

Abstract
Purpose
Glioblastoma multiforme (GBM) is the most lethal form of brain cancer with a median survival of only 12–15 months. Current standard treatment consists of surgery followed by chemoradiation. The poor survival of GBM patients is due to aggressive tumor invasiveness, an inability to remove all tumor tissue, and an innate tumor chemo- and radioresistance. ATM, ataxia telangiectasia (A-T) mutated, is an excellent target for radiosensitizing GBM because of its critical role in regulating the DNA damage response and p53, among other cellular processes. As a first step toward this goal, we recently showed that the novel ATM kinase inhibitor KU-60019 reduced migration, invasion, growth, and potently radiosensitized human glioma cells in vitro.
Experimental Design
Using orthotopic xenograft models of GBM, we now show that KU-60019 is also an effective radiosensitizer in vivo. Human glioma cells expressing reporter genes for monitoring tumor growth and dispersal were grown intra-cranially, and KU-60019 was administered intra-tumorally by convection-enhanced delivery or osmotic pump.
Results
Our results demonstrate that the combined effect of KU-60019 and radiation significantly increased survival of mice 2–3 fold over controls. Importantly, we show that glioma with mutant p53 is much more sensitive to KU-60019 radiosensitization than genetically matched wild-type glioma.
Conclusions
Taken together, our results suggest that an ATM kinase inhibitor may be an effective radiosensitizer and adjuvant therapy for patients with mutant p53 brain cancers.
Introduction
Each year in the United States about 7 out of 100,000 people are diagnosed with high-grade glioma, classified by the World Health Organization (WHO) as Grade III and IV gliomas. Grade IV is also referred to as glioblastoma multiforme (GBM). Hallmark characteristics of GBM include uncontrolled cell proliferation, diffuse infiltration, and resistance to apoptosis (1, 2). These characteristics, at least in part, account for GBM's poor prognosis and resistance toward radio- and chemotherapy, and a median survival of just 12–15 months (3). Primary GBM (type 2) arises de novo with no prior symptoms or evidence of lower grade tumor and occurs most frequently in individual 60 years or older. Secondary GBM (type 1) occurs in younger patients from lower grade brain tumors. Type 2 constitutes the majority (>90%) of all GBMs whereas the remaining are type 1. It has been suggested that genetic mutations and amplifications affecting signaling pathways may account, in part, for GBM's aggressive nature (4, 5). GBMs are also classified into four different subgroups based on gene expression profiling; 1) classical, 2) mesenchymal, 3) neural, and 4) pro-neural (6, 7). Type 2 GBM is mostly found in the classical subgroup with EGFR amplifications and PTEN deletions, as well as some with mutations in p53 (8, 9). On the other hand, type 1 is usually found in the pro-neural subgroup with frequent mutations in PDGFR, IDH1, and p53 (6). The frequency of p53 mutations in type 1 GBM is 65% or greater whereas classical GBM harbors p53 mutations 30% of the time (10).
Mutation of the ATM (ataxia telangiectasia (A-T) mutated) gene is the underlying cause of the rare autosomal recessive disease A-T with an associated extreme radiosensitivity (11). In addition to ATR (A-T and Rad3-related kinase) and DNA-PK (DNA-dependent protein kinase), ATM plays a major role in regulating the DNA damage response (DDR), in particular after ionizing radiation (12). Importantly, radiation activates DNA damage checkpoints and increase DNA repair capacity of cancer stem cells leading to glioma radioresistance and tumor recurrence (13). Therefore, inhibitors of the DDR represent very attractive therapeutic agents worthy of exploration as potential adjuvants to standard GBM therapy (4, 14, 15).
Currently, standard treatment of GBM is surgical resection followed by temozolomide and radiation (16). This therapy results in a median survival of just over one year; thus, it is critical that more effective treatments are developed and explored (17). The last few years have seen the advancement of small molecule inhibitors that disrupt ATM function and sensitize tumor cells to radiation and chemotherapeutic agents (14). One of the first specific ATM inhibitors developed was KU-55933, which binds competitively to the ATP binding pocket of the ATM kinase (18). Another ATM inhibitor, CP466722, was also described recently (19). KU-60019 is an improved, second-generation analogue of KU-55933 >30-fold more effective at blocking radiation-induced phosphorylation of key ATM targets and possesses an impressive ability to radiosensitize human glioma cells in vitro (20). In addition, we also demonstrated that KU-60019 inhibited glioma cell migration and invasion (20), perhaps providing a second level of tumor growth control in between radiation fractions (21). However, the ability of KU-60019, or any other ATM inhibitor to radiosensitize tumors in vivo, has not yet been reported.
We demonstrate herein that orthotopic gliomas are radiosensitized with the ATM kinase-specific inhibitor, KU-60019, resulting in significant increases in the survival of mice. Importantly, p53 mutant glioma is much more sensitive to the combination of KU-60019 and radiation resulting in impressive extended survival, and in some cases, the apparent curing of treated mice.
Materials and Methods
Reagents
Antibodies used in both western blotting and/or immunohistochemistry were anti-p(S1981)-ATM (Millipore); anti-ATM (GeneTex); anti-Ki67 (Novus Biologicals); 53BP1 (BD Biosciences); p53 (Cell Signalling); GAPDH (Millipore); mouse anti-γ-H2AX (Millipore); anti-p-(S824)-KAP1 (Abcam); Alexa Fluor 594 goat anti-mouse IgG, and Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen). Matrigel (BD Biosciences) was used for tumor implantation. KU-60019 was obtained from AstraZeneca.
Cells, cell culture, and treatments
Human glioblastoma U1242/luc-GFP cells have been described previously (20). U87/luc-DsRed and U87/luc-DsRed-p53(281G) cells were generated by infection of U87 cells with Fluc-DsRed2 lentivirus (22) followed by cell sorting, and subsequent infection with LNCX-p53 (281G) retrovirus and selection with G418. U1242/luc-GFP-p53(281G) cells were generated the same way. Cell characterization included the ability to form intracranial tumors in athymic mice, and quantitative reverse transcriptase-PCR expression profiling (U1242). Cells were not tested and authenticated by an external service provider. Pooled populations of cells were used in all instances. Irradiations were performed using an MDS Nordion Gammacell 40 (ON, Canada) research irradiator with a Cs-137 source delivering at a dose rate of 1.05 Gy/min.
For the continuous KU-60019 exposure growth assay, cells were counted on a hemocytometer and seeded in a 12-well plate. After cells were allowed to grow for 48 hr, culture medium was removed, and medium containing KU-60019 (3 μM) or DMSO vehicle was added to quadruplicate wells and returned to the incubator. Seven days after initial exposure to drug, each well was trypsinized and counted on a Beckman Coulter counter, and total number of cells for each well was calculated. Counts from quadruplicate wells were averaged, and plotted.
