

Abstract
Recent clinical trials have shown promise in the use of chimeric antigen receptor(CAR)-transduced T cells; however, augmentation of their activity may broaden their clinical utility and improve their efficacy. We hypothesized that, since CAR action requires proteins essential for TCR signal transduction, deletion of negative regulators of these signaling pathways would enhance CAR signaling and effector T cell function. We tested CAR activity and function in T cells that lacked one or both isoforms of diacylglycerol kinase (dgk) expressed highly in T cells, dgkα and dgkζ, enzymes that metabolize the second messenger diacylglycerol (DAG) and limit Ras/ERK activation. We found that primary murine T cells transduced with CARs specific for the human tumor antigen mesothelin demonstrated greatly enhanced cytokine production and cytotoxicity when co-cultured with a murine mesothelioma line that stably expresses mesothelin. Additionally, we found that dgk-deficient CAR-transduced T cells were more effective in limiting the growth of implanted tumors, both concurrent with and after establishment of tumor. Consistent with our studies in mice, pharmacologic inhibition of dgks also augments function of primary human T cells transduced with CARs. These results suggest that deletion of negative regulators of TCR signaling enhances the activity and function of CAR-expressing T cells and identify dgks as potential targets for improving the clinical potential of CARs.
Keywords: diacylglycerol kinase, chimeric antigen receptor, T cell receptor, tumor, mesothelin
Introduction
Elicitation of T cell effector responses requires signal transduction through the T cell antigen receptor (TCR), a protein complex that binds antigenic peptide presented by major histocompatibility complexes (MHC), as well as through co-stimulatory receptors such as CD28. The effector responses generated from TCR signal transduction differ across individual T cell subsets that are classified according to the expression of cell surface molecules (1). Expression of the surface molecule CD8, for instance, identifies a subset of T cells that respond to antigenic peptides presented in the binding groove of MHC class I. CD8+ T cells are responsible for the recognition and elimination of cells that express antigens derived from intracellular pathogens, such as viruses and intracellular bacteria, and also mutated or embryonic proteins generated by cells that have undergone malignant transformation. Although the extent to which CD8+ T cells are capable of controlling the development and progression of tumorigenesis remains uncertain, it is clear that deficiency of these cells increases the potential for the development of malignancy and that enhanced function of these CD8+ T cells can impart robust anti-tumor responses in both animal model systems and patients (2,3). It is also clear in a number of models that although there may be an initial, potent CD8+ T cell response, this response is often insufficient to fully protect from tumors (4). Mechanisms underlying this failure include (i) the lack of specific antigens with sufficient avidity for the TCR expressed by tumors, (ii) the absence of co-stimulatory ligands expressed by antigen-presenting cells (APCs) within tumor-draining lymph nodes, and (iii) direct suppression of T cell responses within the tumor microenvironment mediated by inhibitory secreted factors such as TGFβ, prostaglandin E2, or adenosine, as well as inhibitory cells, such as regulatory T cells (5).
The potential for effective responses by CD8+ T cells in some instances of incurable malignancies, such as metastatic melanoma, has led to significant interest in defining ways to manipulate these cells to generate more potent responses as well as responses against a more diverse array of tumors. One promising approach has focused on engineering T cells to express chimeric antigen receptors (CARs). CARs are transmembrane fusion proteins that consist of an extracellular antibody domain capable of binding to a specific tumor antigen coupled to intracellular signaling domains from TCR and co-stimulatory components (6). In principle, CARs provide several advantages over the endogenous receptors of T cells. First, the engineered ligand-binding segment of CARs arises from an antibody, obviating the need for MHC presentation. Second, the antibody-binding component of the CAR can be chosen to be both specific and highly sensitive to antigens expressed selectively by tumor cells, increasing avidity of the T cell-tumor interaction and minimizing the potential for destruction of normal “bystander” host cells. Third, engagement of the CAR by ligand stimulates both TCR and co-stimulatory signaling modules, eliminating a requirement for expression of co-stimulatory ligands by tumor-draining antigen-presenting cells. CAR-expressing T cells that come into contact with tumor cells expressing the antigen of interest have been shown to develop functional responses that lead to tumor cell lysis and cytokine production.
There has been considerable success in the use of CARs in animal models (6,7), and recently, CAR-expressing T cells have been shown to be effective in patients to treat refractory chronic lymphocytic leukemias (30,31). Although T cells engineered to express CARs are capable of overcoming some limitations of the endogenous immune system to combat tumors (e.g. CARs are not MHC restricted and hence will lyse tumor cells that have downregulated MHC expression), CAR-expressing T cells still lack intrinsic programming to overcome perhaps the most important component that limits CD8+ T cell anti-tumor responses: the inhibitory tumor microenvironment. We hypothesized that deletion of proteins that limit the strength of TCR signal might overcome this obstacle of impaired CD8+ T cell anti-tumor immunity and impart significantly enhanced anti-tumor functioning in CAR-expressing CD8+ T cells.
For our studies, we chose to target an inhibitor of diacylglycerol (DAG), an essential second messenger that is created by the cleavage of phosphatidyl (4,5) inositol bisphosphate (PIP2) by phospholipase Cγ1 (PLCγ1) after PLCγ1 is phosphorylated and activated by the protein tyrosine kinases that are recruited to the stimulated TCR (1). DAG activates signaling molecules leading to several second messenger cascades, most notably the Ras/ERK pathway that is known to be essential for T cell activation (7). After its generation, DAG is actively metabolized into phosphatidic acid by one of two isoforms of diacylglycerol kinases present within T cells, dgkα or dgkζ (8). Previously, we and others observed that deletion of either dgk isoform potentiates DAG-mediated Ras and ERK activation and augments TCR-induced cytokine production and T cell proliferation (9–11). We have found further that deletion of dgkζ results in improved CD8+ T cell responses by augmenting signaling via the TCR when mice are challenged with a transplantable subcutaneous tumor (12). However, neither the absence of dgkα or dgkζ is sufficient to enable a completely successful anti-tumor response. We speculated therefore that combining CAR therapy with dgk deficiency might boost the ability of T cells to respond to a tumor challenge. Herein, we report the results of our studies demonstrating this augmentation, suggesting that such a combined therapeutic approach may have utility in future clinical trials.
