

Abstract
DNA cytosine methyltransferases regulate the expression of the genome through the precise epigenetic marking of certain cytosines with a methyl group, and aberrant methylation is a hallmark of human diseases including cancer. Targeting these enzymes for drug design is currently a high priority. We have utilized ab initio quantum mechanical/molecular mechanical (QM/MM) molecular dynamics (MD) simulations to extensively investigate the reaction mechanism of the representative DNA methyltransferase HhaI (M.HhaI) from prokaryotes, whose overall mechanism is shared with the mammalian enzymes. We obtain for the first time full free energy profiles for the complete reaction, together with reaction dynamics in atomistic detail. Our results show an energetically preferred mechanism in which nucleophilic attack of cytosine C5 on the S-adenosyl-L-methionine (AdoMet) methyl group is concerted with formation of the Michael adduct between a conserved Cys in the active site with cytosine C6. Spontaneous and reversible proton transfer between a conserved Glu in the active site and cytosine N3 at the transition state was observed in our simulations, revealing the chemical participation of this Glu residue in the catalytic mechanism. Subsequently, the β-elimination of the C5 proton utilizes as base an OH− derived from a conserved crystal water that is part of a proton wire water channel, and this syn β-elimination reaction is the rate-limiting step. Design of novel cytosine methylation inhibitors would be advanced by our structural and thermodynamic characterization of the reaction mechanism.
Keywords: DNA cytosine methylation mechanism, QM/MM-MD simulation, Epigenetics
The DNA methyl transferases play critical roles in many biological functions. They are key players in regulating gene expression1: in embryonic development2, 3, in X-chromosome inactivation4, in genomic imprinting5 and, overall, in epigenetic mechanisms that transmit genetic information without altering the actual base sequence of the DNA through regulation of the methylation status6-8. Aberrant methylation is a feature of cancers and other diseases9-11. Methyl transferase activity is impaired by DNA damage resulting from environmental carcinogens, notably benzo[a]pyrene12, 13 and methylation status has an important impact on the reactivity of DNA with benzo[a]pyrene metabolites14. The design of methylation inhibitors is an active current research frontier15-19. Understanding the mechanism for the methyl transfer reaction in atomistic detail would advance drug design and could provide novel opportunities for regulating gene expression through control of the methylation process.
We focus here on the very well-studied prokaryotic cytosine methyltransferase HhaI (M.HhaI) which carries out the enzymatic process for methylation of cytosine C5 in DNA. Like all methyltransferases it uses S-adenosyl-L-methionine (AdoMet) as the methyl donor, and flipping of the target base into a pocket of the enzyme. It has been established based on numerous investigations that the catalytic process entails nucleophilic attack of cysteine 81 (Cys81) of this enzyme on the cytosine C6 to form a covalent adduct via Michael addition; this adduct promotes the nucleophilicity of the cytosine C5 for attack on the AdoMet methyl group20, and subsequently, the C5 proton is abstracted via a β-elimination reaction6, 21. The mammalian enzymes employ a similar mechanism6, 8, 22, which has recently been substantiated with a crystal structure of a productive covalent DNMT1-DNA complex23. It is notable that this crystal structure reveals a covalent adduct between a conserved cysteine and C6 of the target cytosine, as had been observed crystallographically with the prokaryotic enzyme, including some examples containing incorporated nucleoside inhibitor drugs in place of the cytosine24-26. It is also worth noting that DNMT1 is a maintenance methyltransferase, and in this case the mechanism for maintenance and de novo methyltransferases such as M.HhaI are similar; hence, new mechanistic insights could be relevant to drug design for inhibition of both methyltransferases.
Current DNA methyltransferase inhibitors generally fall into two categories: nucleoside analogues and non-nucleoside inhibitors. The nucleoside analogues become incorporated in the DNA and may function by inhibiting methylation or β-elimination, but they form the Michael adduct and thus acting as covalent inhibitors. Two nucleoside analogue inhibitors are currently in use, azacytidine and decitabine, employed to treat myelodysplastic syndrome. The non-nucleoside inhibitors bind DNA methyltransferases and exert their inhibitory effects through a variety of different mechanisms27.
At present, a full atomistic, thermodynamic and dynamic characterization of the chemical reaction process remains to be determined. Questions that remain not fully resolved include whether the methylation reaction is concerted or stepwise, the specific roles of certain key amino acids in the active site, the nature of the base that abstracts the C5 proton, the role of waters in the chemical process, and the energetics and dynamics of the bond forming and breaking events. Previous computational studies28, 29 for this enzyme system did not obtain free energies or take the enzyme dynamics into account, and either used the semi-empirical DFTB approach as the quantum mechanical method28 or treated the heterogeneous enzyme environment with an implicit continuum solvent model29.
We have utilized Born-Oppenheimer ab initio QM/MM-MD calculations to investigate the mechanism for the methyl transfer reaction in M.HhaI; the chemically reacting moieties are described by the ab initio QM method, the surrounding enzyme and aqueous environment are treated explicitly by classical MM, and the enzyme active site dynamics and those of the surroundings are simulated on an equal footing. This approach now has the power to robustly elucidate the full course of the chemical steps in enzyme mechanisms30-36. We explored many mechanistic issues that remain under consideration, and thoroughly investigated a variety of plausible reaction schemes. Our extensive computational investigations resulted in a preferred methyl transfer mechanism for M.HhaI. This mechanism involves nucleophilic attack of deprotonated Cys81 thiolate on the cytosine C6, concerted with methyl transfer. Subsequently, an OH−, derived through a proton wire to surface water and involving crystal and solvent waters, serves as the base to abstract the C5 proton. Our results provide complete free energy profiles for the reaction mechanism, reaction barriers and transition state and intermediate structures for both methylation and proton abstraction steps; our extensive investigations of possible bases support the previous mechanistic suggestion28 that the OH− is likely to be the proton-abstracting base, and that proton abstraction is chemically rate-determining. We obtain atomistic and dynamic views of the bond breaking and forming processes, which reveal the intricate dynamic interplay between formation of the Michael adduct and the methyl transfer step. We also resolve uncertainties in the role of Glu119 in the active site: it forms a hydrogen bond with cytosine N3 at the reactant and at the transition state where the proton spontaneously and reversibly oscillates between being bonded to Glu and to cytosine N3. Hence, the chemical participation of the Glu in the reaction mechanism is manifested. Thus, our study for the first time fully characterizes the HhaI methyltransferase reaction, provides new molecular insights on experimental data20, 24, 37, and more broadly is very likely applicable to the critical human cytosine methylation enzymes that are key in governing epigenetic inheritance, since mammalian DNMT1 relies on a similar chemical mechanism23.
