43 mg (84%). Mp > 300 °C. (+)-ESMS: m/z 674.1 ([MNa]+). Anal. Calcd (Found) for C28H31FeN6O9·1.5H2O: C, 49.57 (49.62); H, 5.05 (5.12); N, 12.39 (12.17). IR: ε 1640 m, 1621 m, 1550 s, 1509 s, 1458 m, 1352 m, 1290 m, 1232 s cm−1.
Fe[3,4-LI(Me-3,2-HOPO)2(TAMmeg)] (16)
A 51 mg portion of the ligand was used, yielding 49 mg (87%). Mp > 300 °C. (−)-ESMS: m/z 736.2 ([M]−). Anal. Calcd (Found) for C32H36FeKN6O11·0.25KOH·2MeOH: C, 47.83 (47.53); H, 5.22 (4.87); N, 9.84 (9.47); K, 5.72 (5.90). IR: ε 1642 m, 1622 m, 1553 s, 1515 s, 1457 m, 1362 m, 1292 m, 1234 s cm−1.
Fe[3,4-LI(Me-3,2-HOPO)(TAMmeg)2] (17)
A 61 mg portion of the ligand was used, yielding 59 mg (79%). Mp > 300 °C. (+)-ESMS: m/z 900.2 ([K2M-H]+). Anal. Calcd (Found) for C36H41FeK2N6O13·6H2O: C, 42.90 (42.76); H, 5.30 (5.17); N, 8.34 (8.00). IR: ε 1615 m, 1549 s, 1522 s, 1433 m, 1313 m, 1237 m, 1197 s cm−1.
Fe[3,4-LI(TAMmeg)] (18)
A 58 mg portion of the ligand was used, yielding 61 mg (81%). Mp > 300 °C. (−)-ESMS: m/z 907.3 ([MH2]−). Anal. Calcd (Found) for C40H46FeK3N6O15·4.5MeOH: C, 45.75 (45.72); H, 5.52 (5.21); N, 7.19 (6.87). IR: ε 1614 s, 1545 s, 1433 s, 1317 m, 1197 s cm−1.
Fe[5-LIO(TAMmeg)2(TAM)] (19)
A 59 mg portion of the ligand was used, yielding 60 mg (83%). Mp > 300 °C. (−)-ESMS: m/z 974.1 ([MK2]−). Anal. Calcd (Found) for C38H42FeK3N6O16·6H2O: C, 40.75 (40.99); H, 4.86 (4.82); N, 7.50 (7.33). IR: ε 1611 s, 1541 s, 1439 s, 1235 m, 1196 s, 1106 m cm−1.
Electrochemical Measurements
The potentiostat used was supplied by Bio-Analytical Systems (BAS, model 100a). All experiments used a three-electrode cell composed of the following electrodes: Pt wire auxiliary, Ag/AgCl reference, and Hg working (Princeton Applied Research (PAR) model 303a in hanging-mercury-drop-electrode (HMDE) mode, small drop size). The silver electrode was combined with the PAR 303a assembly by insertion via the sample-addition port. Drop knocking and solution purging by the PAR 303a was controlled remotely from the potentiostat; a fresh Hg drop was used for each scan. The reference was calibrated against a SCE electrode (previously calibrated at +241 mV vs. NHE).17 The experimental potential of the reference used in this work was determined to be +189 ± 2 mV vs. NHE.
Aqueous solutions of the ferric complexes were prepared at 5 × 10−5 M Fe and 1 × 10−4 M ligand and contained 0.01 M ammonium acetate buffer with addition of KOH or HCl to give the desired pH. The ionic strength was set to 0.1 M by addition of KCl. These solutions were deoxygenated by stirred purging with Ar gas for 20 min prior to the acquisition of square wave voltammograms. For each scan the square wave modulation amplitude was 25 mV, the step potential was 2 mV, 16-point current sampling was selected, and the square wave frequency was varied over the range 5–50 Hz in order to test for reversibility of the system. Reversibility was evaluated by comparing the digitized current-potential response to the theoretical response as described for square wave voltammetry.18 According to eq. 1, a plot of the function ε against potential, E, has an inverse slope of 2.3RT/nF (59/n mV) and a horizontal intercept at E = E°. Terms are defined as follows: cosh is the hyperbolic cosine function, Esw is the square wave modulation amplitude, εI is the square wave difference current, εIp is the peak difference current, and the physical constants n, F, R, and T have their usual meanings.
