

Abstract
Mechanism studies of a mild palladium catalyzed decarboxylation of aromatic carboxylic acids are described. In particular, reaction orders and activation parameters for the two stages of the transformation were determined. These studies guided development of a catalytic system capable of turnover. Further evidence reinforces that the second stage, protonation of the aryl palladium intermediate, is the rate-determining step of the reaction. The first step, decarboxylative palladation is proposed to occur through an intramolecular electrophilic palladation pathway, which is supported by computational and mechansim studies. In contrast to the reverse reaction (C-H insertion), the data support an electrophilic aromatic substitution mechanism involving a stepwise intramolecular protonation sequence for the protodepalladation portion of the reaction.
Introduction
Aromatic Decarboxylation
One particularly difficult transformation in organic synthesis is the decarboxylation of aromatic systems. Often, the chemistry to form aromatic systems takes advantage of the reactivity of carboxylates, which frequently results in their incorporation as carboxylate substituents in the products.1,2,3,4,5,6,7 In contrast to aliphatic systems, the lack of stabilization in the intermediates formed during aromatic decarboxylation renders the process unfavorable.
Thus, most aromatic decarboxylation available methods utilize very forcing conditions. A classical procedure involves heating the aromatic acid to high temperatures in the presence of a strong protic acid.8,9 This method proceeds through ipso protonation of the aromatic ring, requires temperatures of at least 100 °C, and is generally limited to electron rich systems. Another classical procedure involves heating the aromatic acid in the presence of a copper catalyst and quinoline.10,11,12,13 In this case, decarboxylation occurs through coordination of the copper catalyst to the aromatic ring, which helps stabilize the resulting anion upon loss of carbon dioxide. While the copper/quinoline method encompasses a broader substrate scope than the strong protic acid protocol, high temperatures (>160 °C) are required. A milder, indirect aromatic decarboxylation method is the Barton protocol.14,15 In this three-step approach, decarboxylation occurs via an initial ester formation from the carboxylic acid, followed by decarboxylative radical bromination, and finally bromine hydrogenolysis. More recently, silver salts have been shown to catalyze aromatic decarboxylation. This method offers a broad substrate scope but is limited by higher temperatures (120 °C).16,17,18 Also, microwave heating and hydrothermal decarboxylations have been studied.19,20,21,22,23 However, these methods also require high temperatures, generally >190 °C.
Decarboxylative Couplings
The ability of metal catalysts to participate in decarboxylation processes provides a novel avenue to aryl metal intermediates that can be utilized in a variety of processes including Heck couplings, Ullman couplings, and other cross-couplings.24,25,26,27,28,29,30,31 We were inspired by a report from Myers et al.32,33,34 detailing a palladium catalyzed decarboxylative Heck coupling under mild conditions. Based on those initial reports, our group reported35 the mildest and most efficient method for aromatic decarboxylation utilizing catalytic amounts of a palladium complex. Herein, we describe the full details of the development of the catalytic aromatic protodecarboxylation as well as accompanying mechanistic studies.
Results and Discussion
Preliminary Studies
Our efforts toward a mild aromatic decarboxylation reaction began by investigating the use of hydride and proton sources to reduce the corresponding aryl palladium intermediates formed during processes such as the decarboxylative Heck coupling. Initial experiments rapidly established that aromatic decarboxylation utilizing a palladium species was viable. Specifically, naphthoic acid 1 underwent clean decarboxylative palladation upon reaction with one equivalent of Pd(O2CCF3)2 and Ag2CO3 at 90 °C to afford aryl palladium intermediate 2. Subseqent treatment of the intermediate 2 with dihydrogen at room temperature yielded the protodecarboxylated product 3 in 99% yield (Scheme 1).
Scheme 1.
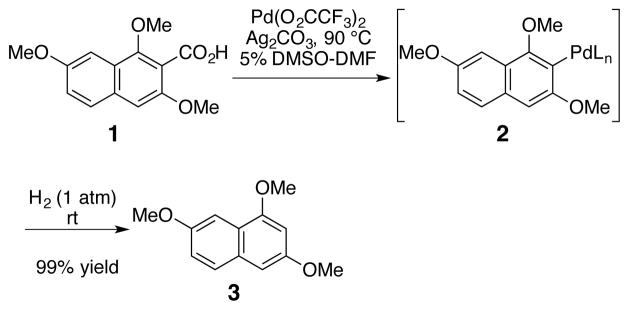
Preliminary Palladium-Promoted Decarboxylation
Application of these conditions to an advanced biaryl 4 provided a high yield (75%) of decarboxylated product 5 (Scheme 2). Moreover, no racemization of the biaryl axis was seen under these milder conditions (90 °C vs. 180 °C with Cu/quinoline). By excluding silver carbonate from the reaction, the yield of 5 was improved to 87%. In all cases when palladium was excluded no decarboxylation was observed.
Scheme 2.
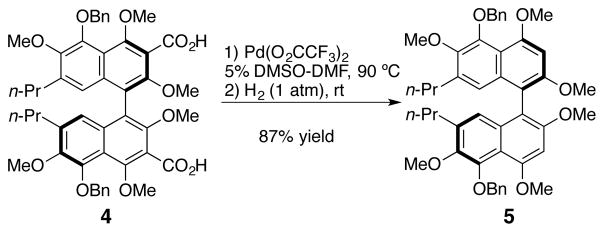
Decarboxylation in the Absence of Ag2CO3
Palladium Promoted Process
Hydride/Proton Sources
In addition to requiring a full equivalent palladium, the conditions described above (Scheme 2) were incompatible with more complex aryls. In particular, biaryls with more highly functionalized C7, C7′-groups suffered from decomposition under the reaction conditions. As a result, alternative hydrogen sources were surveyed for the reduction of the aryl palladium intermediate. To facilitate rapid screening, a GC assay was developed with model substrate 6 (Eq 1). The results are summarized in Table 1.
Table 1.
| Entry | H source (equiv) | Solvent | T (°C) | Time (h) | Conv. (%)b |
|---|---|---|---|---|---|
| 1 | H2 (1 atm) | 5% DMSO-DMF | rt | 3 | 43 |
| 2 | NaOMe (5.0) | 20% DMSO-DMF | 90 | 24 | 81 (79)c |
| 3 | HSi(Et)3 (1.4) | 5% DMSO-DMF | rt | 24 | 59 |
| 4 | HSi(i-Pr)3 (1.4) | 5% DMSO-DMF | rt | 24 | 77 (82)d |
| 5 | HSi(Ph)3 (1.4) | 5% DMSO-DMF | rt | 24 | 65 |
| 6 | PMHS/KF (1.4) | 5% DMSO-DMF | rt | 0.5 | 95 |
| 7 | NaBH4 (2.0) | 5%DMSO-DMF | rt | 2 | 85 |
| 8 | BH3•THF (2.0) | 5%DMSO-DMF | rt | 2 | 83 |
| 9 | NaCO2H (2.0) | MeOH | 70 | 5 | 64 (84)e |
| 10 | NH4CO2H (2.0) | MeOH | 70 | 24 | 65 |
| 11 | Mg/NH4OAc (1.2) | MeOH | 70 | 5 | 71 |
Reaction conditions: initial concentration [6] = 0.11 M and [Pd(O2CCF3)2] = 0.13 M, 3.0 mL solvent, 70 °C for 1 h, then hydrogen source at specified temperature.
GC monitoring of the formation of 8 with biphenyl as internal standard.
79% conversion after 24 h with K2CO3/MeOH.
82% conversion after 24 h with 3 equiv HSi(i-Pr)3.
84% conversion after 24 h with 5% DMSO-DMF.
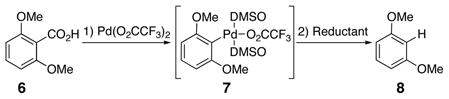 |
(1) |
Under the initial conditions of a dihydrogen atmosphere, conversion to product was sluggish (43% after 3 h; Table 1, entry 1). Alkoxides can act as hydride sources via a β-hydride elimination mechanism in the reduction of aryl halides with palladium.36 When sodium methoxide, or potassium methoxide generated in situ from potassium carbonate and methanol, were used for the reduction of 7, approximately 80% conversion was achieved (entry 2). As these results were encouraging, other hydride sources for the reduction of the aryl palladium intermediate were investigated.
Hydrosilanes are common, mild hydride sources for the reduction of aryl halides and were surveyed in the protodepalladation.37,38,39 Good conversion to product (47–77%) was observed with a wide range of hydrosilanes (Table 1, entries 3–5). In general, the steric hindrance of the hydrosilane displayed a minimal effect on the reduction of the aryl palladium intermediate. Increasing the amount of hydrosilane had the greatest benefit in the reaction and improved the conversion from 77 to 82% (Table 1, entry 4). Even higher conversions (>95%) could be achieved using polymethylhydrosiloxane (PMHS) in combination with KF (entry 6).
Conventional hydride reagents were also effective in the reduction of 7. For example, sodium borohydride and a BH3•THF complex gave high conversion to the protodepalladated product (83–87%, Table 1, entries 7 and 8). A variety of other hydride sources have been used in the literature for the reduction of aryl halides and triflates and were surveyed here.40,41,42 The use of sodium formate in refluxing methanol furnished 64% conversion (entry 9). Employing a 5% DMSO-DMF solvent mixture with sodium formate instead of MeOH increased the conversion to 84% without additional heating. Ammonium formate in refluxing methanol also facilitated decarboxylation and gave a moderate conversion to product (65%, entry 10). Magnesium metal and ammonium acetate in methanol43 has been proposed to mediate the reduction of aryl triflates through a radical pathway and also gave a high conversion in the decarboxylation reaction (71% conversion, entry 11).
The especially mild nature of the palladium promoted decarboxylation combined with the array of usable reductants allows these processes to be compatible with complex substrates. For example, a number of these conditions proved to be suitable for the decarboxylation of advanced intermediates in the total syntheses of hypocrellin A,44,45 (+)-phleichrome,46 and other perylenequinone natural products.47,48
From the results in the palladium promoted reactions, it was clear that a number of reducing agents could be employed to cleanly and mildly reduce the aryl palladium species (i.e., 7) to the corresponding arene (see Table 1). Presumably, an aryl palladium hydride intermediate forms, which undergoes reductive elimination to generate the product and palladium(0). Attempts to add on oxidant to reoxidze the palladium(0) to palladium(II) were unsuccessful. For example, 20 mol% of palladium(II) trifluoroacetate with sodium methoxide, sodium formate, or triisopropylsilane in with (Cu(O2CCF3)2) provided <10% of the desired product. Indeed, all of conditions resulted in formation of palladium(0) and the rapid production of palladium black. Therefore, the development of conditions that would permit palladium reoxidation in the presence of such reducing agents seemed problematic. However, an a key observation from a control reaction revelaed that the protodecarboxylation occurs very slowly in presence of Pd(O2CCF3)2 alone, without any reducing agent. With the prospect of a decarboxylation that did not require a change in oxidation state of palladium, efforts turned to understanding this process further.
Mechanistic Studies
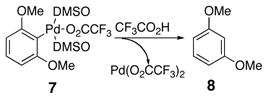
The overall reaction process proposed for the decarboxylation with one equivalent of Pd(O2CCF3)2 alone is comprised of two fundamental parts (Scheme 3). First, the aromatic acid is transformed by means of a palladium(II) source into an aryl palladium(II) species accompanied by the release of carbon dioxide (6 to 7). Second, the aryl palladium intermediate undergoes protonation to yield the protodecarboxylated product (8). Since the second stage of the decarboxylation reaction occurs very slowly without the addition of exogenous acid, intermediate 7 can be generated in pure form, and the two stages of the reaction can be analyzed separately. By further understanding each of the steps, it should be possible to regenerate the initial palladium(II) species to create a decarboxylation process catalytic in palladium.
Scheme 3.
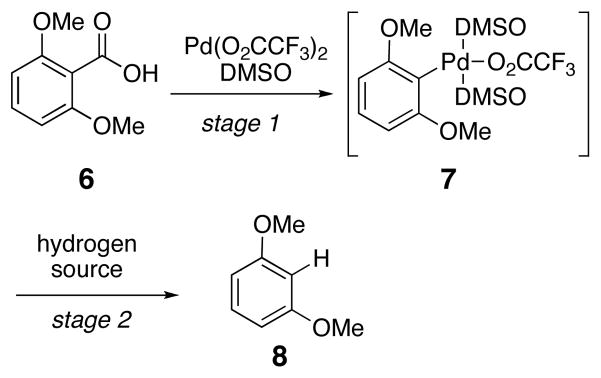
Steps in the Palladium Mediated Aromatic Decarboxylation
Decarboxylative Palladation
In the first stage of the reaction, referred to as decarboxylative palladation, two steps occur. The palladium catalyst initially associates with two DMSO ligands and undergoes carboxyl exchange of one of the trifluoroacetate ligands with the aromatic acid to form an aryl carboxylate palladium species (9, Scheme 4). This aryl carboxylate palladium intermediate (9) forms very rapidly at ambient temperature, but heating is necessary to promote decarboxylative palladation. Notably, the leaving group on palladium has little effect on the facility of this process, since adducts form equally rapidly from Pd(OAc)2 or Pd(O2CCF3)2. Coordination of the ortho-methoxy group as discussed later in this manuscript may contribute to ease of this exchange. To initiate the subsequent decarboxylation, dissociation of a DMSO ligand has been proposed.34 This event allows formation of a transition structure incorporating a square planar palladium(II). Association of the electrophilic palladium center with the nucleophilic arene carbon generates an intermediate that undergoes extrusion of carbon dioxide to form aryl palladium intermediate 7.