Materials and Methods
Mice
Mice deficient in dgkα, dgkζ, or both backcrossed to C57Bl/6 have been described previously (9,10). C57Bl/6 mice containing a transgene for the OVAp T cell receptor (OT-I mice) were obtained from the Jackson Laboratories. Dgkζ-deficient CD45.2 CD90.2 OT-I mice were created by backcrossing these two strains. All experiments were performed in mice 6–12 weeks old. Animal maintenance and experimentation were performed in accordance with the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Listeria infection and EL4-ova tumor model experiment
Splenic CD8+ T cells were isolated from wild type or dgkζ-deficient CD45.2 CD90.2 OT-I mice by flow cytometry (CD8+CD44lo) as described (12). 20,000 cells were transferred i.v. into CD45.2, CD90.1 recipient mice subsequently infected i.v. with 5000 cfu Listeria-ova 24 hours after T cell transfer. One week later, CD45.2+ donor cells were isolated from spleens of recipient mice according to the manufacturer’s instructions (Miltenyi Biotec), and 1.5×106 of isolated cells were transferred i.v. into CD45.1, CD90.2 mice that had been inoculated with 2.5×105 EL4-ova tumor cells, a murine lymphoma line that stably expresses ovalbumin (13), in the right flank 2 weeks prior. Tumors were barely palpable at time of T cell transfer. One week later, mice were euthanized, tumor size was measured, and T cells from spleens and tumors were analyzed.
T cell transduction
MesoCAR, a fusion protein that contains the antigen-binding region of an antibody specific for the human tumor antigen mesothelin fused with CD8a transmembrane domain, CD3ζ, and the costimulatory domain of 4-1BB, has been described previously (14). cDNA encoding mesoCAR was subcloned into the MIGR retrovirus (15), which also expresses green fluorescent protein using an internal ribosomal entry site. The sequence of anti-mesothelin Fv was provided by Ira Pastan (National Cancer Institute, Bethesda, MD) (16). Infective particles were generated from the supernatants of 293T cells transfected with retroviral vector plasmid and helper plasmids using Lipofectamine 2000 (Invitrogen), as previously described (17). Primary murine T cells were isolated as suggested by the manufacturer (Miltenyi Biotec) from the spleens of wild type or dgk-deficient mice and incubated in 24-well plates (4×106 cells/well in 2 mL T cell media with 100 U/mL IL-2) coated with α–CD3 (1 μg/mL) and α-CD28 (2 μg/mL). After 48 hours, cells (1×106 cells/well) were mixed with retrovirus (1 mL crude viral supernatant) in a 24-well plate coated with Retronectin (50 μg/mL; Clontech) and centrifuged without braking at room temperature for 30 minutes at 1200g. After overnight incubation, cells were expanded with 50 U/mL of IL-2 for 48 hours.
Coating beads with recombinant human mesothelin
Target antigens were chemically crosslinked to tosylactivated 4.5 μm Dynabeads (Invitrogen, #140-13), using the manufacturers’ instructions. In brief, 4×107 beads were incubated 16–18 hours at 37°C in the presence of 20 μg of recombinant human mesothelin (RayBiotech, #230-00043) in 0.1M sodium phosphate buffer (pH 7.4) with shaking. After incubation, beads were washed and resuspended in PBS containing 0.5% BSA to a final volume of 1 mL.
Evaluation of CAR T cell effector functions
i) Cytotoxicity and IFN ELISA
A stable cell line of the mouse mesothelioma line AE17 expressing human mesothelin subsequently engineered to express luciferase has been described (14,18). Cytokine release assays were performed by co-culture of T cells with target cells at the described ratios, in triplicate, in 96-well round bottom plates in 200-μL. After 18 hours, cell lysis was determined from the detection of luciferase from the remaining cells using a previously described assay (14). An ELISA Kit (Biolegend) was used to measure IFN-γ.
ii) WINN assay
1×106 mesothelin-expressing TC1 cells, a murine non-small cell lung cancer line with well-established use in the WINN assay (19), were co-injected into the right flank along with 2×105 CAR-transduced T cells (routinely 50% of which were gfp-positive, and thus transduced with CAR). 10 days later, mice were euthanized, and tumor volume was assessed.
iii) IV transfer of CAR-T cells in mice with pre-existing tumor
C57Bl/6 mice were inoculated subcutaneously with 2×106 AE17meso cells. 7 days later, at which point tumors were approximately 100 mm3, mice were injected with 1×107 CAR-transduced T cells i.v. by tail vein. Tumor development was monitored by caliper measurement of tumor diameter over an additional 10 days. Each volumetric determination was derived from the formula 0.52a2b, with a representing the minor axis and b representing the major axis. Alternately, mice were sacrificed at 3 or 6 days after transfer, and the presence of T cells within spleen or tumor was determined by evaluating for gfp expression within T cell subsets by flow cytometry.
iv) Expression of cytotoxic markers following CAR activation
2×106 mesoCAR-T cells derived from mouse splenocytes replete or deficient in dgks were placed in individual wells of a 24-well plate with or without 2×106 mesothelin-coated beads for 18 hours, at 37°C in the presence of 30 U/mL of IL2. After incubation, T cells were stained for the presence of the surface markers TRAIL (eBioscence, #12-5951-82) or FasL (eBioscience, #12-5911-81), or the intracellular markers granzyme B (BD, #51-2090KZ) or perforin (eBioscience, #12-9392-82), using protocols described by the manufacturer. Flow cytometry histograms of marker expression were evaluated from cells that were positive for gfp (indicating expression of CAR) and CD8, and negative for CD4.
T cell immunoblotting and CD69 upregulation
To assess for Erk phosphorylation, 1×106 mesoCAR-transduced T cells were incubated either with mesothelin- or albumin-coated beads in a 1:4 ratio (cells:beads), or with α–CD3ε antibody at 2.5 μg/mL final concentration for indicated time points. Lysates were prepared and immunoblotted for phosphorylated Erk, total Erk, or tubulin (antibodies all from Cell Signaling) as previously described (12). Alternately, protein-bead stimulations were allowed to proceed for 5 hours, and then the surface upregulation of CD69 was determined by flow cytometry.
Primary human CAR-T cell assays
Primary human T cells were obtained from the University of Pennsylvania Clinical Cell Production Facility and mock-infected or transduced with lentivirus expressing mesoCAR as previously described (14). 5×106 T cells were subsequently added to 24-well plates that had been seeded with 5×105 cells either from the EM human mesothelioma line or a stable derivative cell line, EM-meso, engineered to express high levels of mesothelin, in a 24-well dish. After 96 hours of co-incubation (which included the addition of another 3×105 EM or EM-meso cells at the midpoint of co-incubation), cells were resuspended, and T cells were isolated via Lymphoprep density gradient separation (Axis-Shield) as suggested by the manufacturer. Cells were assessed for viability using Trypan Blue, and 1×105 live T cells were co-cultured with 5×103 EM-meso-luc cells expressing luciferase in 96-well plates in the presence or absence of the dgk inhibitors DGK1 (R59022) or DGK2 (R59949) (Sigma) at 5 μg/mL. After 18 hours, remaining tumor cells were washed and lysed and luminescence was evaluated. Cell lysate determinations were corroborated with visual estimate of remaining numbers of tumor cells.
