

Abstract
Detection and interpretation of adverse signals during preclinical and clinical stages of drug development inform the benefit-risk assessment that determines suitability for use in real-world situations. This review considers some recent signals associated with diabetes therapies, illustrating the difficulties in ascribing causality and evaluating absolute risk, predictability, prevention, and containment. Individual clinical trials are necessarily restricted for patient selection, number, and duration; they can introduce allocation and ascertainment bias and they often rely on biomarkers to estimate long-term clinical outcomes. In diabetes, the risk perspective is inevitably confounded by emergent comorbid conditions and potential interactions that limit therapeutic choice, hence the need for new therapies and better use of existing therapies to address the consequences of protracted glucotoxicity. However, for some therapies, the adverse effects may take several years to emerge, and it is evident that faint initial signals under trial conditions cannot be expected to foretell all eventualities. Thus, as information and experience accumulate with time, it should be accepted that benefit-risk deliberations will be refined, and adjustments to prescribing indications may become appropriate.
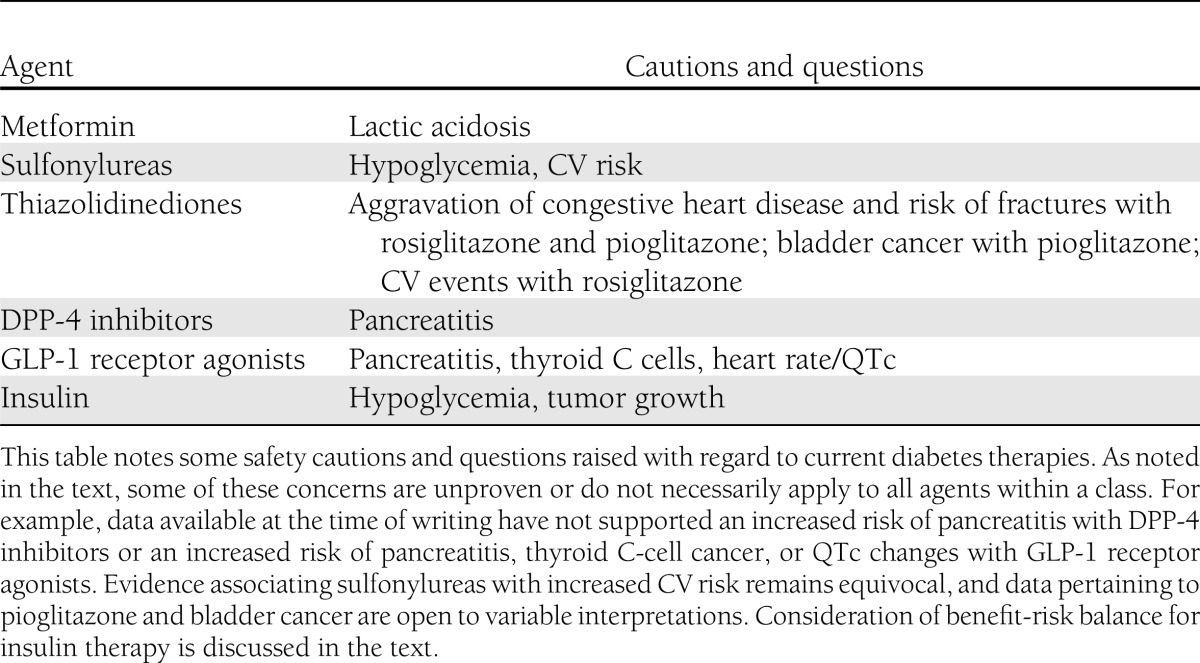
Development of a new pharmacotherapy typically follows a sequence of preclinical and clinical stages (Table 1). Completion of phase 3 marks the accumulation of clinical experience used to prepare an application for marketing authorization by a regulatory agency such as the Food and Drug Administration (FDA) or European Medicines Agency (EMA). This application should provide evidence for an adequately favorable benefit-risk balance for the intended use (1). Detection and interpretation of adverse properties during the development program are critical components of the authorization decision and guide the labeling and postmarketing obligations. The main safety questions raised about diabetes therapies are summarized in Table 2. This review examines the interpretation of adverse signals associated with recently approved blood glucose–lowering agents.
Table 1.