Clonogenic radiosurvival experiments were carried out as described (18, 20, 21). Briefly, U87 and U1242 cells and derivatives were diluted, seeded on 10-cm or 6-cm dishes, and after the cells attached, treated with KU-60019 (3 μM) or not in the medium for 1 hr prior to irradiation. Cells were incubated overnight, and the medium changed to non-drug containing medium 16 hr post-irradiation. Cells were incubated a further 14 days, stained with crystal violet, and colonies consisting of ≥50 cells were counted by eye. Radiosensitization by KU-60019 was determined by fitting radiation survival curves using the linear quadratic equation, and dose enhancement ratios (DERs) were determined by dividing the D37 dose of control cells by the D37 dose of treated cells. A DER greater than 1 indicates radiosensitization.
Luciferase assay were carried out using a PerkinElmer Precisely (2103 Envision™ Multilabel plate reader) and luciferase kit from Perkin Elmer and used as recommended by the manufacturer. For the radiosurvival assay, U87/luc-DsRed+/−p53-281G cells were serially diluted in 96-well microplates and irradiated with the indicated doses. Three days later luciferase assay was carried out to determine viable cell numbers. The mouse glioma cells were derived from GBM of “Mut6” (hGFAP-Cre; p53 f/−; NF1 f/+; PTEN f/+) mice (23). Tumors were harvested and enriched for stem-like cells by growing them as neurospheres in serum free media supplemented with EGF and bFGF (24). Cells were plated in 96-well plates and treated with KU-60019 at the indicated concentrations and irradiated 3 hrs later. After 6 days, cell viability was determined by CellTiter-Glo (Promega). Bioluminescence imaging (BLI) was performed on a Caliper-IVIS-200 after s.c. injection of D-Luciferin (Gold Biotechnology, St. Louis, MO) at 30 mg/mL in PBS.
Flow cytometry
Flow cytometry was carried out as described previously (25). Cells were fixed in 100% methanol and permeabilized in 1% Triton X-100/casein. Cell cycle distribution was analyzed by 7-amino-actinomycin D (7-AAD) and propidium iodide staining (5 mg/mL, 0.1% Triton X-100/PBS). BrdU flow cytometry was carried out as recommended by the manufacturer (BD Biosciences) as described (26). Briefly, U87 and U87-281G cells were irradiated with 10 Gy or left untreated. After 16 hr BrdU was added to the medium (10 μM final), and 5 hr later cells isolated for further processing. Flow cytometry was performed on a BD Biosciences FACS Canto flow cytometer at the VCU Flow Cytometry Core Facility. Data was analyzed using the FACSDiva software.
Imaging and microscopy
Confocal fluorescence microscopy was performed as previously described (25). Cells were grown on Lab-Tek (Naperville, IL) glass slides coated with Matrigel. After treatment, cells were fixed with 3% paraformaldehyde, permeabilized with 0.5% Triton-X 100 in phosphate-buffered saline (PBS) and blocked with casein/0.1% goat serum prior to exposure to primary antibodies followed by secondary antibodies Alexa 488 goat anti-rabbit or goat anti-mouse 546 Fab fragment (Invitrogen) at 1:500 dilution, and nuclei counterstained with DAPI (1 mg/ml). Cells were imaged and analyzed using a Zeiss LSM 510 Meta imaging system in the VCU Microscopy Facility.
Following treatment with irradiation and KU-60019, animals were transcardially perfused with 3% paraformaldehyde followed by post-fixing of brains and treatment with 30% sucrose until no longer buoyant. Brains were cut coronally through the injection site, and the hindbrain embedded in Tissue-Tek OCT Compound (Sakura, Torrance, CA), frozen on dry ice, and stored at −20°C. Cryosectioning was performed on a Leica CM1850 cryostat with 6-μm thickness and slides were stored frozen until staining. Sections were rinsed in PBS, followed by pretreatment in sodium citrate antigen unmasking solution (0.01 M sodium citrate; 0.05% Tween-20; pH 6.0) and boiled followed by aldehyde reduction with NaBH4. Sections were permeabilized with 1% Triton-X-100/PBS and blocked with 10% normal goat serum in Triton-X-100/PBS prior to the exposure to primary antibodies followed by secondary antibodies [Alexa Fluor-594 goat anti-mouse IgG or Alexa Fluor-488 goat anti-rabbit IgG (Invitrogen)] and DAPI. H&E staining was performed on frozen sections fixed in 3% formaldehyde and stained with modified Mayer's Hematoxylin and Eosin Y. After dehydration with ethanol, sections were cleared with Histoclear (National Diagnostics #HS-200), and coverslips mounted on sections with Permount (Fisher #SP15-100). Sections were imaged and analyzed using a Zeiss LSM 510 Meta imaging system at 63× magnification in the Massey Cancer Center Flow Cytometry and Imaging Facility or using an Applied Imaging Ariol SL-50 system (Applied Imaging Corp., San Jose, CA).
To ensure tumor take and equal tumor size distribution throughout the treatment groups all mice injected with U1242/luc-GFP or U87/luc-DsRed cells were imaged using BLI typically at 5, 16 and 32 days post-cell injection using a Caliper IVIS-200. If necessary, later images were also acquired. Fixed and unfixed brains were imaged on a Zeiss SV11 stereomicroscope (Zeiss, Jena, Germany) with a Hamamatsu digital CCD camera C4742-95-12NRB (Hamamatsu, Bridgewater, NJ, USA) and AttoArc2/HB 100 Arc variable intensity microscopy illuminator system (Zeiss) using excitation/emission filters (GFP; 470/525, DsRed; 546/575), and a Caliper IVIS-200 system using GFP filters. Images were captured using AxioVision 3.1 or Living Image 4.0 software, and processed with Adobe® PhotoShop® 5.0.
Animals and animal procedures
All procedures were carried out in accordance with protocol AM10197 approved by the Virginia Commonwealth University IACUC. All mice were subjected to a light/dark cycle of 12 hr with water and food available ad libitum. Athymic nude female mice (5–6 weeks old) were purchased from NCI-Frederick, Gaithersburg, MD. Mice weighing between 15–25 g were anesthetized i.p. with a mixture of ketamine and xylazine (10 mg/kg; 1 mg/kg) in saline, and placed on a heating pad. A 1-cm sagital incision was made, and the skull exposed. A 1-mm burr hole was made 2-mm left lateral and 1-mm posterior to the bregma. A small pocket was created in the brain by inserting a 26-gauge needle with a beveled edge to a depth of 3-mm. This location was found optimal for the insertion of the Brain Infusion Kit 3 (ALZET, Cupertino, CA) cannula connected to an osmotic pump and was therefore used throughout these experiments. U1242/luc-GFP, U87/luc-DsRed or derivatives thereof were trypsinized, washed, and re-suspended in PBS and Matrigel (BD Biosciences, San Jose, CA), and loaded into a Hamilton syringe fitted with a 26-gauge blunt tip needle. The needle was inserted to a depth of 3-mm and 1 μl of cells was injected over a 5 minute period. After injection, the needle was removed over a 5 minute period.