Computational methods
Initial preparation
The initial structure of the enzyme-reactant complex was constructed based on a ternary crystal structure of the DNA methyltransferase M.HhaI with S-adenosyl-L-methionine (AdoMet) and DNA (PDB ID38: 6MHT) (Figure 1)39. The critical methylation target sequence is with C* as the target for methylation by AdoMet. In this structure, the 04′ of the target cytosine had been replaced by a sulfur atom (4′-thio-2′-deoxycytidine) in an effort to inhibit the methylation reaction. In addition, the DNA was hemi-methylated, with a methyl group on C’. Despite the presence of the 4′ thiol, partial reaction did take place and two sets of coordinates were presented for the methyl group and the sulfur of Cys81: in one set, the methyl remained on the AdoMet and the sulfur on Cys81, while in the second set the methyl had transferred to the cytosine C5 and the sulfur of Cys81 was partially bonded to C6 of cytosine. We selected the former set of coordinates for our study, and we remodeled the sulfur atom on the sugar as the natural oxygen. Since we wished to investigate de novo methylation, requiring a prechemistry system containing unmethylated DNA and AdoMet, we replaced the methyl on C’ with a hydrogen atom. No crystal structure of such a prechemistry system without mutation appears to be yet available for M.HhaI; the structure we selected for this study had the highest resolution (2.05Å) of any available M.HhaI crystal structure containing the AdoMet. The molecular modeling was performed with Discovery Studio (Accelrys Software, Inc.). Hydrogen atoms were added to this model of the enzyme-substrate complex by the LEAP module of the AMBER 10 simulation package40. The protonation states of charged residues were computed by the H++41, 42 and pdb2pqr43 programs. In addition, we considered the potential H-bonding network, solvent exposure of the ionizable residues, potential steric clashes if the proton was added, and preservation of the crystal structure in assigning protonation states. The assignments that we made are given in Table S1 (Supporting Information).
Figure 1.
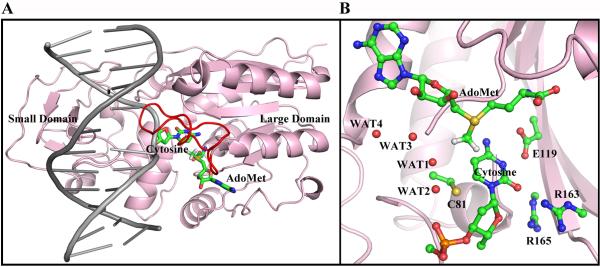
(A) Ternary structure of HhaI methyltransferase (PDB ID38: 6MHT). The flipped out cytosine and the cofactor AdoMet are colored by atom. The protein is pink. The large and small domains are indicated. The DNA is gray and the catalytic loop is red. (B) The active site, including the target cytosine, catalytic Cys81 from the catalytic loop, Glu119, Arg163 and Arg165 from the large domain and the cofactor AdoMet are shown, and crystal waters are indicated. The active site structure was remodeled from the crystal structure as described in Methods.
The initial model was subjected to 4 ns of MD simulations using AMBER 1040. We employed the Amber99SB44-46 force field with modification for DNA by parmbsc047. Bond length, bond angle, torsional and Van der Waals parameters for the methyl donor AdoMet were taken from Markham et al.48. Partial atomic charges for the AdoMet were calculated by using Hartree-Fock quantum mechanical calculations with 6-31G* basis set49, 50 without geometry optimization, employing the Gaussian 03 package from Gaussian, Inc51. The charges were then fitted to each atomic center with the RESP algorithm52. The fitted charges are shown in Table S2 (Supporting Information). The structure was neutralized by 20 Na+ counterions and was solvated with a periodic rectangular box of TIP3P water53, 54 with 10 Å buffer around the enzyme-substrate complex. The total number of atoms in the system was ~ 60453, of which ~ 54432 were water molecules. Details of the MD protocols are given in Supporting Information. The final snapshot from the stable 4ns trajectory was utilized for the subsequent QM/MM calculations. PyMOL (Schrödinger, LLC) was employed to make molecular images and movies.
Born-Oppenheimer Ab Initio QM/MM-MD simulation
In the QM/MM calculations, the enzyme substrate model prepared as described above was partitioned into QM subsystem and MM subsystem, in which all components that participated in chemical reactions are included in the QM region, as illustrated in Figure S1 of Supporting Information. The QM subsystem was treated by the hybrid density functional B3LYP55-57 with a medium split valence basis set and polarization functions 6-31G*. The QM/MM interface was described by a pseudobond approach58-60. All other atoms were described classically. To reduce computational cost for the MM calculations during the QM/MM simulation process, spherical boundary conditions were utilized: the C5 atom of cytosine at the active site was selected as the center and atoms that were 20.0 Å away from the C5 atom were fixed during the simulation. Solvent water molecules with distance from the C5 atom greater than 30.0 Å were removed. The prepared system had ~ 12779 atoms in total, which included 6001 protein atoms, 2258 water molecules and four Na+ counterions. All QM/MM and QM/MM-MD calculations were carried out with modified versions of the Q-Chem61 and Tinker programs62. After the partition of the QM and MM subsystem, the entire reactant system was minimized first by an iterative optimization procedure. Then an iterative minimization procedure with the reaction coordinate driving method63 was employed to map out a minimum energy path with ab initio QM/MM calculations. For each determined structure along the reaction path, an MD simulation of the MM subsystem with the MM force field was further carried out for 500ps with the frozen QM subsystem. The resulting snapshots were used as starting structures for Born-Oppenheimer ab initio QM/MM-MD simulations with umbrella sampling30, 64, 65 that applies a harmonic potential to constrain the reaction coordinate (RC) at successive values. In order to ensure sufficient overlap between the successive windows, force constants in the range of 40 to 100 kcal·mol−1· Å−2 were employed. We sampled 30ps for each window. For these biased QM/MM-MD simulations, the Beeman algorithm66 was used to integrate the Newton equations of motion with a time step of 1fs. Cutoffs of 18 and 12 Å were employed for electrostatic and van der Waals interactions between the MM atoms, respectively. There was no cutoff for electrostatic interactions between the QM and MM regions. Configurations of 25ps were collected for data analysis after a 5ps QM/MM-MD equilibration. Finally, the probability distributions of each window were determined and pieced together with the weighted histogram analysis method (WHAM)67-69 to obtain free energy profiles. This computational protocol (Scheme 1) has been demonstrated to be feasible and successful in several enzyme investigations30-36.
Scheme 1.

Computational protocol
Results
We have made an extensive exploration of the M.HhaI catalyzed methyl transfer reaction, utilizing state-of-the-art ab initio QM/MM-MD simulations. We investigated many reaction schemes. For each scheme, we calculated the minimum energy path or the two-dimensional minimum potential energy surface, employing the reaction coordinate driving method and B3LYP (6-31G*) QM/MM computations. The most promising schemes derived from the one or two-dimensional searches were further investigated with B3LYP (6-31G*) QM/MM-MD simulations with umbrella sampling. Numerous mechanistic paths could be examined through this hierarchy of strategies. We were motivated to apply this robust methodology to this enzyme system because it is a well-studied representative of DNA methyltransferases, which share catalytic mechanistic features6, 8 with mammalian DNMT123. The extensive literature including kinetic studies20, 37, 70, X-ray crystal structures24, 25, 39, 71, 72 and studies of mutant enzymes73-75 have implicated conserved residues76, 77 Cys81, Glu119, Arg163, and Arg165 in the catalytic process. Cys81 is understood to form a covalent bond with cytosine C6 to activate the cytosine C5 for nucleophilic attack on the methyl group of cofactor AdoMet6, 20, 21. However, the literature contains varied perspectives on certain key mechanistic issues.