| (1) |
Solution Thermodynamics
Ligands protonation and complex formation constants were determined using procedures and equipment following previous descriptions.7,8,11
Titration solutions and equipment
Corning high performance combination glass electrodes (response to [H+] was calibrated before each titration)19 were used together with either an Accumet pH meter or a Metrohm Titrino to measure the pH of the experimental solutions. Metrohm autoburets (Dosimat or Titrino) were used for incremental addition of acid or base standard solutions to the titration cell. The titration instruments were fully automated and controlled using LabView software.20 Titrations were performed in 0.1 M KCl supporting electrolyte under positive Ar gas pressure. The temperature of the experimental solution was maintained at 25 °C by an external circulating water bath. UV-Visible spectra for incremental titrations were recorded on a Hewlett-Packard 8452a spectrophotometer (diode array). Solid reagents were weighed on a Metrohm analytical balance accurate to 0.05 mg. All titrant solutions were prepared using distilled water that was further purified by passing through a Millipore Milli-Q reverse osmosis cartridge system. Titrants were degassed by boiling for 1 h while being purged under Ar. Carbonate-free 0.1 M KOH was prepared from Baker Dilut-It concentrate and was standardized by titrating against potassium hydrogen phthalate using phenolphthalein as an indicator. Solutions of 0.1 M HCl were similarly prepared and were standardized by titrating against sodium tetraborate to Methyl Red endpoint. Stock solutions of EDTA were obtained by dissolving EDTA (Fischer) in freshly degassed Milli-Q water. Stock solutions of ferric ion were obtained by dissolving solid FeCl3.6H2O in standardized 0.1 M HCl. The actual ferric ion concentration was determined by titration with a standardized EDTA solution to Variamine Blue endpoint.21
Incremental Titrations
Initial titration solutions were assembled from the constituent reagents in ratios determined previously by modeling using estimated formation constants and the modeling program Hyss.22,23 The solutions were incrementally perturbed by the addition of either acid (HCl) or base (KOH) titrant, followed by a time delay for equilibration (90 seconds for protonation studies; 2 hours for EDTA competition titrations). All titrations were conducted in pairs: first a forward titration from low to high pH, then a reverse titration back to low pH. The data for the two titrations comprising each experiment were pooled for calculation of formation constants when reversibility was achieved. All absorbance measurements used for calculation of formation constants were less than 1.05 absorbance units.
Protonation Constants: Potentiometric Titrations
The protonation constants of 3,4-LI(Me-3,2-HOPO) and 3,4-LI(Me-3,2-HOPO)2(TAMmeg) were determined by potentiometric titration. Solutions were assembled from a weighed portion of ligand and the supporting electrolyte solution, with resulting ligand concentrations between 0.2 and 0.5 mM. An average of 60 – 90 data points were collected in each pair of titrations (forward and back), each data point consisting of a volume increment and a pH reading over the pH range 4 to 10. Refinement of the protonation constants was accomplished using the program Hyperquad,24 which allows simultaneous nonlinear least squares refinement of the data from multiple titration curves.
Protonation Constants: Spectrophotometric Titrations
The protonation constants of all other ligands were determined by spectrophotometric titration due to solubility issues. Solutions were assembled from a weighed portion of compound and the supporting electrolyte solution, with resulting ligand concentrations between 50 and 100 µM. Constant buffering of the solution was assured by the addition of NH4Cl, Hepes and Mes buffers (500 µM). An average of 40 – 50 data points were collected in each pair of ligand titrations (forward and back), each data point consisting of a pH measurement and an absorbance spectra over the pH range 4 to 10. Nonlinear least squares refinement of the protonation constants was accomplished using the program pHab.25
Formation Constants: EDTA Competition Spectrophotometric Titrations
Each EDTA competition experiment consisted of a pair of titrations: forward vs. KOH and reverse vs. HCl. At the lower pH end of the titration, the colorless iron-EDTA complex dominated, but as the pH was raised, the solution turned pink to red, corresponding to the gradual formation of the iron-experimental ligand complex. Solutions were assembled from a weighed portion of ligand and the supporting electrolyte solution, with resulting ligand concentrations of about 0.1 mM. The EDTA was present in a ten fold excess and the iron in a 0.9 fold default over the ligand in order to prevent formation of insoluble iron species. In each incremental experiment, an average of 24 points was collected over a period of 48 hours (~2 hr equilibration time per point). Each data point consisted of a pH measurement and an absorbance spectrum over at least 80 different wavelengths between 400 and 650 nm over the pH range 3 to 7. The data was imported into the refinement program pHab25 and analyzed by non-linear least-squares refinement.