For studies with TGFβ, 1×105 primary human T cells that had been mock infected or transduced with lentivirus-expressing mesoCAR were co-incubated with 5×103 EM-meso-luc cells in the presence of indicated concentrations of TGFβ for 18 hours, and cell numbers were determined as described above.
Results
Dgkζ-deficient activated CD8+ T cells demonstrate enhanced response to tumor
We have previously shown that naïve dgkζ-deficient CD8+ T cells specific for ovalbumin (OT-I cells) are better able to control tumor growth and undergo activation than naïve wild type (wt) OT-I cells after transfer into mice bearing EL4-ovalbumin (EL4-ova)-expressing tumors (12). However, in those studies, implanted tumors were not completely eradicated. We wondered if the effect of dgkζ deletion would be improved if, instead of naïve T cells, we made use of activated T cells that could potentially confer a more robust anti-tumor response. Additionally, this approach would more closely mirror current clinical trials of adoptive CD8+ T cell tumor immunotherapy that utilize cells pre-activated prior to transfer. To generate uniform populations of activated cells, we transferred naïve OT-I cells sufficient or deficient in dgkζ into congenically marked mice and then infected the recipient animals with Listeria engineered to express ovalbumin. One week later, antigen-experienced (CD44-high) donor OT-I cells were recovered from spleens and transferred into EL4-ova tumor-bearing mice. Initially, we noted that expansion of naïve dgkζ-deficient OT-I cells was more robust when compared with naïve wt OT-I cells in response to the antigenic challenge with Listeria-ova (Fig. 1A), as in other dgkζ-deficient CD8+ T cell models of acute infection (9,12); however, there was no difference in activation phenotype of the recovered cells as assessed by CD44 expression between the two different genotypes (data not shown). After transfer of equal numbers of wt or dgkζ-deficient effector cells into EL4-ova tumor-bearing mice, we found that whereas wt OT-I cells conferred no appreciable anti-tumor effect, tumors in mice treated with dgkζ-deficient activated OT-I cells were significantly (p=0.05) reduced in size compared to untreated animals (Fig. 1B). Dgkζ–deficient effector cells also persisted in increased numbers within the spleen of host animals (Fig. 1C) and were observed in larger quantities within the tumors of host animals (Fig. 1D). These data demonstrate that deficiency of dgkζ confers enhanced anti-tumor potential in pre-activated T cells. As in our previous study with naïve dgkζ-deficient OT-I CD8+ T cells, however, we found that transfer of activated dgkζ-deficient CD8+ T cells was insufficient to completely eradicate tumors, suggesting that the strategy of targeting dgkζ alone is insufficient in curtailing the progression of established tumors.
Figure 1. Dgkζ-deficient activated CD8+ T cells demonstrate enhanced tumor responses in vivo.

(A) 20,000 naïve (CD44lo) CD8+ wt or dgkζ-deficient OT-I cells were injected i.v. into congenically marked (CD90.1) mice. 24 hours later, the mice were injected i.v. with 5000 cfu Listeria-ova, and one week later mice were euthanized, and the presence of donor OT-I T cells (CD90.2+, ova tetramer+) were assessed (n=5, quantitation of 1 of 3 representative experiments is shown). CD90.2+ cells from (A) were isolated magnetically and 1×106 cells were injected i.v. into CD45.1+ mice bearing two week old subcutaneous EL4-ova tumors. One week later, mice were euthanized and assessed for (B) tumor size, (C) persistence of donor (CD45.2+, ova tetramer+) T cells, and (D) tumor-infiltrating donor T cells. “No T cell” mice did not receive donor T cells, and CD45.2 cells were not detected in any organ tissue (data not shown). (B = data from 3 pooled experiments, C and D are data from 1 of 3 representative experiments, n=5 in each group).
Deletion of dgkζ enhances functional responses of T cells downstream of chimeric antigen receptors
Given that deletion of dgkζ conferred enhanced activity of CD8+ T cells against established tumors but did not appear to be curative, we wondered whether inhibition of dgk function might augment other approaches shown to have efficacy in enhancing T cell responses against tumor. Therefore, we next designed studies to test the impact of dgk deficiency on effector function of CAR-expressing T cells. Our first experiments tested whether dgkζ loss would augment functional responses after ligation of CARs, similar to the augmentation of TCR-induced functions that we have demonstrated previously (9). For this analysis, we made use of mesoCAR, a fusion protein that has high affinity for the human tumor antigen mesothelin, present on human mesothelioma, pancreatic, and ovarian cancer, coupled to the signaling motifs of the TCR CD3ζ chain and the inducible T cell co-stimulatory receptor 4-1BB. Wt or dgkζ-deficient activated OT-1 cells were transduced with mesoCAR-expressing retrovirus, resulting in approximately 50% transduction efficiency (Fig. 2A). Transduction did not affect the activation state of the T cells, as assessed by expression of CD44, or expression of the endogenous TCR, as assessed with tetramer specific for OT-I (Fig. 2A). MesoCAR transduced dgkζ-deficient and wt OT-1 cells were then compared in their ability to produce IFNγ and mediate target cell lysis after incubation with AE17ova-meso, a murine cell line engineered to express both ovalbumin and human mesothelin. Transduced OT-I cells lacking dgkζ displayed enhanced IFNγ production and enhanced cytotoxicity after incubation with mesothelioma cell lines (Fig. 2B and 2C), indicating that deletion of dgkζ enhances the function of CAR-transduced CD8+ T cells against AE17 cells that express both ova and mesothelin.
Figure 2. Enhanced chimeric antigen receptor (CAR) effector function in dgkζ-deficient CD8+ T cells.