Osmotic pumps (ALZET M2002, M1002, M1007D), containing KU-60019, were implanted 7 days post-cell injection to allow continuous drug delivery to the tumor site for up to 19 days (5.28 μl/day; 100 μl total volume) followed by radiation. An incision was made to expose the burr hole previously made for cell injection and cleaned to remove all bone wax. The pump was inserted and the cannula tip was positioned into the burr hole and glued into place. All radiations were carried out using a MDS Nordion Gammacell 40 research irradiator with a 137-Cs source delivering a dose rate of 1.05 Gy/min to the head only.
Convection enhanced delivery (CED) of KU-60019 was done on day 6, 9 and 12 post-cell injection. A guide screw with a stylet (PlasticsOne, Inc, Roanoke, VA) was inserted into the burr hole to facilitate cell placement and subsequent drug infusions and the incision closed using vet-glue (GLUture Topical Tissue Adhesive, Abbott Laboratories, Chicago, IL, USA). Next, 12.5 μl of 250 μM of KU-60019 in PBS was directly infused into the tumor via a cannula inserted into the guide-screw by CED using a BeeHive pump (BeeHive™, Bioanalytical Systems, West Lafayette, Indiana) set at rate of 0.4 μl/minute. Vehicle alone (PBS) was infused into mice without KU-60019 treatment. Immediately following surgery, mice either received 3 Gy to the head or left untreated. For subsequent treatments, the skin was opened and the stylet removed prior to repeating the infusion followed by irradiation.
Statistics
Mouse survival studies were modeled using Kaplan-Meier curves. Statistical analyses were performed using log-rank test and mean survival in days was reported with standard deviation (SD) for all treatment groups. Clonogenic radiosurvival experiments were analyzed by two-way analysis of variance with radiation survival fraction as the response to cell type and radiation dose along with interaction. Least-squared means of the treatment difference were obtained from the model and tested with ANOVA F-test. All statistical analyses were done in SAS v9.2.
Results
ATM kinase inhibitor KU-60019 radiosensitizes human glioma and mouse glioma stem cells
To confirm and validate the target for KU-60019 as the ATM kinase, U1242 and U87 cells were exposed to KU-60019 and radiation and extracts prepared for western blotting with p-(S1981) ATM antibody. S1981 is a well-established radiation-induced auto-phosphorylation site (27). Both cell lines increased p-(S1981) ATM levels after exposure to radiation and KU-60019 suppressed this increase demonstrating that ATM kinase was inhibited (Fig. 1A). Furthermore, U1242 and U87 cells were similarly radiosensitized in vitro in the presence of low levels of KU-60019 (Fig. 1B). U1242 cells show high DNA repair capacity (curved slope with low α/β ratio), whereas U87 cells have a straighter curve and thus lower repair capacity. Nevertheless, KU-60019 was able to radiosensitize both cell lines to a similar extent. When p53 null glioma cells isolated from a spontaneous mouse GBM model were grown as neurospheres and tested they were also found to be radiosensitized by KU-60019. Altogether, KU-60019 is an effective and specific radiosensitizer of established human glioma cells and primary mouse glioma stem cells in vitro in agreement with earlier studies (20, 21, 28, 29).
Figure 1.
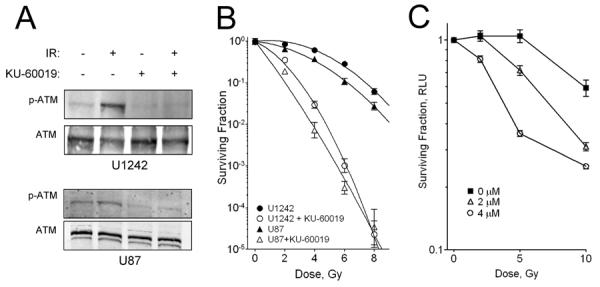
KU-60019 inhibits the ATM kinase and radiosensitizes human glioma cells in vitro. A, Western blotting. U1242 and U87 cells were exposed to 3 μM KU-60019 (−0.5 hr) or left untreated and then irradiated with 5 Gy or not and then processed at 0.5 hr for western blotting with anti-p(S1981)-ATM antibody and normalized to ATM protein with anti-ATM antibody. B, Survival assay. Human glioma U1242 and U87 cells were serially diluted, plated [plating efficiency: 0.20 (U87); 0.22 (U1242)], and exposed to KU-60019 (3 μM) or not and then irradiated with 2, 4, 6, or 8 Gy followed by radiosurvival colony-forming assay. Data points, surviving colonies plotted as fraction of control; error bars, SD; N ≥ 3. Where error bars are not seen they are obscured by symbols. At 8 Gy, KU-60019 radiosensitization of U87 and U1242 cells was highly significant (P < 0.0001). Dose-enhancement ratios - U1242: 3.2; U87: 3.0. Radiobiological parameters - U87 α/β ratio: 11.7 Gy; U87 + KU-60019: 34.8 Gy; U1242: 1.8 Gy; U1242 + KU-60019: 3.4 Gy. C. Survival assay of mouse glioma stem cells. Cells were grown as neurospheres, trypsinized, and seeded in quadruplicate in a microtiter plate followed by exposure to KU-60019 and radiation as described in the Materials and Methods. At 10 Gy, KU-60019 radiosensitization was highly significant (P < 0.0001).
Human U1242/luc-GFP glioma cells form invasive tumors in mouse brain and show radiation dose-dependent increases in radiation-induced repair foci
One characteristic phenotype of human glioma is its invasiveness, which is rarely seen when human xenografts established from tissue culture cell lines are propagated as orthotopic tumors in mice. The U1242 human glioma cell line is unusual in that it has a stable, invasive, and very aggressive phenotype when grown in nude mice (30, 31). We expressed the reporter genes luciferase and GFP, or DsRed, from lentivirus to follow tumor growth. While U1242 cells spread invasively in the brain with high mitotic index, similar to human disease, U87 cells form encapsulated tumors (Fig. 2, and Supplementary Fig. S1). We chose to test KU-60019 on U1242 tumors in our initial experiments because these cells recapitulate the pathology of the human disease to some extent. ATM phosphorylates many proteins in response to radiation and DNA damage (32). Among the best characterized targets are H2AX, KAP1, and 53BP1, all linked to processes associated with DNA double-strand break repair. When mice growing intracranial tumors were irradiated, we were able to document dose-dependent increases in γ-H2AX, p-(S824)-KAP1, and 53BP1 repair foci immediately following radiation (Fig. 3A–C), and in a time course (Fig. 3D). These results demonstrate that invasive human glioma U1242 cells show evidence of radiation-induced DDR, permitting the analysis of KU-60019-dependent ATM inhibition in mice.