Data Treatment
All equilibrium constants were defined as cumulative formation constants, εmlh according to eq. 2, where the ligands are designated as L. Stepwise protonation constants, Kan, may be derived from these cumulative constants according to eq. 3 (describes proton association constants). In the case of the competition experiments, the equilibration of iron between the HOPO/TAM ligands and EDTA was calculated by including the proton association and iron formation constants for EDTA26 as fixed parameters in the refinements. Because of the hexacoordinate nature of all ligands, it was assumed that ternary (i.e., mixed EDTA-Fe-L) complexes were not formed. This assumption was supported by spectral data showing that the band shapes and εmax values of the LMCT transitions of the Fe-L complexes were unchanged in the presence and absence of EDTA.
| (2) |
| (3) |
Each pair of titrations (i.e., forward titration against KOH and reverse titration against HCl) were combined for simultaneous refinement. For potentiometric titrations, both the proton and ligand concentrations were refined, only the proton concentration was allowed to vary in the spectrophotometric studies, and all other concentrations were held at estimated values determined from the volume of standardized stock or the weight of ligand (measured to 0.01 mg). Refined concentrations were within 5% of the analytical values. For spectral titrations, all species formed with the ligands L were considered to have significant absorbance to be observed in the UV-Vis spectra.
Results
Ligand and Ferric Complex Synthesis
The benzyl protected precursors Me-3,2-HOPOBn and Bn(MeTAM) acid chlorides as well as Me-3,2-HOPOBn and BnTAMmeg thiazolides were synthesized following standard procedures.8,14,16 The TAM moiety could also be functionalized by addition of 2-methoxyethylamine under typical amide coupling conditions (Scheme 1). Chromatographic purification of this protected di-substituted TAM moiety (1) followed by deprotection of the methoxy group afforded the mono-substituted BnTAMmeg acid (2) in 88% yield. The corresponding acid chloride was prepared in situ and directly used for the next coupling reaction.
Scheme 1.
Functionalization of the 2,3-dihydroxy-terephthalamide moiety.
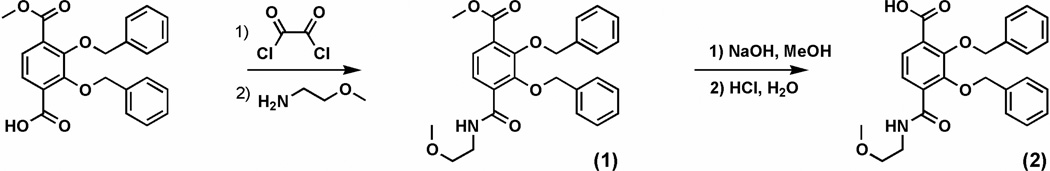
Coupling of spermidine with Me-3,2-HOPOBn and BnTAMmeg acid chlorides yield the respective benzyl-protected homogeneous 3,4-LI(Me-3,2-HOPO)Bn (3) and 3,4-LI(TAMmeg)Bn (5) as shown in Scheme 2. However, the thiazolide activated species only react with aliphatic primary amines, whereas the acid chlorides react with both primary and secondary amines.14 Therefore, selectivity can be achieved by the reaction of spermidine with 2 equivalents of Me-3,2-HOPOBn thiazolide or BnTAMmeg thiazolide to afford the mono amines 3,4-LI-bis(Me-3,2-HOPO)Bn (7) and 3,4-LI-bis( TAMmeg)Bn (10) (Scheme 3). Further immediate coupling with the acid chlorides led to the mixed benzyl-protected precursors 3,4-LI[(Me-3,2-HOPO)2(TAMmeg)]Bn (8) and 3,4-LI[(Me-3,2-HOPO)(TAMmeg)2]Bn (11). Purification of all these benzyl-protected species was easily achieved through silica gel column chromatography, eluting with appropriate gradients of methanol (1 to 10%) in dichloromethane. Deprotection of the benzyl groups under strongly acidic conditions provided the two homogeneous 3,4-LI(Me-3,2-HOPO) (4) and 3,4-LI(TAMmeg) (6) ligands as well as the two mixed 3,4-LI(Me-3,2-HOPO)2(TAMmeg) (9) and 3,4-LI(Me-3,2-HOPO)(TAMmeg)2 (12) ligands in quantitative yields. This synthetic flexibility allows the spermidine scaffold to be selectively attached to Me-3,2-HOPO and TAMmeg binding units, tuning the affinity of the resulting ligands for iron along with potential for in vivo iron chelation.