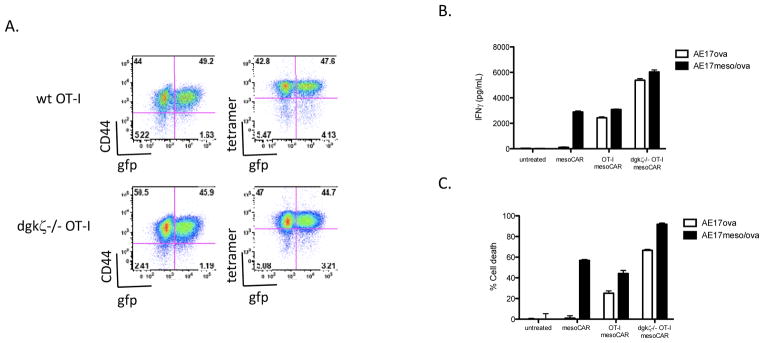
CD8+ wt or dgkζ-deficient OT-I T cells were isolated and transduced with mesoCAR retrovirus expressing gfp from an internal ribosomal entry site (IRES). (A) Cells were evaluated for expression of gfp, CD44 as a marker of activation, and ovalbumin tetramer to assess TCR expression. (B and C) MesoCAR-transduced wt or dgkζ-deficient OT-I cells were incubated with 5000 AE17 ovalbumin cells or AE17 cells expressing both mesothelin and ovalbumin at a ratio of 40:1 in a 96-well plate for 24 hours, and the presence of IFNγ (B) or luciferase (cytotoxicity) (C) in cell supernatants was assessed. Calculated estimates of cytotoxicity were confirmed by visual evaluation of cell culture wells. One of three representative experiments is shown; each well was performed in triplicate. (B and C: p<0.0001 between dgk/AE17ova and wt/AE17ova or dgk/AE17ovameso and wt AE17ovameso. B: p=0.0327 between dgk/AE17ova and dgk/AE17ovameso, p=0.002 between wt/AE17ova and wt/AE17ovameso. C: p<0.0001 between dgk/AE17ova and dgk/AE17ovameso, p=0.007 between wt/AE17ova and wt/AE17ovameso)
Combined deletion of dgkζ and dgkα markedly enhances T cell responses after stimulation of mesoCAR
The finding that deletion of dgkζ enhanced mesoCAR T cell functional responses suggested that these two strategies may be used together for potentiating CD8+ T cell tumor responses. However, these initial experiments were complicated by the fact that the target cells expressed antigens for both TCR (ovalbumin) and CAR (mesothelin). Thus, to avoid potentially confounding our results with ovalbumin-specific TCRs, we continued our experiments in non-TCR transgenic animals. Further, as it is well appreciated that an additional isoform of dgk, dgkα, operates in a similar fashion as dgkζ in T cells and may blunt the effects of targeting dgkζ alone in augmenting T cell function, we intercrossed dgkα and dgkζ-deficient mice to generate animals deficient in both dgk isoforms to study CAR-T cells generated from double knockout (DKO) mice. Naïve T cells were isolated from wt, dgkα−/−, dgkζ−/−, or DKO mice and infected with retrovirus encoding mesoCAR under high IL2 concentrations that favored CD8+ T cell growth (cells were 85% CD8+ T cells at the end of incubation). As observed with deletion of dgkζ in OT-1 cells, deletion of either dgkα or dgkζ in this population of cells expressing the mesoCAR receptor conferred enhanced cytokine production and cytotoxicity when the T cells were incubated with tumor cells expressing mesothelin (Fig. 3A and 3B). Strikingly, DKO cells demonstrated profoundly enhanced effector functions compared to cells with deletion of either dgk isoform alone or to wt cells. The enhanced cytotoxicity observed in these cell lines was mesothelin-specific, since mesoCAR-transduced DKO T cells did not lyse cells AE17 cells expressing an unrelated antigen (AE17ova cells, Fig. 3C).
Figure 3. Deletion of both T cell dgk isoforms significantly enhances CAR-T cell effector functions.

T cells were isolated from wt, dgkα−/−, dgkζ−/−, or DKO mice, transduced with mesoCAR, and assessed for IFNγ production and cytotoxicity of target cells at indicated ratios as described in Figure 2. In part C, a ratio of 40:1 was used for experimental (AE17ovameso) or control (AE17ova) cell lines. One of three representative experiments is shown; each data point was performed in triplicate (p-values for all mesoCAR-expressing constructs < 0.0001 for AE17ova and AE17ovameso).
We next evaluated whether the changes in signal transduction that we have previously observed downstream of the TCR in dgk-deficient T cells e.g. enhanced Ras/Erk/AP-1 signaling (9, 10), were also present downstream of CAR. To that end, we developed a means to stimulate mesoCAR T cells that did not require mesothelin-expressing cells, since these cells express their own Ras-signaling molecules, such as Erk, that could interfere with identifying changes specific to T cells after stimulation. For these studies, we used tosylactivated beads coated with ovalbumin (as a control) or beads coated with mesothelin to stimulate the mesoCAR-expressing cells. Whereas phosphorylation of Erk was not observed during incubation with control beads, Erk phosphorylation could be readily detected during incubation with mesothelin-coated beads or, as expected, after stimulation of the TCR complex through CD3ε (Fig. 4A). Moreover, activation of Erk by mesothelin beads required expression of mesoCAR, since activation of T cells transduced with control retrovirus (MIGR) was not observed (Fig. 4A, right lanes). To test whether deletion of dgks enhanced Erk activation downstream of mesoCAR, we then repeated this experiment with mesoCAR-transduced T cells derived from DKO mice. Similar to the enhanced activation of Erk known downstream of the TCR in T cells deficient in dgks, loss of dgks augmented the activation of Erk downstream of mesoCAR (Fig. 4B). We extended our analysis by investigating the upregulation of CD69 in mesothelin-stimulated wt or DKO T cells, as CD69 expression is controlled by activation of the transcription factor AP-1 following Ras/Erk signaling (9). Consistent with the biochemical enhancement of Erk activation observed in DKO transduced T cells, upregulation of CD69 was also increased in mesoCAR DKO T cells compared with wt cells (Fig. 4C), confirming a role for dgks in regulation of this pathway downstream of CAR. Together, these data suggest that dgk influences CAR signaling in a manner similar to the TCR and that the combination of CAR expression and dgk deletion could represent an effective strategy for augmenting CD8+ T cell anti-tumor responses.
Figure 4. Enhanced CAR signaling in dgk-deficient CD8+ T cells.

(A) 1×106 CAR-transduced (MesoCAR) or vector control (MIGR) CD8+ T cells were incubated with 4×106 albumin-coated beads (alb) or mesothelin coated beads (meso or M) or 2.5 μg/mL α-CD3 (α-CD3ε or α) for the indicated times and assessed for phosphorylated (α-pERK) or total Erk (α-ERK) by immunoblotting. (B) Levels of pERK, total ERK, and actin were assessed after stimulation of 1×106 wt or dgkα−/−dgkζ−/− (double knockout; DKO) CD8+ CAR-T cells with 4×106 mesothelin coated beads (M) or 2.5 μg/mL α-CD3 (α) for 15 minutes. Each immunoblot is a representative experiment from at least three independent iterations. (C) 1×106 wt or DKO-deficient CD8+ CAR T cells were incubated with 4×106 beads coated with albumin (alb) or mesothelin (meso) for 5 hours and surface expression of CD69 was assessed by flow cytometry.