Scheme 4.
Decarboxylative Palladation
Decarboxylative Palladation Kinetics
The reaction of 9 to 7 could be conveniently monitored by 1H NMR spectroscopy in DMSO-d6. Treatment of 6 with 1.2 equivalents of palladium(II) trifluoroacetate, at ambient temperature and in the absence of added acid resulted in rapid carboxyl exchange to yield aryl carboxylate palladium species 9. Upon heating to 70 °C, 9 underwent clean decarboxylative palladation to afford complete conversion to the aryl palladium intermediate 7 (Scheme 4) within one hour as illustrated in Figure 1a. Protodepalladation was slow under these conditions resulting in negligible formation of 8 (≤3%). Kinetic analysis (Figure 1b) revealed that decarboxylative palladation is a first-order process with respect to 9 (k = 8.8 × 10−4 s−1, t1/2 = 13 min) in agreement with prior reports.35,49
Figure 1.
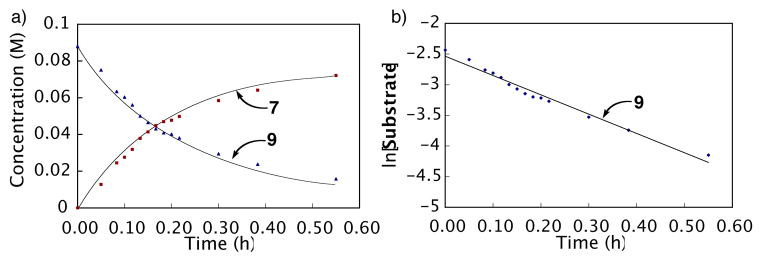
Decarboxylative palladation as monitored by 1H NMR spectroscopy (Substrate = 9). Reaction conditions: initial concentration [6] = 0.09 M and [Pd(O2CCF3)2] = 0.11 M, 0.75 mL DMSO-d6, 70 °C. Equation of the line for b is y = −2.499x – 2.641. R2 = 0.964.
Acid Effect on Decarboxylative Palladation
As illustrated in Scheme 4, one equivalent of trifluoroacetic acid is released upon ligand exchange of the aromatic acid substrate with palladium(II) trifluoroacetate. In order to determine what effect, if any, this released acid might have on the course of the reaction, the rate of the decarboxylative palladation was measured with added acid. Due to the volatility of trifluoroacetic acid under the reaction conditions (70 °C), methanesulfonic acid was used as an acid source for this measurement. As shown in Figure 2, the decarboxylative palladation of 6 was five times slower when 10 equivalents of methanesulfonic acid were added (k = 1.7 × 10−4 s−1, t1/2 = 1.2 h) compared to when no added acid was used (k = 8.8 × 10−4 s−1, t1/2 = 13 min). Thus, added acid inhibits the decarboxylative palladation process.
Figure 2.

Acid effect on the decarboxylative palladation of 6 as monitored by 1H NMR spectroscopy. ■ [6] = 0.08 M, [Pd(O2CCF3)2] = 0.10 M, CH3SO3H (10 equiv), 0.75 mL DMSO-d6, 70 °C; ◆ [6] = 0.09 M, [Pd(O2CCF3)2] = 0.11 M, 0.75 mL DMSO-d6, 70 °C.
The inhibition of the decarboxylative palladation step by acid could occur in several ways. For example, extra acid could protonate 9, coordinate to 9, or displace a DMSO ligand from 9. Any of these changes could alter the inherent decarboxylation rate. Alternatively, added acid may affect the equilibrium that gives rise to 9 (Scheme 5). If the acid source has a strong affinity for palladium, then the effective concentration of 9 may be much lower giving rise to slower rates.
Scheme 5.
Effect of Trifluoroacetic Acid on the Ligand Exchange
The equilibrium in Scheme 5 was monitored using 1H NMR spectroscopy.50 Addition of exactly one equivalent of palladium(II) trifluoroacetate to compound 6 results in the formation of aryl palladium carboxylate species 9, as well as a small amount of bisaryl palladium carboxylate species 10.34 Upon treatment with 10 equivalents of trifluoroacetic acid, the equilibrium shifts back in favor of the carboxylic acid (6). Therefore, in the presence of added acid the effective concentration of 9 is decreased and could account for the slower rate of decarboxylative palladation found in Figure 2.
Decarboxylative Palladation Eyring Studies
A dramatic increase in the decarboxylative palladation reaction rate occurred as the temperature was increased from 50 °C (k = 7.0 × 10−5 s−1, t1/2 = 2.8 h) to 110 °C (k = 2.0 × 10−2 s−1, t1/2 = 35 sec). At all temperatures, the disappearance of adduct 9 was found to occur via a first-order process. Even at 70 °C, the decarboxylative palladation was essentially completed within one hour indicating that this portion of the overall aromatic protodecarboxylation (stage 1, Scheme 3) is facile and not overall rate-determining.
The application of transition state theory51 afforded good linear correlation (Figure 3) and indicated that the mechanism remained unchanged over this range of temperatures. From this data the activation enthalpy (ΔH‡ = 23 kcal/mol) and activation entropy (ΔS‡ = −6.7 cal/K•mol) for the decarboxylation of 9 to 7 were determined. At 298 K, the free energy of activation (ΔG‡) was determined to be 25 kcal/mol.
Figure 3.
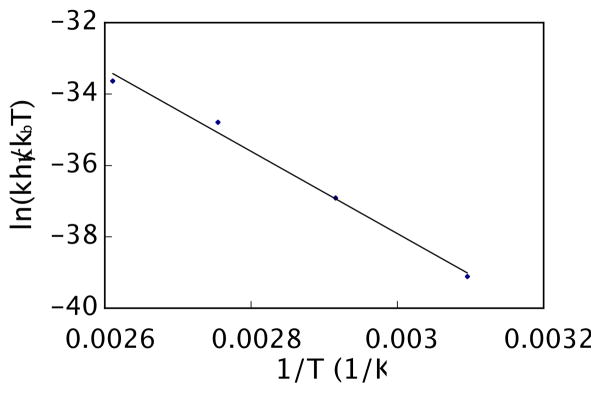
Eyring Plot for Decarboxylative Palladation. Reaction conditions: initial concentration [6] = 0.09 M and [Pd(O2CCF3)2] = 0.11 M, 0.75 mL DMSO-d6 at four different temperatures (50, 70, 90, and 110 °C). Equation of the line is y = 11520x – 3.345. R2 = 0.999.
Mechanism of Decarboxylative Palladation
Scheme 6 depicts some possible mechanisms for the decarboxylation of aryl palladium carboxylate 9 to aryl palladium 7. Insertion into the carboxylate CO bond to generate an acyl palladium species (path A, Scheme 6) is improbable since CO2 rather than CO is generated in the related decarboxylative Heck coupling.32 Protonation of the aromatic ring of aryl palladium carboxylate 9 followed by expulsion of CO2 (path B, Scheme 6) is also unlikely as aryl palladium 7 would require rearomatization through loss of hydrogen. However, this cation intermediate has been shown (see below) to favor rearomatization through loss of palladium, which would not give rise to the isolated product 7.
Scheme 6.

Potential Decarboxylative Palladation Mechanisms
Path C includes an intramolecular electrophilic palladation of the aryl palladium carboxylate resulting in loss of aromaticity via TS1 followed by a concerted decarboxylation rearomatization (TS2). Subsequent general base or intramolecular assisted proton transfer would yield the product. There is significant support for electrophilic palladation in other palladium processes.52,53,54,55 Path C begins with carboxyl exchange of the substrate with a trifluoroacetate ligand to yield an intermediate arylpalladium carboxylate (9), which correlates with 1H NMR observations.34 The initial aryl palladium carboxylate is in equilibrium between several forms (9, 11, 12), and it is most likely that the nonreactive chelated form (11) is the most stable. The carbonyl must become nearly perpendicular to the arene (i.e., 12) resulting in a loss of conjugation via an η1 or η2 complex.56
Intermolecular electrophilic palladation (path D) would not require a perpendicular orientation of the carboxylate to the arene. The rate-determining step for path D would likely be the electrophilic palladation that also results in loss of aromaticity. 1H NMR evidence indicates that ligand exchange between substrate and the palladium(II) trifluoroacetate to 9 is fast and favorable (see Scheme 5) which points against path D. However, there are many examples where observable species do not lie along the actual reaction path.57 Furthermore, the required palladium cation (LnPd(O2CCF3)+) is known to form under these conditions.58,59,60,61 Overall our experimental observations and computational efforts described below support mechanism C as the operative pathway.
In pathway C, loss of aromaticity in TS1 would lead to a large activation enthalpy. However, TS2 would also be expected to have significant activation enthalpy due to the multiple bond-forming and bond-breaking events. Therefore, the measured calculated activation enthalpy (ΔH‡ = 23 kcal/mol) is consistent with either transition state (TS1 or TS2) being the rate determining step.
Decarboxylative Palladation Hammett Study
A Hammett study was investigated to distinguish between TS1 and TS2 of path C.62 From a Hammett study, the reaction constant, ρ, can be determined using σpara parameters from Anslyn and Dougherty; a negative ρ value would indicate charge buildup in the transition state.51 The rate of decarboxylative palladation with para-substituted benzoic acid derivatives (6a-c, R = H, Me, or Cl) was measured and the reaction constant, ρ, was determined to be −1.87 (Figure 4). Similar reaction constants have been observed in other systems that undergo electrophilic palladation mechanisms like TS1.55 This negative ρ value is consistent with the formation of a zwitterionic species (13, Scheme 6). In contrast, TS2 involves a loss of positive charge on the aromatic ring compared to 13 resulting in a positive ρ value if TS2 was rate-determining.63 In addition, decarboxylative palladation occurred at room temperature when the very electron-rich 2,4,6-trimethoxybenzoic acid (6d, R = OMe) was treated with palladium(II) trifluoroacetate in DMSO-d6 (74% conversion after 4 hours, k = 1.3 × 10−4 s−1, t1/2 = 1.5 h), again supporting the electrophilic palladation TS1 proposed above as the rate determing step.
Figure 4.

Hammett Plot of Decarboxylative Palladation. The appropriate substrate was mixed with Pd(O2CCF3)2 (1.2 equiv) in 0.75 mL DMSO-d6 and heated to 50 °C with 1H NMR monitoring. Reaction conditions: initial concentration [6a-c] = 0.066 M and [Pd(O2CCF3)2] = 0.079 M, 0.75 mL DMSO-d6. Equation of the line: y = −1.867x + 0.0029. R2 = 0.999
Comparison of the experimentally determined activation entropy (−6.7 cal/K•mol) with estimated entropies for TS1 and TS2 is also consistent with the results above. TS1 has no molecularity change, but the transition state is organized, and an entropically disfavored process is anticipated (~−10 cal/K•mol).51 Further, TS1 leads to a cationic intermediate suggesting some organization of DMSO in the outer sphere of the transition state. However, work by Suga and later Privalov has shown the energy of solvent reorganization for DMF and DMSO to be minimal for aryl cationic and palladium mediated processes.64,65 TS2 gives rise to two molecules from a single molecule precursor leading to a large favorable entropy that would be offset to some extent by the disfavorable entropy associated with the organized transition state (~10 cal/K•mol).51 As discussed above, solvent reorganization should not play a major role in mechanism TS2 and would not change the estimated entropy.
Decarboxylative Palladation Computational Studies
To gain insight at the mechanism, computations were carried out using B3LYP/6–31G(d) (SDD for Pd) in dichloromethane with a CPCM solvation model.66 As shown in Figure 5, the dissociation of DMSO to form intermediate 12 is uphill in energy by 9.5 kcal/mol, which then forms complexed intermediate 13 with an overall barrier of 12.9 kcal/mol. Subsequent C-C cleavage via TS-2 proceeds with a barrier of 16.4 kcal/mol leading to square planar intermediate. Release of CO2 to 7 is expected to drive the reaction forward.67,68
Figure 5.

Relative free energies (kcal/mol; 298.15 K) calculated using B3LYP/6–31G(d) (SDD for Pd) in dichloromethane (CPCM;UFF). Selected distances are in Ångstroms.
Figure 5 indicates that TS-2 is the rate-determining step; however the energy difference is very small and other methods explored (B3PW91/SDD-gas) also predict TS-1 to be competitive and in some cases even the highest energy transition state (see Supporting Information). The entropic cost associated with TS-1 and TS-2 (with respect to 12) is −3.0 and −0.7 cal/K•mol, respectively. Thus, the activation entropy value for TS-1 is in better agreement with our experimentally determined activation entropy value (−6.7 cal/K•mol). Comparison of Figure 5 with work done by Liu67 and Su68 on monosubstituted benzoic acids undergoing decarboxylative palladation shows that bis-ortho-substituted arenes decarboxylate with lower ΔG‡ and that the ipso-carbon palladium bond is much shorter promoting intramolecular electrophillic palladation of 13 before decarboxylation. Overall, it appears that pathway C is operative in the decarboxylative palladation. For the electron rich systems here, the bulk of the evidence supports TS1 from path C as rate-determining step, but the rate-determing will shift from TS1 to TS2 depending on the substitution of the ring.