We have therefore investigated a number of mechanistic possibilities for the methyl transfer reaction. (M1): Concerted methyl transfer and covalent bonding of Cys81–S− with C6 of cytosine (Figure 2)28. (M2): Stepwise methyl transfer; covalent bonding of Cys81–S− with C6 of cytosine followed by methyl transfer (Figure S3 of Supporting Information)20. (M3): Stepwise methyl transfer catalyzed by proton transfer from Glu119 to N3 of cytosine; the protonation promotes S–C6 covalent bond formation and is concerted with it; methyl transfer follows (Figure S4 of Supporting Information)78. (M4): Deprotonation of Cys81–SH before the methyl transfer reaction; Cys81–SH is deprotonated by the non-bridging phosphate oxygen on the 3′ side of the target cytosine, via a mediating water29; then the concerted methyl transfer reaction proceeds (Figure S5 of Supporting Information). (M5): Direct nucleophilic attack of the C5 on the methyl group of AdoMet, but with Cys81 protonated (Cys81–SH). This mechanism tests the possibility that the role of Cys81 is to electrostatically foster the activation of C5 for the methyl transfer step, without forming a covalent bond with cytosine C6 (Figure S6 of Supporting Information). Our motivation (M4 and M5) was that the protonation state of the cysteine in the reactant complex is an open question29, 37.
Figure 2.

(A) Mechanism for the methyl transfer reaction catalyzed by M.HhaI involving concerted methyl transfer and covalent bonding of Cys81–S− with C6 of cytosine. Key structures of the obtained mechanism with bond distances are shown. Blue dashed lines indicate hydrogen bonds. Red dashed lines denote key distances. (B) Free energy profile. The potential energy profile and bond length analyses are shown in Figure S2 of Supporting Information. (C) Distance between cytosine N3 and HOε2 of Glu119 (black) and the distance between cytosine N3 and Oε2 of Glu119 (red) in the QM/MM-MD simulation; there are two configurations at the transition state 1 (TS1) with Glu119 hydrogen oscillating between being covalently linked to cytosine N3 and Glu119. These are circled in 2A. See Movie S1 of Supporting Information.
For the cytosine C5 proton β-elimination, we investigated several possibilities for the proton-abstracting base. (E1): The leaving Cys81–S− directly abstracts the proton (Figure S7 of Supporting Information)79. (E2): Cys81–S− abstracts the proton through a water bridge (Figure S8 of Supporting Information)29. (E3): The non-bridging phosphate oxygen on the 3′ side of the target cytosine abstracts the proton, via a two-water bridge (Figure S9 of Supporting Information)24. (E4): A proton wire water channel provides a OH− as the base (Figure 3A)28.
Figure 3.

Mechanism for the proton β-elimination reaction catalyzed by M.HhaI. (A) A proton wire provides a OH− as the base to abstract the cytosine C5 proton (H5). The chain of water molecules connecting to surface bulk water is shown by dashed lines. WAT1 and WAT3 are at the sites of the respective crystal waters shown in Figure 1B and WAT1 provides the OH−. (B) The mechanism with the key structures and distances denoted. The blue dashed lines are hydrogen bonds. Red dashed lines denote key distances. (C) Free energy profile. See Movie S2 of Supporting Information.
Our wide surveys have indicated that the energetically most favored mechanism entails methyl transfer that is concerted with covalent bonding of Cys81–S− and cytosine C6, and that the base for proton abstraction is an OH− provided by a proton wire through a water channel. The mechanism also reveals a dynamic proton transfer between Glu119 and cytosine N3 during the transition state of the methyl transfer step.
Methyl transfer is concerted with Michael addition and catalyzed by proton transfer from Glu119 in the transition state
We have determined that the first, methyl transfer step in our preferred mechanism for the reaction is a nucleophilic attack of the C5 on the methyl group of AdoMet, with concerted catalytic attack by the Cys81–S− group at the C6 position of the target cytosine to form a 5-methyl-6-Cys-81-S-5,6-dihydrocytosine intermediate (Figure 2A). It is noteworthy that the Cys81–S−, which is highly nucleophilic, attacks the cytosine C6 spontaneously. A free energy profile for the methyl transfer step was determined by ab initio QM/MM-MD simulation and umbrella sampling as shown in Figure 2B, determined by employing 21 umbrella windows along the reaction coordinate, each simulated for 30ps with B3LYP (6-31G*) QM/MM-MD calculations. The calculated free energy activation barrier for the methylation reaction is 15.8 ± 0.3 kcal/mol. Unrestrained 1 - 2ps QM/MM MD simulations of 39 snapshots from the transition state structures showed that they relaxed to the reactant and intermediate with almost equal probability, indicating that a real transition state had been located (Figure S10 of Supporting Information). The intermediate 1 (I1) is an energetically very stable state, which has a much lower free energy (−30.4 kcal/mol) than the reactant state.
We obtained the active site geometry of the determined reactant, transition state, and intermediate in the initial methylation step, as shown in Figure 2A and Movie S1 of Supporting Information. In the reactant state, the Cys81–S−, is 3.2Å away from the C6 of the cytosine ring, and is well positioned for nucleophilic attack on C6 to form a covalently bonded adduct between Cys81–S− and cytosine C6. The methyl donor AdoMet is also well situated for an in-line nucleophilic attack by C5 of the activated cytosine, with a CH3–C5 distance of 3.3Å. At the transition state, the AdoMet S–CH3 distance has stretched from its normal covalent bond distance of 1.8Å to 2.2Å, and the CH3–C5 distance has shortened to 2.3Å. In addition, the distance between cytosine C6 and Cys81–S− has shortened to 2.2Å. The 30ps QM/MM-MD simulation of the transition state structure showed fluctuations in the S–C6 distance that occasionally reached ~ 3Å (Figure S11 of Supporting Information), indicating that this bond can episodically reverse and reform, consistent with experimental evidence37. As described below, the Glu119 carboxylic acid hydrogen is hydrogen bonded to N3 of cytosine in the reactant and transition state. Notably, the 30ps QM/MM-MD simulation for the transition state showed that the Glu119 carboxylic acid hydrogen spontaneously fluctuates between being positioned on the Glu119 with a hydrogen bond to cytosine N3 and being covalently bonded to the cytosine N3, with constant heavy atom to heavy atom distance (Figure 2C). When the methyl group has completely transferred to the cytosine, we obtain the energetically very stable intermediate 1 (I1).