Deletion of both dgks enhances activity of T cells downstream of CARs against tumor in vivo
We next sought to determine whether deletion of dgk isoforms conferred enhanced anti-tumor responses in vivo making use of the WINN assay. For these experiments, mice were injected with a mixture of TC1meso cells, a murine non-small cell lung cancer line (19), along with mesoCAR-expressing wt T cells or mesoCAR T cells lacking dgkα, dgkζ, or both dgk isoforms. Although mice that received wt mesoCAR-transduced T cells or T cells lacking a single isoform of dgk were unable to completely control the growth of mesotheliomas, DKO T cells eradicated the mesotheliomas (Fig. 5A), indicating that, as suggested by in vitro studies, targeting dgk generates meaningful enhancement of CD8+ T cells against tumor. Pronounced differences were also noted when AE17meso cells were used as target cells in the WINN assay (data not shown). While this experiment offered proof of principle that DKO T cells conferred enhanced in vivo activity against mesothelioma, it did not directly assess whether deletion of dgk isoforms would be capable of limiting the growth of established tumors, which is more representative of how CAR-T cells would be utilized clinically. To that end, AE17meso cells were injected into the flanks of mice, and tumors were allowed to develop to approximately 100 mm3 in size before intravenous administration of CAR-T cells. Under these conditions, whereas wt mesoCAR-transduced T cells were ineffective at limiting tumor growth, mesoCAR-transduced T cells deficient in either dgk isoform significantly (p<0.05) decreased the rate of tumor growth (Fig. 5B), an effect increased by the deletion of both dgk isoforms. In addition to enhanced effector function, this effect could in part be related to the increased number of dgk-deficient mesoCAR T cells in tumor-bearing mice, since quantitative differences in T cells between mesoCAR wt and dgkζ T cells were appreciated 6 days after transfer (Fig. 5C); however, mesoCAR T cells of any genotype were not detected at day 10 or later timepoints under the experimental conditions used.
Figure 5. Dgk-deficient mesoCAR-transduced T cells control mesothelioma in vivo.

(A) 1×106 TC1meso cells were co-injected subcutaneously with 2×105 wt mesoCAR-transduced T cells or mesoCAR-transduced T cells lacking one or both (DKO) of the indicated dgk. 10 days later, mice were euthanized and tumors were measured. 1 of 2 representative experiments (n=4–5) is shown. p value of wt vs DKO mesoCAR T cells is 0.05. (B) 2×106 AE17meso cells were injected into flanks of C57Bl/6 mice. One week later, 1×107 CAR-transduced T cells of indicated genotype were injected i.v. into mice and tumors were measured at indicated time points (n=5 in each genotype, p value for DKO CAR-T cells vs wt CAR-T cells = 0.0141 at day #10 post-transfer). (C) Alternately, mice were sacrificed 3 and 6 days after T cell transfer of indicated genotypes as in (B) and the presence of mesoCAR T cells in tumor (top) or spleen (bottom) was determined (n=3 in each group, 1 of 2 representative experiments. p value = 0.0082 (tumor) and 0.0461 (spleen) at day 6; day 3 results did not differ significantly).
Inhibition of dgks confers enhanced antitumor responses to human T cells
Although the demonstration of enhanced mesoCAR function in murine T cells lacking dgk isoforms provides a rationale for targeting dgks to augment T cell responses against tumors, an important next step is to establish a role for dgks in CAR-expressing primary human T cells. Over the course of our studies with primary human T cells transduced with mesoCARs, we have noted that these cells develop reduced functional responsiveness after extended co-culture with mesothelin-expressing tumor cells. As shown, 96 hours of co-incubation of mesoCAR T cells with EM-meso cells, a mesothelioma line engineered to express high levels of mesothelin, results in significant impairment of mesoCAR T cell lysis of target cells upon re-culture (Fig. 6A, left panel). This effect is reminiscent of various models of antigen-induced anergy, as impaired cytotoxicity is not generated after co-culture of mesoCAR T cells with parental EM cells that do not express mesothelin (Fig. 6A). Since we and others have previously shown that deletion of dgks mitigates the induction of anergy (10, 11), we hypothesized that inhibition of dgks might also diminish the impaired cytotoxicity observed in our assay. To test this, we incubated mesoCAR T cells with EM-meso cells for 96 hours, and then assessed their ability to lyse target cells in the presence of dgk inhibitors R59022 (DGK1 inhibitor) or R59949 (DGK2 inhibitor). We observed that the addition of either dgk inhibitor was sufficient to reverse the impaired cytotoxicity present in mesothelin-exposed mesoCAR T cells (Fig. 6A, center and right panels), indicating that, similar to our findings in mice, inhibition of dgk function appears to augment anti-tumor activity of primary human T cells expressing CARs. These data also suggest that in addition to augmenting TCR (or CAR)-mediated signaling, blockade of dgk may enhance T cell anti-tumor responses by mitigating antigen-induced unresponsiveness of the effector cells.
Figure 6. Dgk inhibitors enhance the cytotoxic capacity of impaired human mesoCAR-transduced T cells.

(A) MesoCAR-transduced primary human cells were left unexposed or exposed to a human tumor line that does not express mesothelin (EM) or expresses high levels of mesothelin (EM-meso) for 96 hours. 105 T cells were then isolated and recultured with 5×103 luciferase-expressing EM-meso cells for 18 hours in the absence or presence of dgk inhibitors R59022 (DGK1 Inhibitor) or R59949 (DGK2 Inhibitor) and cell death of target cells was assessed by luciferase release (Data from triplicate wells of one of three representative experiments is shown. p value of EM-meso exposed T cells to EM exposed T cells = 0.004 in no inhibitor group. p value of EM-meso exposed T cells in the absence of inhibitor to DGK1 inhibitor = 0.006 or DGK2 inhibitor = 0.003). (B) Lysis of 5×103 EM-meso cells was assessed during incubation with no T cells or 105 untransduced or mesoCAR-transduced primary human T cell for 18 hours in the presence or absence of the indicated concentration of TGFβ (Data from triplicate wells of one of three representative experiments. p-value of mesoCAR T cells between 0 and 10 pg/mL = 0.05, 0 and 100 pg/mL = 0.025, and 0 and 1000 pg/mL = 0.017).