Protodepalladation
The second stage of the decarboxylation reaction (stage 2, Scheme 3) entails cleavage of the aryl-palladium bond via a protonation event (Eq 2). This protodepalladation occurs when the substrate 6 is treated with 1.2 equivalents of palladium(II) trifluoroacetate (Scheme 3); no further acid or hydride source is necessary. From the balanced chemical reaction, it is clear that the proton must arise from the original carboxylic acid hydrogen. For this step of the overall process, the proton source is generated in the very first ligand exchange step from trifluoroacetic acid (6 to 9 in Scheme 4).
 |
(2) |
Protodepalladation Kinetics
Compound 7 can be conveniently generated by heating aromatic acid 6 with palladium(II) trifluoroacetate in DMSO-d6 for 1 h at 80 °C (stage 1, Scheme 3). With this intermediate in hand, we set out to investigate the protonation process in detail with the aim of developing an aromatic protodecarboxylation process catalytic in palladium. Heating of 7 that had been generated in situ in DMSO-d6 resulted in very slow formation of protodepalladated product 8 (32% conversion after 18 h at 80 °C). Reasoning that the proton source (trifluoroacetic acid, bp 72 °C) was volatilizing into the headspace of the reaction vessel, we undertook further studies with a less volatile acid source. Thus, excess methanesulfonic acid was added to in situ derived 7, and the formation of the product 8 was monitored by gas chromatography (GC) and 1H NMR spectroscopy.
Even with added acid, the process was relatively slow (~40% complete after 12 h at 65 °C). In order to probe the reaction order of this process further, 1H NMR (Figure 6 and Figure 6b) and GC analyses50 were undertaken over at least three half-lives. These experiments yielded almost identical results (1H NMR: k = 1.3 × 10−5 s−1, t1/2 = 14 h; GC: k = 1.4 × 10−5 s−1, t1/2 = 14 h) and definitively indicated that protodepalladation is a first-order process with respect to aryl palladium intermediate 7.69
Figure 6.
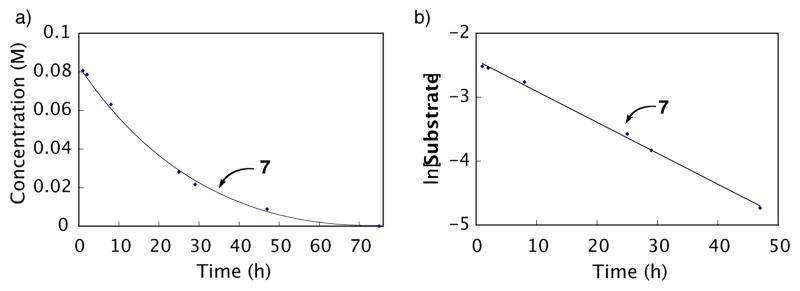
Protodepalladation as monitored by 1H NMR spectroscopy (Substrate = 7). Compound 6 was mixed with Pd(O2CCF3)2 in 0.75 mL DMSO-d6 and heated to 80 °C for 1 h. After cooling, a 1H NMR spectrum was taken, CH3SO3H was added, and 1H NMR monitoring was continued at 65 °C. Reaction conditions: initial concentration [6] = 0.08 M and [Pd(O2CCF3)2] = 0.10 M, 10 equiv CH3SO3H, 0.75 mL DMSO-d6. Equation of the line for b is y = 0.048x – 2.424. R2 = 0.998.
Acid Effect on Protodepalladation
Since it was also prudent to understand the role of acid in the final step of the decarboxylation reaction, a 1H NMR experiment was conducted to examine the effect of acid concentration in the conversion of 7 to 8. Protonation occurred much slower when 1 equivalent (k = 5.3 × 10−7 s−1, t1/2 = 365 h) or 3 equivalents of methanesulfonic acid was added (k = 3.3 × 10−6 s−1, t1/2 = 58 h) as opposed to the standard 10 equivalents (k = 1.3 × 10−5 s−1, t1/2 = 14 h). While this data appeared to fit a first order dependence in acid, a Lineweaver-Burk plot70 indicated that saturation behavior was possible.50 Unfortunately, experiments to confirm saturation behavior with higher concentrations of acid were unfruitful due to broadening in the 1H NMR spectra. In order to distinguish between introduction of acid in the depalladation transition state (general acid, first order in acid) vs. protonation prior to depalladation (saturation in acid), the acid concentration was examined with substoichiometric amounts of palladium (see Catalytic Decarboxylation Section below) and saturation behavior was confirmed.
Effects of Solvent on Reaction Rate
Numerous solvents were screened during the optimization of the reaction, and a 5% DMSO-DMF mixture proved superior to either DMSO or DMF alone, as had been observed by Myers for the decarboxylative Heck coupling (2-fold increase with 5% DMSO-DMF vs. DMSO).71 This result is surprising as the rate-determining step is different for this process compared to the decarboxylative Heck reaction34 (protodepalladation vs. decarboxylative palladation). Furthermore, the protodepalladation was subject to a more dramatic decrease in conversion when DMSO was used in place of the 5% DMSO-DMF mixture. With 5% DMSO-DMF (k = 3.5 × 10−4 s−1, t1/2 = 33 min), protodepalladation occurs 24 times faster than in DMSO alone (k = 1.4 × 10−5 s−1, t1/2 = 14 h) (Figure 7).
Figure 7.

Solvent effect on the protonation of 7 as monitored by GC: ◆ DMSO; ■ 5% DMSO-DMF. Reaction conditions: initial concentration [6] = 0.10 M and [Pd(O2CCF3)2] = 0.12 M, CH3SO3H (10 equiv), 65 °C.
Evidently, the rate determining protodepalladation is much more sensitive to the solvent composition than the decarboxylation step that is rate-determining in the decarboxylative Heck coupling.34 Since DMSO is known to act as a ligand for palladium, the presence of a small amount of DMSO clearly allows the formation of a new palladium species with superior reactivity. On the other hand, the use of DMSO as the singular solvent likely saturates the palladium center thereby impeding access of ligands/substrates necessary for the reaction to proceed.72 A scenario consistent with these results is outlined in Scheme 7. If coordination of a second molecule of acid is necessary for protodepalladation to occur (see below), then excess DMSO would act as an inhibitor of the reaction. By the same argument, the addition of further acid will increase the concentration of the key precursor 14 thereby accelerating the reaction. In such a preequilibrium (specific acid catalysis), the acid species should exhibit saturation kinetics while DMSO will display more complex behavior consistent with that observed above (slower rates above and below the critical threshold concentration needed to allow coordination of one DMSO ligand to palladium).
Scheme 7.
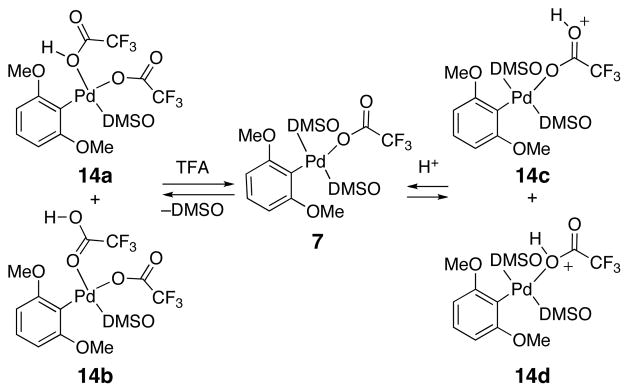
Effect of DMSO on Protonation Intermediates/Transition States
Protodepalladation Eyring Studies
The effect of temperature on the rate-determining protodepalladation step was also determined. Aryl palladium intermediate 7 was preformed by heating substrate 6 with 1.2 equivalents of palladium(II) trifluoroacetate. At this point, 10 equivalents of methanesulfonic acid were added to initiate the protodepalladation and the reaction was monitored by 1H NMR spectroscopy at various temperatures. In all cases, clean formation of product 8 was observed via a first-order process. The protodepalladation step was rate-determining at all temperatures and, in practical terms, a temperature of at least 70 °C (k = 2.4 × 10−5 s−1, t1/2 = 7.9 h) was required to achieve a reasonable rate of reaction.
An Eyring analysis provided ΔH‡ (26 kcal/mol) and ΔS‡ (−4.6 cal/K•mol) values for the protodepalladation of 7 to 8. Therefore at 298 K, the free energy of activation (ΔG‡) was found to be 27 kcal/mol. As expected, these results reinforce protonation of the aryl palladium intermediate (7) as the rate-determining step of the overall decarboxylation reaction (decarboxylative palladation: ΔG‡298 = 25 kcal/mol vs. protodepalladation: ΔG‡298 = 27 kcal/mol). Based on the relatively small differences between the two barriers, it is entirely possible that the rate-determining step may shift for different substrates. As we detail in the substrate scope below, it became apparent di-ortho-methoxy substitution on the arene carboxylic acid provides the most efficient reaction and that non-coordinating ortho-substituents are disfavorable overall. Even though such groups would facilitate protodepalladation by relief of strain, the initial decarboxylative palladation becomes less favorable.
Mechanism of Protodepalladation
A number of possible mechanisms can be proposed to account for the protonation reaction (Scheme 8). In the first pathway (A), an intermolecular protonation of the aromatic ring occurs in a concerted manner through either a four- (A1) or six-membered (A2) transition state.
Scheme 8.

Potential Protodepalladation Mechanisms
In a second pathway (B), the protonation occurs in a stepwise intermolecular manner, yielding a Wheland intermediate73 15. Then, loss of palladium and rearomatization affords the final product.
Alternatively, protonation could occur through an intramolecular pathway (C or D). In these pathways, formation of a protonated palladium species is proposed via coordination of a further trifluoroacetic acid molecule (14a or 14b) or through a proton transfer (14c or 14d). In path C, a concerted intramolecular protonation of the aromatic ring through a four- (C1) or six-membered (C2) transition state would yield the product and a palladium species.
Path D would also occur through a precoordinated intermediate (14a-d), but in a stepwise intramolecular fashion. In this case, a four- (D1) or six-membered (D2) intramolecular protonation transition state would yield the same protonated intermediate 15 from pathway B. This Wheland intermediate (15) could subsequently undergo rearomatization in a similar manner to afford the protodepalladated product.
Distinguishing between mechanisms A, B and C, B can be achieved by considering the effect of acid on protodepalladation. Paths A and B should display first-order kinetics with respect to acid since the introduction of acid occurs in the transition state (general acid catalysis). As stated previously, it has been shown that acid exhibits saturation behavior in the protodepalladation step, so neither path A nor B is likely to be the protodepalladation mechanism. On the other hand, path C and D would require the formation of the precoordinated intermediate, 14a-d, in order to achieve protodepalladation (specific acid catalysis) and is consistent with the observed saturation behavior of acid.
While this type of protonation has not been studied in the literature74, the reverse reaction (C-H activation) has been widely explored.75,76,77,78,79,80,81,82 In the reverse reaction (C-H activation), five types of mechanisms have been proposed:83,84,85,86,87,88,89 oxidative insertion,84,88 carbopalladation (Heck-like),83 electrophilic aromatic substitution (SEAr),53,55 σ-bond metathesis (concerted metallation deprotonation),86,87,89 and intermolecular proton abstraction (SE3).86,87,89 Both the oxidative insertion and carbopalladation mechanisms have received the least support in the literature and do not mirror the proposed mechanisms for protodepalladation. Since intermolecular paths A and B have been discounted based on saturation behavior with acid, the reverse of the intermolecular proton abstraction mechanism is also unlikely to be operative in the protodepalladation.
Conversely, σ-bond metathesis is strongly supported and more likely than SEAr for C-H insertion. The reverse of σ-bond metathesis would closely resemble pathway C in that a concerted transition state is taking place. However, an electrophilic aromatic substitution type mechanism requires the formation of a Wheland intermediate in the transition state, which is similar to pathway D. A Hammett study62 was used to further distinguish between pathways C and D (see below following benzoic acid substrate scope). Based on the negative ρ value of the Hammett study, it is likely that protodepalladation proceeds through a transition state with positive charge buildup51; this finding is most consistent with path D. While it is difficult to discern between paths D1 and D2, most of the experimental data in the literature for C-H activation mechanisms favor six-membered transition states as opposed to four-membered transition states.85,87
The results above are consistent with protodepalladation occuring by pathway D, stepwise intramolecular protonation. Comparison of the experimentally determined activation entropy (−4.6 cal/K•mol) with estimated entropies for the transition states of pathway C and D are also consistent with the results above. In pathway C, the formation of two product molecules from a single intermediate is entropically highly favorable. A high degree of organization required in the transition state, including solvation, would offset this trend, leading to a somewhat entropically favored (~10 cal/K•mol) process overall.51 In pathway D, the transition states here differ from those of paths C1 or C2 in that D1 and D2 retain the carbon-palladium bond and are unimolecular. Overall, the activation entropy for paths D1 and D2 should be disfavorable due to rigidity and solvent organization (~−10 cal/K•mol).51
Catalytic Decarboxylation
Analysis of the Reaction Cycle
From the palladium promoted results in Table 1, it was clear that a number of reducing agents could be employed to cleanly and mildly effect an overall protodecarboxylation reaction. However, these reagents resulted in reduction of the palladium during the protodepalladation step. Since palladium(II) is required for the initial decarboxylative palladation step, reoxidants such as Cu(O2CCF3)2 were studied, but failed to close the catalytic cycle. Thus, it was necessary to identify a hydrogen source that would be compatible with the decarboxylative palladation was necessary.