Stages in the development of a new drug
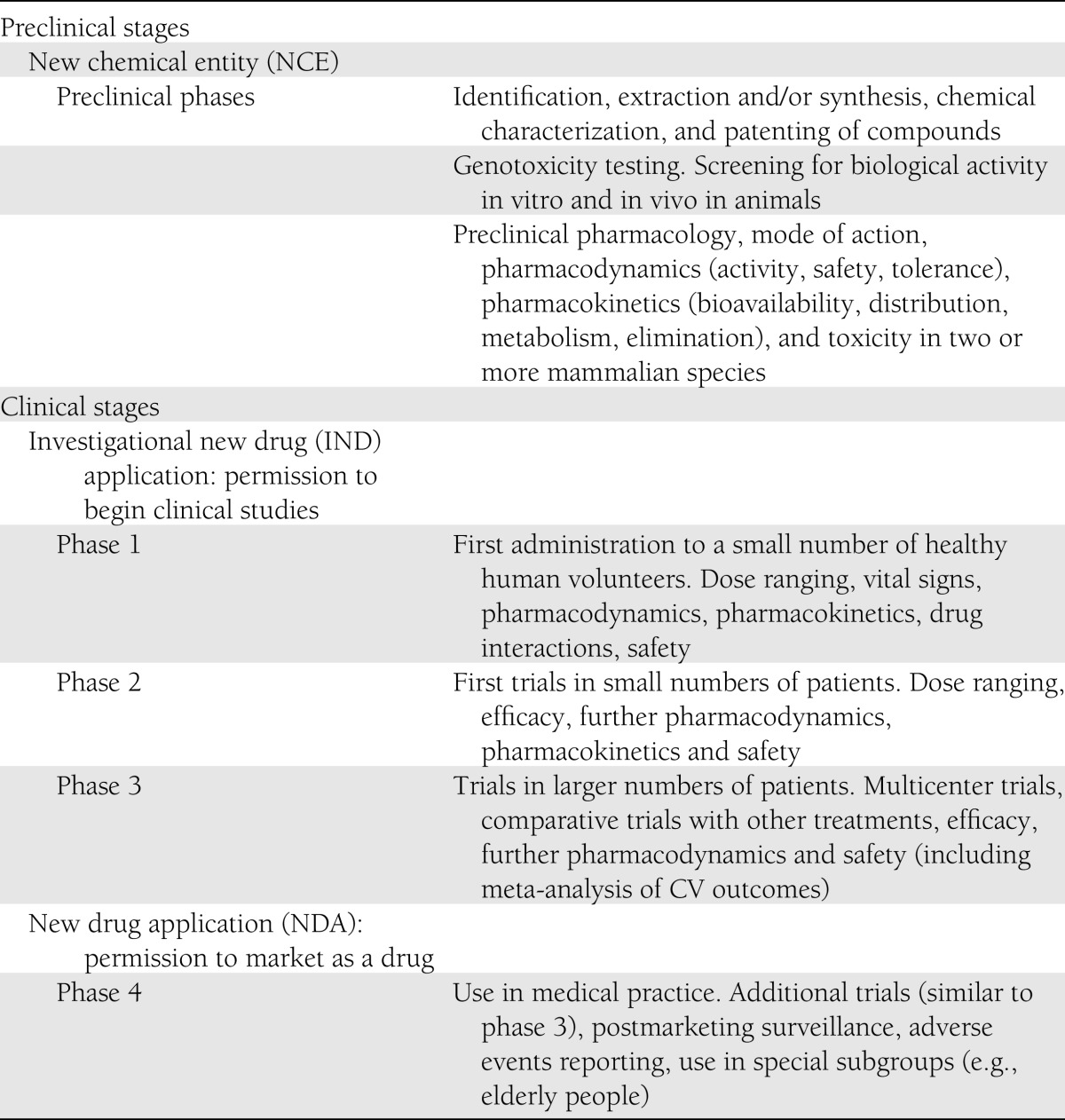
Table 2.
Safety cautions and questions raised about current diabetes therapies
PREAPPROVAL CLINICAL TRIALS
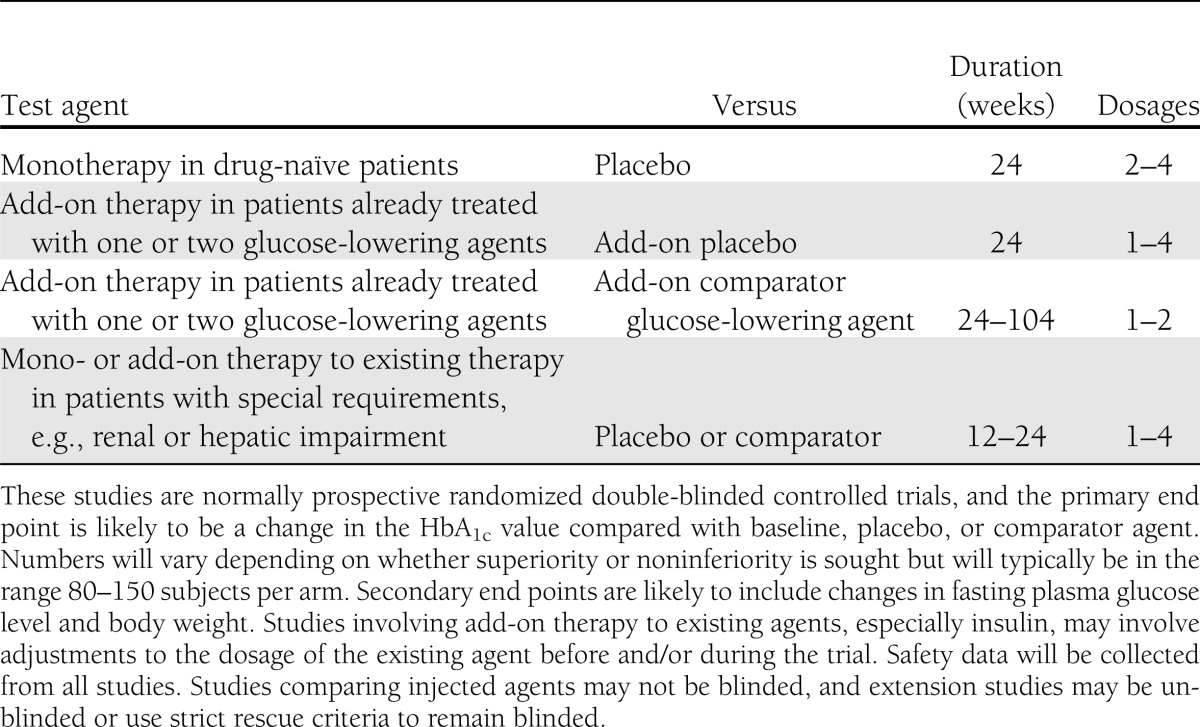
The journey from molecule to medicine is likely to take at least 10 years for a new class of agent and to cost ∼$500 million (1–3). If the many unsuccessful drug discovery studies and early development programs are factored in, the average approved drug probably reflects $1.3–1.8 billion of investment, so there is a strong incentive to secure marketing authorization once an agent has progressed through phase 3 (2,3). For a new diabetes medication, phase 3 usually takes ∼3 years and consumes up to 90% of the total development costs; hence, an agent that is entered into phase 3 has invariably passed very thorough scrutiny of phase 2 efficacy and safety data and an assessment of marketing potential. Phase 3 customarily involves a minimum of 1,500 patients treated with the test agent (Table 3), generating 1,000–3,000 patient-years of exposure and a database from which to consider benefits and risks for a real-world setting (4,5).
Table 3.
Typical phase 3 development program of clinical trials for an oral blood glucose–lowering medication for type 2 diabetes
DETERMINING BENEFITS AND RISKS
Although it is well recognized that early and sustained reductions of HbA1c markedly reduce microvascular complications and may contribute to reduced macrovascular risk, about one-half of diabetic patients still do not achieve or maintain sufficient glycemic control to avoid substantial long-term morbidity (6–10). New types of glucose-lowering agents are particularly required for type 2 diabetes because of the multivariable mix of genetic and environmental factors that conspire to create a progressive and highly heterogeneous natural history, with at least eight major organ systems being etiopathogenically implicated (11).
With regard to benefit, it is unlikely that an agent will be submitted for marketing authorization if it does not meet recognized approvable efficacy criteria. The generally accepted efficacy surrogate is a reduction in HbA1c that is commensurate with the baseline HbA1c of the patient population studied (greater improvements of glycemic control expected if higher baseline hyperglycemia), taking into account the concomitant health issues within that population and