Dgk-deficient T cells demonstrate reduced sensitivity to TGFβ
Following our observation that inhibition of dgks reverses the antigen-induced inactivation of CAR-expressing T cells, we asked whether deletion of dgk might also reduce sensitivity to other inhibitory influences of T cells. One such inhibitor, TGFβ, is of particular relevance, since secretion of this cytokine by tumor cells has been shown to actively inhibit CD8+ effector T cell responses against tumors (20). Further, TGFβ is speculated to mediate its effects, in part, by dampening the Ras/Erk signal transduction pathway, which is known to be affected by dgks (21,22). We initially examined whether TGFβ could lead to inhibition of cytotoxicity of human mesoCAR T cells incubated with EM-meso cells. The addition of TGFβ resulted in diminished cytotoxicity by transduced mesoCAR human T cells (Fig. 6B), at levels of TGFβ similar to that present in culture media of EM-meso cells (81.31 pg/mL/24hrs/106 cells; data not shown). Next, we tested if deletion of dgks could diminish the effects of TGFβ. Murine mesoCAR T cells replete or deficient in dgks were incubated with AE17meso cells as described previously, and cytotoxicity and IFNγ production were assessed in the presence or absence of TGFβ. As demonstrated before, deletion of either or both dgk isoforms resulted in mesoCAR T cells with enhanced function when compared with wt mesoCAR T cells (Fig. 7A and 7B, white bars). Addition of TGFβ at lower (1 ng/mL) or higher (10 ng/mL) concentrations of TGFβ were found to reduce cytotoxicity and IFNγ secretion in wt mesoCAR T cells; however, these functions were not affected in mesoCAR T cells deficient in either or both dgks (Fig. 7A and 7B). These data suggest that deletion of dgks confers relative resistance to TGFβ for mesoCAR T cells. The finding of relative insensitivity to inhibitory stimuli appears not to be solely restricted to TGFβ, since greater functional responses were also observed by dgk-deficient mesoCAR-transduced T cells in the presence of the inhibitory stimuli prostaglandin E2 and adenosine (Supplemental Figure 1), although to a lesser extent than that observed with TGFβ. Collectively our data suggest that deletion of dgks augments effector function of CAR-expressing CD8+ T cells not only by augmenting signaling through the CAR itself but also by reducing sensitivity of the effector cells to physiologically relevant inhibitory signals.
Figure 7. Dgk-deficient T cells are less inhibited by TGFβ.

1×105 naïve (CD44lo) mesoCAR-transduced T cells of the indicated genotype were incubated with 5000 AE17meso target cells at a ratio of 40:1 in the absence or presence of TGFβ at the indicated concentration. (A) IFNγ and (B) cytotoxicity of target cells at 24 hours was assessed as described in Figure 2. One of three representative experiments is shown. Each data point was performed in triplicate (p < 0.0001 between wt and all dgk-deficient T cells treated with TGFβ).
Increased FasL and TRAIL expression in dgk-deficient T cells
Since TGFβ expression suppresses mediators of CD8+ T cell cytotoxicity (23), such as perforin, granzyme B, FasL and TRAIL, and since CAR T cell cytotoxicity is mediated through these effector molecules (reviewed in 24), we hypothesized that dgk-deficient CAR cells would show greater upregulation of these cytotoxic mediators when compared with wt T cells and that this upregulation may be the basis for the enhanced cytotoxicity observed in dgk-deficient mesoCAR T cells. Wt mesoCAR T cells or mesoCAR T cells deficient in one or both dgk isoforms were exposed to mesothelin-coated beads in the presence of IL2 for 18 hours. Expression of FasL, TRAIL, granzyme B, and perforin were then evaluated by flow cytometry. As predicted, mesoCAR T cells deficient in one or both dgks demonstrated enhanced expression of the cytotoxic cell surface proteins FasL and TRAIL when compared with wt mesoCAR T cells (Supplemental Figure 2). In contrast, no difference was observed in the expression of the intracellular cytotoxic proteins granzyme B- or perforin-expressing in cells lacking dgks (Supplemental Figure 3). These data suggest that FasL and TRAIL may help facilitate the augmented cytotoxicity observed in dgk-deficient mesoCAR T cells.
Discussion
Current strategies aimed at augmenting T cell immune responses against malignancy have focused either on assisting the initial activation, or priming, of naïve T cells, or on potentiating the effects of activated, or primed, T cells. For instance, antibodies that activate CD40 on APCs upregulate co-stimulatory molecules that help facilitate priming of naïve cells (25–27). In contrast, antibodies that block the T cell inhibitory cell surface molecule CTLA-4 minimally impact naïve T cells but significantly enhance proliferation and effector function of primed T cells (28,29). Inhibition of proteins that negatively regulate signal transduction downstream of the TCR has garnered recent attention as a potential strategy for augmenting T cell responses to malignancy at the time of T cell priming. For instance, deletion of Casitas-B-lineage lymphoma b (cbl-b), an E3 ubiquitin ligase responsible for the degradation of several proteins important in TCR signal transduction, results in T cells with a decreased requirement of co-stimulation at the time of activation and enhanced anti-tumor activity of naïve T cells (30–33).
We had previously demonstrated, similar to cbl-b, that deletion of dgkζ enhanced the effector functions of naïve CD8+ T cells (13). Although deletion of cbl-b and dgkζ both result in changes downstream of the TCR, dgkζ and dgkα; likely act directly to regulate the threshold for activation of T cells downstream of the TCR. Under a currently posited model of TCR signaling, the interplay of two Ras activating proteins, the guanine nucleotide-exchange factors (GEFs) SOS and RasGRP1, determine whether the threshold for Ras activation is met within a T cell after TCR engagement, an event required for T cell activation (34). In this model, TCR ligation results in the production of DAG, which binds and activates RasGRP1, and generates small amounts of active Ras. If enough active Ras is generated, it is able to bind an allosteric Ras-binding site on SOS, activating SOS and facilitating generation of most of the active Ras within activated T cells. In a manner largely consistent with this model, we had previously found that deletion of dgkζ resulted in a decreased threshold of T cell activation, a finding that correlated with enhanced responses in naïve CD8+ T cells.
In the studies presented herein, we found that deletion of dgks also has profound effects on activated T cells. After uniform activation of naïve wt or dgkζ-deficient ovalbumin-specific T cells with Listeria-ova and then transfer into mice with subcutaneous ovalbumin-expressing EL4 lymphoma, we found that dgkζ-deficient T cells showed enhanced activity against tumor and increased persistence, both within the spleen and the tumor itself. This finding suggests that alteration of T cell threshold plays an important role at multiple stages with the T cell life cycle and identifies dgkζ as a means to simultaneously target both naïve and activated populations of effector T cells.
The role of dgks in limiting the effector function of activated CD8+ T cells makes dgks a potential target for CAR-expressing T cells, a strategy gaining traction in the clinical treatment of human malignancies. In current clinical trial protocols, human T cells are transduced with lentivirus-expressing CARs in the presence of CD3 and CD28 or CD3 and 41BB (CD137), a process that induces T cell division and activation (35). However, it is now clear that additional strategies will be necessary to harness the full potential of CAR-T cells, especially in the treatment of solid malignancies. Although clinical trials in CLL appear promising (35,36), earlier work with CAR-transduced T cells in solid malignancies, such as ovarian cancer (37) and renal cell carcinoma (38), were less encouraging, with an absence of objective tumor response and the lack of T cell persistence. In the studies described here, we evaluated whether dgks represent a possible strategy for augmenting CAR-expressing T cells.