Scheme 2.

Synthesis of the homogeneous 3,4-LI(Me-3,2-HOPO) and 3,4-LI(TAMmeg) ligands.
Scheme 3.

Synthesis of the mixed 3,4-LI(Me-3,2-HOPO)2(TAMmeg) and 3,4-LI(Me-3,2-HOPO)(TAMmeg)2 ligands.
In order to continue previous studies,9 another new iron chelator, which takes advantage of the bifunctionality of 2,3-dihydroxyterephthalamide by linking two terminal TAMmeg binding groups to a central TAM unit, was synthesized (Scheme 4). The di-substituted benzyl-protected TAM-thiazolide was coupled to the 5-LIO(TAM) diamine as described earlier9 to permit the following reaction of the free amines with two mono-substituted benzyl-protected TAM-thiazolide units. The benzyl protecting groups are cleaved by treatment with strong acidic conditions to produce the new ligand, 5-LIO(TAMmeg)2(TAM) (14), in an overall yield of 85%.
Scheme 4.

Synthesis of 5-LIO(TAMmeg)2(TAM).
The iron complexes of all ligands were prepared by reaction of a methanolic solution of each ligand with ferric acetylacetonate, followed by addition of base when necessary. Concentration of the complex solutions and precipitation from diethyl ether afforded the analytically pure ferric complexes.
Electrochemistry
The reduction potentials of the ferric complexes were determined by square wave voltammetry at a hanging drop mercury electrode (HDME). A set of voltammograms are illustrated for the Fe[5-LIO(TAMmeg)2(TAM)] complex in Figure 2, upper panel. Reversibility of the voltammetric waves was verified by transforming the current-potential response according to eq. 1 to give the function ε, as plotted in Figure 2, lower panel. This function was linear within a potential window of 100 mV about the standard reduction potential (E°) for all frequencies (10–50 Hz). The value for E° determined from this plot was in perfect agreement with the peak potential, Ep. This reversible response for the complexes was confined to a mildly alkaline pH region (more alkaline as more TAM units were incorporated in the ligand); at lower pH the E° values were no longer constant and moved to more positive values with distortion of the waveform. At higher pH values, the waveform also became disorted and the peak currents diminished as the solution became decolorized due to hydroxide competition for ferric ion. The reduction potentials are listed in Table 1. The corresponding square wave voltammograms for all 3,4-LI ligands are shown in the supporting information (Figures S3–S6) as well as a representative cyclic voltammogram for the Fe[5-LIO(TAMmeg)2(TAM)] complex (Figure S7) obtained in similar experimental conditions.
Figure 2.

Upper panel: Square wave voltammograms at the HMDE of 5 × 10−5 M Fe[5-LIO(TAMmeg)2(TAM)] in 0.01 M ammonium acetate buffer, pH 9.0, ionic strength 0.1 M (KCl), Esw = 25 mV, Estep = 2 mV, frequencies given in hertz. Lower panel: Conversion of the voltammograms to the ε function, according to eq. 1.
Table 1.
