

Abstract
Designing an optimal HIV-1 vaccine faces the challenge of identifying antigens that induce a broad immune capacity. One factor to control the breadth of T cell responses is the surface morphology of a peptide-MHC complex. Here, we present an in silico protocol for predicting peptide-MHC structure. A robust signature of a conformational transition was identified during all-atom molecular dynamics, which results in a model with high accuracy. A large test set was used in constructing our protocol and we went another step further using a blind test with a wild-type peptide and two highly immunogenic mutants, which predicted substantial conformational changes in both mutants. The center residues at position five of the analogs were configured to be accessible to solvent, forming a prominent surface, while the residue of the wild-type peptide was to point laterally towards the side of the binding cleft. We then experimentally determined the structures of the blind test set, using high resolution of X-ray crystallography, which verified predicted conformational changes. Our observation strongly supports a positive association of the surface morphology of a peptide-MHC complex to its immunogenicity. Our study offers the prospect of enhancing immunogenicity of vaccines by identifying MHC binding immunogens.
1. Introduction
The ability of human immunodeficiency virus-1 (HIV-1) to evade immune detection as well as the presence of many circulating variants of the virus poses a significant challenge in the development of prophylactic vaccines (Gaschen et al., 2002). Numerous studies demonstrated that the breadth of T cell responsiveness is more important than the magnitude during HIV infections (Currier et al., 2011; Frahm et al., 2006; Frahm et al., 2004; Geldmacher et al., 2007; Honeyborne et al., 2007; Rolland et al., 2008). In this context, diverse T cell receptor (TCR) repertoires have been convincingly shown to be advantageous in elimination of viral variants during infections of HIV-1 (Altfeld et al., 2001; Douek et al., 2002), simian immunodeficiency virus (SIV) (Price et al., 2004), and hepatitis C virus (HCV) (Douek et al., 2002; Meyer-Olson et al., 2004; Price et al., 2004). One of the characteristics that might control diversity of the T cell repertoire is the surface morphology of the peptide-major histocompatibility complex (pMHC). Typically, peptides of 8–10 amino acids long can fit well in the MHC molecule, and in such occasion, several experiments have demonstrated that surface prominence is associated with the breath of T cell responsiveness (La Gruta et al., 2008; Probst-Kepper et al., 2004; Stewart-Jones et al., 2003; Turner et al., 2005). For example, the influenza MP peptide, which forms a flat, featureless conformation in the peptide-binding cleft showed a highly conserved TCR usage in most of human subjects (Stewart-Jones et al., 2003). A direct association between the prominence of the peptide and the degree of TCR diversity has been demonstrated in an influenza infection study in mice (La Gruta et al., 2008). The pMHC surface that is structurally protruding has the potential to functionally engage more diverse TCR molecules than would a flat pMHC surface (Stewart-Jones et al., 2003). In addition, many immunodominant epitopes have been shown to be prominent from the center of the MHC molecule (Liu et al.; Meijers et al., 2005). Therefore, a prominent morphology is hypothesized to be related with optimal T cell responsiveness.
Accurately determining the structure of pMHC can help to predict robust T cell reactivity by determining which peptides tend to be prominent from the binding cleft. In this manner, in-silico prediction of pMHC conformation would be a valuable tool for vaccine design because experimentally determining each structure for the thousands of candidates would be excessively time consuming and costly. In parallel with recent remarkable advances in computational methodology, in silico research on protein modeling has achieved a significant success in obtaining the detailed information on folding including thermodynamic and kinetic (Freddolino et al., 2010; Lindorff-Larsen et al., 2011; Piana et al., 2012; Shaw et al., 2010). For instance, the free energy landscape along the reaction coordinate has been accurately estimated from long-time simulation trajectories of repetitive conformational transitions (Best and Hummer, 2005; Dellago et al., 1998; Piana et al., 2012). Still, in more complex systems including pMHC, it is not an easy task to obtain “de novo” structure prediction comparable to high-resolution X-ray crystallography, mainly due to the difficulty of determining the energetically favored state out of a vast pool of local minima (Floudas, 1999; Levinthal, 1969). Reports of the X-ray structures of many peptides bound to MHC molecules reveal a common architecture of N- and C- terminal ends anchoring to the binding pocket (Falk et al., 1991; Matsumura et al., 1992). However, the central region of peptides highly varies allowing a fine control of TCR recognition (Collins et al., 1994; Madden et al., 1993), which makes it difficult to predict the surface morphology of bound peptides in high precision.
There have been many attempts to predict pMHC conformation, including docking algorithms (Desmet et al., 1997; Leach, 1994), protein threading (Bui et al., 2006), all-atom molecular dynamics (MD) simulations (Fagerberg et al., 2006), and hybrid approaches between docking algorithms and MD simulations (Kish-Catalone et al., 2006). These efforts have been less than perfectly successful to identify the correct structure, showing a considerable variation in the degree of accuracy. Overall, the target structure was correctly predicted when it was homologous to the structure of the model template. The outcome of MD simulations was desirable simply because the ensemble of initial structures was sampled from the X-ray crystal structure itself. These limitations motivated us to develop a robust identifier to predict the docking structure of pMHC molecules even when the target conformation is different from that of the template.
Here we present an accurate computational tool that will help to predict robust TCR reactivity by predicting which peptides are likely to be prominent from the peptide-binding cleft. Our protocol started with preparing a starting configuration using homology modeling. We then employed simulated annealing molecular dynamics simulations with the AMBER force field to generate an ensemble of potential pMHC conformations. Lastly, we used all-atom MD simulations with the AMBER force field to generate a minimized energy structure. In particular, during the MD simulations, we monitored the simulation trajectory to find conformational transitions. If there were transitions, we chose the most frequent of the post-transition conformations as the final conformation used for prediction of the structure. Our structure prediction was developed using a large test set of nonameric peptides bound to the MHC class I molecule, HLA-A*0201. The prediction algorithm was used to perform a blind prediction for three unknown pMHC structures. We then experimentally determined the structures of the blind test set using high resolution of X-ray crystallography.
2. Materials and methods
2.1. Simulated annealing protocol
The structure built by the MODELLER was first heated from 300K to 1500K during 80 ps then equilibrated at 1500K during 80 ps. Thereafter, the system was cooled for 800 ps, resulting in a temperature of 300K and cooled again for 40 ps to 283 K. Langevin dynamics was performed using the sander module of the AMBER9 with a time step of 2 fs. The SHAKE algorithm was implemented for proton covalent bonds and a cutoff distance of 10 Å was introduced for Lennard-Jones interactions. Solvent effects were treated implicitly by adapting the pairwise Generalized Born model as parameterized by Tsui and Case (Tsui and Case, 2000).