After establishing a retroviral system to efficiently transduce murine T cells, we found, as with TCR signaling, that deletion of dgkζ augmented Erk activation, a phosphorylation event that occurs downstream of DAG formation, after CAR ligation. Deletion of dgkζ was also found to augment CAR-dependent effector functions, since these cells exhibited enhanced cytokine production and target cell killing relative to their wt counterparts. Deletion of both T cell isoforms of dgk resulted in even greater enhancement of effector functions of mesoCAR-transduced cells and resulted in control of tumor in vivo in tumor-bearing mice. These results are encouraging for ongoing clinical trials, since murine studies of CAR-transduced T cells have accurately predicted clinical outcomes in past trials (6,35,39).
Although CARs are uniquely positioned to deal with the limited presence of antigen and co-stimulation found within the tumor environment, they do not address a third issue relating to T cell response to tumor: inhibitory stimuli. In these studies, we uncovered deletion of dgks as a novel strategy for enhancing T cell activity in the presence of inhibitory stimuli. Specifically, we found that prolonged exposure to antigen or the tumor microenvironment inhibitors PGE2, adenosine and TGFβ were less able to suppress CD8+ effector functions in T cells that lacked one or both T cell isoforms of dgk. TGFβ is thought to be a key mediator of tumor-mediated inhibition, since it is secreted by a variety of tumors, and inhibition of TGFβ signaling, through the expression of a dominant negative receptor, results in enhanced tumor-specific activity of cytotoxic lymphocytes (40,41). In fact, the amount of TGFβ produced by human cancers, such as prostate cancer, inversely correlates to a patient’s overall prognosis (42,43). The enhanced Ras activation imparted by the loss of dgks might explain how dgk-deficient lymphocytes develop insensitivity to TGFβ. Since TGFβ is known to result in the reduced phosphorylation of Itk, a Tec kinase important in PLCγ1 activation (44), and since PLCγ1 is the protein directly responsible for DAG generation in T cells, one could envision that deletion of dgks might directly subvert this TGFβ-induced signaling alteration. Future studies will address the potential means by which loss of dgks confers insensitivity to TGFβ, including direct effects, such as enhanced Ras activation downstream of TCR, and indirect effects, such as increased expression of cytotoxic proteins including FasL and TRAIL.
We believe these findings could be translated clinically. We have demonstrated that pharmacologic inhibition of dgks augment the efficacy of human CAR T cells under inhibitory in vitro conditions. This preliminary finding suggests that dgks play an important role in human T cells and that dgks may represent attractive clinical targets in augmenting CAR T cell-based therapies. We are currently extending our preliminary data in human T cells by developing model systems in which dgk activity is suppressed through decreased dgk expression (e.g., through expression of shRNA specific for dgks), or inhibited dgk function (e.g., through expression of dominant negative forms of dgks). Of course, as one develops more potent CAR T cells, issues of toxicity may become relevant. We could not assess toxicity in our model system, since the CAR T cells are specific for human mesothelin and do not react with an endogenous mouse protein. However, we have developed a second model in our laboratory using CARs specific for the murine antigen mouse fibroblast activation protein (FAP), an antigen overexpressed on cancer-associated fibroblasts. In preliminary studies, we have observed enhanced anti-tumor efficacy using dgkζ-deficient FAP-CAR T cells in tumor-bearing mice, without evidence of enhanced toxicity (data not shown). Despite this initial indication that dgks can be targeted safely, careful attention to toxicity will be required if dgk-knockdown of CAR T cells is moved into clinical trials. One approach that our group has used when introducing CAR T cells with augmented function is to begin the trial using T cells transduced with short-lived CAR mRNA (45), thus mitigating the potential for chronic CAR-induced autoimmunity.
Our data support the notion that combining CAR expression, which improves targeting of T cells to tumors and drives an initial stimulatory response, with inhibition of proteins known to blunt the effectiveness of the TCR signal may synergize to drive an effector response. The additional value of creating effector T cells resistant to the inhibitory environment generated by the tumor is also likely to contribute to the enhanced efficacy observed in this combined approach. Collectively, our data suggest that targeting dgks, as one means to blunt an endogenous inhibitory response, could be a useful mechanism to improve CAR-based strategies in the treatment of human malignancy.
Supplementary Material
Footnotes
The authors report no potential conflict of interest for the data presented in this manuscript.
SMA and GAK contributed material support, helped write the manuscript, helped analyze and interpret data, and supervised the studies.
Contributor Information
Matthew J. Riese, Email: matthew.riese@bcw.edu.
Liang-Chuan S. Wang, Email: wanli@mail.med.upenn.edu.
Edmund K. Moon, Email: edmund.moon@uphs.upenn.edu.
Rohan P. Joshi, Email: rjoshi@mail.med.upenn.edu.
Anjana Ranganathan, Email: anjana.ranganathan@uphs.upenn.edu.
Carl H. June, Email: cjune@exchange.upenn.edu.