We analyzed hydrogen bonding interactions between certain active site amino acid residues and substrate. Glu119, Arg163 and Arg165 are conserved residues76, 77. Mutating Glu11973 and Arg16575 reduces the overall catalytic rate by several orders of magnitude. While there are no mutation studies for Arg163, it has been implicated as playing a role in the methylation process24, 28, 29, 75. These amino acids may be essential for maintaining the target cytosine in the flipped-out position and/or they may play a role in the chemical reaction directly73. We monitored hydrogen bonding interactions between these residues and the substrate for the key states in our preferred mechanism. Hydrogen bond occupancies are shown in Figure 4 and time-dependence of distances and angles are given in Figure S12 of Supporting Information. We find close and stable hydrogen bonds between Glu119 and cytosine N3 as well as N4 in the reactant state; in the transition state the Glu119 carboxylic acid hydrogen spontaneously and reversibly transfers to N3 as described above (Figure 2C); it is present as a hydrogen bond in the intermediate state. This Glu119 was protonated in the reactant state for all investigated mechanisms because crystal structures of M.HhaI complexed with unmethylated or hemimethylated DNA together with S-adenosyl-L-homocysteine (AdoHcy) show heavy atom to heavy atom distances between Glu119 carboxylic acid oxygen (Oε2) and cytosine N3 that strongly suggest hydrogen bonding, with distances of 3.22 Å, 2.75 Å and 2.97 Å for the three obtained structures24. Protonation allows hydrogen bonding with or proton transfer to cytosine N3. The stable hydrogen bond we observed explains the increase in pKa in the enzyme, as the energetic cost of protonation of the Glu119 is compensated by the formation of the hydrogen bond80. Hydrogen bonding between Arg163 and cytosine O2 is weak in all three states but stabilizing electrostatic interactions from near-hydrogen bonding orientations are present in all states (Figure S12 of Supporting Information). Arg165 also maintains hydrogen bonds or electrostatic interactions that are near-hydrogen bonding with cytosine O2 and 4′ in the reactant, transition state and intermediate.
Figure 4.
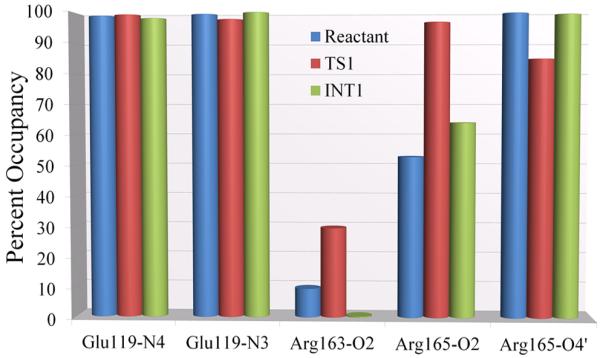
Hydrogen bond occupancies along the 30ps QM/MM-MD simulation for each key state in the methyl transfer step. Hydrogen bond criteria: heavy atom to heavy atom distance less than 3.4Å and heavy atom – H – heavy atom angle of 180° ± 45°.
Other mechanistic possibilities for the methyl transfer that were investigated are fully detailed in Supporting Information and Figures S3 - S6 of Supporting Information. They were all unfavorable, providing unstable intermediates or much higher energy barriers.
β-elimination uses a crystal water-derived OH− as base
The nature of the base and mechanism for abstraction of the C5 proton has been the subject of considerable interest. We thoroughly explored four different possibilities, detailed above, beginning with the intermediate 1 (I1) from the methyl transfer step (Figure 2A). One possibility for the base, suggested by Zhang and Bruice28, is that the base is a nearby OH−; a solvent water channel mediates the proton interchange to provide the OH−, and it was pointed out that the production of this OH− would cost about 12 kcal/mola. Of the mechanisms that we explored, this mechanism provided the lowest free energy profile. Moreover, we determined that the OH− could be provided by a proton wire to bulk water. Using 10ns molecular dynamics simulations, we observed a stable channel of water emanating from the approximate position of WAT1 (Figure 1B) to the enzyme surface and bulk water. The channel is shown in Figure 3A for a random snapshot of the MD. It is noteworthy that WAT1 and WAT3 in Figure 3A are in positions of crystal waters24, 39 and they remained there stably throughout the MD simulation. Other crystal and solvent waters may provide different proton wire channels. WAT1 is conserved in a number of crystal structures of M.HhaI24, 75, 81. We replaced the water at the WAT1 position with a OH−, and the system is referred to as intermediate 2 (I2). The mechanism for proton abstraction through OH− is shown in Figure 3B and Movie S2 of Supporting Information. Our obtained free energy profile using B3LYP (6-31G*) QM/MM-MD simulations with 30 umbrella sampling windows, each calculated for 30ps is shown in Figure 3C. A barrier of 8.7 ± 0.9 kcal/mol was obtained. Together with the 12 kcal/mol required to produce the OH−, the barrier is 20.7 kcal/mol, making the proton abstraction as the rate limiting step.
Figure 3B shows that in the intermediate state 2 (I2), the OH− is 3.2Å away from the C5 proton, while in the transition state, the distance has shortened to 1.4Å, and the C5 proton has begun to leave the C5 with a distance of 1.3Å. In the intermediate and transition state, the bond between cytosine C6 and Cys81–S− remains intact (1.9Å) until the C5–H5 bond is broken, and then the Cys81 detaches and releases the methylated cytosine and AdoHcy. The complete bond length analysis for the process which reveals this dynamic is shown in Figure S13 of Supporting Information. Our hydrogen bond analyses (Figure 5) show that as in the methylation step, hydrogen bonding or electrostatic interactions due to near-hydrogen bonding orientations (Figure S12 of Supporting Information) are present in intermediate, transition state and product except for Glu119 in the product. In this case, cytosine N3 remains hydrogen bonded through a water but cytosine N4 is no longer close to Glu119, initiating the release of the methylated cytosine.
Figure 5.
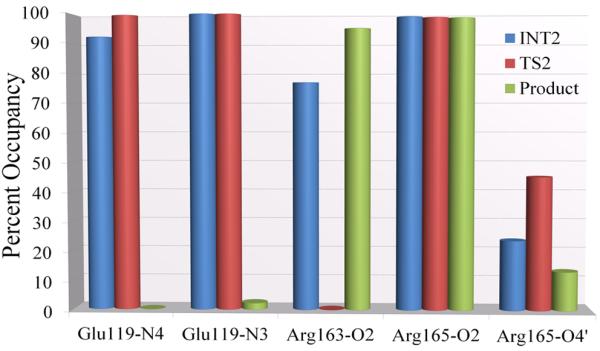
Hydrogen bond occupancies along the 30ps QM/MM-MD simulation for each key state in the β-elimination step. Hydrogen bond criteria are the same as in Figure 4.