Electrochemical Data for the Five New Ligands.
| 3,4-LI (Me-3,2-HOPO) |
3,4-LI (Me-3,2-HOPO)2 (TAMmeg) |
3,4-LI (Me-3,2-HOPO) (TAMmeg)2 |
3,4-LI (TAMmeg) |
5-LIO (TAMmeg)2 (TAM) |
|
|---|---|---|---|---|---|
| E° (mV vs NHE) | −276 ± 3 | −327 ± 2 | −553 ± 2 | −1032 ± 4 | −773 ±2 |
| Reversible pH range | 7–9 | 7.5–9.5 | 8–10 | 8.5–10.5 | 8–10.5 |
Protonation Constants
These were determined by potentiometric titrations for the two ligands 3,4-LI(Me-3,2-HOPO) and 3,4-LI(Me-3,3-HOPO)2(TAMmeg). The relatively high concentration required to ensure precise pH buffering precluded the simultaneous collection of spectral data. The limited solubility in acidic aqueous solution of the ligands incorporating more than one TAM unit, 3,4-LI(Me-3,2-HOPO)(TAMmeg)2, 3,4-LI(TAMmeg) and 5-LIO(TAMmeg)2(TAM), necessitated the use of spectrophotometric titrations (Figure S-1, Supporting Information). For each set of data, the reversibility of the protonation steps was confirmed by simultaneous refinement of forward (against KOH) and reverse (against HCl) titration. Figures 3 and 4 indicate the close agreement obtained between experimental observations and the numerical analyses. For each ligand, three sequential protonation equilibria (Table 2) were assigned to sequential removal of one proton from each of the three binding groups. Additional protonation equilibria were observed due to the presence of a second removable proton on each TAM binding unit and were defined with a constant value of 11.1 for the refinements.7
Figure 3.

Potentiometric titration curves for the two ligands 3,4-LI(Me-3,2-HOPO) and 3,4-LI(Me-3,2-HOPO)2(TAMmeg). Symbols give observed pH measurements, and lines give the calculated fits.
Figure 4.

Spectrophotometric titrations of the ligands 3,4-LI(Me-3,2-HOPO)(TAMmeg)2, 3,4-LI (TAMmeg) and 5-LIO(TAMmeg)2(TAM). Symbols give observed absorbance measurements at a given wavelength, and lines give the calculated fits.
Table 2.
Ligand Protonation and Ferric Complex Formation Constants. Figures in parentheses give the uncertainty determined from the standard deviation between three independent titrations. pM values indicate the negative log value of the free iron(III) concentration at pH 7.4, 1 µM [Fe], 10 µM [L].
| log εmlh |
||||||
|---|---|---|---|---|---|---|
| Species | m l h | 3,4-LI (Me-3,2-HOPO) |
3,4-LI (Me-3,2-HOPO)2 (TAMmeg) |
3,4-LI (Me-3,2-HOPO) (TAMmeg)2 |
3,4-LI (TAMmeg) |
5-LIO (TAMmeg)2 (TAM) |
| LH | 0 1 1 | 8.41 (5) | 11.10 | 11.10 | 11.10 | 11.10 |
| LH2 | 0 1 2 | 15.03 (6) | 18.98 (2) | 22.20 | 22.20 | 22.20 |
| LH3 | 0 1 3 | 20.85 (6) | 25.48 (1) | 30.61 (2) | 33.30 | 33.30 |
| LH4 | 0 1 4 | 30.95 (3) | 37.32 (8) | 41.64 (3) | 40.89 (6) | |
| LH5 | 0 1 5 | 43.23 (6) | 48.75 (3) | 47.54 (1) | ||
| LH6 | 0 1 6 | 54.86 (3) | 52.85 (4) | |||
| FeL | 1 1 0 | 25.67 (2) | 30.65 (2) | 36.25 (2) | 43.78 (2) | 44.99 (6) |
| pM | 25.5 | 27.3 | 28.7 | 32.5 | 34.4 | |
Iron Coordination Chemistry