References
- 1.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turcotte S, Rosenberg SA. Immunotherapy for metastatic solid cancers. Adv Surg. 2011;45:341–60. doi: 10.1016/j.yasu.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007 Jun;117(6):1466–76. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011 Mar 25;331(6024):1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 5.Quezada SA, Peggs KS, Simpson TR, Allison JP. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev. 2011 May;241(1):104–18. doi: 10.1111/j.1600-065X.2011.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009 Aug;17(8):1453–64. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dower NA, Stang SL, Bottorff DA, Ebinu JO, Dickie P, Ostergaard HL, et al. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat Immunol. 2000 Oct;1(4):317–21. doi: 10.1038/79766. [DOI] [PubMed] [Google Scholar]
- 8.Topham MK, Prescott SM. Diacylglycerol kinase zeta regulates Ras activation by a novel mechanism. J Cell Biol. 2001 Mar 19;152(6):1135–43. doi: 10.1083/jcb.152.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong X-P, Hainey EA, Olenchock BA, Jordan MS, Maltzman JS, Nichols KE, et al. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat Immunol. 2003 Sep;4(9):882–90. doi: 10.1038/ni958. [DOI] [PubMed] [Google Scholar]
- 10.Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, et al. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006 Nov;7(11):1174–81. doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- 11.Zha Y, Marks R, Ho AW, Peterson AC, Janardhan S, Brown I, et al. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat Immunol. 2006 Nov;7(11):1166–73. doi: 10.1038/ni1394. [DOI] [PubMed] [Google Scholar]
- 12.Riese MJ, Grewal J, Das J, Zou T, Patil V, Chakraborty AK, et al. Decreased diacylglycerol metabolism enhances ERK activation and augments CD8+ T cell functional responses. J Biol Chem. 2011 Feb 18;286(7):5254–65. doi: 10.1074/jbc.M110.171884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell. 1988 Sep 9;54(6):777–85. doi: 10.1016/s0092-8674(88)91043-4. [DOI] [PubMed] [Google Scholar]
- 14.Moon EK, Carpenito C, Sun J, Wang L-CS, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011 Jul 15;17(14):4719–30. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993 Sep 15;90(18):8392–6. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chowdhury PS, Pastan I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat Biotechnol. 1999 Jun;17(6):568–72. doi: 10.1038/9872. [DOI] [PubMed] [Google Scholar]
- 17.Jordan MS, Sadler J, Austin JE, Finkelstein LD, Singer AL, Schwartzberg PL, et al. Functional hierarchy of the N-terminal tyrosines of SLP-76. J Immunol. 2006 Feb 15;176(4):2430–8. doi: 10.4049/jimmunol.176.4.2430. [DOI] [PubMed] [Google Scholar]
- 18.Tchou J, Wang L-C, Selven B, Zhang H, Conejo-Garcia J, Borghaei H, et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat. 2012 Jun;133(2):799–804. doi: 10.1007/s10549-012-2018-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeLong P, Tanaka T, Kruklitis R, Henry AC, Kapoor V, Kaiser LR, et al. Use of cyclooxygenase-2 inhibition to enhance the efficacy of immunotherapy. Cancer Res. 2003 Nov 15;63(22):7845–52. [PubMed] [Google Scholar]
- 20.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010 Aug;10(8):554–67. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamura M, Kitaura J, Enomoto Y, Lu Y, Nishimura K, Isobe M, et al. Transforming growth factor-β-stimulated clone-22 is a negative-feedback regulator of Ras/Raf signaling: Implications for tumorigenesis. Cancer Sci. 2012 Jan;103(1):26–33. doi: 10.1111/j.1349-7006.2011.02108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.di Bari MG, Lutsiak MEC, Takai S, Mostböck S, Farsaci B, Semnani RT, et al. TGF-beta modulates the functionality of tumor-infiltrating CD8+ T cells through effects on TCR signaling and Spred1 expression. Cancer Immunol Immunother. 2009 Nov;58(11):1809–18. doi: 10.1007/s00262-009-0692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bedi A, Chang X, Noonan K, Pham V, Bedi R, Fertig EJ, et al. Inhibition of transforming growth factor-β enhances the in vivo antitumor efficacy of epidermal growth factor receptor-targeted therapy. Mol Cancer Ther. 2012 Aug 27; doi: 10.1158/1535-7163.MCT-12-0101-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cartellieri M, Bachmann M, Feldmann A, Bippes C, Stamova S, Wehner R, et al. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J Biomed Biotechnol. 2010;2010:956304. doi: 10.1155/2010/956304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998 Jun 4;393(6684):478–80. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 26.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998 Jun 4;393(6684):480–3. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 27.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998 Jun 4;393(6684):474–8. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 28.Chambers CA, Sullivan TJ, Truong T, Allison JP. Secondary but not primary T cell responses are enhanced in CTLA-4-deficient CD8+ T cells. Eur J Immunol [Internet] 1998 Oct;28(10):3137–43. doi: 10.1002/(SICI)1521-4141(199810)28:10<3137::AID-IMMU3137>3.0.CO;2-X. Available from: http://onlinelibrary.wiley.com/doi/10.1002/(SICI)1521-4141(199810)28:10%3C3137::AID-IMMU3137%3E3.0.CO;2-X/abstract. [DOI] [PubMed] [Google Scholar]
- 29.Gajewski TF, Fallarino F, Fields PE, Rivas F, Alegre ML. Absence of CTLA-4 lowers the activation threshold of primed CD8+ TCR-transgenic T cells: lack of correlation with Src homology domain 2-containing protein tyrosine phosphatase. J Immunol. 2001 Mar 15;166(6):3900–7. doi: 10.4049/jimmunol.166.6.3900. [DOI] [PubMed] [Google Scholar]
- 30.Chiang JY, Jang IK, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest. 2007 Apr;117(4):1029–36. doi: 10.1172/JCI29472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loeser S, Loser K, Bijker MS, Rangachari M, van der Burg SH, Wada T, et al. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J Exp Med. 2007 Apr 16;204(4):879–91. doi: 10.1084/jem.20061699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stromnes IM, Blattman JN, Tan X, Jeevanjee S, Gu H, Greenberg PD. Abrogating Cbl-b in effector CD8(+) T cells improves the efficacy of adoptive therapy of leukemia in mice. J Clin Invest. 2010 Oct;120(10):3722–34. doi: 10.1172/JCI41991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallner S, Gruber T, Baier G, Wolf D. Releasing the brake: targeting Cbl-b to enhance lymphocyte effector functions. Clin Dev Immunol. 2012;2012:692639. doi: 10.1155/2012/692639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das J, Ho M, Zikherman J, Govern C, Yang M, Weiss A, et al. Digital signaling and hysteresis characterize ras activation in lymphoid cells. Cell. 2009 Jan 23;136(2):337–51. doi: 10.1016/j.cell.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011 Aug 10;3(95):95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011 Aug 25;365(8):725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006 Oct 15;12(20 Pt 1):6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006 May 1;24(13):e20–2. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 39.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 2009 Mar 3;106(9):3360–5. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001 Oct;7(10):1118–22. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 41.Bollard CM, Rössig C, Calonge MJ, Huls MH, Wagner H-J, Massague J, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002 May 1;99(9):3179–87. doi: 10.1182/blood.v99.9.3179. [DOI] [PubMed] [Google Scholar]
- 42.Wikström P, Stattin P, Franck-Lissbrant I, Damber JE, Bergh A. Transforming growth factor beta1 is associated with angiogenesis, metastasis, and poor clinical outcome in prostate cancer. Prostate. 1998 Sep 15;37(1):19–29. doi: 10.1002/(sici)1097-0045(19980915)37:1<19::aid-pros4>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 43.Eastham JA, Truong LD, Rogers E, Kattan M, Flanders KC, Scardino PT, et al. Transforming growth factor-beta 1: comparative immunohistochemical localization in human primary and metastatic prostate cancer. Lab Invest. 1995 Nov;73(5):628–35. [PubMed] [Google Scholar]
- 44.Chen C-H, Seguin-Devaux C, Burke NA, Oriss TB, Watkins SC, Clipstone N, et al. Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J Exp Med. 2003 Jun 16;197(12):1689–99. doi: 10.1084/jem.20021170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010 Nov 15;70(22):9053–61. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.