Other mechanistic possibilities that we investigated for the proton abstraction had much higher energy barriers. These are detailed in full in Supporting Information and Figures S7 - S9 of Supporting Information.
Discussion
We investigated extensively the methylation reaction catalyzed by M.HhaI, exploring many mechanistic possibilities; we used for the first time, state-of-the-art ab initio QM/MM-MD methods in which the chemically reacting moieties are treated by high level ab initio QM methods while the surrounding enzyme is treated by classical MM. A key feature of our approach is that the dynamics of the enzyme active site and its surroundings are treated on an equal footing. The free energy profile along the reaction coordinate is obtained from a series of biased simulations63, which are employed to enhance the sampling of lower probability states, with the weighted histogram analysis method (WHAM)67-69. This advanced approach provides a first-principle QM description of the chemical reaction. Furthermore, it adequately accounts for the biological environment. Importantly, it takes a balanced account of the fluctuations of the reaction active site and the surrounding enzyme system. Mechanisms of various enzymes that are consistent with experimental data have been determined with this powerful method30-36.
We investigated mechanistic issues that have been of significant interest, but are not definitively resolved. For the methyl transfer step of the reaction, we explored whether the formation of the Michael adduct between conserved Cys81–S− and cytosine C6 and the transfer of the methyl group are concerted or stepwise, as there are conflicting views20, 28, 29, 78. We also investigated the possibility that the conserved residue Glu119 donates a proton to cytosine N3 to promote the formation of the covalent adduct78. In addition, we considered the possibility that the Cys81–SH thiol is deprotonated to the Cys81–S− thiolate through proton transfer to a non-bridging phosphate oxygen29. Also, we investigated a mechanism in which the Cys81–SH, the thiol protonated state--which cannot form a covalent adduct--might provide non-bonded electrostatic stabilization to facilitate the methyl transfer. For the proton elimination step, we considered the following potential bases: (1) the leaving thiolate of Cys81 acts as the base either directly, or (2) through a water29; (3) the 3′ non-bridging phosphate oxygen of the target cytosine acts as the base via two waters; and (4) an OH− derived from a crystal water acts as the base, utilizing a water channel as suggested by Zhang and Bruice28. While our extensive investigations provided a clearly favored mechanism, it remains a possibility, as always in computational investigations of reaction energy surfaces, that there are other pathways that were not found.
Our energetically preferred mechanism (Movie S1, Supporting Information) involves a methylation reaction in which spontaneous attack of Cys81–S− to form a Michael adduct with cytosine C6 is concerted with methyl transfer, in agreement with Zhang and Bruice28. Our bond length analyses (Figure S11 of Supporting Information) show that the covalent bond between Cys81–S− and cytosine C6 forms rapidly, and QM/MM-MD simulations show that the bond can form and break reversibly, consistent with kinetic studies37. An important finding is the observation of proton transfer from Glu119 to cytosine N3, spontaneously and reversibly, during the transition state for the methylation step; this transfer indicates the catalytic participation of Glu119 in the chemical reaction, as proposed by Verdine78. The conserved Glu119 is protonated and hydrogen bonds with cytosine N3 and N4 throughout the whole reaction (Figure 4 and Figure 5) until the release of the product. The increase in pKa of the Glu in the enzyme is explained by the stable hydrogen bond to cytosine N3, as its protonation allows the formation of the hydrogen bond80. The hydrogen bonds between Glu119 and cytosine provide electrostatic support for the mechanism, particularly by withdrawing electrons in the reactant to make C6 more positive for attack by Cys81–S−, and these hydrogen bonds have been strongly suggested by crystal structures24.
For the proton elimination step (Movie S2, Supporting Information), our favored mechanism utilizes as base a OH− that has migrated to the active site through a proton wire involving a channel of waters between the active site and bulk water. Simulations have shown that proton transfer between adjacent water molecules in a proton wire is spontaneous and very fast82. The key water that provided the OH− is in the position of a crystal water (Figure 1B and Figure 3A), and other crystal waters may participate in the proton wire. We found that the proton abstraction step is rate-limiting when factoring in the energetic cost, about 12 kcal/mola, of generating the OH− through the proton wire. This is chemically reasonable since the proton abstraction involves a syn β-elimination, the proton leaves on the same face of the cytosine ring as the Cys81–S−, which is sterically crowded and hence slow83. Furthermore, the difficult syn-elimination requires a strong base for the proton abstraction, which supports the OH− as base in our preferred mechanism. The total barrier of 20.7 kcal/mol for this rate limiting step is in good agreement with measured kcat values for the overall methyl transfer reaction (0.02 S−1 to 0.09 S−1 20, 84, 85, which corresponds to 19.0 ~ 20.0 kcal/mol according to transition state theory: (where kcat is the catalytic rate-constant, kB is the Boltzmann constant, h is the Planck constant, R is the universal gas constant, T is the temperature (300K) and ΔG‡ is the free energy of activation).
The roles of the conserved residues Glu119, Arg163 and Arg165 are further elucidated in our hydrogen bond analyses. All of them provide stabilizing hydrogen bonding (Figures 4 and 5) or near-hydrogen bonding electrostatic interactions (Figure S12 of Supporting Information) throughout both the methyl transfer and β-elimination steps except for Glu119 in the final product, which has moved away to initiate the release. In addition to stabilizing the flipped out position of the substrate cytosine in the enzyme reactive site pocket73, 75, their electrostatic impact in the reactant state in withdrawing electrons from the target cytosine makes C6 more positive for attack by Cys81–S−. Furthermore, we demonstrated that the Glu119 carboxylic acid proton is reversibly transferred to cytosine N3 in the transition state for methylation and thus this residue participates in the chemical reaction78.
Conclusion
We have utilized state-of-the-art ab initio QM/MM-MD simulations, exploring multiple mechanistic possibilities, to fully characterize the reaction mechanism of a representative DNA cytosine methyltransferase. From prokaryotes to mammals, all of these enzymes share mechanistic features. They play critical roles in governing epigenetically the function of the genome. Furthermore, their function is misregulated in many human diseases and targeting them pharmacologically is currently a very important research direction. Molecular understanding of the mechanism is required to effectively target cytosine methyltransferases with aberrant activity. Our results provide for the first time a complete structural and thermodynamic characterization of the full reaction profile. We provide an atomistic characterization of the reaction dynamics, and a determination of the roles of key protein residues in the active site. We show how a conserved Glu residue chemically promotes the methyl transfer reaction during the transition state, and how a conserved crystal water provides a critical OH− base needed for β-elimination of the C5 proton, via a proton wire through a water channel. The complete depiction of the structural, dynamic and thermodynamic elements in the cytosine methyltransferase reaction should facilitate the design of therapeutic inhibitors, particularly novel and more potent transition state analog inhibitors that are of interest for drug design as transition states become fully characterized86.