additional benefits of the agent (12). The favorable–unfavorable boundary of benefits and risks will reflect whether efficacy is comparable with or better than existing therapies, achieved by a new mode of action that can be substituted for or add compatibly to these therapies, or assist subpopulations and comorbid conditions inadequately served by existing therapies. Set against this, we have the frequency and severity of apparent adverse effects, their predictability, potential avoidance, minimization or rectification, and practicalities of identifying and containing risk during real-world usage. Beyond this, theoretical adverse effects might be contemplated with regard to mode of action or other known pharmacodynamic or pharmacokinetic properties (4,5,12).
Preregistration trials are mostly designed and powered for a primary efficacy end point, customarily a reduction in HbA1c over 6 months at two or more dosage strengths (Table 3). As durability of efficacy receives more attention, extended randomized controlled trials are envisioned, with greater use of add-on rescue therapy to enable prolonged double blinding, and greater acceptance of post hoc analyses may be required. However, there will always be a desire for more drug exposure to assess risk, especially that pertaining to more vulnerable subpopulations. This raises the issues of preregistration meta-analyses of trial data and the role of postmarketing studies.
Relative risk against a placebo or comparator (or sometimes compared with a separate study of similarly disposed patients) provides a convenient indicator for interpreting frequency and severity of adverse signals, but absolute risk is perhaps more likely to interest the patient and prescriber. Figure 1 illustrates the absolute risk (per 1,000 patient-years) for fatal and major comorbid complications of diabetes alongside various reported adverse events attributed to diabetes pharmacotherapies (13–28). This perspective hopefully will rationalize the inevitable limitations of a preregistration trial database as a first step for ongoing surveillance to identify uncommon, rare, or slowly emerging adverse properties, including long-term risks of fatal, life-threatening, or permanently disabling events (13). Tolerability and quality of life may not be strictly within the remit of safety, but they are issues that affect adherence and impinge on benefit-risk deliberations.
Figure 1.