Figure 2.

Human U1242/luc-GFP glioma cells form invasive and highly mitotic tumors in the brains of athymic mice. A, U1242 tumors are invasive. Brains were removed and imaged on a Zeiss SV11 stereomicroscope with a Hamamatsu digital CCD camera variable intensity microscopy illuminator system, and a Caliper IVIS-200 system using GFP filters, respectively. Subsequently, brains were embedded in OCT, sectioned in 6-μm sections, and stained with H & E. B, High mitotic index of intra-cranial U1242/luc-GFP tumors. Frozen sections were stained with anti-Ki67 antibody, followed by goat anti-mouse-Alexa 594, and counterstained with DAPI. GFP is the signal from the U1242/luc-GFP cells. The mitotic index (percentage Ki67+/GFP+ vs. Ki67−/GFP+ cells) was estimated at >50%.
Figure 3.

Human U1242/luc-GFP gliomas show dose-dependent increases in γ-H2AX, p-(S824)-KAP1, and 53BP1 foci formation after radiation. Athymic female mice growing U1242/luc-GFP intra-cranial tumors were exposed to 0, 5 or 10 Gy followed by perfusion with fixative immediately, 1, or 2 hr post-irradiation. Brains were post-fixed, embedded in OCT and sectioned on a cryostat in 6-μm sections. Sections were stained with anti-p-(S139)-H2AX (A and B), p-(S824)-KAP1 (C), and 53BP1 (D) antibodies, respectively, followed by goat anti-mouse-Alexa 594. Nuclei were counterstained with DAPI. Section (A) shows GFP+ U1242/luc-GFP tumor whereas (B – D) do not. Instead, the characteristic elongated shape of the U1242 cells is clearly seen after DAPI staining.
KU-60019 radiosensitizes orthotopic glioma xenografts and significantly extends the survival of mice
When KU-60019 is systemically administered either intra-peritoneally or orally, plasma levels only reach the low micromolar range, which is barely able to radiosensitize flank tumors (21). The blood-brain-barrier (BBB) and blood-tumor-barrier (BTB) represent major obstacles in the effective treatment of brain tumors (33). To reach meaningful drug concentrations of KU-60019 within the tumor, the BBB/BTB need to be bypassed or drugs administered locally. Both osmotic pumps, as well as clinically-utilized convection-enhanced delivery (CED), partially bypass the BBB/BTB and deliver drugs directly to the tumor to improve efficacy and reduce potential systemic toxicity (33, 34). We first explored the use of commercially available osmotic pumps to deliver KU-60019 at effective levels to the brain. Preliminary results with a dye acting as a surrogate for KU-60019 suggested that efficient spread occurred (Supplementary Fig. S2). When mice harboring U1242/luc-GFP tumors were treated with KU-60019 administered from an osmotic pump followed by 2 Gy of irradiation, we were able to demonstrate a complete elimination of tumor cells on day 20 by bioluminescence imaging (BLI) 7 days after radiation (Fig. 4A, top), a finding that was confirmed by GFP imaging of the extracted brain (Fig. 4A, bottom). This result suggested that osmotic pumps administer KU-60019 to sufficient levels to radiosensitize U1242 gliomas.
Figure 4.
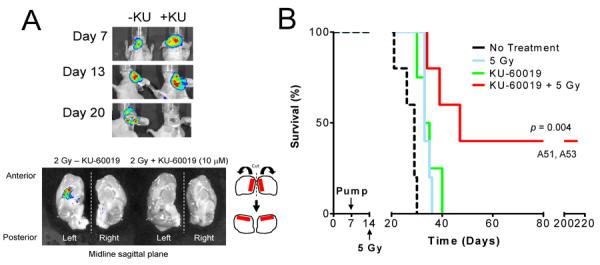
Radiosensitization of orthotopic U1242/luc-GFP tumors with KU-60019. Athymic female mice were implanted with U1242/luc-GFP cells. Osmotic pumps (ALZET M2002) were filled with KU-60019 or PBS, connected to a cannula (Brain infusion kit 3; ALZET), inserted in the skull at a depth of 3 mm at the site of cell injection, and the pump placed under the skin on the back of the mouse. The M2002 pump delivers the drug at a rate of 0.5 μl/hr over 14 days. A, Mice with tumors were imaged by BLI 7 days post-cell injection followed by pump implantation with KU-60019 (10 μM) (+KU) or not (-KU). Tumor growth was determined at 13 days by BLI followed by 2 Gy of cranial irradiation to both mice. Seven days later (day 20) mice were imaged by BLI, euthanized, and the brains removed for GFP imaging using Caliper IVIS-200. Tumor was evident in the brain receiving radiation alone, but not in the brain receiving KU-60019 and radiation. B, Radiosensitization of U1242/luc-GFP tumors with KU-60019 delivered by intra-tumoral osmotic pump. Cells were injected followed by implantation of osmotic pumps as in (A) except that pumps (ALZET M1007D) were inserted 7 days after cell implantation and irradiation (single dose of 5 Gy) was done 7 days (day 14) later. Tumor growth was monitored by BLI. Two mice (A51 and A53), in the KU-60019 and radiation group that survived for >200 days and were apparently healthy when euthanized. Mean survival: no treatment; 27 ± 4 days (N = 5), 5 Gy; 34 ± 1 days (N = 5), KU-60019; 35 ± 4 days (N = 4), and KU-60019 + 5 Gy; 109 ± 94 days (N = 5). A significant 75 day growth-delay between the KU-60019 and radiation and radiation alone groups was noted (P = 0.00402 vs. all treatments).
Encouraged by this marked result, we then designed an experiment to determine the effect of KU-60019 and radiation on the survival of mice. Mice with U1242/luc-GFP intracranial tumors were implanted with osmotic pumps loaded with KU-60019 7 days after cell implantation, followed by a single dose of radiation on day 13. Mice treated with KU-60019 and radiation survived for 109 days, significantly longer (P = 0.004 vs. all other treatments) than mice treated with KU-60019 alone (35 days), radiation alone (34 days), or after no treatment (27 days) (Fig. 4B). However, we noticed a considerable variation in the survival of the mice in the KU-60019 and radiation group. Some mice barely showed a survival benefit whereas others (e.g., A51 and A53) showed complete responses and survived for more than 200 days. Despite initially having tumors, at the time euthanasia these mice showed no signs of the disease (Supplementary Fig. S3). We suspected these differences in survival were a result of variable drug delivery throughout the brain because of insufficient ability of this type of pump to apply positive pressure. Therefore, we decided to administer KU-60019 in a manner used clinically, i.e., by CED, immediately prior to irradiation. We also decided to repeat this procedure three times in line with clinical practice and possibly achieve a better therapeutic response, while using a low dose of radiation to maximize detection of the KU-60019 effect.