Hydroxypyridinone and terephthalamide ligands are especially good chelators for iron at low pH because of their relatively high acidity. Acid concentrations of ~ 10 M are required to liberate iron completely. Thus, competition methods are necessary for the determination of equilibrium constants, because iron-HOPO/TAM complexes cannot be significantly dissociated by standard titrimetric methods. The use of EDTA is because of its well-known stability constants and lack of absorbance in the visible region.26 The spectral data sets from the competition titrations are illustrated in Figure S-2, Supporting Information. These experiments exploit the broad ligand-to-metal-charge-transfer (LMCT) bands which increase in intensity as iron exchanges from the colorless EDTA complex to the red FeL complex. The quality of fit in the nonlinear least squares fit to the spectral data is illustrated in Figure 5. All metal complexes are purple-red to red in color. For the 3,4-LI based ligands, the LMCT bands are better defined as more HOPO units are incorporated; indeed 3,4-LI(Me-3,2-HOPO) exhibits a visible transition centered at 430 nm (ε = 4900 M−1 cm−1) and a second, less intense, at 532 nm (ε = 4800 M−1 cm−1). The bimodal nature of the LMCT bands is characteristic of HOPO complexes.27 More surprisingly, the complex formed with 5-LIO(TAMmeg)2(TAM) also shows two very well resolved LMCT bands at 445 nm (ε = 7000 M−1 cm−1) and 525 nm (ε = 5500M−1 cm−1) as well as a strong π ε π* transition shifted from the UV region (as for all the other complexes) to lower energies at 370 nm (ε = 11000 M−1 cm−1).
Figure 5.

EDTA-competition spectrophotometric titrations of ligands. Symbols give observed absorbance measurements at a given wavelength (highest energy LMCT band), and lines give the calculated fits.
Discussion
Ligand Acidity and Iron Formation Constants
The five hexadentate ligands investigated in this study incorporate a combination of the two metal-binding subunits Me-3,2-HOPO and TAMmeg. Both hydroxypyridinone and terephthalamide units are effective for binding iron at comparable concentrations. However, terephthalamide units are more basic28 and so are especially effective at higher (physiological) pH. Indeed, the sum of the first three pKa’s (Table 2) can be compared for the three ligands 3,4-LI(Me-3,2-HOPO)2(TAMmeg), 3,4-LI(Me-3,2-HOPO)(TAMmeg)2, and 3,4-LI(TAMmeg): they are respectively 19.85, 21.03, and 21.56. The acidity of the ligand is reduced as a hydroxypyridinone unit is replaced by a terephthalamide unit on the same backbone.
As expected, the corresponding trend is observed in the iron complex formation constants: the log ε110 value increases with the number of TAM units. These systematic trends become even more apparent when evaluated as the parameter pM(FeIII).29 Calculation of this parameter provides a convenient comparison of ligands of varying acidity and/or denticity. In the present series, the pM values of Table 2 indicate that the incorporation of additional TAM units on a same backbone leads to ligands with significantly increased affinity for iron (Figure 6). The same trend was observed for the combination of these binding units on the tripodal TREN scaffold. 8
Figure 6.