We imposed two types of restraints. First, a 20 kcal/mol harmonic energy barrier was assigned to each of the receptor atoms of the α1 and α2 domains to limit the motion of the receptor. Second, the distances of the four hydrogen bonds between the peptide and the receptor were maintained during the simulation as we found that these four hydrogen bonds were conserved across all 18 complexes of our test set. The conserved ones are (i) OG1 atom of THR 143 and C-terminal oxygen of peptide, (ii) NE1 atom of TRP 147 and O atom of peptide at position 8, (iii) OH atom of TYR 159 and O atom of peptide at position 1, and (iv) OH atom of TYR 171 and N-terminal nitrogen of peptide. The simulated annealing cycle was repeated by assigning random velocity to each atom of the p-MHC complex and an ensemble of a total of 100 p-MHC conformations was obtained for each p-MHC complex.
2.2. Molecular dynamics simulation
With an initial configuration obtained from the simulated annealing protocol, Langevin dynamics simulations were performed at 283 K during 10 ns using the pmemd module of the AMBER9. The parm99 force field parameters were used with the pairwise Generalized Born implicit solvent model as parameterized by Tsui and Case (Tsui and Case, 2000). Prior to the dynamics run, the receptor was relaxed by steepest descent energy minimization, followed by conjugate gradient energy minimization. The same restraints that were used in the simulated annealing protocol were implemented. All bonds involving hydrogen atoms were constrained using the SHAKE algorithm. A nonbonded cutoff distance of 12 Å and a time step of 2 fs were used.
2.3. X-ray crystallization data collection
The X-ray crystal data have been deposited in the PDB with accession codes 3V5D, 3V5H, and 3V5K. The X-ray crystal data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID and 22-BM beamlines at the Advanced Photon Source, Argonne National Laboratory. Peptide/HLA-A*0201 complexes were made by folding in vitro as described in Ref. 51. Briefly, A2.1 heavy chains (amino acids 1–275) and beta-2-microglobulin were produced as inclusion bodies. Synthesized peptides were added to the folding buffer by rapid dilution. Protein folded over the course of 36–48 hrs at 10°C, was concentrated and then purified by gel filtration chromatography. Complete removal of salts is critical to crystallization so the gel filtration buffer was exchanged into 25 MES pH 6.5. Crystallization of pMHC (15–20mg/ml) occurred at 10–16% PEG 8000 (Fluka) 25mM MES pH6.5 by hanging drop, vapor diffusion. The best crystals were obtained by microseeding. Single plate-like crystals of various thickness were harvested from the drops and transferred to artificial mother liquor and then rapidly transferred to a similar solution supplemented with 10% glycerol for cryoprotection. Crystals were plunged into liquid nitrogen and stored until used for data collection. Diffraction data were collected at APS beamlines 22-ID for the KVAELVWFL or 22-BM for the KVAELVHFL and KVAEIVHFL.
2.4. X-ray crystallization structure solution and refinement
The highest resolution data for the wild-type structure were very mosaic and difficult to index. These data were processed using XDS (Kabsch, 2010). The data for the two analogs were processed with HKL2000(Otwinowski and Minor, 1997). Data were further scaled and a subset of data tagged for cross-validation using Rfree (Brunger, 1992) in CCP4 program suite (Winn et al.). The phases were determined by molecular replacement using Phaser (McCoy et al., 2007) using one copy of the heavy chain and beta-2-microglobulin from an unrelated A*0201 structure 1EEZ without a bound peptide and with an average temperature factor. After first fitting of the correct peptide in coot (Emsley et al., 2010), the structure was refined with autoBuster (http://www.globalphasing.com/buster/manual/autobuster/manual/index.html) using local non-crystallographic symmetry restraints (autoncs), TLS refinement with the peptide-binding domain and peptide as one unit, the a3 domain (amino acids 184–275) and beta-2-microglobulin as two other units. Since there are two copies of the complex in the asymmetric unit, this generates 6 TLS domains. After a few rounds of refinement and manual intervention with coot, water molecules were added with arp-warp (Lazmin et al., 2001). The ARP/WARP suite for automated construction and refinement of protein models and well-defined glycerol molecules were added to the model and refined using autoBuster with the WAT option.
3. Results
3.1. Test set selection
In developing the protocol, we used a test set of 18 X-ray structures (Borbulevych et al., 2005; Buslepp et al., 2001; Kuhns et al., 1999; Lee et al., 2004; Martinez-Hackert et al., 2006; Sharma et al., 2001; Webb et al., 2004) of the nonameric peptides bound to the MHC class I molecule HLA-A*0201, in the Protein Data Bank (see Table 1). The test set consisted of 9 HIV peptides, 8 tumor-associated peptides, and 1 synthetic peptide. Comparison of the bound receptors showed the backbone atom root mean square deviation (RMSD) differences of 0.80 (± 0.31) Å on average with the largest displacement, 1.22 Å (Figure 1A). Binding of different peptides to the receptor, HLA-A*0201, does not induce significant conformational changes of the pocket, which provides a basis for a rigid approximation of the MHC molecule in our simulations. The bound configurations of the 18 peptides were compared and most of the peptide backbones were similar to each other except one, 2V2X, which is notably different (Figure 1B). Our test set exhibits substantial variations in the side chain directions, leading to a versatile surface configuration of the pMHC complexes (Figure 2). The binding specificity of peptide-HLA-A*0201 interactions within the test set was analyzed using LIGPLOT (Wallace et al., 1995). The data clearly identified four conserved hydrogen bonds across all of the 18 pMHC complexes (Figure 1C). We utilized the concurrence of the hydrogen bonds to restrict the distances among the peptide and receptor atoms during our simulations.
Table 1.
The test set of peptides restricted by MHC class I molecule HLA-A*0201. We used pMHC complex 2V2W (sequence SLYNTVATL) as a common template for homology modeling. The PDB code, amino acid sequence, the all-atom RMSD between X-ray structure and predicted one by the homology modeling (MODELLER), and the all-atom RMSD between X-ray structure and the most abundant conformation obtained from simulated annealing trials.
| PDB code | Sequence | All-Atom RMSD (Å) to X-ray | |
|---|---|---|---|
| Homology modeling | Simulated annealing | ||
| 1I7U | ALWGFVPVL | 1.65 | 2.12 |
| 1QR1 | IISAVVGIL | 1.35 | 1.45 |
| 1EEY | ILSALVGIV | 1.36 | 1.33 |
| 1EEZ | ILSALVGIL | 1.53 | 1.51 |
| 1S9W | SLLMWITQC | 1.87 | 2.02 |
| 1S9X | SLLMWITQA | 1.92 | 2.04 |
| 1S9Y | SLLMWITQS | 1.74 | 2.26 |
| 1TVB | ITDQVPFSV | 2.01 | 1.62 |
| 1TVH | IMDQVPFSV | 1.87 | 1.87 |
| 2V2X | SLFNTVATL | 2.36 | 2.18 |
| 1S8D | SLANTVATL | 1.02 | 1.11 |
| 1T1W | SLFNTIAVL | 1.20 | 1.26 |
| 1T1X | SLYLTVATL | 1.28 | 1.38 |
| 1T1Y | SLYNVVATL | 1.33 | 1.42 |
| 1T1Z | ALYNTAAAL | 1.18 | 1.42 |
| 1T20 | SLYNTIATL | 1.35 | 1.45 |
| 2C7U | SLFNTIAVL | 1.13 | 1.19 |
| Average | 1.54 | 1.63 | |
| Standard Deviation | 0.37 | 0.37 | |
Figure 1.