Risk of diabetes complications and adverse events associated with diabetes drug therapies expressed as absolute risk per 1,000 patient-years. Black bars show representative values for risk in diabetic patients, and gray bars indicate excess risk of events in patients treated with specific glucose-lowering medications relative to diabetic patients receiving comparator therapies. Incidence rates are subject to considerable variation with age, duration of diabetes, and concomitant morbidity. Adapted with permission from Bergenstal et al. (13). Data were derived from references 13–28. ACCORD, Action to Control Cardiovascular Risk in Diabetes; CHF, congestive heart failure; PVD, peripheral vascular disease; UKPDS, U.K. Prospective Diabetes Study.
Because diabetes is for life, comorbid conditions are rife, and therapies mostly last for much longer than the trial periods, the rigorous safety cautions for a new drug approval often require a comprehensive postmarketing risk management program to accompany standard pharmacovigilance monitoring (5). These can include small studies in special populations and crucial attention to issues of suspicion from preregistration signals, such as trends in biomarkers together with safety-first labeling restrictions. Accepting a caveat for the unpredictable, black box warnings can serve a valuable precautionary role to minimize misuse.
HYPOGLYCEMIA
Treatment with antidiabetic drugs commonly produces mild hypoglycemic symptoms, especially with insulin, sulfonylureas, or combinations involving these classes (29). Signs of moderate or severe hypoglycemia and extra risks associated with patient unawareness or use in vulnerable groups, such as the elderly or renally impaired, are well rehearsed in product labeling and education packages. Nevertheless propensity for hypoglycemia forms an integral part of the calculation of acceptable risk for all diabetes pharmacotherapies (12). Where clinically significant risk is anticipated with combination therapy, this is typically minimized by recommending down-titration of the existing agent to coincide with the addition of the new agent. The interpretation of hypoglycemic events is usefully assisted by mode-of-action studies, particularly for agents that raise in-sulin concentration, to check whether the already-impaired counterregulatory capability in diabetic hypoglycemic states is further compromised (30).
Unfortunately, the quantification of hypoglycemic risk in clinical trials continues to lack conformity, making it difficult to compare rates of hypoglycemia between studies. Nonsevere hypoglycemia might include symptoms or signs that are self-managed by the patient with or without a blood glucose measurement of <70 mg/dL (<3.9 mmol/L) down to 50 mg/dL (2.8 mmol/L). Severe hypoglycemia is usually distinguished by the involvement of third-party assistance or a blood glucose measurement of <54 mg/dL (<3 mmol/L). However, whether the patient requires (as opposed to requests) this assistance is often vague, and it is sometimes difficult to obtain a blood glucose measurement at these times. A further problem is nocturnal hypoglycemia, which may pass unrecognized unless inconveniently interrupted with a wake-up and a glucose test (29). Thus, small differences in nocturnal hypoglycemia reported during comparator insulin trials cannot be overinterpreted when assessing hypoglycemic risk for licensing purposes (12). A combined analysis of data from several trials can be useful for this purpose, but confirmation requires a continuous glucose monitoring study.
WEIGHT CHANGES
Although increased adiposity with insulin, sulfonylureas, and thiazolidinediones is sometimes overlooked as a risk factor, the escalation in vascular susceptibility that accompanies coexistent diabetes and obesity is a quantifiable and important component of overall risk (31). Additionally, rapid weight gain may signal issues other than adiposity, such as fluid retention with thiazolidinediones (32). The trade-off between improved glycemic control and adipose weight gain is a perennial treatment conundrum for which only individualized advice can be recommended, bearing in mind that dietary measures to counter the weight gain can aggravate susceptibility to hypoglycemia (33). Rapid initial weight loss during a clinical trial may lead to a detection bias for certain tumors, as discussed in a subsequent section of this review (34,35).
CARDIOVASCULAR RISK
Because cardiovascular (CV) diseases are highly prevalent and the most common cause of premature death among individuals with diabetes (Fig. 1), considerable attention has been directed toward CV risk assessment and minimization. However, being able to attribute causality to events that are not unexpected in the diabetic population is not a precise science (7). To illustrate the difficulties, we can reflect on the University Group Diabetes Program (UGDP) study of the 1960s. Although this brought to attention the lactic acidosis issue with phenformin (prompting further investigation that led to the first diabetes drug withdrawal by the FDA in 1978), it also initiated concern about possible detrimental CV effects of sulfonylureas (36). Despite a further 4 decades of very extensive usage and endless clinical trials and database analyses, the overall balance of evidence is still equivocal (13). Previous accusations that insulin is atherogenic have since been dismissed, but they complicated risk assessments in a bygone era (37). They also remind us that treatments that sustain life expectancy among patients in poor general health can result in emergent disease-related morbid conditions that should not be confused with iatrogenic morbid conditions.