Correlation between the pM(FeIII) values and the incorporation of terephthalamide units on a same backbone for the 3,4-LI and 5-LIO series of ligands. The pM value for Fe[5-LIO(Me-3,2-HOPO)2(TAM)] was determined in a previous study.9
The results show that these ligands all form very stable iron complexes in vitro and should be thermodynamically capable of competing with transferrin (pM 23.6)30 for iron in vivo more effectively than desferrioxamine B (pM 26.9).31 In addition, the pM values of the linear 3,4-LI ligands are only 1 to 2 log units lower than their TREN analogs,8 which emphasizes the role of the preorganized backbone but also validates the linear design concept. Moreover, the last linear ligand, 5-LIO(TAMmeg)2(TAM), with a Kf of 1045, possesses one of the highest iron affinities reported for any chelator.8,32–35
Electrochemistry
All of the ferric complexes displayed reversible reduction potentials in slightly alkaline aqueous solutions. The limits of reversibility for each complex (higher pH as TAM units are added for the 3,4-LI ligands) are well predicted by the formation constants of Table 2. At the more acidic limit, the ferrous complex is not stable with respect to proton competition while at basic pH the limit is determined by the point at which ferric ion is removed from the complexes as FeO(OH)-(s).36 Stepwise substitution into the 3,4-LI ligand structure of the HOPO unit for the TAMmeg binding group results in a negative shift of the redox potential by 71 mV (1.2 × 59 mV), 226 mV (3.8 × 59 mV) and 479 mV (8.1 × 59 mV) indicating an induced selectivity for the ferric state by several orders of magnitude. This is consistent with the behavior of 2,3-dihydroxyterephthalamide ligands, which are extremely powerful Fe3+ chelators and to stabilize this oxidation state of the metal.28,37
Conclusion
Preparation of the ligand series examined here was made possible by the inherent selectivity of acid chloride and thiazolide activated species for amide coupling. Systematic replacement of hydroxypyridinone by 2,3-dihydroxyterephthalamide units allowed tuning of the various properties of the five newly synthesized ligands. In the two respective 3,4-LI and 5-LIO series, acidity of the ligand is reduced and affinity for ferric ion is increased with additional TAM units. The selectivity for ferric ion over ferrous ion is also improved as TAM units are incorporated on the spermidine scaffold. These non-tripodal ligands still have a very high affinity for iron and will probably exhibit different behavior in a biological environment. Initial biological investigations confirm that chelation of ferric ion in vivo is improved with the incorporation of TAMmeg binding moieties into mixed linear and tripodal chelators containing Me-3,2-HOPO units.15
Supplementary Material
Acknowledgment
We thank Dr Kristy Clarke-Jurchen for helpful discussions. This research was supported by the National Institutes of Health (NIH Grant DK057814).
Footnotes
Supporting Information Available: Figures showing spectral data obtained for all spectrophotometric titration, solution speciation diagrams, and voltammograms. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jurchen KMC, Raymond KN. Ferric Ion Sequestering Agents. 42. Part 41. Inorg. Chem. 2006 In Press. [Google Scholar]
- 2.Crichton RR. Inorganic Biochemistry of Iron Metabolism. New York, London: Ellis Hardwood; 1991. [Google Scholar]
- 3.Koppenol WH. In: Iron Chelators: New Development Strategies. Badman DG, Bergeron RJ, Brittenham GM, editors. Ponte Vedra Beach, FL: The Saratoga Group; 2000. pp. 3–10. [Google Scholar]
- 4.Hershko C. Reviews in Clinical and Experimental Hematology. 2000;4:337–361. [Google Scholar]
- 5.Hershko C, Konijn AM, Link G. In: Iron Chelators: New Development Strategies. Badman DG, Bergeron RJ, Brittenham GM, editors. Ponte Vedra Beach, FL: The Saratoga Group; 2000. pp. 157–176. [Google Scholar]