Based on the above mechanism studies, we concluded that a version catalytic in the metal species should be accessible without using a reducing agent as the hydrogen source. The overall reaction cycle comprised of the two stages (see Scheme 3) studied above is illustrated in Scheme 9. The first step is an equilibrium carboxyl exchange of a trifluoroacetate ligand with the aromatic acid (6) to form the aryl carboxylate palladium species (9). Next, dissociation of a DMSO ligand triggers a electrophillic intramolecular palladation resulting in loss of carbon dioxide. The resultant aryl palladium intermediate (7) can react with the trifluoroacetic acid released during the first ligand exchange. Rate-limiting proton transfer through a six-membered transition state results in a Wheland intermediate that undergoes depalladation to yield the decarboxylated product (8) and regenerates the active palladium catalyst. The key points are that trifluoroacetic acid is released and consumed during the reaction and that there is no apparent change in palladium oxidation state.
Scheme 9.

Catalytic Cycle for the Aromatic Decarboxylation
In fact, reaction does occur with catalytic palladium(II) trifluoroacetate alone, but very slowly (34% conversion after 24 h with 20 mol% Pd(O2CCF3)2 at 65 °C in 5% DMSO-DMF). If the above analysis is correct, then acceleration of the rate-determining second stage of the reaction is key to creating a useful catalytic process. As discussed above, the first stage of the reaction was inhibited by acid while the second stage appeared to exhibit saturation kinetics with acid. To determine if acid saturation kinetics occurs in the rate-determining step under catalytic conditions, reaction of 6 was monitored by GC with 0.15 equivalents of palladium(II) trifluoroacetate at several different trifluoroacetic acid concentrations. As expected, a first order dependence in acid is not observed at high concentrations (Figure 9a) and the rate of the reaction remains nearly constant. A Lineweaver-Burk plot70 (Figure 9b) confirmed that saturation in acid is occurring, which further supports the proposed mechanism for protodepalladation (Scheme 8, pathway D). In general, addition of modest amounts of exogenous acid resulted in an overall rate enhancement.
Figure 9.
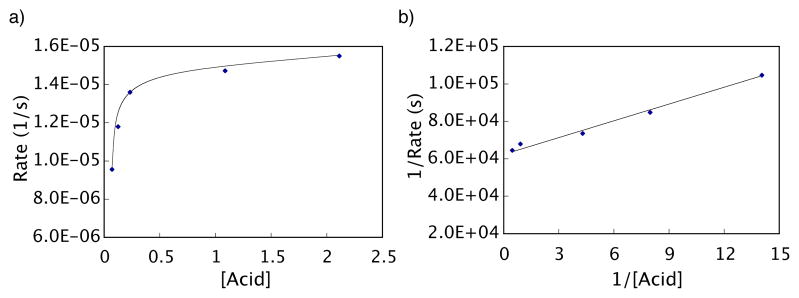
Acid concentration effect on the decarboxylation of 6 as monitored by GC. Reaction Conditions: [6] = 0.11 M, [Pd(O2CCF3)2] = 0.016 M, CF3CO2H a) Reaction at 5 different acid concentrations (4.3, 7.7, 14.3, 67.7, and 134.3 equivalents with respect to palladium). b) Lineweaver-Burk plot70 (double reciprocal plot) confirming saturation kinetics. Equation of the line is y = 2861.6x + 63192. R2 = 0.986.
Acid Sources with Catalytic Pd
Since the addition of modest amounts of trifluoroacetic acid provided a significant rate enhancement in the decarboxylation reaction, a variety of protic acids (10 equiv) were screened. Using 20 mol% of palladium(II) trifluoroacetate in 5% DMSO-DMF at 70 °C, the protodecarboxylation of 6 was assessed by GC (Table 2). As expected, trifluoroacetic acid gave high conversion to decarboxylated product (86%, entry 1) after only 24 hours. Longer reaction times yielded only slightly higher conversion.
Table 2.
Hydrochloric acid, which has a lower pKa than trifluoroacetic acid but cannot form a 4 or 6 member transition state (path D, Scheme 8, D1 or D2) during protodepalladation, failed to provide any conversion to the decarboxylated product at either 10 equivalents (entry 2) or 2 equivalents. Both nitric acid and methanesulfonic acid were slightly less efficient than trifluoroacetic acid (entries 3 and 4; 61 and 76%, respectively), despite their increased acidity. Carboxylic acids gave variable results with p-nitrobenzoic acid (Table 2, entry 5) giving rise to a very rapid rate. Unfortunately, the p-nitrobenzoic acid reactions led to a number of byproducts and were difficult to purify. Formic acid (96%) did not yield any significant amount of product (entry 6), perhaps due to reduction of the palladium(II) species via decarboxylation of the formic acid. Acetic acid acted slightly more slowly than trifluoroacetic acid, but still provided a moderate conversion (66%, entry 7). Nevertheless, no direct correlation between pKa and reaction rate was discerned. Due to convenience and uniformly good conversions, trifluoroacetic acid was chosen as the proton source for further studies on the aromatic decarboxylation reaction.
Palladium Order Under Catalytic Conditions
To determine if the order in palladium remained the same for the catalytic vs. promoted reacitons (see above), reactions at different catalyst loadings were examined (Figure 10a). A first order correlation was observed (Figure 10b) and supports a similar reaction mechanism for both processes. While no decarboxylation was observed in the absence of palladium catalyst, good rates were observed even with 10 mol% catalyst loading (62% conversion after 24 h). Attempts to extend the catalyst lifetime by adding a stoichiometric oxidant, such as Cu(O2CCF3)2, were unsuccessful.
Figure 10.
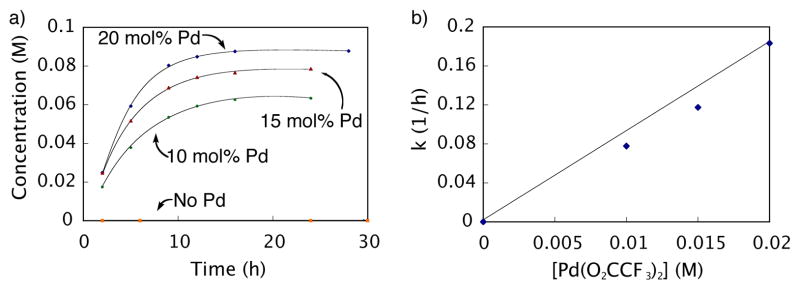
Effect of catalyst loading on the decarboxylation of 6 as monitored by GC: ◆ = 20 mol% Pd(O2CCF3)2, ▲ = 15 mol% Pd(O2CCF3)2, ● = 10 mol% Pd(O2CCF3)2, and ■ = No Pd(O2CCF3)2. Reaction conditions: initial concentration [6] = 0.10 M and [Pd(O2CCF3)2] = 0.020, 0.015, and 0.010 M respectively, CF3CO2H (10 equiv), 3.0 mL 5% DMSO-DMF, 70 °C. a) Reaction run at 4 different palladium concentrations. b) Rate vs. [Pd(O2CCF3)2]. Equation of the line is y = 8.928x – 0.005. R2 = 0.986.
Solvent Screen
A number of solvents were screened in the decarboxylation reaction (Table 3). Using the standard 5% DMSO-DMF solvent mixture provided ~81% conversion of decarboxylated product (entry 1). The use of a stringently dried 5% DMSO-DMF mixture slightly increased the conversion to 85% (entry 2). Therefore, it was concluded that small amounts of water do not significantly inhibit the decarboxylation reaction. The use of either DMSO or DMF alone gave only moderate conversions to product (entries 3 and 4; 32 and 53%, respectively). Since DMSO acts as a ligand throughout the reaction and must be able to undergo facile ligand exchanges, an excess of DMSO would slow the overall rate of reaction significantly. However, DMSO can stabilize Pd(II) intermediates and slow the formation of palladium black. Therefore, a small amount of DMSO dramatically improves the reaction.92,93,94,95 While DMF may also act as a ligand96,97 for palladium in the decarboxylation reaction, it is not as effective at stabilizing the reactive intermediates. In light of this analysis, it was surprising that the decarboxylated product was obtained with only slightly lower conversion in xylenes (73%, entry 5). However, the rapid formation of a black precipitate was noted, and the reaction slowed noticeably.
Table 3.
| Entry | Solvent | Conv. (%)b |
|---|---|---|
| 1 | 5% DMSO-DMF | 81 |
| 2 | 5% DMSO-DMFc | 85 |
| 3 | DMSO | 32 |
| 4 | DMF | 53 |
| 5 | xylenes | 73 |
| 6 | MeOHd | 43 |
| 7 | MeOH | 70 |
| 8 | EtOH | 56 |
| 9 | CF3CH2OH | 58 |
| 10 | i-PrOH | 81 |
Reaction conditions: initial concentration [6] = 0.10 M and [Pd(O2CCF3)2] = 0.020 M, 10 equiv CF3CO2H, 3.0 mL solvent, 70 °C, 24 h.
GC monitoring with biphenyl as internal standard.
Distilled solvents.
No added CF3CO2H.
Protic solvents were examined as they could serve as alternative proton sources during the reaction. The use of methanol as the solvent, without additional trifluoroacetic acid, resulted in 43% conversion (entry 7) confirming this hypothesis. Adding 10 equivalents of trifluoroacetic acid to methanol increased the conversion to 70% (entry 8). Other protic solvents, such as ethanol, trifluoroethanol, and isopropanol, produced similar conversion of decarboxylated products in the presence of 10 equivalents of trifluoroacetic acid (entries 9–11; 56, 58, and 81%, respectively). The reactions in alcoholic solvents proceeded rapidly and then stalled due to the immediate formation of a black precipitate, presumably a palladium aggregate. While alcoholic solvents appear very favorable with respect to the protodecarboxylation process, β-hydride elimination serves as a reducing source decomposing the palladium(II) catalyst. The addition of an oxidant such as Cu(O2CCF3)2 did not prevent the decomposition of palladium with alcoholic solvents. As a consequence, the 5% DMSO-DMF solvent mixture appeared to provide the optimum of reactivity and catalyst stabilization.
Metal Sources
All attempts to perform the decarboxylation under these conditions using other metal sources, including Ag(O2CCF3), Zn(O2CCF3)2, Cu(O2CCF3)2, In(O2CCF3)3, Ni(O2CCF3)2, Ag(OSO2CF3), Cu(OSO2CF3)2, Zn(OSO2CF3)2, and In(OSO2CF3)3, were unsuccessful. Transmetallation from palladium to another aryl metal species was also attempted to facilitate protonation and potentially reduce the amount of palladium required. However, use of the metal sources above in concert with Pd(O2CCF3)2 failed to provide any benefit.
Palladium(II) acetate was found to be an acceptable alternative for the decarboxylation reaction with methanesulfonic acid. Reaction of 6 with 20 mol% of palladium(II) acetate and 10 equivalents of methanesulfonic acid at 80 °C yielded a comparable reaction rate (k = 2.2 × 10−5 s−1) to that of palladium(II) trifluoroacetate under the same conditions (k = 1.5 × 10−5 s−1).50 This result stands in contrast to the decarboxylative Heck reaction,32 which does not proceed well with palladium(II) acetate. Since ligand exchange occurs readily with exogenous acid, the use of either palladium(II) source in the presence of methanesulfonic acid should yield the same electrophilic palladium intermediate and result in similar rates of reaction. The intermediacy of an electrophilic palladium intermediate was further supported by the inferior reactivity observed when palladium(II) acetate and acetic acid (69% conversion after 24 h) was used in lieu of palladium(II) trifluoroacetate and trifluoroacetic acid (100% conversion after 24 h). In order to limit the number of different palladium species present in the reaction while retaining a highly electrophilic palladium source, a combination of palladium(II) trifluoroacetate and trifluoroacetic acid was employed for further studies.
Ligand Studies
Based upon the mechanisms discussed above, there appears to be at least one coordination site available for a ligand in both the decarboxylative palladation and protodepalladation steps. DMSO has been proposed to act in this capacity under the typical reaction conditions. To determine whether other ligands can function more effectively than DMSO, a series of phosphine and phosphine oxides were examined in addition to pyridine and bis-sulfoxide. The decarboxylation was slightly inhibited in the presence of 20 mol% triphenylphosphine, independent of the solvent (5% DMSO-DMF, isopropanol, or xylenes) (Table 4, entries 1–3). The addition of triarylphosphine ligands to palladium(II) acetate or palladium(II) trifluoroacetate results in rapid formation of palladium(0) complexes.98,99,100 This reduction of palladium(II) complexes is especially facile in DMF with excess phosphine ligand.100 In fact, further increasing the amount of triphenylphosphine in the decarboxylation reaction to one equivalent led to a much greater inhibition of the reaction (entries 4–6). The bidentate phosphine ligands, 1,3-bis(diphenylphosphino)propane and 1,2-bis(diphenylphosphino)ethane, displayed a similar effect on the conversion (Table 4, entries 7–9). While one equivalent of a bidentate phosphine with respect to palladium promotes only slow reduction of palladium(II) complexes to palladium(0) complexes, larger quantities of the bidentate phosphine ligand will quickly reduce palladium(II) to palladium(0).99 As a result, the decarboxylation reaction was only slightly inhibited at low concentration of bidentate phosphine (entries 7 and 8) whereas at a higher concentration the reaction suffered from even lower conversion (entry 9). The weak coordination of phosphine oxides to palladium(II) catalysts can help prevent precipitation of palladium black101 and could assist in the protonation via a similar intramolecular six-membered transition state as pathway D2 (Scheme 8). In fact, addition of 5 equivalents of trioctylphosphine oxide gave only a minor reduction in the conversion (entry 10, 71%).