To compensate for the relatively small volume that can be infused by CED over a reasonable time, the concentration of KU-60019 was increased to 250 μM. Using these conditions, drug distribution in the mouse brain was determined by LC-MS analysis and shown to be concentrated around the infusion site spreading 1–2 millimeters longitudinally (Supplementary Table S1). KU-60019 reached peak concentrations of ~5 μM, above those needed to inhibit ATM (21). Mice with U1242/luc-GFP intracranial tumors were infused with KU-60019 by CED on day 6, 9 and 12, immediately followed each time with 3 Gy of radiation (Fig. 5A). Mice treated with KU-60019 and radiation survived for 60 days, significantly longer (P = 0.00238 vs. all other treatments) than mice treated with KU-60019 alone (38 days), radiation alone (38 days), or after no treatment (35 days). Increased survival after KU-60019 and radiation was reflected in the reduction of tumor growth determined by BLI on day 5, 16, and 30 (Supplementary Fig. S4). To ensure that the levels of drug delivered into the brain were sufficient to block ATM kinase activity, we examined the ability of the drug to inhibit γ-H2AX foci formation (25, 35). We found that KU-60019 was able to inhibit foci formation in both tumor (elongated nuclei) as well as mouse brain (round nuclei with characteristic, large nucleoli) (Fig. 5B and C). A similar reduction was seen when p-(S824)-KAP1 foci was examined (Supplementary Fig. S5). These results suggest that CED is able to effectively administer KU-60019 and penetrate diseased brain to radiosensitize the tumor. In addition, normal brain showed a similar response to radiation and KU-60019 in what appeared to be a sub-population of cells, but presently, we do not know what these cells are or the impact on the brain. Nevertheless, a significant prolongation of survival was seen with KU-60019 and radiation at a radiation dose that produced negligible effects on survival when used as a single agent.
Figure 5.

KU-60019 delivered by CED radiosensitizes human orthotopic gliomas. A, KU-60019 radiosensitization. Human glioma U1242/luc-GFP cells were injected intra-cranially and tumor growth determined by BLI. Six days after cell injection, KU-60019 (250 μM) in 12.5 μl was infused by CED directly into the tumor followed immediately by 3 Gy of radiation to the head. This procedure was repeated again on day 9 and 12. Mean survival: no treatment; 35 ± 8 days (N = 5), 3 × 3 Gy; 38 ± 8 days (N = 7), 3 × KU-60019; 38 ± 8 days (N = 6), and 3 × (KU-60019 + 3 Gy); 60 ± 19 days (N = 6). The KU-60019 and radiation group survived significantly longer (P = 0.00238 vs. all treatments) with a growth delay of 22 days over radiation alone. Note that radiation alone did not increase survival compared to no treatment. (B and C) KU-60019 blocks repair foci formation in tumor and mouse brain. Human glioma U1242/luc-GFP cells were injected intra-cranially and allowed to form tumors as determined by BLI. At ~21 days the mice were infused with KU-60019 (250 μM) and immediately exposed to either 0 or 5 Gy of radiation followed by cardiac perfusion with fixative. Brains were post-fixed, imbedded in OCT and sectioned in 6-μm sections. Sections were stained with anti-p(S139)-H2AX antibody followed by goat anti-mouse-Alexa 594, counterstained with DAPI, and imaged by confocal microscopy at 63×. Insets show individual nuclei. B, KU-60019 inhibits DDR in tumor. Glioma cells (elongated nuclei) show γ-H2AX foci after irradiation, which is blocked by KU-60019. C, Radiation response in mouse brain is inhibited by KU-60019. Mouse brain cells (small, round nuclei with distinct, large nucleoli) forms γ-H2AX foci, which is also inhibited by KU-60019.
p53 mutant gliomas treated with KU-60019 are more radiosensitive than their isogenic, p53 wild-type counterparts
To explore the ability of KU-60019 to radiosensitize other human gliomas we turned to U87 cells, which have been used extensively in the past in similar studies. Furthermore, U87 cells express wild-type p53 making it possible to manipulate p53 status and demonstrate possible effects on radiosensitization and the response to KU-60019. To generate U87/luc-DsRed cells with mutant p53 we infected with a retrovirus expressing the p53-281G allele (36), and selected for G-418 resistance resulting in constitutive expression of the p53-281G protein in untreated cells (Fig. 6A). These cells were then characterized in response to KU-60019 and radiation. As expected, the U87-281G cells have an impaired G1/S checkpoint after radiation, are significantly more sensitive to KU-60019 and radiation alone (20, 21), and to KU-60019 and radiation combined relative to the parental U87 cells (Fig. 6B–E). Radiation followed by BrdU labeling showed that whereas entry into S was reduced by almost 50% (28.2% to 14.4% BrdU+ cells) in U87 cells, the levels in U87-281G cells were barely reduced (25.1% to 23.2% BrdU+) (Fig. 6B). Similar reductions were seen when cells were examined by fluorescence microscopy (Supplementary Fig. S6). Thus, the G1/S checkpoint is intact in U87 cells and impaired in U87-281G cells as expected (37). In addition, both U87 and U87-281G cells showed distinct G2/M arrest after radiation, which was more clearly seen with U87-281G cells because of the disrupted G1/S checkpoint.
Figure 6.