- 6.Bergeron RJ, McManis JS, Wiegand J, Weimar WR, Brittenham GM. In: Iron Chelators: New Development Strategies. Badman DG, Bergeron RJ, Brittenham GM, editors. Ponte Vedra Beach, FL: The Saratoga Group; 2000. pp. 253–292. [Google Scholar]
- 7.Cohen SM, O'Sullivan B, Raymond KN. Inorg. Chem. 2000;39:4339–4346. doi: 10.1021/ic000239g. [DOI] [PubMed] [Google Scholar]
- 8.Jurchen KMC, Raymond KN. accepted Inorg. Chem. 2005 [Google Scholar]
- 9.Jurchen KMC, Raymond KN. J. Coord. Chem. 2005;58:55–80. [Google Scholar]
- 10.Liu ZD, Hider RC. Coord. Chem. Rev. 2002;232:151–171. [Google Scholar]
- 11.Xu J, O'Sullivan B, Raymond KN. Inorg. Chem. 2002;41:6731–6742. doi: 10.1021/ic025610+. [DOI] [PubMed] [Google Scholar]
- 12.O'Sullivan B, Xu J, Raymond Kenneth N. In: Iron Chelators: New Development Strategies. Badman DG, Bergeron RJ, Brittenham GM, editors. Ponte Vedra Beach, FL: The Saratoga Group; 2000. pp. 177–208. [Google Scholar]
- 13.Yokel RA, Fredenburg AM, Durbin PW, Xu J, Rayens MK, Raymond KN. J. Pharm. Sci. 2000;89:545–555. doi: 10.1002/(SICI)1520-6017(200004)89:4<545::AID-JPS12>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 14.Xu J, Durbin PW, Kullgren B, Ebbe SN, Uhlir LC, Raymond KN. J. Med. Chem. 2002;45:3963–3971. doi: 10.1021/jm010564t. [DOI] [PubMed] [Google Scholar]
- 15.Durbin PW, Kullgren B, Ebbe SN, Abergel RJ, Jurchen KMC, Raymond KN. N. Manuscript in preparation. 2005 [Google Scholar]
- 16.Hou ZG, Whisenhunt DW, Xu JD, Raymond KN. J. Am. Chem. Soc. 1994;116:840–846. [Google Scholar]
- 17.Ives DJG, Janz GJ. Reference Electrodes, Theory and Practice. New York: Academic Press; 1961. [Google Scholar]
- 18.Aoki K, Maeda K, Osteryoung JJ. Electroan. Chem. 1989;272:17–28. [Google Scholar]
- 19.Gans P, O'Sullivan B. Talanta. 2000;51:33–37. doi: 10.1016/s0039-9140(99)00245-3. [DOI] [PubMed] [Google Scholar]
- 20.LABVIEW. 5.0.1 ed. Austin, TX: National Instruments Corp.; [Google Scholar]
- 21.Ueno K, Imamura T, Cheng KL. Handbook of Organic Analytical Reagents. 2 ed. Boca Raton, FL: CRC Press; 1992. [Google Scholar]
- 22.Alderighi L, Gans P, Ienco A, Peters D, Sabatini A, Vacca A. Coord. Chem. Rev. 1999;184:311–318. [Google Scholar]
- 23.Alderighi L, Gans P, Ienco A, Peters D, Sabatini A, Vacca A. HYSS. Leeds, U.K. and Florence, Italy: 1999. [Google Scholar]
- 24.Gans P, Sabatini A, Vacca A. HYPERQUAD. Leeds, U.K. and Florence, Italy: 2000. [Google Scholar]
- 25.Gans P, Sabatini A, Vacca A. Ann. Chim. (Rome) 1999;89:45–49. [Google Scholar]
- 26.Smith RM, Martell AE. Critical Stability Constants. Vol. 1–4. New York: Plenum; 1977. [Google Scholar]
- 27.Meyer M, Telford JR, Cohen SM, White DJ, Xu J, Raymond KN. J. Am. Chem. Soc. 1997;119:10093–10103. [Google Scholar]
- 28.Garrett TM, Miller PW, Raymond KN. Inorg. Chem. 1989;28:128–133. [Google Scholar]
- 29.pM is the negative logarithm of free iron concentration in equilibrium with complexed and free ligand, at physiological pH (pH 7.4) with 1 micromolar total iron concentration and 10 micromolar total ligand concentration
- 30.Raymond KN, Telford JR. In: Bioinorganic Chemistry An Inorganic Perspective of Life. Kessissoglou DP, editor. Vol. 459. The Netherlands: Kluwer Academic Publishers; 1995. pp. 25–37. [Google Scholar]
- 31.Evers A, Hancock RD, Martell AE, Motekaitis RJ. Inorg. Chem. 1989;28:2189–2195. [Google Scholar]
- 32.Harris WR, Carrano CJ, Raymond KN. J. Am. Chem. Soc. 1979;101:2213–2214. [Google Scholar]
- 33.Kiggen W, Voegtle F, Franken S, Puff H. Tetrahedron. 1986;42:1859–1872. [Google Scholar]
- 34.Loomis LD, Raymond KN. Inorg. Chem. 1991;30:906–911. [Google Scholar]
- 35.Stutte P, Kiggen W, Voegtle F. Tetrahedron. 1987;43:2065–2074. [Google Scholar]
- 36.Baes CF, Mesmer RE. The hydrolysis of cations. New York: Wiley; 1976. [Google Scholar]
- 37.Garrett TM, McMurry TJ, Hosseini MW, Reyes ZE, Hahn FE, Raymond KN. J. Am. Chem. Soc. 1991;113:2965–2977. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.