Table 4.
| Entry | Ligand (equiv) | Solvent | Conv. (%)b |
|---|---|---|---|
| 1 | PPh3 (0.2) | 5% DMSO-DMF | 65 |
| 2 | PPh3 (0.2) | i-PrOH | 56 |
| 3 | PPh3 (0.2) | xylenes | 78 |
| 4 | PPh3 (1.0) | 5% DMSO-DMF | 3 |
| 5 | PPh3 (1.0) | i-PrOH | 8 |
| 6 | PPh3 (1.0) | xylenes | 39 |
| 7 | dppe (0.2) | xylenes | 75 |
| 8 | dppp (0.2) | xylenes | 66 |
| 9 | dppp (1.0) | xylenes | 41 |
| 10 | P(O)(octyl)3 (5.0) | 5% DMSO-DMF | 71 |
| 11 | Pyridine | 5% DMSO-DMF | No Prod |
| 12 | Bis-sulfoxidec | 5% DMSO-DMF | 66 |
Reaction conditions: initial concentration [6] = 0.10 M and [Pd(O2CCF3)2] = 0.020 M, 10 equiv CF3CO2H, 3.0 mL solvent, 70 °C, 24 h.
GC monitoring of the formation of 8 with biphenyl as internal standard.
Pd(OC2CCF3)2 was replaced with Pd(PhSOCH2CH2SOPh)(OAc)2.
Pyridine has been shown to minimize the formation of palladium black and could be substituted for DMSO with palladium (II), but its addition shut down the reaction process (entry 11, no prod).102 Also, palladium bis-sulfoxide with DMSO has been shown to be a stabilizer for palladium (II), but replacement of Pd(O2CCF3)2 with Pd(PhSOCH2CH2SOPh)(OAc)2 was not as successful as the original conditions (66%, entry 12 vs. 85%, entry 2 in Table 3).103 Overall, none of the ligands or DMSO surrogates surveyed resulted in any improvement of the decarboxylation reaction.
Substrate Scope
From the optimization studies described above, the general conditions for the aromatic decarboxylation reaction were determined to be 20 mol% palladium(II) trifluoroacetate and 10 equivalents trifluoroacetic acid in 5% DMSO-DMF with heating at ~70 °C. Under these conditions and with careful isolation of the volatile decarboxylated product (8a), a quantitative yield was obtained for 6a (Table 5, entry 1). In the absence of the palladium catalyst, substrate 6a failed to decarboxylate (Table 5, entry 1), proving that palladium is necessary in the reaction.
Table 5.
Benzoic Acid Derivatives in Catalytic Decarboxylationa
| |||
|---|---|---|---|
| Entry | Substrate | Product | Yield (%)b |
| 1 |
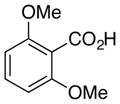 6a, R = H |
 8a, R =H |
Quant.c |
| 2 |
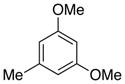
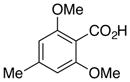 6b, R = Me |
 8b, R = Me |
80 |
| 3 |
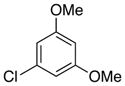
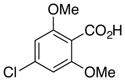 6c, R = Cl |
 8c, R = Cl |
89 |
| 4 |
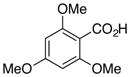 6d, R = OMe |
 8d, R = OMe |
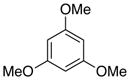
99d |
| 5 |
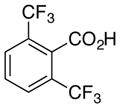 6e, R = H |
 8e, R = H |
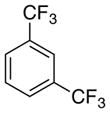
No Prod |
| 6 |
 6f, R = H |
 8f, R = H |
No Prode |
Reaction conditions: [6a-f] = 0.060 M and [Pd(O2CCF3)2] = 0.013 M, 10 equiv CF3CO2H, 3.0 mL 5% DMSO-DMF, 70 °C.
Isolated yield.
No reaction was seen in the absence of Pd(O2CCF3)2.
In the absence of Pd(O2CCF3)2 only 3% conversion to 8d was observed after 1 h at 70 °C (~40% conversion after 21.5 h).
~18% conversion by 1H NMR with stoichiometric palladium at 100 °C.
A variety of para-substituted benzoic acid derivatives (6b-d) were treated under the optimized decarboxylation conditions to give comparable yields of the decarboxylated products (Table 5, entries 2–4). While decarboxylation of 2,6-dimethoxy-4-methylbenzoic acid (6b) in 5% DMSO-DMF was completed in approximately 24 hours, the decarboxylation of 4-chloro-2,6-dimethoxybenzoic acid (6c) was about twice as slow under the same conditions (~48 h). Furthermore, the decarboxylation of 2,4,6-trimethoxybenzoic acid (6d) was extremely rapid under the standard reaction conditions (~100% conversion after 1 h at 70 °C). As expected, 6d failed to decarboxylate at an appreciable rate in the absence of palladium (~3% after 1 h at 70 °C), despite the fact that very electron rich substrates can decarboxylate spontaneously when heated in the presence of strong acid.8
The rate acceleration seen with the highly electron rich 2,4,6-trimethoxybenzoic acid (6d) warranted a further Hammett study. If the proposed electrophilic aromatic substitution mechanism depicted in path D (Scheme 8) for the rate-determining protodepalladation step is valid, then a large negative ρ value should be observed.104 On the other hand, if protodepalladation occurs through a σ-bond metathesis type mechanism (path C), a large positive ρ value would be expected.105 From Figure 11, a ρ = −4.03 (using σ+ parameters from Anslyn and Dougherty)51 was measured, which is consistent with a buildup of positive charge in the transition state. Therefore, the proposed protodepalladation mechanism, via a Wheland intermediate (path D, Scheme 8), is further supported.
Figure 11.

Hammett Plot for Catalytic Aromatic Decarboxylation. The appropriate substrate was mixed with Pd(O2CCF3)2 (0.2 equiv) and CH3SO3H (10 equiv) in 0.75 mL DMSO-d6 and heated to 70 °C with 1H NMR monitoring. Reaction conditions: initial concentration [6a-d] = 0.088 M and [Pd(O2CCF3)2] = 0.018 M, 10 equiv CH3SO3H, 0.75 mL DMSO-d6. Equation of the line is y = −4.025x − 0.0677. R2 = 0.979.
As expected from the Hammett studies above, a very electron poor substrate, 2,6-bistrifluoromethylbenzoic acid (6e, Table 5, entry 5) failed to yield any significant amount of decarboxylated product, even with a full equivalent of palladium and stronger heating (120 °C). Other substrates of intermediate electronic character, such as 2-fluoro-6-methoxybenzoic acid (6f, Table 5, entry 6) and 2,4,6-trimethylbenzoic acid (16), also provided very poor conversion to the desired products (<20%) at elevated temperatures (100 °C) with a full equivalent of palladium. Compounds 6e and 16 also cannot form a dative bond with palladium as in proposed intermediate 11 (Scheme 6, path C), which may decrease their reactivity. The decarboxylation of 16 may compete with lactonization from C-H insertion into the ortho-methyl substituent.30 However, reacting the potassium salt of 2,4,6-trimethylbenzoic acid (16a) with an equivalent of palladium afforded 42% yield of the decarboxylated product (17, Scheme 10).
Scheme 10.

Decarboxylation of Potassium Salt of 2,4,6-Trimethylbenzoic Acid
Mono-ortho-substituted benzoic acids suffered from poor yields and considerable byproduct formation. It has been previously shown that a secondary pathway in which palladium undergoes C-H insertion ortho to the carboxylate group is a major byproduct with similar mono-ortho-substituted substrates.32,33 A number of groups have exploited this ortho-directing ability of carboxylate groups.106,107,108,109,110,111,112 Under the protodecarboxylation conditions, both 2,5-dimethoxybenzoic acid and 2,3,4-trimethoxybenzoic acid led to a significant amount of the ortho-palladation byproduct and very little conversion to the decarboxylated products. At this time, no conditions have been found to completely prevent the C-H insertion pathway. However, decarboxylation of 2,4,5-trimethoxybenzoic acid (18) with catalytic palladium at 70 °C afforded 25% yield of the desired product (19, Scheme 11). Again, the yield could be increased by treating the potassium salt of 2,4,5-trimethoxybenzoic acid (18a) with an equivalent of palladium to afford 55% yield of the decarboxylated product (19, Scheme 11).
Scheme 11.
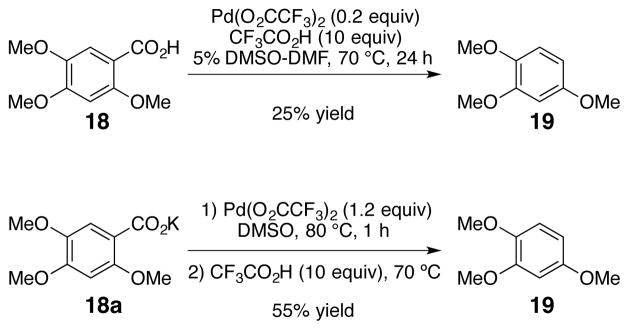
Decarboxylation of Mono-ortho-substituted Benzoic Acids
Nevertheless, it would seem that electron rich di-ortho-substituted substrates are necessary for efficient decarboxylation. In addition to preventing C-H insertion and facilitating protonation of the aryl palladium intermediate, it is possible that the ortho-substituents also coordinate to the palladium and stabilize the reactive intermediates.
Based on the parameters defined above, electron rich naphthoic acids were subjected to the catalytic decarboxylation conditions. As predicted, these substrates underwent facile decarboxylation resulting in good yields (71–75%) of the decarboxylated products (Table 6, entries 1–4). Once again, in the absence of palladium, no decarboxylation was observed (entry 3). Surprisingly, naphthoic acid 26 failed to yield greater than 15% conversion to product, even with an equivalent of palladium at elevated temperatures (120 °C) (entry 5). Since 26 fails whereas the electronically similar 1 succeeds, one might infer that the C1-methyl causes a steric gearing interaction thereby blocking effective reaction of the carboxylate. However, the fact that 24 with a C1-methyl and one additional distal methoxy relative to 26 succeeds indicates that electron donating groups on the ring also play a role by the enhancing the reactivity.
Table 6.
Other Aromatic Acid Substrates in Catalytic Decarboxylationa
| |||
|---|---|---|---|
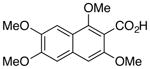
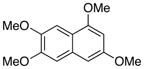
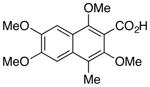
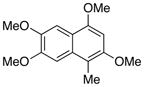
| Entry | Substrate | Product | Yield (%)b |
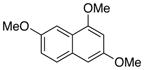
| 1 |
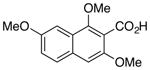 1 |
 3 |
71c |
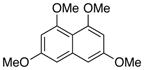
| 2 |
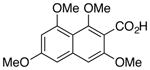 20 |
 21 |
72 |
| 3 |
 22 |
 23 |
74d |
| 4 |
 24 |
 25 |
75c |
| 5 |
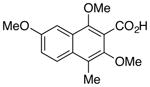 26 |
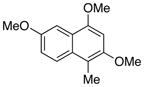 27 |
No Prode |
| 6 |
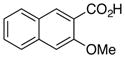 28 |
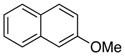 29 |
No Prod |
| 7 |
 30 |
 29 |
88 |
Reaction conditions: [Naphthoic Acid] = 0.065 M and [Pd(O2CCF3)2] = 0.013 M, 10 equiv CF3CO2H, 3.0 mL 5% DMSO-DMF, 70 °C.
Isolated yield.
80 °C.
No reaction was seen in the absence of Pd(O2CCF3)2.
~15% conversion by 1H NMR with stoichiometric palladium at 120 °C.
Akin to substrates such as 2,5-dimethoxybenzoic acid and 2,3,4-trimethoxybenzoic, mono-ortho-substituted naphthoic acid (28) failed to decarboxylate under the reaction conditions (Table 6, entry 6). However, it was hypothesized that 2-methoxy-1-naphthoic acid (30) would undergo facile decarboxylation even though C-H insertion is possible into the aromatic ring adjacent to the carboxylate. Placement of the carboxylic acid at the benzylic position should provide stabilization of the reactive intermediate, since the Wheland intermediate only disrupts the aromaticity of one ring (30a) vs. both aromatic rings (28a, Figure 12). Supporting this analysis and the proposed mechanism, clean decarboxylation of 30 occurred to afford 88% yield of decarboxylated product 29 (Table 6, entry 7).
Figure 12.