Supplementary Material
Acknowledgements
This work used computational resources of the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant MCB060037 to S.B. We also gratefully acknowledge computational resources provided by the Multi-purpose High Performance Computing resource of New York University (NYU-ITS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Funding The research reported in this publication was supported by National Institutes of Health (NIH) [R01-CA-75449 and R01-CA-28038 to S.B., and R01-GM079223 to Y.Z.], and NSF [CHE-CAREER-0448156 to Y.Z.]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Abbreviations
- QM/MM-MD
quantum mechanical/molecular mechanical - molecular dynamics
- M.HhaI
methyltransferase HhaI
- AdoMet
S-adenosyl-L-methionine
- DNMT1
DNA methyltransferase 1
- AdoHcy
S-adenosyl-L-homocysteine
Footnotes
In pure water, [H2O] is 55.6 M, [OH−] is 10−7 M at pH = 7.0. Thus, the probability of observing an OH− versus a water molecule is 10−7/55.6. Therefore, the free energy difference ΔG between OH− and H2O at pH = 7.0 is: ΔG = −RTlnKeq= −0.5961×ln(10−7/55.6) = 12.0 kcal/mol.
Conflict of interest statement. None declared.
Supporting Information Additional details regarding MD simulation protocol and other tested mechanisms. Protonation state of charged residue assignments (Table S1), and force field parameters (Table S2). Figures S1 - S14 and Movies S1 - S2 as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mueller WC, von Deimling A. Gene regulation by methylation. Recent Results Cancer Res. 2009;171:217–239. doi: 10.1007/978-3-540-31206-2_13. [DOI] [PubMed] [Google Scholar]
- 2.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 3.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 4.Panning B, Jaenisch R. DNA hypomethylation can activate Xist expression and silence X-linked genes. Genes Dev. 1996;10:1991–2002. doi: 10.1101/gad.10.16.1991. [DOI] [PubMed] [Google Scholar]
- 5.Hore TA, Rapkins RW, Graves JA. Construction and evolution of imprinted loci in mammals. Trends Genet. 2007;23:440–448. doi: 10.1016/j.tig.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem. 2002;3:274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 7.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 8.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 9.Robertson KD. DNA methylation and human disease. Nat. Rev. Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 10.Kulis M, Esteller M. DNA methylation and cancer. Adv. Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 11.Kobow K, Blumcke I. The emerging role of DNA methylation in epileptogenesis. Epilepsia. 2012;53:11–20. doi: 10.1111/epi.12031. [DOI] [PubMed] [Google Scholar]
- 12.Subach OM, Baskunov VB, Darii MV, Maltseva DV, Alexandrov DA, Kirsanova OV, Kolbanovskiy A, Kolbanovskiy M, Johnson F, Bonala R, Geacintov NE, Gromova ES. Impact of benzo[a]pyrene-2′-deoxyguanosine lesions on methylation of DNA by SssI and HhaI DNA methyltransferases. Biochemistry. 2006;45:6142–6159. doi: 10.1021/bi0511639. [DOI] [PubMed] [Google Scholar]
- 13.Lukashevich OV, Baskunov VB, Darii MV, Kolbanovskiy A, Baykov AA, Gromova ES. Dnmt3a-CD is less susceptible to bulky benzo[a]pyrene diol epoxide-derived DNA lesions than prokaryotic DNA methyltransferases. Biochemistry. 2011;50:875–881. doi: 10.1021/bi101717b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guza R, Kotandeniya D, Murphy K, Dissanayake T, Lin C, Giambasu GM, Lad RR, Wojciechowski F, Amin S, Sturla SJ, Hudson RH, York DM, Jankowiak R, Jones R, Tretyakova NY. Influence of C-5 substituted cytosine and related nucleoside analogs on the formation of benzo[a]pyrene diol epoxide-dG adducts at CG base pairs of DNA. Nucleic Acids Res. 2011;39:3988–4006. doi: 10.1093/nar/gkq1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medina-Franco JL, Caulfield T. Advances in the computational development of DNA methyltransferase inhibitors. Drug Discov. Today. 2011;16:418–425. doi: 10.1016/j.drudis.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Ren J, Singh BN, Huang Q, Li ZF, Gao Y, Mishra P, Hwa YL, Li JP, Dowdy SC, Jiang SW. DNA hypermethylation as a chemotherapy target. Cell. Signal. 2011;23:1082–1093. doi: 10.1016/j.cellsig.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Xu F, Mao C, Ding Y, Rui C, Wu L, Shi A, Zhang H, Zhang L, Xu Z. Molecular and Enzymatic Profiles of Mammalian DNA Methyltransferases: Structures and Targets for Drugs. Curr. Med. Chem. 2010;17:4052–4071. doi: 10.2174/092986710793205372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhe-Paganon S, Syeda F, Park L. DNA methyl transferase 1: regulatory mechanisms and implications in health and disease. Int. J. Biochem. Mol. Biol. 2011;2:58–66. [PMC free article] [PubMed] [Google Scholar]
- 19.Ceccaldi A, Rajavelu A, Ragozin S, Senamaud-Beaufort C, Bashtrykov P, Testa N, Dali-Ali H, Maulay-Bailly C, Amand S, Guianvarc’h D, Jeltsch A, Arimondo PB. Identification of Novel Inhibitors of DNA Methylation by Screening of a Chemical Library. ACS Chem. Biol. 2013;8:543–548. doi: 10.1021/cb300565z. [DOI] [PubMed] [Google Scholar]
- 20.Wu JC, Santi DV. Kinetic and catalytic mechanism of HhaI methyltransferase. J. Biol. Chem. 1987;262:4778–4786. [PubMed] [Google Scholar]
- 21.Sankpal UT, Rao DN. Structure, function, and mechanism of HhaI DNA methyltransferases. Crit. Rev. Biochem. Mol. Bio. 2002;37:167–197. doi: 10.1080/10409230290771492. [DOI] [PubMed] [Google Scholar]
- 22.Cheng XD, Blumenthal RM. Mammalian DNA methyltransferases: A structural perspective. Structure. 2008;16:341–350. doi: 10.1016/j.str.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song J, Teplova M, Ishibe-Murakami S, Patel DJ. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 2012;335:709–712. doi: 10.1126/science.1214453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Gara M, Klimasauskas S, Roberts RJ, Cheng XD. Enzymatic C5-cytosine methylation of DNA: Mechanistic implications of new crystal structures for HhaI methyltransferase-DNA-AdoHcy complexes. J. Mol. Biol. 1996;261:634–645. doi: 10.1006/jmbi.1996.0489. [DOI] [PubMed] [Google Scholar]