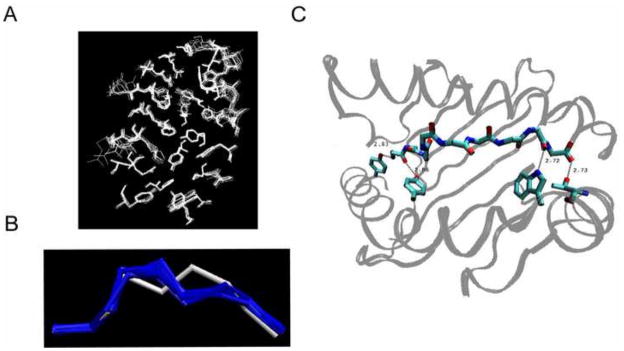
Test set comparison. A. Superimposed 18 binding pockets of the MHC class molecule HLA-A*0201, obtained by X-ray crystallography. Each pocket represents the bound conformation to each of 18 peptides. The receptor residues within 5 Å from the bound peptide are considered only. B. Superimposed conformations of the 18 epitope backbones restricted by HLA-A*0201, obtained by X-ray crystallography. The peptide 2V2X is represented by white and the other 17 peptides are represented by blue. C. Four conserved hydrogen bonds between the peptide backbone and the receptor side chain.
Figure 2.

Test set prediction. Superposition of the bound conformations of the antigenic peptides obtained from the X-ray crystal structures (cyan) with the MHC removed, the homology modeled peptides from MODELLER (orange), and the most frequent conformation obtained from simulation annealing trials (red).
3.2. Homology modeling
We built homology-based models using three different software programs: MODELLER (Marti-Renom et al., 2000; Sali and Blundell, 1993), ROSETTA (Chivian and Baker, 2006; Raman et al., 2009), and PRIME (McRobb et al.; Yoo and Medina-Franco), with the same template choice of pMHC complex 2V2W. Each model structure obtained was superimposed to its crystal structure, and all-atom root mean square deviation (RMSD) between the predicted and experimental peptide conformations was calculated. Here we reported all-atom RMSD by including all atoms of backbone and side-chain residues, excluding hydrogen atoms. We found that the MODELLER generally performed best; the mean of all-atom RMSD (excluding hydrogens) was 1.54(± 0.37) Å from MODELLER, 3.11(± 0.22) Å from ROSETTA, and 3.10(± 0.19) Å from PRIME, respectively (Table 1). Figure 2 shows the superposition of the predicted peptide conformations from MODELLER with the actual conformations from the X-ray crystal structures. Not too surprisingly, all of the homology models showed strong dependence on the level of similarity between the target and template structures. When the template had a peptide backbone configuration similar to the test peptide configuration, MODELLER did very well. However, it was a much more challenging problem when the target peptide structure deviated from the template peptide structure. For peptide 2V2X, the modeling programs failed to predict the correct backbone conformation, resulting in the all-atom RMSD of 2.36 Å (MODELLER).
Furthermore and more importantly, MODELLER failed to choose the optimal template when it was given a set of template options that included the correct peptide backbone. For each peptide, we used four or five different templates, which were the representatives of the ensemble obtained through the simulated annealing runs. The prediction by the MODELLER was highly dependent on each initial template. The all-atom RMSD between the template structure and the X-ray structure was significantly correlated with that between the prediction and the X-ray structure (correlation coefficient=0.99 and F=2884.9 and p<0.001). Moreover, when the MODELLER used multiple templates, we observed that it was not guaranteed to select the correct template. For the 2V2X, the MODELLER failed to choose the template with the smallest all-atom RMSD to the X-ray structure (1.3 Å) although that was one of the five templates we used. Instead, the MODELLER predicted the conformation, which deviates from the X-ray with the all-atom RMSD of 2.07 Å. We concluded that employing multiple templates was not able to fix the template dependence of the MODELLER. Therefore, standard homology modeling approaches are not sufficient to generate high quality models for pMHC complexes.
3.3. Simulated annealing using all-atom molecular dynamics simulations
We next took a comprehensive approach to overcome the limitations of homology modeling by employing all-atom molecular dynamics simulations with the AMBER force field. We generated an ensemble of peptide conformations by adopting a simulated annealing scheme which is one of the effective methods that are able to overcome energy barriers and explore conformational space (Frenkel and Smit, 2002). Each pMHC complex, which was initially configured by the MODELLER, was heated up to 1500K and then slowly cooled down to room temperature, thereby guiding the complex to an energy minimized structure. The entire procedure was optimized using the trials of several different annealing schemes. For example, when the complex was heated up to 800K, conformational changes in side chains were predominant, while heating the complex to 1500K permitted motions of not only side chains, but also of the backbone of the peptide. A linear cooling schedule was found to be more effective than an exponentially decreasing temperature, especially, when the initial starting configuration was not similar to the correct one.
An ensemble of 100 simulated annealed structures was then analyzed using clustering algorithms to correctly select the most abundant configuration from the ensemble. Several algorithms including Bayesian, hierarchical, k-means, and self-organizing maps (SOM) (Shao et al., 2007) were tested along with the centroid structure of the ensemble. We used pairwise all-atom RMSD values among 100 simulated annealed structures for each clustering method and chose the conformation which was closest to the centroid of the major cluster. When we compared the conformation selected by each clustering method, the Bayesian method slightly performed better than others: The all-atom RMSD between the predicted and the X-ray, averaged over 17 complexes, was 1.63(± 0.37) Å for Bayesian, 1.69(± 0.40) Å for hierarchical, 1.70(± 0.39) Å for k-means, 1.68(± 0.35) Å for SOM, and 1.71(± 0.40) Å for the centroid, respectively. Figure 2 compares the respective X-ray crystal structures with the conformations predicted by the process using MODELLER, followed by simulated annealing trials and choosing the final model by grouping using Bayesian statistics. Surprisingly, the predictions based on simulated annealing followed by Bayesian clustering were comparable to those of the MODELLER (Table 1). The most abundant configuration within the ensemble generated by the simulated annealing does not necessarily represent the correct configuration. The backbone of the major configuration within the 2V2X ensemble was incorrectly predicted, as it was from MODELLER’s prediction. Only 10% of the ensemble of structures from simulated annealing had the correct backbone topology based on the crystal structure. Results indicated that the simulated annealing was able to sample a broad range of configurations including backbone rotations of bound peptides. However, the most abundant structure within the ensemble was not consistent with the X-ray crystal structure when the starting backbone configuration of the simulated annealing was not consistent with that of the X-ray crystal structure.