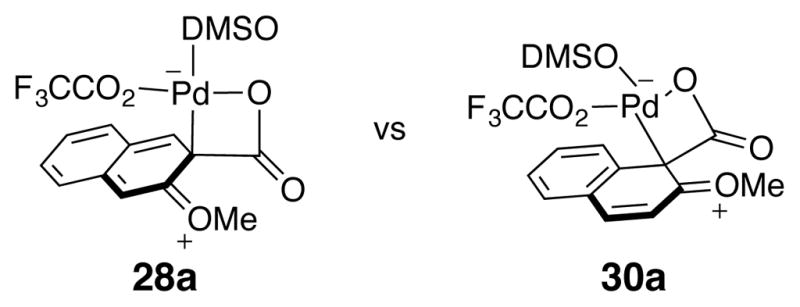
Comparison of Wheland Intermediates 28a and 30a
Conclusions
In summary, a full study of the optimization of the palladium-mediated decarboxylation reaction has been presented. The palladium promoted decarboxylation has proven to be especially mild when combined with an array of usable reductants, making this process combatible with complex substrates. The determination of the kinetic reaction orders for both stages of the aromatic decarboxylation reaction combined with measurement of the activation parameters provides evidence that protodepalladation is the rate-determining step of the reaction. However, the small free energy difference between the two steps (~2 kcal/mol) implies that changes in the substrates can shift the rate-determining step. It is proposed that the decarboxylative palladation occurs through an intramolecular electrophilic palladation followed by decarboxylation; this hypothesis is supported by results from a Hammett study, an Eyring study, and computational work. Furthermore, protodepalladation is proposed to take place through an electrophilic aromatic substitution mechanism via a stepwise intramolecular protonation sequence. This result is especially noteworthy in light of the importance of the reverse process (C-H activation), which under many conditions involves a substantially different concerted σ-bond metathesis. The presence of an SEAr mechanism here, which involves a Wheland intermediate, shows that different mechanistic pathways can be accessed under different reaction conditions. Thus, these results provide a starting point to modify protodepalladation and C-H activation reaction conditions to favor different mechanisms and encompass different substrate classes.
These mechanistic and kinetic results have provided a greater understanding of the aromatic decarboxylation reaction and allowed a reaction with catalytic palladium to be developed. Under catalytic conditions, saturation behavior in protic acid was confirmed, and a Hammett study further supported the proposed protodepalladation mechanism. This transformation is especially useful for hindered aromatic carboxylic acids (bis-ortho-substituted). Future work will focus on broadening the substrate scope of the catalytic aromatic decarboxylation reaction.
EXPERIMENTAL SECTION
General experimental considerations are provided in the Supporting Information. Starting material compounds 1, 4, 20, 22, 24 were made following our previous report.35
General Procedure for GC Reaction Monitoring
To a solution of 6a (60 mg, 0.33 mmol) in the appropriate solvent (3.0 mL), was added palladium (II) trifluoroacetate (110 mg, 0.33 mmol) and biphenyl (51 mg, 0.33 mmol). The reaction mixture was then heated at 70 °C for 1 h. To this solution was added the appropriate hydrogen/hydride source at the temperature specified. At regular intervals, aliquots (~100 μL) were removed. To this aliquot was added 1 M HCl and EtOAc. The solution was then shaken and the organic phase was filtered through SiO2 (10% MeOH/CH2Cl2). The sample was then analyzed by gas chromatography (GC) using the conditions outlined in the Supporting Information.
General Procedure for 1H NMR Reaction Monitoring
To a solution of 6a (12 mg, 0.066 mmol) in DMSO-d6 (0.75 ml) was added palladium(II) trifluoroacetate (26 mg, 0.079 mmol). A 1H NMR spectrum was then taken. The mixture was then warmed to the indicated temperature and spectra were taken periodically throughout the course of the reaction. If a hydrogen/hydride source was being studied the reaction was then cooled to room temperature, a 1H NMR spectrum was taken, the hydrogen/hydride source was added and the mixture was heated to the indicated temperature.
Computational Methods
All calculations were carried using B3LYP113,114,115 functionals using 6–31G(d) basis for main group elements and SDD for Pd in solvent (dichloromethane) using CPCM116 solvation model. All stationary points were characterized as minima or transition state structures using frequency analysis.
1,3,7-Trimethoxynaphthalene (3). General Procedure A
To a solution of 1 (10 mg, 0.038 mmol) in 5% DMSO-DMF (0.5 mL) was added palladium(II) trifluoroacetate (14.3 mg, 0.043 mmol) and Ag2CO3 ( 10.5 mg, 0.038 mmol). The mixture was then heated to 90 °C for 1 h. After cooling, 1 N HCl was added and the mixture was extracted with EtOAc. The organic phase was washed with H2O and brine, dried over Na2SO4 and concentrated. This material was then dissolved in THF (1.0 mL) and stirred under a H2 atmosphere for 5 min. After filtration through Celite®, the material was chromatographed in 20% EtOAc/Hexanes to afford 3 (8.2 mg) in 99% yield as a brown amorphous solid; mp 69–71 °C; 1H NMR (500 MHz, CDCl3) δ 7.59 (d, J = 8.9 Hz, 1H), 7.47 (d, J = 2.6 Hz, 1H), 7.13 (dd, J = 8.9, 2.6 Hz, 1H), 6.70 (d, J = 2.1 Hz, 1H), 6.52 (d, J = 2.2 Hz, 1H), 3.98 (s, 3H), 3.91 (s, 3H), 3.89 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 156.6, 156.0, 155.7, 130.2, 128.1, 122.3, 119.5, 100.9, 98.2, 98.1, 55.7, 55.5, 55.4; IR (film) 2988, 2945, 2907, 1737, 1606 cm−1; HRMS (CI) m/z: [M]+ calcd for C13H14O3 218.0943, found 218.0939.
5,5′-Bis-benzyloxy-2,2′,4,4′,6,6′-hexamethoxy-7,7′-dipropyl-[1,1′]-binaphthalene (5)
General procedure A was used with the following alterations: Compound 4 was employed and Ag2CO3 was excluded in the first step. Compound 5 (27 mg) was obtained in 99% yield as a resin; 1H NMR (500 MHz, CDCl3) δ 7.62 (d, J = 7.0 Hz, 4H), 7.43 (t, J = 7.3 Hz, 4H), 7.37 (t, J = 7.3 Hz, 2H), 6.76 (s, 2H), 6.64 (s, 2H), 5.10 (m, 4H), 3.99 (s, 6H), 3.92 (s, 6H), 3.74 (s, 6H), 2.46–2.53 (m, 4H), 1.35–1.42 (m, 4H), 0.75 (t, J = 7.2 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 157.1, 154.9, 148.8, 147.2, 138.9, 137.0, 133.7, 129.0, 128.5, 127.8, 122.1, 116.5, 112.5, 95.9, 76.2, 61.4, 57.5, 56.1, 32.8, 23.6, 14.0; IR (film) 2933, 1338 cm−1; HRMS (ESI) m/z: [M+Na]+ calcd for C46H50O8Na 753.3403, found 753.3427.
4-Chloro-2,6-dimethoxybenzoic acid (6c)
Procedure adapted from previously reported method to synthesize the ester varient.117 A solution of 1-chloro-3,5-dimethoxybenzene (8c) (840 μL, 5.8 mmol) in THF (40 mL) was cooled to −20 °C. To this solution was added n-butyllithium (2.6 mL, 6.4 mmol). The solution was stirred for 1 h at −20 °C and quenched with powdered CO2 (29 g, 670 mmol). The mixture was allowed to warm to room temperature and concentrated under reduced pressure. The mixture was then diluted with saturated Na2CO3 and extracted twice with Et2O. The aqueous layer was acidified with 6 M HCl and extracted twice with CHCl3. The CHCl3 organic extracts were dried with Na2SO4 and concentrated to yield 6c in 5% yield (60 mg) as a white powder: mp 179–181 °C; 1H NMR (500 MHz, CDCl3) δ 6.60 (s, 2H), 3.86 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 170.1, 158.3, 137.8, 110.4, 105.2, 56.5; IR (film) 3398, 2950, 2873, 2657, 2587, 2541, 1699, 1591, 1460, 1406 cm−1; HRMS (FT-MS) m/z: [M-OH] − calcd for C9H8ClO3 199.0162, found 199.0157.
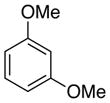
1,3-Dimethoxybenzene (8a).118 General Procedure B
To a solution of 6a (R = H) (200 mg, 1.1 mmol) in 5% DMSO-DMF (10 mL), palladium(II) trifluoroacetate (73 mg, 0.22 mmol) was added. To this solution was added CF3CO2H (820 μL, 11 mmol). The reaction mixture was heated at 70 °C for 24 h. After cooling, the mixture was diluted with Et2O, and washed sequentially with 1 M HCl, saturated NH4Cl three times, H2O three times, and brine. The organic extracts were dried over Na2SO4 and concentrated at ambient temperature and pressure by slow evaporation on the bench-top. The resulting residue was chromatographed in 20% Et2O/Pentanes to afford 8a (R = H) (152 mg) in quantitative yield as a pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.19 (t, J = 8.2 Hz, 1H), 6.52 (dd, J = 2.4, 8.2 Hz, 2H), 6.48 (t, J = 2.4 Hz, 1H), 3.80 (s, 6H).
3,5-Dimethoxytoluene (8b).119
General procedure B was employed with the following alterations: Compound 6b (R = Me) was employed. Compound 8b (R = Me) (134 mg) was isolated in 80% yield as a pale yellow oil; 1H NMR (300 MHz, CDCl3) δ 6.35-6.34 (m, 2H), 6.30-6.29 (m, 1H), 3.78 (s, 6H), 2.31 (s, 3H).
1-Chloro-3,5-dimethoxybenzene (8c).120
General procedure B was employed with the following alterations: Compound 6c (R = Cl) was employed. Compound 8c (R = Cl) (169 mg) was isolated in 89% yield as a pale yellow oil; 1H NMR (300 MHz, CDCl3) δ 6.53 (m, 2H), 6.36 (m, 1H), 3.78 (s, 6H).
1,3,5-Trimethoxybenzene (8d).121
General procedure B was employed with the following alterations: Compound 6d (R = OMe) was employed. Compound 8d (R = OMe) (183 mg) was isolated in 99% yield as a white solid; 1H NMR (300 MHz, CDCl3) δ 6.10 (s, 3H), 3.77 (s, 9H).
1,3,5-Trimethylbenzene (17).122 General Procedure C
To a solution of 2,4,6-trimethylbenzoic acid (16) (300 mg, 1.8 mmol) in MeOH (15 mL) was added potassium tert-butoxide (210 mg, 1.8 mmol). The suspension was stirred at room temperature for 4 h and then concentrated by rotary evaporation to afford 16a (370 mg) in quantitative yield and was used without further purification.
To a solution of 16a (100 mg, 0.49 mmol) in DMSO (3.0 mL), palladium(II) trifluoroacetate (200 mg, 0.59 mmol) was added. This solution was heated to 80 °C for 24 h. Then trifluoroacetic acid (370 μL, 4.9 mmol) was added and the solution was heated at 70 °C overnight. After cooling, the mixture was diluted with Et2O and washed three times with saturated NH4Cl, three times with H2O, and once with brine. The organic extracts were dried over Na2SO4 and concentrated at ambient temperature and pressure by slow evaporation on the bench-top. The resultant residue was chromatographed in 50% Et2O/Pentane to afford 17 (25 mg) in 42% yield as a brown amorphous solid; 1H NMR (500 MHz, CDCl3) δ 6.90 (s, 3H), 2.43 (s, 9H).
1,2,4-Trimethoxybenzene (19).123
General procedure B was employed with the following alterations: Compound 18 was employed. Compound 19 (46 mg) was isolated in 25% yield as a yellow oil; 1H NMR (500 MHz, CDCl3) δ 6.78 (d, J = 8.7 Hz, 1H), 6.51 (d, J = 2.8 Hz, 1H), 6.39 (dd, J = 8.7, 2.8 Hz, 1H), 3.85 (s, 3H), 3.83 (s, 3H), 3.76 (s, 3H).
General procedure C was used to form 19 from 18a with the following alterations: the initial heating time before addition of trifluoroacetic acid was 30 min. Compound 19 (102 mg) was isolated in 55% yield as a yellow oil.
1,3,7-Trimethoxy-4-methyl-2-naphthoic acid (26)
To a solution of methyl 1,3-dihydroxy-7-methoxynaphthalene-2-carboxylate (200 mg, 0.85 mmol) in 3.9 mL of THF:H2O (1.0:0.3), n-Bu4NBr (28 mg, 0.085 mmol) and KOH (478 mg, 8.4 mmol) was added. To this solution dimethyl sulfate (810 μL, 8.4 mmol) was added and the mixture was stirred at room temperature for 4 h. The reagents were quenched with 30% NH4OH for 30 min. The resultant mixture was extracted with Et2O and washed three times with 30% NH4OH and one time with H2O and brine. The organic phase was dried over Na2SO4 and concentrated. The resulting residue was chromatographed in 5% EtOAc/Hexanes to afford methyl 1,3,7-trimethoxy-4-methyl-2-naphthoate (31) (48 mg) in 19% yield; mp 38–40 °C; 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 9.2 Hz, 1H), 7.39 (d, J = 2.7 Hz, 1H), 7.22 (dd, J = 9.2, 2.7 Hz, 1H), 4.00 (s, 3H), 3.99 (s, 3H), 3.93 (s, 3H), 3.82 (s, 3H), 2.53 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.5, 157.4, 151.3, 150.1, 130.1, 126.2, 126.1, 121.6, 120.0, 101.3, 62.9, 62.6, 55.5, 52.6, 11.3; IR (film) 2999, 2949, 2872, 2845, 1733, 1602 cm−1; HRMS (CI) m/z: [M]+ calcd for C16H18O5 290.1154, found 290.1156.