Interpreting adverse CV signals was brought to the fore with rosiglitazone, which after almost 10 years of postmarketing experience, created sufficiently ambiguous adverse event data that the drug was withdrawn in Europe, but the FDA settled for tighter labeling (38,39). Although relevant CV event data were available in the GlaxoSmithKline clinical study register website and received updated evaluation by regulatory agencies (40), it was a meta-analysis in the New England Journal of Medicine that attracted attention (26). With data from 42 randomized trials, excluding six studies in which there were no CV events, this analysis reported that rosiglitazone was associated with an increased odds for myocardial infarction (MI) (odds ratio 1.43, 95% CI 1.03–1.98) and CV death (1.64, 0.98–2.74). Expressed as events/total patients for rosiglitazone versus control groups, these odds ratios corresponded to 86/14,372 versus 72/11,634 for MI and 39/10,936 versus 22/9,509 for CV deaths. The analyses have been repeated and debated extensively elsewhere (41,42) and will not be reiterated further here, but some generic lessons for interpreting signals during development programs are highlighted. When the FDA advisory committee initially deliberated the new drug application for rosiglitazone, there were data on CV events from five randomized trials. The occurrence of MI was 6/1,967 and 3/793 for the rosiglitazone and control groups, respectively, corresponding to 0.30 and 0.37%, respectively. The occurrence of CV deaths was 2/1,967 and 0/793 for the rosiglitazone and control groups, respectively. So there was no obvious signal, and biomarker data tended to be positive, although fluid retention, edema, risk of heart failure, and weight gain were dutifully considered. At the time, attention was distracted toward the liver because another thiazolidinedione, troglitazone, was in the spotlight for idiosyncratic hepatotoxicity (43). With regard to the approval of thiazolidinediones, Europe evaluated these agents a little later and made approvals for second-line indication (unless metformin was not appropriate) and excluded New York Heart Association class I–IV (vs. first-line approval and exclusion of New York Heart Association class III and IV by the FDA).
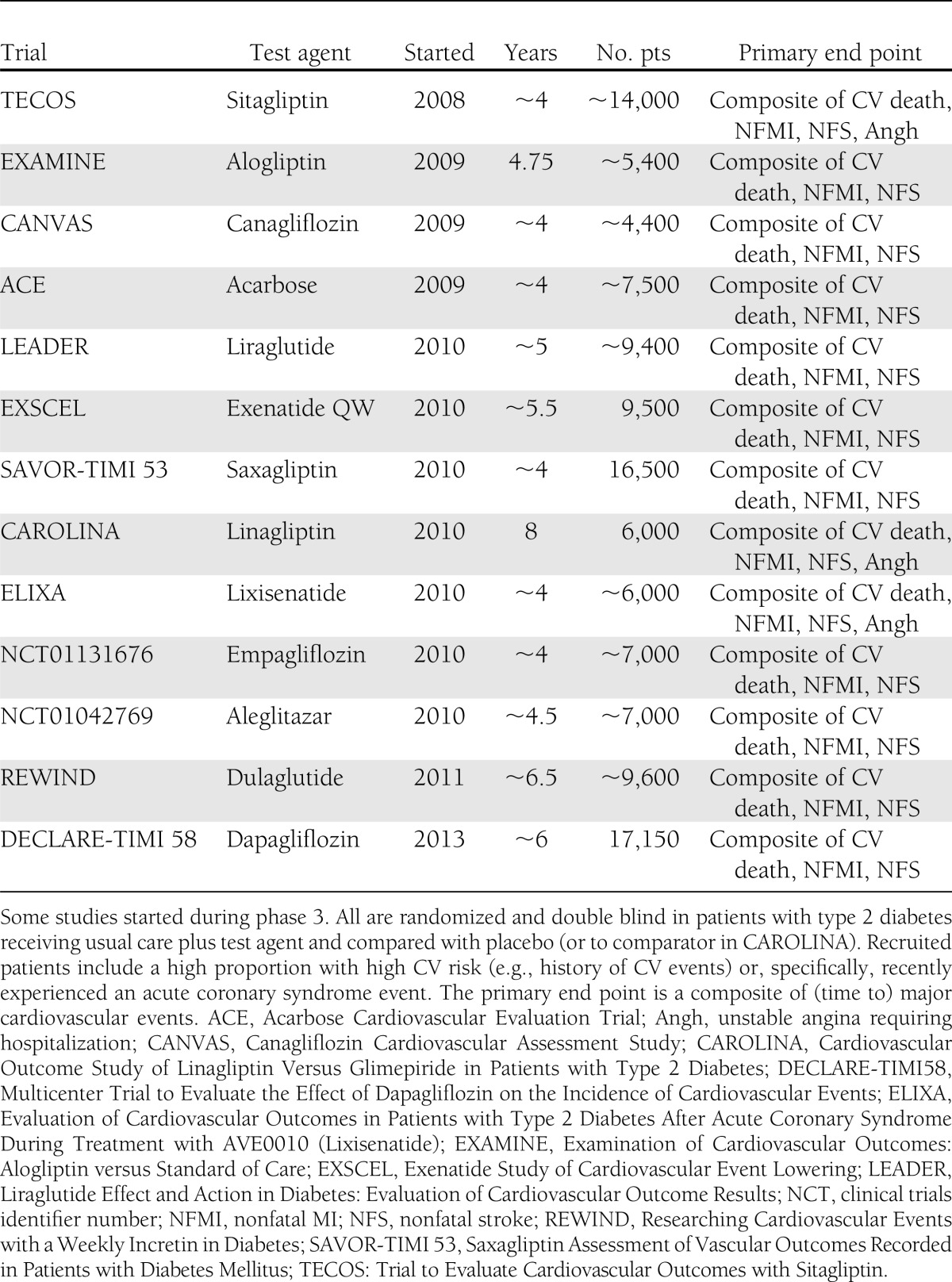
The rosiglitazone experience prompted the FDA to issue new guidance about CV risk (44). This requires a meta-analysis of important CV events in phase 2/3 to achieve an upper 95% CI of <1.3 to qualify for approval without requiring a postmarketing CV trial, provided that overall benefit and risk support approval (Fig. 2). If the upper 95% CI is >1.8, additional phase 3 safety studies are required before resubmission for marketing authorization. If the upper CI lies between 1.3 and 1.8 and approval is otherwise appropriate, then a postmarketing CV events study generally will be necessary and required to show an upper 95% CI of <1.3. In practice, each sponsor of recently approved drugs has elected to undertake such a study (or have been encouraged to do so by the FDA), even if the phase 2/3 CV events conform to an upper 95% CI <1.3. Indeed, such postmarketing studies appear to be almost obligatory because no sponsor company would wish for its product to be disadvantaged in years to come if its competitors can claim hard end point CV data from large purpose-designed trials. These studies, which are currently ongoing, are summarized in Table 4 (45). Although they are mostly event driven, differences in estimated event rates and duration, selection of major CV events, power calculations, types of statistics (superiority vs. noninferiority), and use of placebo, an active comparator, or both make direct comparisons between studies difficult.