- 25.Klimasauskas S, Kumar S, Roberts RJ, Cheng XD. Hhal Methyltransferase Flips Its Target Base out of the DNA Helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 26.Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. Zebularine: A novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J. Mol. Biol. 2002;321:591–599. doi: 10.1016/S0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fahy J, Jeltsch A, Arimondo PB. DNA methyltransferase inhibitors in cancer: a chemical and therapeutic patent overview and selected clinical studies. Expert. Opin. Ther. Pat. 2012;22:1427–1442. doi: 10.1517/13543776.2012.729579. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Bruice TC. The mechanism of M.HhaI DNA C5 cytosine methyltransferase enzyme: a quantum mechanics/molecular mechanics approach. Proc. Natl. Acad. Sci. USA. 2006;103:6148–6153. doi: 10.1073/pnas.0601587103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zangi R, Arrieta A, Cossio FP. Mechanism of DNA methylation: the double role of DNA as a substrate and as a cofactor. J. Mol. Biol. 2010;400:632–644. doi: 10.1016/j.jmb.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 30.Hu P, Wang SL, Zhang YK. Highly Dissociative and Concerted Mechanism for the Nicotinamide Cleavage Reaction in Sir2Tm Enzyme Suggested by Ab Initio QM/MM Molecular Dynamics Simulations. J. Am. Chem. Soc. 2008;130:16721–16728. doi: 10.1021/ja807269j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu RB, Wang SL, Zhou NJ, Cao ZX, Zhang YK. A Proton-Shuttle Reaction Mechanism for Histone Deacetylase 8 and the Catalytic Role of Metal Ions. J. Am. Chem. Soc. 2010;132:9471–9479. doi: 10.1021/ja103932d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou YZ, Wang SL, Zhang YK. Catalytic Reaction Mechanism of Acetylcholinesterase Determined by Born-Oppenheimer Ab Initio QM/MM Molecular Dynamics Simulations. J. Phys. Chem. B. 2010;114:8817–8825. doi: 10.1021/jp104258d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith GK, Ke ZH, Guo H, Hengge AC. Insights into the Phosphoryl Transfer Mechanism of Cyclin-Dependent Protein Kinases from ab Initio QM/MM Free-Energy Studies. J. Phys. Chem. B. 2011;115:13713–13722. doi: 10.1021/jp207532s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lior-Hoffmann L, Wang LH, Wang SL, Geacintov NE, Broyde S, Zhang YK. Preferred WMSA catalytic mechanism of the nucleotidyl transfer reaction in human DNA polymerase kappa elucidates error-free bypass of a bulky DNA lesion. Nucleic Acids Res. 2012;40:9193–9205. doi: 10.1093/nar/gks653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sirin GS, Zhou YZ, Lior-Hoffmann L, Wang SL, Zhang YK. Aging Mechanism of Soman Inhibited Acetylcholinesterase. J. Phys. Chem. B. 2012;116:12199–12207. doi: 10.1021/jp307790v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rooklin DW, Lu M, Zhang YK. Revelation of a Catalytic Calcium-Binding Site Elucidates Unusual Metal Dependence of a Human Apyrase. J. Am. Chem. Soc. 2012;134:15595–15603. doi: 10.1021/ja307267y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svedruzic ZM, Reich NO. The mechanism of target base attack in DNA cytosine carbon 5 methylation. Biochemistry. 2004;43:11460–11473. doi: 10.1021/bi0496743. [DOI] [PubMed] [Google Scholar]
- 38.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Iype L, Jain S, Fagan P, Marvin J, Padilla D, Ravichandran V, Schneider B, Thanki N, Weissig H, Westbrook JD, Zardecki C. The Protein Data Bank. Acta. Crystallogr. Sect. D-Biol. Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 39.Kumar S, Horton JR, Jones GD, Walker RT, Roberts RJ, Cheng X. DNA containing 4′-thio-2′-deoxycytidine inhibits methylation by HhaI methyltransferase. Nucleic Acids Res. 1997;25:2773–2783. doi: 10.1093/nar/25.14.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Case DA, D. TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, W. RC, Zhang W, Merz KM, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, W. KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Yang L, T. C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman a. P. A. AMBER. Vol. 10. University of California; San Francisco: 2008. [Google Scholar]
- 41.Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:W368–371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anandakrishnan R, Onufriev A. Analysis of basic clustering algorithms for numerical estimation of statistical averages in biomolecules. J. Comput. Biol. 2008;15:165–184. doi: 10.1089/cmb.2007.0144. [DOI] [PubMed] [Google Scholar]
- 43.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 45.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A 2nd Generation Force-Field for the Simulation of Proteins, Nucleic-Acids, and Organic-Molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 46.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins:Structure Function and Bioinformatics. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, Laughton CA, Orozco M. Refinenement of the AMBER force field for nucleic acids: Improving the description of alpha/gamma conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Markham GD, Norrby PO, Bock CW. S-Adenosylmethionine conformations in solution and in protein complexes: Conformational influences of the sulfonium group. Biochemistry. 2002;41:7636–7646. doi: 10.1021/bi025682c. [DOI] [PubMed] [Google Scholar]
- 49.Petersson GA, Bennett A, Tensfeldt TG, Allaham MA, Shirley WA, Mantzaris J. A Complete Basis Set Model Chemistry .1. The Total Energies of Closed-Shell Atoms and Hydrides of the 1st-Row Elements. J. Chem. Phys. 1988;89:2193–2218. [Google Scholar]
- 50.Petersson GA, Allaham MA. A Complete Basis Set Model Chemistry .2. Open-Shell Systems and the Total Energies of the 1st-Row Atoms. J. Chem. Phys. 1991;94:6081–6090. [Google Scholar]
- 51.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery J,JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. revision D.01 Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 52.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges - the Resp Model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 53.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 54.Price DJ, Brooks CL. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004;121:10096–10103. doi: 10.1063/1.1808117. [DOI] [PubMed] [Google Scholar]
- 55.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 56.Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1998;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 57.Becke AD. Density-Functional Thermochemistry .3. The Role of Exact Exchange. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 58.Zhang Y, Lee T, Yang W. A pseudobond approach to combining quantum mechanical and molecular mechanical methods. J. Chem. Phys. 1999;110:46–54. [Google Scholar]