To a solution of methyl 1,3,7-trimethoxy-4-methyl-2-naphthoate (31) (48 mg, 0.17 mmol) in MeOH (2.0 mL), concentrated NaOH (2.0 mL) was added. The solution was stirred at room temperature for 24 h. The reaction mixture was then acidified with concentrated HCl and then extracted with CH2Cl2 three times. The organic extracts were then washed with brine and dried over Na2SO4, filtered, and concentrated to afford 26 (38 mg) in 83% yield as a white solid; mp 114–117 °C; 1H NMR (500 MHz, CDCl3) δ 7.88 (d, J = 9.2 Hz, 1H), 7.42 (d, J = 2.6 Hz, 1H), 7.25 (dd, J = 9.2, 2.6 Hz, 1H), 4.08 (s, 3H), 3.95 (s, 3H), 3.91 (s, 3H), 2.55 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.0, 157.6, 152.2, 150.1, 130.5, 126.3, 126.1, 122.0, 120.5, 119.8, 101.5, 63.3, 62.9, 55.6, 11.4; IR (film) 3563, 2999, 2941, 2868, 2845, 1710, 1602, 1509 cm−1; HRMS (ESI) (m/z): [M+Na]+ calcd for C15H16NaO5 299.0895; found 299.0883.
General Procedure D was employed for the synthesis of compounds 3, 21, 23, 25 and 29 in Table 6.
1,3,6,8-Tetramethoxynaphthalene (21).124 General Procedure D
To a solution of 20 (11 mg, 0.038 mmol) in 5% DMSO-DMF (0.5 mL), palladium(II) trifluoroacetate (2.5 mg, 0.0076 mmol) was added. To this solution was added trifluoroacetic acid (28 μL, 0.38 mmol). The reaction mixture was heated at 70 °C for 24 h. After cooling, the mixture was diluted with Et2O, and washed sequentially with 1 M HCl, saturated NH4Cl, H2O, and brine. The organic extracts were then dried over Na2SO4 and concentrated at ambient temperature and pressure by slow evaporation on the bench-top. The resulting residue was chromatographed in 10% Et2O/Pentane to afford 21 (6.8 mg) in 72% yield as a black crystalline solid; 1H NMR (500 MHz, CDCl3) δ 6.63 (d, J = 2.2 Hz, 2H), 6.37 (d, J = 2.2 Hz, 2H), 3.92 (s, 6H), 3.88 (s, 6H).
1,3,6,7-Tetramethoxynaphthalene (23)
Compound 23 (7.0 mg) was isolated in 74% yield as a dark brown amorphous solid; mp 120–125 °C; 1H NMR (500 MHz, CDCl3) δ 7.45 (s, 1H), 7.02 (s, 1H), 6.66 (d, J = 2.1 Hz, 1H), 6.42 (d, J = 2.2 Hz, 1H), 3.99 (s, 3H), 3.98 (s, 3H), 3.96 (s, 3H), 3.89 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 157.3, 155.7, 150.4, 147.4, 130.8, 116.1, 105.9, 101.5, 97.7, 96.1, 56.0, 55.9, 55.6, 55.5; IR (film) 2999, 2937, 2833, 1633, 1610, 1513 cm−1; HRMS (ESI) m/z: [MH]+ calcd for C14H17O4 249.1127, found 249.1137.
2,4,6,7-Tetramethoxy-1-methylnaphthalene (25)
Compound 25 (7.5 mg) was isolated in 75% yield as an orange amorphous solid; mp 134–140 °C; 1H NMR (500 MHz, CDCl3) δ 7.49 (s, 1H), 7.12 (s, 1H), 6.57 (s, 1H), 4.02 (s, 3H), 4.00 (s, 3H), 3.996 (s, 3H), 3.92 (s, 3H), 2.43 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 153.9, 150.3, 147.4, 130.1, 116.3, 110.9, 103.8, 102.8, 101.5, 93.4, 57.6, 56.0, 55.9, 55.7, 10.6; IR (film) 2991, 2930, 2833, 1629, 1598, 1513, 1494 cm−1; HRMS (CI) m/z: [M]+ calcd for C15H18O4 262.1205, found 262.1204.
2-Methoxynaphthalene (29).125
Compound 29 (5.3 mg) was isolated in 88% yield as a yellow Solid; 1H NMR (300 MHz, CDCl3) δ 7.78-7.72 (m, 3H), 7.47-7.41 (m, 1H), 7.36-7.31 (m, 1H), 7.17-7.14 (m, 2H), 3.93 (s, 3H);
Supplementary Material
Figure 8.
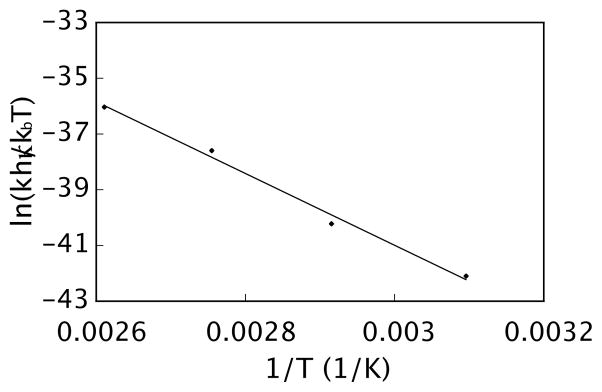
Eyring Plot for Protodepalladation. Compound 6 was mixed with Pd(O2CCF3)2 in 0.75 mL DMSO-d6 and heated to 80 °C for 1 h. After cooling, a 1H NMR spectrum was taken, CH3SO3H was added, and 1H NMR monitoring was continued at four different temperatures (50, 70, 90, and 110 °C). Reaction conditions: initial concentration [6] = 0.08 M and [Pd(O2CCF3)2] = 0.10 M, 10 equiv CH3SO3H, 0.75 mL DMSO-d6. Equation of the line is y = −12885x − 2.337. R2 = 0.992.
Acknowledgments
This research was supported by the National Institutes of Health (CA-109164) and the the National Science Foundation (CHE-1213230). Partial instrumentation support was provided by the NIH for MS (1S10RR023444) and NMR (1S10RR022442) and by the NSF for an X-Ray Diffractometer (CHE-0840438). We are grateful to 3D Pharmaceuticals (C.A.M.), the Division of Organic Chemistry of the ACS (C.A.M.) for graduate fellowships, and the NIH (Grant GM087605) for support (O.G). This work used the Extreme Science and Engineering Discovery Environment (XSEDE; TG-CHE120052).
Footnotes
GC and 1H NMR data for reaction monitoring and kinetic studies, 1H and 13C NMR spectra for new compounds. Additional details on computations, including coordinate and energies for computed structures and full Gaussian citation. This material is free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nakajima M, Miyoshi I, Kanayama K, Hashimoto S, Noji M, Koga K. J Org Chem. 1999;64:2264–2271. [Google Scholar]
- 2.Mulrooney CA, Li XL, DiVirgilio ES, Kozlowski MC. J Am Chem Soc. 2003;125:6856–6857. doi: 10.1021/ja027745k. [DOI] [PubMed] [Google Scholar]
- 3.Giles RGF, Green IR, van Eeden N. Eur J Org Chem. 2004:4416–4423. [Google Scholar]
- 4.Brimble MA, Houghton SI, Woodgate PD. Tetrahedron. 2007;63:880–887. [Google Scholar]
- 5.DiVirgilio ES, Dugan EC, Mulrooney CA, Kozlowski MC. Org Lett. 2007;9:385–388. doi: 10.1021/ol062468y. [DOI] [PubMed] [Google Scholar]
- 6.Huang XA, Xue JA. J Org Chem. 2007;72:3965–3968. doi: 10.1021/jo070241q. [DOI] [PubMed] [Google Scholar]
- 7.Krohn K, Vukics K. Synthesis. 2007:2894–2900. [Google Scholar]
- 8.Olah GA, Laali K, Mehrotra AK. J Org Chem. 1983;48:3360–3362. [Google Scholar]
- 9.Horper W, Marner FJ. Phytochemistry. 1996;41:451–456. [Google Scholar]
- 10.Shepard AF, Winslow NR, Johnson JR. J Am Chem Soc. 1930;52:2083–2090. [Google Scholar]
- 11.Cohen T, Schambach RA. J Am Chem Soc. 1970;92:3189–3190. [Google Scholar]
- 12.Cohen T, Berninger RW, Wood JT. J Org Chem. 1978;43:837–848. [Google Scholar]
- 13.Pulgarin C, Tabacchi R. Helv Chim Acta. 1988;71:876–880. [Google Scholar]
- 14.Barton DHR, Lacher B, Zard SZ. Tetrahedron Lett. 1985;26:5939–5942. [Google Scholar]
- 15.Barton DHR, Lacher B, Zard SZ. Tetrahedron. 1987;43:4321–4328. [Google Scholar]
- 16.Goosen LJ, Linder C, Rodriguez N, Lange PP, Fromm A. Chem Commun. 2009:7173–7175. doi: 10.1039/b912509d. [DOI] [PubMed] [Google Scholar]
- 17.Cornella J, Sanchez C, Banawa D, Larrosa I. Chem Commun. 2009:7176–7178. doi: 10.1039/b916646g. [DOI] [PubMed] [Google Scholar]
- 18.Seo S, Taylor JB, Greaney MF. Chem Commun. 2010:8270–8272. doi: 10.1039/c2cc33306f. [DOI] [PubMed] [Google Scholar]
- 19.Frederiksen LB, Grobosch TH, Jones JR, Lu SY, Zhao CC. J Chem Res, Synop. 2000:42–43. [Google Scholar]
- 20.Matsubara S, Yokota Y, Oshima K. Org Lett. 2004;6:2071–2073. doi: 10.1021/ol0492602. [DOI] [PubMed] [Google Scholar]
- 21.Sharma A, Kumar R, Sharma N, Kumar V, Sinha AK. Adv Synth Catal. 2008;350:2910–2920. [Google Scholar]
- 22.Goossen LJ, Manjolinho F, Khan BA, Rodriguez N. J Org Chem. 2009;74:2620–2623. doi: 10.1021/jo802628z. [DOI] [PubMed] [Google Scholar]
- 23.Tilstam U. Org Process Res Dev. 2012;16:1449–1454. [Google Scholar]
- 24.Rodriguez N, Goossen LJ. Chem Soc Rev. 2011;40:5030–5048. doi: 10.1039/c1cs15093f. [DOI] [PubMed] [Google Scholar]
- 25.Dzik WI, Lange PP, Goossen LJ. Chem Sci. 2012;3:2671–2678. [Google Scholar]
- 26.Cornella J, Larrosa I. Synthesis. 2012;44:653–676. [Google Scholar]
- 27.Becht JM, Le Drian C. J Org Chem. 2011;76:6327–6330. doi: 10.1021/jo200344w. [DOI] [PubMed] [Google Scholar]
- 28.Shen Z, Ni Z, Mo S, Wang J, Zhu Y. Chem Eur J. 2012;18:4859–4865. doi: 10.1002/chem.201103438. [DOI] [PubMed] [Google Scholar]
- 29.Zhou J, Wu G, Zhang M, Jie X, Su W. Chem Eur J. 2012;18:8032–8036. doi: 10.1002/chem.201200829. [DOI] [PubMed] [Google Scholar]
- 30.Lee JM, Chang S. Tetrahedron Lett. 2006;47:1375–1379. [Google Scholar]
- 31.Wang C, Piel I, Glorius F. J Am Chem Soc. 2009:4194–4195. doi: 10.1021/ja8100598. [DOI] [PubMed] [Google Scholar]
- 32.Myers AG, Tanaka D, Mannion MR. J Am Chem Soc. 2002;124:11250–11251. doi: 10.1021/ja027523m. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka D, Myers AG. Org Lett. 2004;6:433–436. doi: 10.1021/ol0363467. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka D, Romeril SP, Myers AG. J Am Chem Soc. 2005;127:10323–10333. doi: 10.1021/ja052099l. [DOI] [PubMed] [Google Scholar]
- 35.Dickstein JS, Mulrooney CA, O’Brien EM, Morgan BJ, Kozlowski MC. Org Lett. 2007;9:2441–2444. doi: 10.1021/ol070749f. [DOI] [PubMed] [Google Scholar]
- 36.Zask A, Helquist P. J Org Chem. 1978;43:1619–1620. [Google Scholar]
- 37.Murata M, Suzuki K, Watanabe S, Masuda Y. J Org Chem. 1997;62:8569–8571. doi: 10.1021/jo971143f. [DOI] [PubMed] [Google Scholar]
- 38.Rahaim RJ, Maleczka RE. Tetrahedron Lett. 2002;43:8823–8826. [Google Scholar]
- 39.Yamanoi Y. J Org Chem. 2005;70:9607–9609. doi: 10.1021/jo051131r. [DOI] [PubMed] [Google Scholar]
- 40.Helquist P. Tetrahedron Lett. 1978:1913–1914. [Google Scholar]
- 41.Cacchi S, Ciattini PG, Morera E, Ortar G. Tetrahedron Lett. 1986;27:5541–5544. [Google Scholar]
- 42.Logan ME, Oinen ME. Organometallics. 2006;25:1052–1054. [Google Scholar]
- 43.Sajiki H, Mori A, Mizusaki T, Ikawa T, Maegawa T, Hirota K. Org Lett. 2006;8:987–990. doi: 10.1021/ol060045q. [DOI] [PubMed] [Google Scholar]
- 44.O’Brien EM, Morgan BJ, Kozlowski MC. Angew Chem Int Ed. 2008;47:6877–6880. doi: 10.1002/anie.200800734. [DOI] [PubMed] [Google Scholar]
- 45.O’Brien EM, Morgan BJ, Mulrooney CA, Carroll PJ, Kozlowski MC. J Org Chem. 2010;75:57–68. doi: 10.1021/jo901386d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morgan BJ, Dey S, Johnson SW, Kozlowski MC. J Am Chem Soc. 2009;131:9413–9425. doi: 10.1021/ja902324j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mulrooney CA, Morgan BJ, Li X, Kozlowski MC. J Org Chem. 2010;75:16–29. doi: 10.1021/jo9013832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mulrooney CA, O’Brien EM, Morgan BJ, Kozlowski MC. Eur J Org Chem. 2012:3887–3904. doi: 10.1002/ejoc.201200184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Studies with a related substrate as the sodium salt (see ref. 34) also showed a first order dependence in decarboxylation (80 °C, k = 4.8 × 10−3 s−1, t1/2 = 2.5 min).