Genetically matched human glioma differing in p53 status show a significantly different response to KU-60019. U87/luc-DsRed (p53 wild type) cells were infected with a mouse retrovirus expressing the p53-281G allele. A, Western blotting for p53 shows the presence of p53-281G in U87-281G cells but not in the parental U87 cells. B, Cell cycle analysis (BrdU and 7-AAD flow cytometry) of untreated and irradiated (10 Gy) cells shows that the G1/S checkpoint is abrogated in U87-281G whereas parental cells have an intact checkpoint. Both cell populations have intact G2/M checkpoints. C, Growth and survival assay (trypan blue exclusion) after 7 days shows that U87-281G cells grow faster and are also more sensitive to KU-60019 (3 μM) than parental U87 cells. D, Survival assay (luciferase) shows that U87-281G cells are also more radiosensitive than the parental U87 cells (P = 0.0154 at 9 Gy). E, Survival assay (colony forming) shows that both U87 (P =0.0005) and U87-281G (P =0.0014) cells are significantly radiosensitized at 2 Gy by KU-60019. Furthermore, U87-281G cells shows a trend toward being more radiosensitive than parental U87 cells but this difference was not significant (P =0.166). Importantly, U87-281G cells were significantly more sensitive to KU-60019 and 2 Gy than U87 cells (P <0.0001). Plating efficiency: 0.19 (U87); 0.12 (U1242). F, U87 glioma with mutant p53-281G (top panel) is more sensitive to KU-60019 and radiation than parental p53 wild type U87 glioma (bottom panel). Human glioma U87/luc-DsRed (p53 wild type) and matched p53-281G U87/luc-DsRed cells were injected intra-cranially into athymic mice. Mice were treated as described in the legend to Fig. 5. Mice injected with p53-281G U87/luc-DsRed cells showed >160 days of tumor growth-delay after KU-60019 and radiation relative to radiation alone (P = 0.00011 vs. all treatments). All animals had evidence of tumors on day 5 (Supplementary Fig. S8). Mean survival: no treatment; 42 ± 3 day (N = 5), 3 × 3 Gy; 44 ± 6 days (N = 5); 3 × KU-60019; 51 ± 15 days (N = 6), and 3 × (KU-60019 + 3 Gy): >160 ± 49 days (N = 8). One mouse in the KU-60019 and radiation group developed an infection and was euthanized on day 181. Mice injected with the parental U87 cells only showed 16 days of growth-delay after KU-60019 and radiation relative to radiation alone (P = 0.0448). However, KU-60019 and radiation was not significantly different from KU-60019 alone. Mean survival: no treatment; 46 ± 6 days (N = 6), 3 × 3 Gy; 49 ± 10 days (N = 6); 3 × KU-60019; 70 ± 8 days (N = 5), 3 × (KU-60019 + 3 Gy); 65 ± 16 days N = 7).
Next, mice were implanted with either U87 or U87/p53-281G cells followed by treatment with KU-60019 and radiation to determine the effect on survival. Mice with U87 gliomas treated with KU-60019 delivered by CED on day 6, 9 and 12 followed by radiation survived for 65 days whereas mice treated with KU-60019 alone survived for 70 days, radiation alone for 49 days, and untreated mice for 46 days (Fig. 6F bottom). Although mice in the KU-60019 and radiation combination group survived significantly longer than radiation alone mice (P = 0.0448), there was no difference in survival between this group and the KU-60019 alone group. There was a statistical difference in survival between KU-60019 alone and the no treatment groups (P = 0.00019). Thus, U87 glioma is relatively refractory to KU-60019 radiosensitization. On the other hand, mice with U87-281G tumors (Fig. 6F top), treated with the KU-60019 and radiation combination survived >160 days, significantly longer (P = 0.00011 vs. all other treatments) than mice treated with KU-60019 alone (51 days), radiation alone (44 days), or after no treatment (42 days). Similar to the results with mice harboring parental U87 tumors, mice treated with KU-60019 alone showed a trend toward longer survival but this difference was not significant (P = 0.13). Thus, genetically matched p53 mutant U87 tumors dramatically responded to KU-60019 radiosensitization resulting in a subset of mice in this group apparently cured of disease. Additionally, there was a trend of increased survival of mice treated with KU-60019 alone in both the p53 wild-type and 281G mutant sets lending support to the idea that inhibiting ATM might reduce tumor dispersal in between radiation fractions (21).
Before we had verified the p53 status of the U1242 cells as having the R175H mutation, in agreement with previous reports (38, 39), we had already infected these cells with the retrovirus expressing the p53-281G allele and initiated a survival experiment. In support of the data obtained with the U87 p53-matched experiment, we noticed a significant improvement in the response to KU-60019 and radiation with U1242/p53-281G tumors compared to parental U1242 tumors (Supplementary Fig. S7). Mice with U1242/p53-281G tumors treated with KU-60019 followed by radiation, survived for 145 days, significantly longer (P = 0.0004) than mice treated with KU-60019 alone (66 days), radiation alone (58 days), or no treatment (52 days). Also, mice with U1242/p53-281G mutant tumors responded significantly better to treatment than mice with parental U1242 tumors across all treatments (P = 0.0004), corroborating the results with the U87 set that glioma with a p53 mutation is more sensitive to KU-60019 radiosensitization. The mechanism underlying the enhanced response as a result of manipulating p53 status in the U1242 cells is not known but currently under investigation. It is possible that 281G and 175H cooperate in some fashion to make these cells more sensitive to radiosensitization by KU-60019 (40), or, alternatively, 175H might gain some function when 281G is also expressed resulting in a more responsive phenotype.
Discussion
Inhibiting ATM for cancer therapy has been a long-sought goal for radiobiologists and oncologists alike (14). With the advent of highly specific inhibitors for the ATM kinase, this has recently become a reality (18–20). We have demonstrated herein that the ATM kinase inhibitor, KU-60019, was able to radiosensitize gliomas grown orthotopically in mice. Radiosensitization was most striking in U87 gliomas expressing mutant p53, where a large proportion of the mice appeared to have been cured of disease.
There are no known unique ATM phosphorylation sites (32). However, H2AX and KAP1 are considered more specific targets for the ATM kinase at early times after radiation, whereas the related DNA-PK and ATR kinases serve as complementary backups for ATM at later times (20, 41–43). We were able to validate ATM inhibition by KU-60019 in vivo by demonstrating reduced γ-H2AX, as well as p-(S824)-KAP1, foci formation immediately following irradiation in both the tumor and in mouse brain. These results suggest that KU-60019 is able to inhibit the ATM kinase in tissue regardless of being cancerous or not. Since increased survival correlated well with the ability of KU-60019 to suppress foci formation, this suggests that interfering with ATM signaling caused the reduction in tumor growth and prolonged the survival of mice.
We have shown in this study that glioma with mutant p53 is much more sensitive to KU-60019-dependent radiosensitization. This result was not unexpected since several recent studies demonstrated that human lymphomas with double mutations in p53 and ATM, or Chk2, were much more sensitive to chemotherapy than tumors with a single mutation (44–46). Several excellent reviews have discussed therapeutic strategies based on the DDR synthetic lethality principle (47, 48). Additionally, caffeine, a broad-spectrum PIKK inhibitor, was shown to radiosensitize p53 null but not matched p53 wild type cells in vitro (49, 50). The results from our study underscore these previous findings and extend this concept by demonstrating enhanced radiosensitization of p53 mutant glioma with an ATM kinase inhibitor in vivo. Importantly, tumors with normal p53 were found to be protected against chemotherapy (44). Thus, one would assume that normal tissues and cells would be protected as well. It was recently demonstrated that under conditions when the cell cycle kinase Cdk1 was inhibited or depleted, normal, non-transformed cells were protected against DNA damaging drugs while efficiently killing tumor cells (51). We are also encouraged by the result that p53 null mouse glioma stem cells were radiosensitized by KU-60019, a result in agreement with a previous study (29). Therefore, targeting the ATM kinase with a small molecule inhibitor in p53 mutant glioma would be very attractive since it is expected that the glioma would be sensitive to radiation whereas normal tissue would be protected or less affected.