Figure 2.

FDA guidance for industry for a meta-analysis of phase 2/3 data to evaluate CV risk with new glucose-lowering therapies to treat type 2 diabetes. Based on ref. 44. *If overall benefit-risk analysis of the drug supports approval. RR, relative risk.
Table 4.
Postmarketing CV outcome studies with diabetes therapies
Effective use of antihypertensive and lipid-controlling medications has reduced the rates for major adverse CV events among diabetic patients (46), which in turn will affect the spread of CIs in an analysis of CV events, especially in phase 2/3 studies, where the hypothetical wonder drug with no events would defy the statistical analysis. To boost event rates, the recruitment of extra patients at high CV risk may alter the primary–secondary preventive balance of the risk assessment. The importance of time in the generation of CV events must also be borne in mind because events occurring early in trials may more strongly reflect the accumulated risk in the months and years before trial entry rather than reflect the initial effects of the test drug. For example, in a more recent analysis of the rosiglitazone CV event data, it is evident that short-term trials give very different and far-worse outcomes than longer-term studies (47). Moreover, several large trials of glycemic intensification have found that CV event rates are initially worse before they subsequently show benefit (48,49), whereas others suggest a possible association between CV events and the extent and speed of intensification (7).
Dealing with CV changes that are not necessarily included as major adverse cardiac events requires a different approach and is illustrated for some GLP-1 receptor agonists, which tend to slightly increase heart rate (sharp cardiological intake of breath) but decrease blood pressure slightly (possible cardiological sigh of relief). GLP-1 receptors were identified in the vasculature and myocardium, so small bespoke safety studies were warranted. These studies focused on corrected QT (QTc) interval duration, which reassuringly showed no clinically significant alterations (50,51).
CANCER
Whereas CV events are common in diabetes, cancers are uncommon or rare but modestly increased (overall by ∼40%) (52). Detection is often delayed, and the attributing cause is complicated by familial susceptibility; present and prior obesity; history of exposure to carcinogens, including smoking; and comorbid conditions. Allocation factors can also confuse the evidence surrounding malignancies; for example, an uneven randomization for ethnicity, gender, socioeconomic status, and educational or geographical background can significantly influence the occurrence of cancers. Many patients who enter clinical trials will not have previously received the type of vigilant, frequent attention of a health care professional inviting them to report anything different since the last visit. For example, rapid weight loss during the early development trials with orlistat was associated with an increased detection of breast tumors (34,35) and led to extensive studies of possible carcinogenicity, which had negative findings. Additionally, protracted studies confirmed that the risk of breast tumors did not increase with time but rather diminished, suggesting that the combination of weight loss and attentive clinical care in a trial setting conferred a detection bias for breast cancer—a valuable benefit for the patients but a substantial delay for drug development (34,35).