3.4. Detecting conformational transitions for designating the correct structure
We sought out an alternative method for designating the correct structure within the ensemble. We chose four pMHC complexes out of the test set consisting of 2V2X whose backbone structure is incorrectly predicted by the MODELLER and 1S8D, 1I7U, and 1EEY whose backbone structures are correctly predicted by the MODELLER. For each of 100 structures generated by the simulated annealing for each of four complexes, we performed a 10 ns all-atom MD simulation. For each run of 10 ns at room temperature, we sampled every 2 ps and collected 5000 conformations. Then the RMSD trajectory was measured by comparing these 5000 peptide structures with the X-ray crystal structure and asked if a conformational transition occurred by flagging when the change in the all-atom RMSD was greater than 20% of the initial RMSD to the X-ray structure. Using the 20% as a cut-off, allowed us to focus only on major changes rather than minor motions of the side chains. The additional 10 ns MD simulations of the 100 conformations of the 2V2X complex resulted in a total of 20 conformational transitions out of 100 runs. Out of 20 transitions, we found that 19 cases made a sharp transition towards the structure closest to the X-ray crystal and the most prevalent conformation of the post-transition structures showed the all-atom RMSD of 1.37 Å to the X-ray structure. Figure 3A displays a representative case of the transitions. For the 1S8D complex, which is the most accurately built model by the MODELLER, we observed a total of six transitions out of 100 MD simulation runs, and all of those were verified as ones transitioning towards back the structure closest to the X-ray structure (Figure 3A). Likewise, the complex 1I7U and 1EEY showed 5 and 3 transitions, respectively and all of those were towards each major conformer of the simulated annealing, which was closest to each X-ray structure (Figure 3A). Table 2 compares the all-atom RMSDs to the X-ray structure by the MODELLER and the MD simulations for the four complexes. When the MODELLER performs poorly, as in 2V2X, mainly due to the deviation of the target from the template, the signature of the transition in the MD simulations improved the level of prediction significantly. On the other hand, when the MODELLER’s prediction was acceptable, the signature of the transition was slightly worse but comparable to the MODELLER’s prediction. Our observation suggests that the signature of the conformational transition provides a way to fix the template dependence of the MODELLER.
Figure 3.
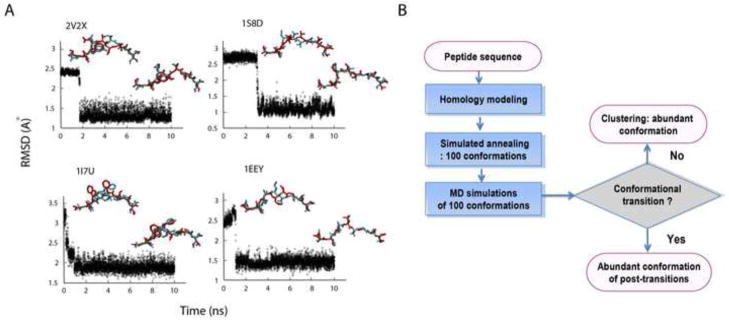
Conformational transition. MD simulations reveal conformational transitions towards the correct configuration. A. The RMSD trajectories of the 2V2X, 1S8D, 1I7U, and 1EEY ligands during each run of the 10 ns MD simulation. Conformations of the pre- and post-transition (red) are compared to the x-ray structure (cyan). B. Summary of our prediction protocol.
Table 2.
The RMSD of 2V2X, 1S8D, 1I7U, and 1EEY between the X-ray structure and the predicted one by the homology modeling or by the MD simulations.
| RMSD (Å) to X-ray | ||
|---|---|---|
| MODELLER | MD Simulations | |
| 2V2X | 2.36 | 1.37 |
| 1S8D | 1.02 | 1.11 |
| 1I7U | 1.66 | 1.95 |
| 1EEY | 1.39 | 1.49 |
3.5. Blind test and confirmation by X-ray crystallography
With our optimized protocol, which was summarized in Figure 3B, we predicted the conformations three nonameric peptides consisting of a tumor-associated wild peptide, KVAELVHFL and two heteroclitic analogs, KVAEIVHFL and KVAELVWFL, restricted by the HLA-A*0201. Our goal was to examine whether our method correctly predicts structures in the absence of any prior information. Following our protocol, 100 structures for each of the three complexes were generated by the simulated annealing method which started from the configuration of the MODELLER’s prediction. Then an additional 10 ns all-atom MD simulation for each of 100 structures was performed and the RMSD trajectory was measured. Here the three structures were unknown, the RMSD values were measured by comparing with the configuration predicted by the MODELLER. The MD simulations identified four and two transitions for the KVAELVHFL and the KVAELVWF out of 100 runs for each complex, respectively. Post-transitional conformations were then collected and the most prevalent conformation was designated as our final prediction. For the peptide, KVAEIVHFL, we did not observe any conformational transitions in the ensemble of 100 conformers and the most frequent of those was selected as a final prediction. Our structural prediction indicated significant differences in the surface morphology among the wild-type peptide and the mutants; for the wild-type peptide, the leucine at position 5 was pointing laterally towards a helix on the receptor, holding a flat surface (Figure 4). On the other hand, the same P5 leucine of the analog, KVAELVWFL, was predicted to point directly towards the T cell receptor, holding a prominent surface (Figure 4). The isoleucine of the analog, KVAEIVHFL, was also predicted to be configured towards the T cell receptor, resulting in a protruding surface (Figure 4). Our prediction of this significant conformational change from the flat surface to the prominent surface by a single amino acid substitution is particularly notable given that the substitution of leucine to isoleucine at position 5 could be expected to be very subtle because the two amino acids are isosteric.
Figure 4.
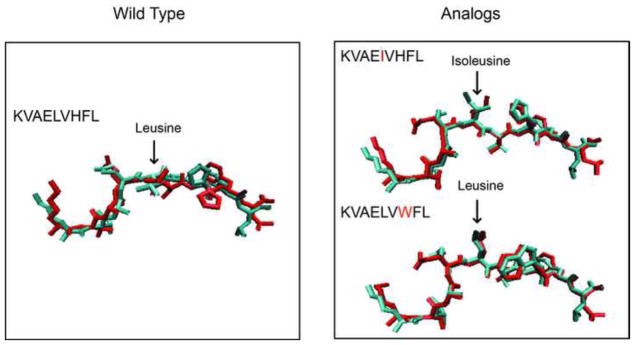
Blind set prediction. A blind test confirms that this procedure correctly predicts structures not in the training ensemble. Comparisons of the predicted conformations by the MD simulations (red) to the X-ray structure (cyan) for the wild peptide (KVAELVHFL) and the two heteroclitic analogs, KVAEIVHFL and KVAELVWFL. The center residue of each peptide is marked by an arrow.
The crystal structures for the three peptides from the blind test, the wild-type peptide, KVAELVHFL, and the two heteroclitic peptides, KVAEIVHFL and KVAELVWFL each bound to HLA-A0201, were determined to 2.0 Å, 1.63 Å, and 2.31 Å resolutions, respectively (Table 3). Interestingly, the crystal structure of the wild peptide is unique when compared to crystal structures of other peptides bound to MHC because the electron density suggested a relatively small set of structures by the peptide and not just a single one (Figure 5 and Figure 6). Most often, the structures either show one conformation or missing density suggesting many potential conformations. Two paths of KVAELVHFL peptide could be interpreted with confidence and were modeled in the final structure. The main difference between the two paths was two inverted orientations for the P5-P6 peptide bond carbonyl, which were common in both asymmetric units as shown in Figure 5 and Figure 6. Both X-ray crystals confirmed the computational prediction that the center residue, leucine, is configured to point laterally towards the receptor, forming a flat surface with the all-atom RMSDs of 2.04 Å and 1.99 Å to the predicted structure, respectively.