Even though U1242 tumors harbor the p53 R175H allele, they were not as sensitive to KU-60019 and radiation as the U87/p53-281G tumors based on survival. We speculate that this is due to the ability of U1242 tumors to spread throughout the brain (see Supplementary Fig. S1), and perhaps evade the drug. On the other hand, U87 tumors are encapsulated and more contained and might thus provide a more accessible, physical target for KU-60019. Thus, it is likely that mice harboring U1242 tumors die from tumor dispersal throughout the parenchyma, whereas mice with U87 tumors die from increased intracranial pressure produced by the tumor. The differential response to KU-60019 and enhanced radiosensitization seen in vivo with p53 mutant gliomas is not reflected in vitro since both U87 (wild type) and U1242 (p53 mutant) are similarly radiosensitized (see Fig. 1). However, these two cell lines are derived from different individuals with many genetic differences in addition to p53 that would likely influence the response to KU-60019 in vitro. Thus, the p53 effect can be seen clearly in vitro when isogenic cells differing only in p53 status are compared as we did with the U87 cells (see Fig. 6C–E). We attribute differences in results between experiments using osmotic pump and CED a reflection of different experimental conditions and duration of drug exposure. Ongoing studies using panels of human and mouse gliomas and primary glioma stem cells with varying p53 status are exploring the use of higher doses of radiation to maximize survival, and identifying potential side effects of KU-60019 on normal brain. Whether KU-60019 radiosensitization seen at higher doses of radiation in vitro would also be observed in vivo, is currently not known. However, at higher radiation doses one would not be able to take full advantage of the radiosensitizer and spare normal tissue since higher radiation doses will likely damage the brain despite having a normal p53. Furthermore, whether treatment with KU-60019 alone provides a survival benefit is currently under investigation.
Local drug delivery increases the therapeutic effect and at the same time reduces general toxicity to treated subjects. Convection-enhanced delivery is becoming a more frequent experimental treatment option in the management of brain tumors but adds complexity and cost to the procedure. In addition, technical difficulties associated with cannula placement verification and the prevention of drug backflow are issues that still need to be resolved (33). While the effect of KU-60019 radiosensitization on tumors is clear, the effect on mouse brain is not known. This issue has not been a primary concern when patients survive for just a little more than a year, at best. We have not observed gross abnormalities on normal mouse brain after treatment with KU-60019 and radiation. Further studies on the response of normal tissue to KU-60019, and brain in particular, with and without radiation, are therefore needed since prolonging survival of subjects would likely expose any detrimental late effects on brain function and cognition due to treatment. However, It is important to point out that all chemo- and radiation therapy induce damage in normal tissues, a side effect of cancer therapy and fact that needs to be balanced against therapeutic gain and quality of life. Delivery of an ATM inhibitor locally by CED should in part alleviate such concern.
Altogether, our results suggest that ATM kinase inhibition may be an effective adjuvant therapy for patients with mutant p53 brain cancers. Since a third of all GBM cases have p53 mutations, this population of patients might be a particularly attractive target group.
Supplementary Material
Translational Relevance.
KU-60019 is a novel, second-generation, and highly specific ATM kinase inhibitor that blocks radiation-induced DNA damage response and potently radiosensitizes human glioma cells in vitro. In addition, KU-60019 alone inhibits glioma cell migration, invasion, and growth, suggesting that critical growth and pro-survival signaling pathways are also under control of ATM. In order to see whether KU-60019 would be an effective radiosensitizer in vivo, we utilized orthotopic human glioma xenograft models varying in p53 status and invasiveness. Our results demonstrate, for the first time, that an ATM inhibitor, KU-60019, and radiation significantly increase survival of mice 2–3 fold over controls. Importantly, we show that glioma with mutant p53 is much more sensitive to KU-60019 radiosensitization than genetically matched wild-type glioma. In conclusion, our results suggest that an ATM kinase inhibitor may be an effective radiosensitizer and adjuvant therapy for patients with mutant p53 brain cancers.
Acknowledgments
We thank Khalid Shah (Harvard University) for virus reagent and Michael Kastan (Duke University) for helpful discussion.
Grant Support This work was supported by National Institutes of Health Grants R01NS064593 and R21CA156995 (to K.V.). L.B.-T. was supported by T32CA113277. S.E.G was supported by American Brain Tumor Association and T32CA085159, A.F.W. by T32CA085159, and J.M.B. by F30CA171893. Shared Resources used in this project were supported by VCU, the Massey Cancer Center (P30CA016059), and the Department of Neurobiology & Anatomy Microscopy Facility (supported in part by P30NS047463).
Footnotes
Disclosure of Potential Conflicts of Interest The authors declare no conflict of interest.
Authors' Contributions Conception and design: L. Biddlestone-Thorpe, M.J. O'Connor, and K. Valerie.
Development of methodology: L. Biddlestone-Thorpe., M. Sajjad, E. Rosenberg, J.M. Beckta, N.C.K. Valerie, D. Gilfor, S.E. Golding, D.G. Temesi, and S.K. Lim.
Acquisition of data: L. Biddlestone-Thorpe, M. Sajjad, E. Rosenberg, B.R. Adams, J.M. Beckta, M. Tokarz, N.C.K. Valerie, A.F. Wagner, A. Khalil, S.E. Golding, D.G. Temesi, K.S. Choe, and S.K. Lim.
Analysis and interpretation of data: L. Biddlestone-Thorpe, E. Rosenberg, J.M. Beckta, N.C.K. Valerie, S.E. Golding, N.D. Mukhopadhyay, D.G. Temesi, A. Lau, K. S. Choe, and K. Valerie.
Writing, review, and/or revision of the manuscript: L. Biddlestone-Thorpe, A. Lau, M.J. O'Connor, and K. Valerie.
Material support: S. Deb, M.J. O'Connor, S.K. Lim, and L.F. Parada.