Pancreatic cancer
Deciding the proportionate level of caution to ascribe to a potential danger signal is never straightforward. Take for example the evidence for a putative link between incretin therapies (GLP-1 receptor agonists and DPP-4 inhibitors) and pancreatitis, pancreatic ductal metaplasia, and pancreatic cancer (28,53–56). Reports of cases of acute pancreatitis in patients receiving incretin therapies generated awareness, which may have prompted further similar reports and an accumulation of cases in the pharmacovigilance databases (57,58). However, the diagnosis of pancreatitis was not always confirmed; severity was highly variable; and although the condition is known to be more common among diabetic patients than among the nondiabetic population, estimated incidence rates have varied widely from <1 to ∼5 per 1,000 patient-years (13,17,57). Given a need to await additional retrospective interrogation of large databases, the labeling advice to discontinue incretins in patients where pancreatitis is suspected would seem proportionate.
It is relevant to note here that pharmacovigilance is an ongoing process to monitor side effects, but a reported event is not necessarily caused by the medicines to which the report relates and should not be interpreted as meaning that a medicine is unsafe to use (57,58). Spontaneous reporting of suspected side effects should not be used as a basis for estimating incidence rates because they provide a numerator without knowing the denominator. Reporting rates are variable for many reasons, events are not confirmed, and confounding factors may not be taken into account (5,58).
An appreciation that pancreatitis presents a risk for pancreatic cancer (59), coupled with an animal study suggesting that incretins might cause pancreatic duct cell proliferation, sparked interest in a potential link between incretins and pancreatic cancer (55,60). Additional studies in several animal species could not confirm the ductal proliferation (61), but the FDA database showed more cases of pancreatic cancer in patients who had received certain incretins compared with other classes of diabetes drugs (62). The problem of attempting numerical analyses from spontaneous adverse event reporting has already been noted, but could this be a signal? Pancreatic cancer is rare at <0.1 per 1,000 patient-years. It is slow to develop to detection, and unlike other cancers, its occurrence tends to decrease with time after diagnosis of diabetes (62). So, numbers and time may preclude an early resolution from postmarketing trials. In general, clinical experience to date does not appear to support an association of incretins with pancreatic cancer (63,64), but the issue has emphasized that procedures for extrapolating preclinical signals to clinical situations and projecting possible clinical signals into future clinical events are still tenuous.
Thyroid cancer
An example of scrambled signals is that of a purported link between liraglutide and thyroid C-cell medullary cancer. Thyroid C cells of rats and mice express high concentrations of GLP-1 receptors and respond to high concentrations of GLP-1 receptor agonists with excess calcitonin secretion and C-cell hyperplasia (65). In clinical studies, liraglutide slightly raised calcitonin concentrations, but human thyroid C cells have a low expression of GLP-1 receptors, and calcitonin concentrations were not elevated to an extent that is considered a signal for medullary C-cell cancer (66). Bearing in mind that such cancers are rare, ∼600–1,000 annually in the U.S., a clinical trial is not an option. Sensibly, the FDA explained its reasoning in a high-profile journal, a move that was similarly useful with regard to metformin and lactic acidosis some 15 years ago (67).
Bladder cancer
Interpreting preclinical and clinical signals suggestive of a bladder cancer risk with pioglitazone is still difficult after more than one decade of accumulated evidence. Animal studies suggested a metabolite that could irritate the urothelium and cause localized hyperplasia (68), and a major clinical trial (PROactive [Prospective Pioglitazone Clinical Trial in Macrovascular Events]) noted an increased occurrence of bladder tumors (25), but no excess was observed after a 6-year observational follow-up (69). Several meta-analyses based around five large cohort studies totaling >2.3 million diabetic patients have shown a small increased risk of bladder tumors (1.17–2.22 fold), with a slightly higher risk (1.3–1.4 fold) in those exposed for >2 years (70,71). Given a bladder cancer incidence of ∼0.5 per 1,000 patient-years in diabetes, these studies indicated an increased risk of ∼0.1 per 1,000 patient-years. However, within these studies, there were difficulties in assessing prior exposure to key risk factors, including smoking, the time taken for these cancers to develop, and variability in detection. Additionally, the risk of some other cancers may have been reduced in patients receiving pioglitazone, leaving no apparent overall cancer imbalance (72). Some countries in Europe decided to discontinue pioglitazone, but the EMA has not endorsed this, preferring to modify the label to recommend avoidance of the drug in patients with active bladder cancer and in those at high risk (73). Thus, we have another example of varying interpretations by different expert groups assessing the same information and weighing the extent of adverse signals and events against the benefits.
Another cancer question has been raised for the sodium glucose cotransporter-2 inhibitor dapagliflozin, which was recently approved in Europe and Australia. Although there was no evidence of mutagenicity or carcinogenicity in preclinical trials and no overall imbalance in malignancies during the development program, there were increases in breast, prostate, and bladder cancers (74). Given the weight loss associated with this agent, the detection bias discussed for orlistat may apply, and the prostate cancers (mostly well advanced) were generally discovered early in the trials when scrutiny of urinary phenomena brought to attention hematuria and urination difficulties. A history of hematuria recently before or at randomization was recorded for 7 of 10 cases of bladder cancer; these cancers were mostly detected too early and were too advanced to have originated after treatment with dapagliflozin. Whether persistently high glucose in the urine could promote growth of preexisting tumors is not entirely excluded, but extensive preclinical studies have not shown a signal, and patients with familial renal glucosuria do not appear to experience detrimental effects of lifelong glucosuria (75).