Table 3.
The statistics of X-ray crystallographic data.
| KVAELVHFL | KVAEIVHFL | KVAELVWFL | |
|---|---|---|---|
| Data Processing | |||
|
| |||
| Space Group | p21 | p21 | p21 |
| Cell Dimensions | 63.2, 90.49, 79.97 | 62.80, 87.60, 79.80 | 62.80, 87.10, 79.70 |
| Angles (°) | 90.00, 90.06, 90.00 | 90.00, 89.96, 90.00 | 90.00, 90.04, 90.00 |
| Molecules/AU | 2 | 2 | 2 |
| Resolution (Å) | 50.0–2.00 (2.11–2.0) | 50.0 – 1.63 (1.66–1.63) | 30.0–2.3 (2.38–2.30) |
| Rmerge (%) | 0.12 (0.41) | 0.049 (0.36) | 0.079 (0.29) |
| <I/s> | 11.3 (4.3) | 22.0 (3.2) | 13.5 (4.3) |
| Unique Reflections | 60582 | 106085 | 35327 |
| Average Redundancy | 7.1 (6.5) | 3.7 (3.1) | 3.9 (4.0) |
| mosaicity | 0.2–0.4 | 0.4–0.6 | 0.4–0.6 |
| Completeness (%) | 0.99 (0.95) | 0.97 (0.72) | 0.94 (0.99) |
| Processing software | XDS | HKL2000 | HKL2000 |
|
| |||
| Refinement | |||
|
| |||
| Resolution Range (Å) | 40.0–2.0 (2.05–2.00) | 16.43–1.63 (1.67–1.63) | 30.0–2.3 (2.38–2.30) |
| Average B-factor (Å2) | 25.4 | 29.6 | 38.6 |
| Number of Reflections | 60,433 (4247) | 105,970 (5285) | 35,266 (1754) |
| Rfac | 0.23 (0.22) | 0.19 (0.22) | 0.19 (0.20) |
| Rfree | 0.26 (0.25) | 0.21 (0.26) | 0.23 (0.27) |
| Number of non-H atoms | 6401 | 6332 | 6332 |
| Ramachandran | |||
| Residues in most favored regions | 711 | 729 | 732 |
| Residues in additional allowed regions | 22 | 22 | 24 |
| Residues in generously allowed regions | 0 | 0 | 0 |
| Residues in disallowed regions | 0 | 0 | 0 |
| ESU based on Luzzati Plot | 0.32 | 0.21 | 0.31 |
| RMSD bond lengths | 0.01 | 0.01 | 0.01 |
| RMSD bond angles | 1.05 | 1.00 | 1.07 |
| Refinement Program | autoBuster | autoBuster | autoBuster |
Figure 5.

The x-ray structures of the pMHC in the blind test set. A. The electron density around the wild peptide (KVAELVHFL) and two mutants (KVAEIVHFL and KVAELVWFL), bound to HLA-A*0201. The asymmetric unit of the crystals contains two copies of pMHC complexes. The electron density for the first copy is shown here and the electron density for the peptide in the second copy is shown in Figure 6. B. Crystallographic models of the three peptides bound to HLA-A*0201. The overlay of the two configurations for the wild peptide is in blue and cyan.
Figure 6.
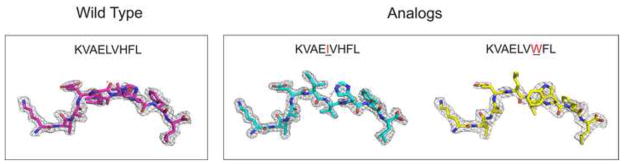
The x-ray structure of the blind test set. The asymmetric unit of the crystals contains two copies of the pMHC complexes. The electron density for the first copy is shown in Figure 5A, the density for the second copy is shown here.
The crystals for the two mutants were also very similar to our predicted structures. For the analog, KVAEIVHFL, the substitution of leucine to isoleucine at P5 caused a large change in structure (Figure 5 and Figure 6): The isoleucine at P5 points directly towards the T cell receptor, holding a prominent configuration, in consistent with our model prediction (all-atom RMSD equals 1.63 Å). Similarly, the X-ray crystal of the KVAELVWFL confirmed a predicted change such that the P5 leucine is accessible to solvent, resulting in an astonishingly similar structure to our prediction with an all-atom RMSD of 1.19 Å. On the other hand, the level of prediction by the MODELLER was not desirable; the central region of the wild-type peptide was configured to point towards the T cell receptor resulting in the all-atom RMSD of 2.40 Å (Figure 7). The predictions by the MODELLER for the two mutants, KVAEIVHFL and KVAELVWFL, showed the all-atom RMSDs of 1.58 Å and 2.57 Å to the X-ray structures, respectively. When the MODELLER’s prediction was accurate, for KVAEIVHFL, we did not observe any conformational transition. On the contrary, when the MODELLER’s predictions were poor, both for KVAELVHFL and KVAELVWFL, the signature of conformational transition considerably improved the accuracy of prediction.
Figure 7.

Blind Test. Comparisons of the predicted conformations by the MODELLER (orange) to the X-ray structure (cyan) for the wild peptide (KVAELVHFL) and the two heteroclitic analogs, KVAEIVHFL and KVAELVWFL.
4. Discussion
We performed structure prediction of nonameric peptides bound to the class I HLA allele A*0201, using a protocol consisting of three steps (see Figure 3B). The first step of homology modeling chose the initial guess relatively close to the X-ray structure when the template used in the homology modeling was similar with the X-ray structure. In the second step of the simulated annealing, we generated an ensemble of peptide conformations. Although the configuration close to the X-ray structure was present within the ensemble, the most abundant configuration within the ensemble did not necessarily represent the correct configuration. The backbone of the major configuration within the 2V2X ensemble was incorrectly predicted, as it was from MODELLER’s prediction. A post-hoc analysis showed that only 10% of the ensemble of structures from simulated annealing had the correct backbone morphology of the crystal structure. One way of designating the correct structure within the ensemble would be calculating the free energy of each configuration and find the configuration of minimum free energy (Frenkel and Smit, 2002; Jarzynski, 1997; Kirkwood, 1935; Park et al., 2003; Torrie and Valleau, 1974). However the calculation is out of reach, since it requires a very long time simulation for each local minimum to obtain a precise estimate on the entropy contribution. The third step of our protocol, monitoring conformational transitions provided a way to overcome this difficulty because we can directly obtain the information about free energy differences between the conformations, not necessarily calculating the absolute free energy value itself. Indeed, information on free energy differences is enough for us to designate the most energetically favored structure.