- 50.See Supporting Information
- 51.For information on transition state theory, see: Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Sausalito: 2006. pp. 365–374. For Hammett study and rho values, see pp 445–453. For entropy estimation, see pp 373, Table 7.1.
- 52.Martin-Matute B, Mateo C, Cardenas DJ, Echavarren AM. Chem-Eur J. 2001;7:2341–2348. doi: 10.1002/1521-3765(20010601)7:11<2341::aid-chem23410>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 53.Park CH, Ryabova V, Seregin IV, Sromek AW, Gevorgyan V. Org Lett. 2004;6:1159–1162. doi: 10.1021/ol049866q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tunge JA, Foresee LN. Organometallics. 2005;24:6440–6444. [Google Scholar]
- 55.Lane BS, Brown MA, Sames D. J Am Chem Soc. 2005;127:8050–8057. doi: 10.1021/ja043273t. [DOI] [PubMed] [Google Scholar]
- 56.Campora J, Gutierrez-Puebla E, Lopez JA, Monge A, Palma P, del Rio D, Carmona E. Angew Chem Int Ed. 2001;40:3641–3644. doi: 10.1002/1521-3773(20011001)40:19<3641::aid-anie3641>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 57.Halpern J. Science. 1982;217:401–407. doi: 10.1126/science.217.4558.401. [DOI] [PubMed] [Google Scholar]
- 58.Amatore C, Carre E, Jutand A. Acta Chem Scand. 1998;52:100–106. [Google Scholar]
- 59.Jia CG, Lu WJ, Oyamada J, Kitamura T, Matsuda K, Irie M, Fujiwara Y. J Am Chem Soc. 2000;122:7252–7263. [Google Scholar]
- 60.Jia CG, Piao DG, Oyamada JZ, Lu WJ, Kitamura T, Fujiwara Y. Science. 2000;287:1992–1995. doi: 10.1126/science.287.5460.1992. [DOI] [PubMed] [Google Scholar]
- 61.Jia CG, Kitamura T, Fujiwara Y. Acc Chem Res. 2001;34:633–639. doi: 10.1021/ar000209h. [DOI] [PubMed] [Google Scholar]
- 62.Hammett LP. J Am Chem Soc. 1937;59:96–103. [Google Scholar]
- 63.From computed ΔG‡ values (B3LYP/6–31G* wih SDD for Pd in CH2Cl2) a ρ value of −0.61 is obtained for TS1 (p-H 12.9 kcal/mol, p-Cl 13.1 kcal/mol) and +0.61 is otained for TS2 (p-H 16.4 kcal/mol, p-Cl 16.2 kcal/mol).
- 64.Suga K, Aoyagui S. Bull Chem Soc Jpn. 1973;46:755–761. [Google Scholar]
- 65.Zierkiewicz W, Privalov T. Organometallics. 2005;24:6019–6028. [Google Scholar]
- 66.All calculations were carried out using GAUSSIAN09. See supporting information for computational details and comparasion with other methods.
- 67.Zhang SL, Fu Y, Shang R, Guo QX, Liu L. J Am Chem Soc. 2009;132:638–646. doi: 10.1021/ja907448t. [DOI] [PubMed] [Google Scholar]
- 68.Xue L, Su W, Lin Z. Dalton Trans. 2010;39:9815–9822. doi: 10.1039/c0dt00491j. [DOI] [PubMed] [Google Scholar]
- 69.At less than 50% conversion, the data accomodated both zero and first order processes. See ref. 34.
- 70.Lineweaver H, Burk D. J Am Chem Soc. 1934;56:658–666. [Google Scholar]
- 71.Studies with a related substrate as the sodium salt (see ref. 34) showed a 2-fold increase with 5% DMSO-DMF (60 °C, k = 1.2 × 10−3 s−1) vs. DMSO alone (60 °C, k = 5.5 × 10−4 s−1).
- 72.1H NMR spectroscopy studies by Stahl of Pd(TFA)2 and DMSO show unbound DMSO around the Pd when greater than 2 equiv of DMSO to Pd is used, see: Diao T, White P, Guzei I, Stahl SS. Inorg Chem. 2012;51:11898–11909. doi: 10.1021/ic301799p.
- 73.Wheland GW. J Am Chem Soc. 1942;64:900–908. [Google Scholar]
- 74.For computational studies on silver and copper catalyzed decarboxylations, see: Xue LX, Su W, Lin Z. Dalton Trans. 2011;40:11926–11937. doi: 10.1039/c1dt10771b.
- 75.Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Chem Soc Rev. 2009;38:3242–3272. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]
- 76.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wencel-Delord J, Drege T, Liu F, Glorius F. Chem Soc Rev. 2011;40:4740–4761. doi: 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]
- 78.Young CS, Dong VM. Chem Rev. 2011;111:1215–1292. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]
- 79.Liu C, Zhang H, Shi W, Lei A. Chem Rev. 2011;111:1780–1824. doi: 10.1021/cr100379j. [DOI] [PubMed] [Google Scholar]
- 80.Kuhl N, Hopkinson MN, Wencel-Delord J, Glorius F. Angew Chem Int Ed. 2012;51:10236–10254. doi: 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]
- 81.Deng Y, Persson AKA, Backvall JE. Chem Eur J. 2012;18:11498–11523. doi: 10.1002/chem.201201494. [DOI] [PubMed] [Google Scholar]
- 82.Yamaguchi J, Yamaguchi A, Itami K. Angew Chem Int Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]
- 83.Toyota M, Ilangovan A, Okamoto R, Masaki T, Arakawa M, Ihara M. Org Lett. 2002;4:4293–4296. doi: 10.1021/ol020187u. [DOI] [PubMed] [Google Scholar]
- 84.Kalyani D, Deprez NR, Desai LV, Sanford MS. J Am Chem Soc. 2005;127:7330–7331. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]
- 85.Davies DL, Donald SMA, Macgregor SA. J Am Chem Soc. 2005;127:13754–13755. doi: 10.1021/ja052047w. [DOI] [PubMed] [Google Scholar]
- 86.Campeau LC, Parisien M, Jean A, Fagnou K. J Am Chem Soc. 2006;128:581–590. doi: 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]
- 87.Lafrance M, Rowley CN, Woo TK, Fagnou K. J Am Chem Soc. 2006;128:8754–8756. doi: 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]
- 88.Hull KL, Lanni EL, Sanford MS. J Am Chem Soc. 2006;128:14047–14049. doi: 10.1021/ja065718e. [DOI] [PubMed] [Google Scholar]
- 89.Garcia-Cuadrado D, de Mendoza P, Braga AAC, Maseras F, Echavarren AM. J Am Chem Soc. 2007;129:6880–6886. doi: 10.1021/ja071034a. [DOI] [PubMed] [Google Scholar]
- 90.Wade LG., Jr . Organic Chemistry. 6. Pearson Prentice Hall; New Jersey: 2006. [Google Scholar]
- 91.For the pKa of acid entries 1–2, 4–5 and 7 in DMSO, see: Bordwell FG. Acc Chem Res. 1988;21:456–463.
- 92.Bancroft DP, Cotton FA, Verbruggen M. Acta Crystallogr, Sect C. 1989;45:1289–1292. [Google Scholar]
- 93.Zhou CX, Larock RC. J Am Chem Soc. 2004;126:2302–2303. doi: 10.1021/ja038687l. [DOI] [PubMed] [Google Scholar]
- 94.Zierkiewicz W, Privalov T. Organometallics. 2005;24:6019–6028. [Google Scholar]
- 95.Brasche G, Garcia-Fortanet J, Buchwald SL. Org Lett. 2008;10:2207–2210. doi: 10.1021/ol800619c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Amatore C, Carre E, Jutand A. Acta Chem Scand. 1998;52:100–106. [Google Scholar]
- 97.Amatore C, Godin B, Jutand A, Lemaitre F. Chem Eur J. 2007;13:2002–2011. doi: 10.1002/chem.200600153. [DOI] [PubMed] [Google Scholar]
- 98.Amatore C, Jutand A, Mbarki MA. Organometallics. 1992;11:3009–3013. [Google Scholar]
- 99.Amatore C, Jutand A, Thuilliez A. Organometallics. 2001;20:3241–3249. [Google Scholar]
- 100.Amatore C, Jutand A, Lemaitre F, Ricard JL, Kozuch S, Shaik S. J Organomet Chem. 2004;689:3728–3734. [Google Scholar]
- 101.Denmark SE, Smith RC, Tymonko SA. Tetrahedron. 2007;63:5730–5738. doi: 10.1016/j.tet.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Steinhoff BA, Guzei IA, Stahl SS. J Am Chem Soc. 2004;126:11268–11278. doi: 10.1021/ja049962m. [DOI] [PubMed] [Google Scholar]
- 103.Chen MS, White MC. J Am Chem Soc. 2004;126:1346–1347. doi: 10.1021/ja039107n. [DOI] [PubMed] [Google Scholar]
- 104.Lowry TH, Richardson KS. Mechanism and Theory in Organic Chemistry. 3. Plenum Press; New York: 1987. [Google Scholar]
- 105.DeYonker NJ, Foley NA, Cundari TR, Gunnoe TB, Petersen JL. Organometallics. 2007;26:6604–6611. [Google Scholar]
- 106.Miura M, Tsuda T, Satoh T, Pivsa-Art S, Nomura M. J Org Chem. 1998;63:5211–5215. [Google Scholar]
- 107.Forgione P, Brochu MC, St-Onge M, Thesen KH, Bailey MD, Bilodeau F. J Am Chem Soc. 2006;128:11350–11351. doi: 10.1021/ja063511f. [DOI] [PubMed] [Google Scholar]
- 108.Giri R, Maugel N, Li JJ, Wang DH, Breazzano SP, Saunders LB, Yu JQ. J Am Chem Soc. 2007;129:3510–3511. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]
- 109.Giri R, Yu JQ. J Am Chem Soc. 2008;130:14082–14083. doi: 10.1021/ja8063827. [DOI] [PubMed] [Google Scholar]
- 110.Mei TS, Giri R, Maugel N, Yu JQ. Angew Chem Int Ed. 2008;47:5215–5219. doi: 10.1002/anie.200705613. [DOI] [PubMed] [Google Scholar]
- 111.Voutchkova A, Coplin A, Leadbeater NE, Crabtree RH. Chem Commun. 2008:6312–6314. doi: 10.1039/b813998a. [DOI] [PubMed] [Google Scholar]
- 112.Maehara A, Tsurugi H, Satoh T, Miura M. Org Lett. 2008;10:1159–1162. doi: 10.1021/ol8000602. [DOI] [PubMed] [Google Scholar]
- 113.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 114.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 115.Vosko SH, Wilk L, Nusair M. Can J Phys. 1980;58:1200. [Google Scholar]
- 116.Barone V, Cossi M. J Phys Chem A. 1998;102:1995. [Google Scholar]
- 117.Ishii H, Sugiura T, Akiyama Y, Ichikawa Y, Watanabe T, Murakami Y. Chem Pharm Bull. 1990;38:2118–2126. [Google Scholar]
- 118.Saa JM, Dopico M, Martonell G, Garcia-Raso A. J Org Chem. 1990;55:991–995. [Google Scholar]
- 119.Trost BM, Pissot-Soldermann C, Chen I, Schroeder GM. J Am Chem Soc. 2004;126:4480–4481. doi: 10.1021/ja0497025. [DOI] [PubMed] [Google Scholar]
- 120.Murphy JM, Liao X, Hartwig JF. J Am Chem Soc. 2007;129:15434–15435. doi: 10.1021/ja076498n. [DOI] [PubMed] [Google Scholar]
- 121.Kreis M, Palmelund A, Bunch L, Madsen R. Adv Synth Catal. 2006;348:2148–2154. [Google Scholar]
- 122.Hunt DF, Sethi SK. J Am Chem Soc. 1980;102:6953–6963. [Google Scholar]
- 123.Mathivanan N, Cozens F, McClelland RA, Steenken S. J Am Chem Soc. 1992;114:2198–2203. [Google Scholar]
- 124.McCulloch MWB, Malcolm WB, Barrow RA. Tetrahedron Lett. 2005;46:7619–7621. [Google Scholar]
- 125.Hsu YC, Datta S, Ting CM, Liu RS. Org Lett. 2008;10:521–524. doi: 10.1021/ol7030334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.