References
- 1.Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain Pathol. 1993;3:255–68. doi: 10.1111/j.1750-3639.1993.tb00752.x. [DOI] [PubMed] [Google Scholar]
- 2.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. The American journal of pathology. 2007;170:1445–53. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 4.Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nature reviews Cancer. 2010;10:319–31. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- 5.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59:1169–80. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- 6.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 8.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–33. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chow LM, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, et al. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer cell. 2011;19:305–16. doi: 10.1016/j.ccr.2011.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–69. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 12.Bensimon A, Aebersold R, Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS letters. 2011;585:1625–39. doi: 10.1016/j.febslet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 13.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 14.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nature reviews Cancer. 2011;11:239–53. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 15.Squatrito M, Holland EC. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer research. 2011;71:5945–9. doi: 10.1158/0008-5472.CAN-11-1245. [DOI] [PubMed] [Google Scholar]
- 16.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 17.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 18.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 19.Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008;68:7466–74. doi: 10.1158/0008-5472.CAN-08-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther. 2009;8:2894–902. doi: 10.1158/1535-7163.MCT-09-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golding SE, Rosenberg E, Adams BR, Wignarajah S, Beckta JM, O'Connor MJ, et al. Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle. 2012;11:1167–73. doi: 10.4161/cc.11.6.19576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arwert E, Hingtgen S, Figueiredo JL, Bergquist H, Mahmood U, Weissleder R, et al. Visualizing the dynamics of EGFR activity and antiglioma therapies in vivo. Cancer research. 2007;67:7335–42. doi: 10.1158/0008-5472.CAN-07-0077. [DOI] [PubMed] [Google Scholar]
- 23.Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer research. 2008;68:3286–94. doi: 10.1158/0008-5472.CAN-07-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 25.Golding SE, Rosenberg E, Neill S, Dent P, Povirk LF, Valerie K. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res. 2007;67:1046–53. doi: 10.1158/0008-5472.CAN-06-2371. [DOI] [PubMed] [Google Scholar]
- 26.Khalil A, Morgan RN, Adams BR, Golding SE, Dever SM, Rosenberg E, et al. ATM-dependent ERK signaling via AKT in response to DNA double-strand breaks. Cell Cycle. 2011;10:481–91. doi: 10.4161/cc.10.3.14713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 28.White JS, Choi S, Bakkenist CJ. Transient ATM kinase inhibition disrupts DNA damage-induced sister chromatid exchange. Sci Signal. 2010;3:ra44. doi: 10.1126/scisignal.2000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raso A, Vecchio D, Cappelli E, Ropolo M, Poggi A, Nozza P, et al. Characterization of glioma stem cells through multiple stem cell markers and their specific sensitization to double-strand break-inducing agents by pharmacological inhibition of ataxia telangiectasia mutated protein. Brain Pathol. 2012;22:677–88. doi: 10.1111/j.1750-3639.2012.00566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amos S, Mut M, diPierro CG, Carpenter JE, Xiao A, Kohutek ZA, et al. Protein kinase C-alpha-mediated regulation of low-density lipoprotein receptor related protein and urokinase increases astrocytoma invasion. Cancer Res. 2007;67:10241–51. doi: 10.1158/0008-5472.CAN-07-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westermark B, Ponten J, Hugosson R. Determinants for the establishment of permanent tissue culture lines from human gliomas. Acta Pathol Microbiol Scand A. 1973;81:791–805. doi: 10.1111/j.1699-0463.1973.tb03573.x. [DOI] [PubMed] [Google Scholar]
- 32.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 33.Biddlestone-Thorpe L, Marchi N, Guo K, Ghosh C, Janigro D, Valerie K, et al. Nanomaterial-mediated CNS delivery of diagnostic and therapeutic agents. Adv Drug Deliv Rev. 2011;64:605–13. doi: 10.1016/j.addr.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buonerba C, Di Lorenzo G, Marinelli A, Federico P, Palmieri G, Imbimbo M, et al. A comprehensive outlook on intracerebral therapy of malignant gliomas. Crit Rev Oncol Hematol. 2011;80:54–68. doi: 10.1016/j.critrevonc.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Adams BR, Golding SE, Rao RR, Valerie K. Dynamic dependence on ATR and ATM for double-strand break repair in human embryonic stem cells and neural descendants. PLoS One. 2010;5:e10001. doi: 10.1371/journal.pone.0010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gualberto A, Aldape K, Kozakiewicz K, Tlsty TD. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5166–71. doi: 10.1073/pnas.95.9.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haas-Kogan DA, Kogan SS, Yount G, Hsu J, Haas M, Deen DF, et al. p53 function influences the effect of fractionated radiotherapy on glioblastoma tumors. Int J Radiat Oncol Biol Phys. 1999;43:399–403. doi: 10.1016/s0360-3016(98)00393-9. [DOI] [PubMed] [Google Scholar]
- 38.Sabharwal N, Holland EC, Vazquez M. Live cell labeling of glial progenitor cells using targeted quantum dots. Ann Biomed Eng. 2009;37:1967–73. doi: 10.1007/s10439-009-9703-4. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Guessous F, Kwon S, Kumar M, Ibidapo O, Fuller L, et al. PTEN has tumor-promoting properties in the setting of gain-of-function p53 mutations. Cancer Res. 2008;68:1723–31. doi: 10.1158/0008-5472.CAN-07-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurova KV, Rokhlin OW, Budanov AV, Burdelya LG, Chumakov PM, Cohen MB, et al. Cooperation of two mutant p53 alleles contributes to Fas resistance of prostate carcinoma cells. Cancer research. 2003;63:2905–12. [PubMed] [Google Scholar]
- 41.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM Phosphorylates Histone H2AX in Response to DNA Double-strand Breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 42.Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–6. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 43.White DE, Negorev D, Peng H, Ivanov AV, Maul GG, Rauscher FJ., 3rd KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer research. 2006;66:11594–9. doi: 10.1158/0008-5472.CAN-06-4138. [DOI] [PubMed] [Google Scholar]
- 44.Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, et al. The combined status of ATM and p53 link tumor development with therapeutic response. Genes & development. 2009;23:1895–909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williamson CT, Muzik H, Turhan AG, Zamo A, O'Connor MJ, Bebb DG, et al. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol Cancer Ther. 2012;9:347–57. doi: 10.1158/1535-7163.MCT-09-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williamson CT, Kubota E, Hamill JD, Klimowicz A, Ye R, Muzik H, et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol Med. 2012;4:515–27. doi: 10.1002/emmm.201200229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bartek J, Jr., Ng K, Bartek J, Fischer W, Carter B, Chen CC. Key concepts in glioblastoma therapy. Journal of neurology, neurosurgery, and psychiatry. 2012;83:753–60. doi: 10.1136/jnnp-2011-300709. [DOI] [PubMed] [Google Scholar]
- 49.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–82. [PubMed] [Google Scholar]
- 50.Powell SN, DeFrank JS, Connell P, Eogan M, Preffer F, Dombkowski D, et al. Differential sensitivity of p53(−) and p53(+) cells to caffeine-induced radiosensitization and override of G2 delay. Cancer Res. 1995;55:1643–8. [PubMed] [Google Scholar]
- 51.Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, et al. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Molecular cell. 2009;35:327–39. doi: 10.1016/j.molcel.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.