The main hypothesis for the signature of transition is that the transition rate from a metastable, local energy minimum conformation to the energetically favored conformation is greater than the reverse transition rate. For instance, in a simple two-state model as illustrated in Figure 8, the forward and backward transition rates are proportional to exp(−Ff/T) and exp(−Fb/T), respectively. Here Ff(Fb) is the free energy barrier of forward (backward) transition and T is the temperature of the system. Thus, the ratio between two rates becomes exp[−(Ff − Fb)/T], which is greater than 1, implying that the forward transition toward the energetically favored conformation is more frequent than the backward transition. We reason that since simulated annealing did not always reliably predict the correct structure, it must sample suboptimal conformations around the initial starting configuration more frequently, not having a chance to sample the optimal conformation most frequently. Additional equilibration of the 10 ns MD simulations for each conformation of the ensemble provides opportunities of transitions among configurations. Because energy barriers of the transitions toward the energetically favored state are smaller than those of the backward transitions, transitions toward the correct structure likely outnumber those out of the correct structure. Therefore, choosing the most abundant of the post-transition conformations is presumed to be the best criteria for predicting thermodynamically favorable configurations. Our approach has some distinctive advantages. First, the longer additional MD simulation time we have, the more accurate transition rates we obtain (Best and Hummer, 2005; Dellago et al., 1998; Piana et al., 2012), and thus the prediction of the energetically favored configuration should become more accurate. Second, we do not apply any approximation except the choice of force field. Thus, as long as the force field is sufficiently good enough, our method guarantees a fine outcome.
Figure 8.
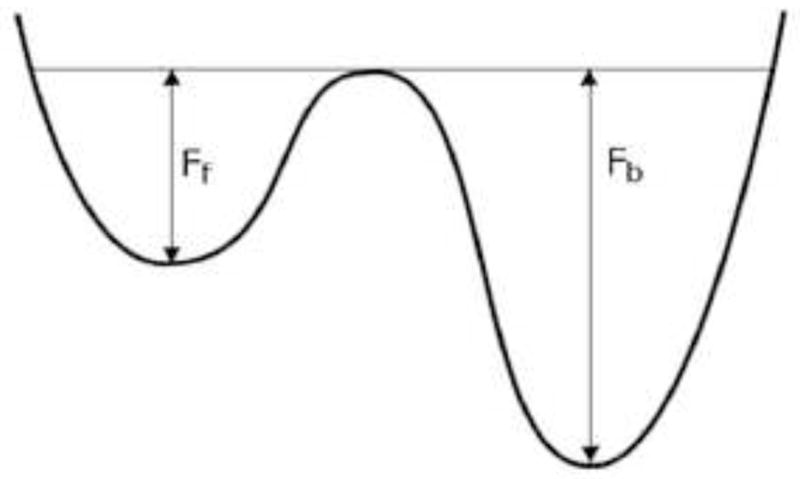
Two state model. Two-state model with the energy barrier of the forward transition, Ff and that of the backward transition, Fb.
Our protocol successfully predicted the conformations of a wild peptide, KVAELVHFL and two heteroclitic analogs, KVAEIVHFL and KVAELVWFL, restricted by the HLA-A*0201. This set of peptides was selected because i) we did not know the crystal structures of the complexes and ii) the two analogs were known as significantly more immunogenic than the wild-type peptide; a single amino acid substitution from the wild-type peptide had been reported to result in the increase of the antigenic sensitivity of T cells up to 107 fold (Tangri et al., 2001). We hypothesized that that increase in immunogenicity would be associated with a substantial change in the surface morphology of the complex. Indeed, our protocol predicted that the substitution causes a large change such that the isoleucine at P5 of KVAEIVHFL points directly towards the T cell receptor, holding a prominent conformation as well (Figure 5). As hypothesized, the more immunogenic mutants KVAELVWFL and KVAEIVHFL shared the characteristic of having a protruding central residue, which potentially contributed to enhanced antigenic sensitivity presumably by facilitating recruitment of diverse T cells. Our observation showed a positive association among immunogenicity and the degree of prominence of the peptide bound to MHC molecule. Additional supporting evidence was proto-oncogene HER-2/neu-derived peptide IISAVVGIL (1QR1) from our test set. This peptide had been shown to be poorly recognized by cytotoxic T cells, resulting in the failure of elimination of tumors (Kuhns et al., 1999). As shown in Figure 2, the surface morphology of the peptide 1QR1 was relatively flat and featureless. Collectively, our observation supports the hypothesis that the surface morphology of the pMHC complex is one of the confounding factors determining immunogenicity of each pMHC complex.
We have demonstrated that our prediction protocol could be a powerful tool for designating MHC binding immunogens. By identifying a robust criterion for selecting the best from the ensemble of candidate conformations, we devised an accurate prediction protocol of identifying highly immunogenic peptides. Our method provides key information of each antigen on its ability to recruit diverse T cell population by characterizing the surface morphology of the HIV-1 peptide and MHC complex. This efficient protocol can be used to prioritize vaccine candidates by addressing the quality of vaccine-induced T cell responses in terms of their ability to recognize diverse circulating HIV-1 strains and suppress viral escapes. Our protocol also has a potential for being applied to screening antigens to optimize vaccines for other pathogenic infections and cancer where the breadth of T cell responses is pivotal for the outcome of the diseases.
The structure of the peptide-MHC complex is important for the breadth of T cells.
We perform molecular dynamics simulations for the peptide-MHC complex.
We probe conformational transitions, achieving high prediction accuracy.
We report three novel peptide-MHC structures showing conformational differences.