- 59.Zhang YK. Improved pseudobonds for combined ab initio quantum mechanical/molecular mechanical methods. J. Chem. Phys. 2005;122:024114. doi: 10.1063/1.1834899. [DOI] [PubMed] [Google Scholar]
- 60.Zhang YK. Pseudobond ab initio QM/MM approach and its applications to enzyme reactions. Theor. Chem. Acc. 2006;116:43–50. [Google Scholar]
- 61.Shao Y, Molnar LF, Jung Y, Kussmann J, Ochsenfeld C, Brown ST, Gilbert ATB, Slipchenko LV, Levchenko SV, O’Neill DP, DiStasio RA, Lochan RC, Wang T, Beran GJO, Besley NA, Herbert JM, Lin CY, Van Voorhis T, Chien SH, Sodt A, Steele RP, Rassolov VA, Maslen PE, Korambath PP, Adamson RD, Austin B, Baker J, Byrd EFC, Dachsel H, Doerksen RJ, Dreuw A, Dunietz BD, Dutoi AD, Furlani TR, Gwaltney SR, Heyden A, Hirata S, Hsu CP, Kedziora G, Khalliulin RZ, Klunzinger P, Lee AM, Lee MS, Liang W, Lotan I, Nair N, Peters B, Proynov EI, Pieniazek PA, Rhee YM, Ritchie J, Rosta E, Sherrill CD, Simmonett AC, Subotnik JE, Woodcock HL, Zhang W, Bell AT, Chakraborty AK, Chipman DM, Keil FJ, Warshel A, Hehre WJ, Schaefer HF, Kong J, Krylov AI, Gill PMW, Head-Gordon M. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006;8:3172–3191. doi: 10.1039/b517914a. [DOI] [PubMed] [Google Scholar]
- 62.Ponder JW. TINKER, software Tools for molecular design. Version 4.2 ed Washington University in St. Louis; St. Louis. WA: 2004. [Google Scholar]
- 63.Zhang YK, Liu HY, Yang WT. Free energy calculation on enzyme reactions with an efficient iterative procedure to determine minimum energy paths on a combined ab initio QM/MM potential energy surface. J. Chem. Phys. 2000;112:3483–3492. [Google Scholar]
- 64.Hu P, Wang SL, Zhang YK. How do SET-domain protein lysine methyltransferases achieve the methylation state specificity? Revisited by Ab initio QM/MM molecular dynamics simulations. J. Am. Chem. Soc. 2008;130:3806–3813. doi: 10.1021/ja075896n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu H, Yang WT. Free energies of chemical reactions in solution and in enzymes with ab initio quantum mechanics/molecular mechanics methods. Annu. Rev. Phys. Chem. 2008;59:573–601. doi: 10.1146/annurev.physchem.59.032607.093618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beeman D. Some Multistep Methods for Use in Molecular-Dynamics Calculations. J. Comput. Phys. 1976;20:130–139. [Google Scholar]
- 67.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Phys. 1992;13:1011–1021. [Google Scholar]
- 68.Souaille M, Roux B. Extension to the weighted histogram analysis method: combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 2001;135:40–57. [Google Scholar]
- 69.Ferrenberg AM, Swendsen RH. New Monte-Carlo Technique for Studying Phase-Transitions. Phys. Rev. Lett. 1988;61:2635–2638. doi: 10.1103/PhysRevLett.61.2635. [DOI] [PubMed] [Google Scholar]
- 70.Merkiene E, Klimasauskas S. Probing a rate-limiting step by mutational perturbation of AdoMet binding in the Hhal methyltransferase. Nucleic Acids Res. 2005;33:307–315. doi: 10.1093/nar/gki175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Gara M, Roberts RJ, Cheng XD. A structural basis for the preferential binding of hemimethylated DNA by HhaI DNA methyltransferase. J. Mol. Biol. 1996;263:597–606. doi: 10.1006/jmbi.1996.0601. [DOI] [PubMed] [Google Scholar]
- 72.Cheng XD, Kumar S, Posfai J, Pflugrath JW, Roberts RJ. Crystal-Structure of the Hhal DNA Methyltransferase Complexed with S-Adenosyl-L-Methionine. Cell. 1993;74:299–307. doi: 10.1016/0092-8674(93)90421-l. [DOI] [PubMed] [Google Scholar]
- 73.Shieh FK, Reich NO. AdoMet-dependent methyl-transfer: Glu(119) is essential for DNA c5-cytosine methyltransferase M.Hhal. J. Mol. Biol. 2007;373:1157–1168. doi: 10.1016/j.jmb.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 74.Youngblood B, Shieh FK, Buller F, Bullock T, Reich NO. S-Adenosyl-L-methionine-dependent methyl transfer: Observable precatalytic intermediates during DNA cytosine methylation. Biochemistry. 2007;46:8766–8775. doi: 10.1021/bi7005948. [DOI] [PubMed] [Google Scholar]
- 75.Shieh FK, Youngblood B, Reich NO. The role of Arg165 towards base flipping, base stabilization and catalysis in M.Hhal. J. Mol. Biol. 2006;362:516–527. doi: 10.1016/j.jmb.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 76.Kumar S, Cheng XD, Klimasauskas S, Mi S, Posfai J, Roberts RJ, Wilson GG. The DNA (Cytosine-5) Methyltransferases. Nucleic Acids Res. 1994;22:1–10. doi: 10.1093/nar/22.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Posfai J, Bhagwat AS, Posfai G, Roberts RJ. Predictive Motifs Derived from Cytosine Methyltransferases. Nucleic Acids Res. 1989;17:2421–2435. doi: 10.1093/nar/17.7.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen L, Macmillan AM, Verdine GL. Mutational Separation of DNA-Binding from Catalysis in a DNA Cytosine Methyltransferase. J. Am. Chem. Soc. 1993;115:5318–5319. [Google Scholar]
- 79.Verdine GL. The flip side of DNA methylation. Cell. 1994;76:197–200. doi: 10.1016/0092-8674(94)90326-3. [DOI] [PubMed] [Google Scholar]
- 80.Cleland WW, Perry AF, Gerlt JA. The low barrier hydrgen bond in enzymatic catalysis. J. Biol. Chem. 1998;273:25529–25532. doi: 10.1074/jbc.273.40.25529. [DOI] [PubMed] [Google Scholar]
- 81.O’Gara M, Horton JR, Roberts RJ, Cheng X. Structures of HhaI methyltransferase complexed with substrates containing mismatches at the target base. Nat. Struct. Biol. 1998;5:872–877. doi: 10.1038/2312. [DOI] [PubMed] [Google Scholar]
- 82.Pomes R, Roux B. Structure and dynamics of a proton wire: A theoretical study of H+ translocation along the single-file water chain in the gramicidin a channel. Biophys. J. 1996;71:19–39. doi: 10.1016/S0006-3495(96)79211-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morrision RT, Boyd RN. Stereochemistry II. Stereoselective and Stereospecific Reactions, in Organic Chemistry. 6th ed Prentice-Hall; New Delhi: 1992. p. 377. [Google Scholar]
- 84.Lindstrom WM, Jr., Flynn J, Reich NO. Reconciling structure and function in HhaI DNA cytosine-C-5 methyltransferase. J. Biol. Chem. 2000;275:4912–4919. doi: 10.1074/jbc.275.7.4912. [DOI] [PubMed] [Google Scholar]
- 85.Vilkaitis G, Merkiene E, Serva S, Weinhold E, Klimasauskas S. The mechanism of DNA cytosine-5 methylation. Kinetic and mutational dissection of Hhai methyltransferase. J. Biol. Chem. 2001;276:20924–20934. doi: 10.1074/jbc.M101429200. [DOI] [PubMed] [Google Scholar]
- 86.Schramm VL. Enzymatic transition states, transition-state analogs, dynamics, thermodynamics, and lifetimes. Annu. Rev. Biochem. 2011;80:703–732. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.