Acknowledgments
We thank T. Allen and H. Stern for stimulating discussions. We thank G. Schatz for reviewing this manuscript. This work was supported by NIH grants R01 AI083115, R01 AI095066, and P30 AI078498. The X-ray crystal data have been deposited in the PDB with accession codes 3V5D, 3V5H, and 3V5K. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID and 22-BM beamlines at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altfeld M, Rosenberg ES, Shankarappa R, Mukherjee JS, Hecht FM, Eldridge RL, Addo MM, Poon SH, Phillips MN, Robbins GK, Sax PE, Boswell S, Kahn JO, Brander C, Goulder PJ, Levy JA, Mullins JI, Walker BD. Cellular immune responses and viral diversity in individuals treated during acute and early HIV-1 infection. J Exp Med. 2001;193:169–80. doi: 10.1084/jem.193.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Hummer G. Reaction coordinates and rates from transition paths. P Natl Acad Sci USA. 2005;102:6732–6737. doi: 10.1073/pnas.0408098102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbulevych OY, Baxter TK, Yu Z, Restifo NP, Baker BM. Increased immunogenicity of an anchor-modified tumor-associated antigen is due to the enhanced stability of the peptide/MHC complex: implications for vaccine design. J Immunol. 2005;174:4812–20. doi: 10.4049/jimmunol.174.8.4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–5. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Bui HH, Schiewe AJ, von Grafenstein H, Haworth IS. Structural prediction of peptides binding to MHC class I molecules. Proteins. 2006;63:43–52. doi: 10.1002/prot.20870. [DOI] [PubMed] [Google Scholar]
- Buslepp J, Zhao R, Donnini D, Loftus D, Saad M, Appella E, Collins EJ. T cell activity correlates with oligomeric peptide-major histocompatibility complex binding on T cell surface. J Biol Chem. 2001;276:47320–8. doi: 10.1074/jbc.M109231200. [DOI] [PubMed] [Google Scholar]
- Chivian D, Baker D. Homology modeling using parametric alignment ensemble generation with consensus and energy-based model selection. Nucleic Acids Res. 2006;34:e112. doi: 10.1093/nar/gkl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins EJ, Garboczi DN, Wiley DC. Three-dimensional structure of a peptide extending from one end of a class I MHC binding site. Nature. 1994;371:626–9. doi: 10.1038/371626a0. [DOI] [PubMed] [Google Scholar]
- Currier JR, Robb ML, Michael NL, Marovich MA. Defining epitope coverage requirements for T cell-based HIV vaccines: theoretical considerations and practical applications. Journal of translational medicine. 2011;9:212. doi: 10.1186/1479-5876-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellago C, Bolhuis PG, Csajka FS, Chandler D. Transition path sampling and the calculation of rate constants. Journal of Chemical Physics. 1998;108:1964–1977. [Google Scholar]
- Desmet J, Wilson IA, Joniau M, De Maeyer M, Lasters I. Computation of the binding of fully flexible peptides to proteins with flexible side chains. FASEB J. 1997;11:164–72. doi: 10.1096/fasebj.11.2.9039959. [DOI] [PubMed] [Google Scholar]
- Douek DC, Betts MR, Brenchley JM, Hill BJ, Ambrozak DR, Ngai KL, Karandikar NJ, Casazza JP, Koup RA. A novel approach to the analysis of specificity, clonality, and frequency of HIV-specific T cell responses reveals a potential mechanism for control of viral escape. J Immunol. 2002;168:3099–104. doi: 10.4049/jimmunol.168.6.3099. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg T, Cerottini JC, Michielin O. Structural prediction of peptides bound to MHC class I. J Mol Biol. 2006;356:521–46. doi: 10.1016/j.jmb.2005.11.059. [DOI] [PubMed] [Google Scholar]
- Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–6. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- Floudas CA. Global optimization in design and control of chemical process systems. Dynamics & Control of Process Systems 1998. 1999;1 and 2:169–178. [Google Scholar]
- Frahm N, Kiepiela P, Adams S, Linde CH, Hewitt HS, Sango K, Feeney ME, Addo MM, Lichterfeld M, Lahaie MP, Pae E, Wurcel AG, Roach T, St John MA, Altfeld M, Marincola FM, Moore C, Mallal S, Carrington M, Heckerman D, Allen TM, Mullins JI, Korber BT, Goulder PJ, Walker BD, Brander C. Control of human immunodeficiency virus replication by cytotoxic T lymphocytes targeting subdominant epitopes. Nat Immunol. 2006;7:173–8. doi: 10.1038/ni1281. [DOI] [PubMed] [Google Scholar]
- Frahm N, Korber BT, Adams CM, Szinger JJ, Draenert R, Addo MM, Feeney ME, Yusim K, Sango K, Brown NV, SenGupta D, Piechocka-Trocha A, Simonis T, Marincola FM, Wurcel AG, Stone DR, Russell CJ, Adolf P, Cohen D, Roach T, StJohn A, Khatri A, Davis K, Mullins J, Goulder PJ, Walker BD, Brander C. Consistent cytotoxic-T-lymphocyte targeting of immunodominant regions in human immunodeficiency virus across multiple ethnicities. J Virol. 2004;78:2187–200. doi: 10.1128/JVI.78.5.2187-2200.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino PL, Harrison CB, Liu Y, Schulten K. Challenges in protein folding simulations: Timescale, representation, and analysis. Nature physics. 2010;6:751–758. doi: 10.1038/nphys1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel D, Smit B. Understanding molecular simulation : from algorithms to applications. 2. Academic Press; San Diego: 2002. [Google Scholar]
- Gaschen B, Taylor J, Yusim K, Foley B, Gao F, Lang D, Novitsky V, Haynes B, Hahn BH, Bhattacharya T, Korber B. Diversity considerations in HIV-1 vaccine selection. Science. 2002;296:2354–60. doi: 10.1126/science.1070441. [DOI] [PubMed] [Google Scholar]
- Geldmacher C, Currier JR, Herrmann E, Haule A, Kuta E, McCutchan F, Njovu L, Geis S, Hoffmann O, Maboko L, Williamson C, Birx D, Meyerhans A, Cox J, Hoelscher M. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J Virol. 2007;81:2440–8. doi: 10.1128/JVI.01847-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honeyborne I, Prendergast A, Pereyra F, Leslie A, Crawford H, Payne R, Reddy S, Bishop K, Moodley E, Nair K, van der Stok M, McCarthy N, Rousseau CM, Addo M, Mullins JI, Brander C, Kiepiela P, Walker BD, Goulder PJ. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific CD8+ T-cell epitopes. J Virol. 2007;81:3667–72. doi: 10.1128/JVI.02689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarzynski C. Nonequilibrium equality for free energy differences. Physical Review Letters. 1997;78:2690–2693. [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood JG. Statistical mechanics of fluid mixtures. Journal of Chemical Physics. 1935;3:300–313. [Google Scholar]
- Kish-Catalone TM, Lu W, Gallo RC, DeVico AL. Preclinical evaluation of synthetic -2 RANTES as a candidate vaginal microbicide to target CCR5. Antimicrob Agents Chemother. 2006;50:1497–509. doi: 10.1128/AAC.50.4.1497-1509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhns JJ, Batalia MA, Yan S, Collins EJ. Poor binding of a HER-2/neu epitope (GP2) to HLA-A2.1 is due to a lack of interactions with the center of the peptide. J Biol Chem. 1999;274:36422–7. doi: 10.1074/jbc.274.51.36422. [DOI] [PubMed] [Google Scholar]
- La Gruta NL, Thomas PG, Webb AI, Dunstone MA, Cukalac T, Doherty PC, Purcell AW, Rossjohn J, Turner SJ. Epitope-specific TCRbeta repertoire diversity imparts no functional advantage on the CD8+ T cell response to cognate viral peptides. Proc Natl Acad Sci U S A. 2008;105:2034–9. doi: 10.1073/pnas.0711682102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazmin VS, Perrakis A, Wilson KS. The ARP/WARP suite for automoated construction and refinement of protein models. Kluwer Academic Publishers; Dordrecht: 2001. [Google Scholar]
- Leach AR. Ligand docking to proteins with discrete side-chain flexibility. J Mol Biol. 1994;235:345–56. doi: 10.1016/s0022-2836(05)80038-5. [DOI] [PubMed] [Google Scholar]
- Lee JK, Stewart-Jones G, Dong T, Harlos K, Di Gleria K, Dorrell L, Douek DC, van der Merwe PA, Jones EY, McMichael AJ. T cell cross-reactivity and conformational changes during TCR engagement. J Exp Med. 2004;200:1455–66. doi: 10.1084/jem.20041251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinthal . In: Mossbauer Spectroscopy in Biological Systems, Proceedings of a meeting held at Allerson house, Monticello, Illinois. Debrunner P, Tsibris JCM, Munck E, editors. University of Illinois Press; Urbana, Illinois: 1969. pp. 22–24. [Google Scholar]
- Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How fast-folding proteins fold. Science. 2011;334:517–20. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- Liu J, Wu P, Gao F, Qi J, Kawana-Tachikawa A, Xie J, Vavricka CJ, Iwamoto A, Li T, Gao GF. Novel immunodominant peptide presentation strategy: a featured HLA-A*2402-restricted cytotoxic T-lymphocyte epitope stabilized by intrachain hydrogen bonds from severe acute respiratory syndrome coronavirus nucleocapsid protein. J Virol. 2010;84:11849–57. doi: 10.1128/JVI.01464-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden DR, Garboczi DN, Wiley DC. The antigenic identity of peptide-MHC complexes: a comparison of the conformations of five viral peptides presented by HLA-A2. Cell. 1993;75:693–708. doi: 10.1016/0092-8674(93)90490-h. [DOI] [PubMed] [Google Scholar]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Martinez-Hackert E, Anikeeva N, Kalams SA, Walker BD, Hendrickson WA, Sykulev Y. Structural basis for degenerate recognition of natural HIV peptide variants by cytotoxic lymphocytes. J Biol Chem. 2006;281:20205–12. doi: 10.1074/jbc.M601934200. [DOI] [PubMed] [Google Scholar]
- Matsumura M, Fremont DH, Peterson PA, Wilson IA. Emerging principles for the recognition of peptide antigens by MHC class I molecules. Science. 1992;257:927–34. doi: 10.1126/science.1323878. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRobb FM, Capuano B, Crosby IT, Chalmers DK, Yuriev E. Homology modeling and docking evaluation of aminergic G protein-coupled receptors. J Chem Inf Model. 2010;50:626–37. doi: 10.1021/ci900444q. [DOI] [PubMed] [Google Scholar]
- Meijers R, Lai CC, Yang Y, Liu JH, Zhong W, Wang JH, Reinherz EL. Crystal structures of murine MHC Class I H-2 D(b) and K(b) molecules in complex with CTL epitopes from influenza A virus: implications for TCR repertoire selection and immunodominance. J Mol Biol. 2005;345:1099–110. doi: 10.1016/j.jmb.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Meyer-Olson D, Shoukry NH, Brady KW, Kim H, Olson DP, Hartman K, Shintani AK, Walker CM, Kalams SA. Limited T cell receptor diversity of HCV-specific T cell responses is associated with CTL escape. J Exp Med. 2004;200:307–19. doi: 10.1084/jem.20040638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Park S, Khalili-Araghi F, Tajkhorshid E, Schulten K. Free energy calculation from steered molecular dynamics simulations using Jarzynski’s equality. Journal of Chemical Physics. 2003;119:3559–3566. [Google Scholar]
- Piana S, Lindorff-Larsen K, Shaw DE. Protein folding kinetics and thermodynamics from atomistic simulation. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1201811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DA, West SM, Betts MR, Ruff LE, Brenchley JM, Ambrozak DR, Edghill-Smith Y, Kuroda MJ, Bogdan D, Kunstman K, Letvin NL, Franchini G, Wolinsky SM, Koup RA, Douek DC. T cell receptor recognition motifs govern immune escape patterns in acute SIV infection. Immunity. 2004;21:793–803. doi: 10.1016/j.immuni.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Probst-Kepper M, Hecht HJ, Herrmann H, Janke V, Ocklenburg F, Klempnauer J, van den Eynde BJ, Weiss S. Conformational restraints and flexibility of 14-meric peptides in complex with HLA-B*3501. J Immunol. 2004;173:5610–6. doi: 10.4049/jimmunol.173.9.5610. [DOI] [PubMed] [Google Scholar]
- Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, Kim D, Kellogg E, DiMaio F, Lange O, Kinch L, Sheffler W, Kim BH, Das R, Grishin NV, Baker D. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins. 2009;77(Suppl 9):89–99. doi: 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland M, Heckerman D, Deng W, Rousseau CM, Coovadia H, Bishop K, Goulder PJ, Walker BD, Brander C, Mullins JI. Broad and Gag-biased HIV-1 epitope repertoires are associated with lower viral loads. PLoS One. 2008;3:e1424. doi: 10.1371/journal.pone.0001424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Shao JY, Tanner SW, Thompson N, Cheatham TE. Clustering molecular dynamics trajectories: Characterizing the performance of different clustering algorithms. Journal of Chemical Theory and Computation. 2007;3:2312–2334. doi: 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- Sharma AK, Kuhns JJ, Yan S, Friedline RH, Long B, Tisch R, Collins EJ. Class I major histocompatibility complex anchor substitutions alter the conformation of T cell receptor contacts. J Biol Chem. 2001;276:21443–9. doi: 10.1074/jbc.M010791200. [DOI] [PubMed] [Google Scholar]
- Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, Wriggers W. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330:341–6. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- Stewart-Jones GB, McMichael AJ, Bell JI, Stuart DI, Jones EY. A structural basis for immunodominant human T cell receptor recognition. Nat Immunol. 2003;4:657–63. doi: 10.1038/ni942. [DOI] [PubMed] [Google Scholar]
- Tangri S, Ishioka GY, Huang X, Sidney J, Southwood S, Fikes J, Sette A. Structural features of peptide analogs of human histocompatibility leukocyte antigen class I epitopes that are more potent and immunogenic than wild-type peptide. J Exp Med. 2001;194:833–46. doi: 10.1084/jem.194.6.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrie GM, Valleau JP. Monte-Carlo Free-Energy Estimates Using Non-Boltzmann Sampling - Application to Subcritical Lennard-Jones Fluid. Chem Phys Lett. 1974;28:578–581. [Google Scholar]
- Tsui V, Case DA. Molecular Dynamics Simulations of Nucleic Acids with a Generalized Born Solvation Model. J Am Chem Soc. 2000;122:2489–2498. [Google Scholar]
- Turner SJ, Kedzierska K, Komodromou H, La Gruta NL, Dunstone MA, Webb AI, Webby R, Walden H, Xie W, McCluskey J, Purcell AW, Rossjohn J, Doherty PC. Lack of prominent peptide-major histocompatibility complex features limits repertoire diversity in virus-specific CD8+ T cell populations. Nat Immunol. 2005;6:382–9. doi: 10.1038/ni1175. [DOI] [PubMed] [Google Scholar]
- Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–34. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- Webb AI, Dunstone MA, Chen W, Aguilar MI, Chen Q, Jackson H, Chang L, Kjer-Nielsen L, Beddoe T, McCluskey J, Rossjohn J, Purcell AW. Functional and structural characteristics of NY-ESO-1-related HLA A2-restricted epitopes and the design of a novel immunogenic analogue. J Biol Chem. 2004;279:23438–46. doi: 10.1074/jbc.M314066200. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 67:235–42. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J, Medina-Franco JL. Homology modeling, docking and structure-based pharmacophore of inhibitors of DNA methyltransferase. J Comput Aided Mol Des. 2011;25:555–67. doi: 10.1007/s10822-011-9441-1. [DOI] [PubMed] [Google Scholar]