Insulin and cancer
The evidence with regard to insulin and cancer is far too extensive and controversial to dissect here (52,76–79). There are so many confounders in most of the studies that it is possible to question and criticize any interpretation. Thus are the shaky foundations upon which adverse events have to be assessed. Insulin is not of itself carcinogenic, but through its interactions with the different isoforms of the insulin receptor and the IGF1 receptor, it stands under suspicion of promoting tumor growth. By way of summary and excluding less solid evidence, it appears that replacement amounts of human insulin do not increase risk. However, this conclusion cannot yet be extended to the massive pharmacological doses needed for highly insulin resistant individuals who likely carry an abundance of other risks and for whom alternative glucose-lowering therapies are not available. Analog insulins in present use do not appear to be any less safe in this context, as most recently reported by the ORIGIN (Outcome Reduction with Initial Glargine Intervention) trial (80). For the record, most circulating glargine is dearginated to a low-affinity binding metabolite (M1) before it meets tissue receptors (79,81). Also recall that the highest endogenous exposure to insulin is in the liver, and this is likely to have been exaggerated during a hyperinsulinemic prediabetes period, whereas subcutaneous administration of insulin alters the disposition of insulin in favor of other tissues. A consensus report of the American Diabetes Association and American Cancer Society reasonably advises that on the balance of current evidence, cancer risk should not be a major factor in choosing between available diabetes therapies for the average patient, although patients with a very high risk for cancer occurrence or recurrence may require more careful consideration (82).
BONE FRACTURES
Thiazolidinediones have been associated with declining bone mineral density and increased risk of bone fractures, especially in older women (13,83–85). Although thiazolidinediones continue to receive careful clinical investigation, they provide a further example of the importance of preclinical mechanistic studies. In vitro and animal data have shown effects of peroxisome proliferator-activated receptor-γ (PPARγ) agonists on the differentiation pathways of various cell types populating bone (86). Although the various biomarkers of bone health can be used effectively for diagnosis and therapeutic monitoring, there are considerable limitations to their predictive value for the individual, and interpretation of small changes during preregistration trials remains uncertain (87,88).
CONCLUSIONS
A particular challenge when interpreting adverse signals in clinical trials is the need to extrapolate to a real-world environment where patient populations may be more diverse, prescribers less specialized, and monitoring less conscientious. Faint signals from preregistration trials can take a decade or more to reveal their clinical consequences and often are unpredictable and confounded by allocation and detection bias, prior exposure to risk factors (known and unknown), and limitations of time and numbers.
The need to revise the label or even withdraw a drug should not be viewed as a failure of foresight but, rather, as the mark of a vigilant and responsive regulatory process. To minimize risk, new medicines have explicit exclusions reinforced with temporary (and sometimes permanent) black box warnings that permit availability to defined patient populations while allowing time for separate controlled studies with more vulnerable or different subpopulations. Mounting pressures to make all clinical trial information available and all databases accessible to everyone will introduce opportunities for further misinterpretation. Transparency requires responsibility, but benefit and risk often are a judgment of probability, and revisions have to be accepted without prejudice as new information emerges. The teachings of the 16th century Swiss-German physician Paracelsus remind us that no chemical is absolutely safe, and the difference between a medicine and a poison can be the dose.
Acknowledgments
C.J.B. has undertaken ad-hoc consultancy for Bristol-Myers Squibb, AstraZeneca, Merck Sharp & Dohme, Novo Nordisk, sanofi-aventis, Janssen, Eli Lilly, Roche, and Takeda; delivered continuing medical education programs sponsored by Bristol-Myers Squibb, AstraZeneca, GlaxoSmithKline, Merck Serono, Merck Sharp & Dohme, Eli Lilly, and Boehringer Ingelheim; and received travel or accommodation reimbursement from AstraZeneca and Bristol-Myers Squibb. No other potential conflicts of interest relevant to this article were reported.
C.J.B. was sole author, researched the data, and prepared the manuscript.
This article is based on a State-of-the-Art lecture delivered at the 72nd Scientific Sessions of the American Diabetes Association, Philadelphia, Pennsylvania, 8–12 June 2012.
C.J.B. served as an expert witness for the FDA, EMA, and Medicines and Healthcare Products Regulatory Agency (MHRA), providing benefit-risk assessments and testimony. He also served as a representative of the European Association for the Study of Diabetes to EMA. His research has contributed to the development of diabetes and obesity medicines with preclinical and clinical studies, and he is well known for his early work on metformin.
Footnotes
See accompanying articles, pp. 1804, 1823, 2118, and 2126.
References
- 1.Bailey CJ. Future drug treatment for type 2 diabetes. In Textbook of Diabetes. 4th ed. Holt R, Cockram C, Flyvberg A, Goldstein B, Eds. Oxford, Blackwell, 2010, p. 1017–1044 [Google Scholar]
- 2.Kaitin KI, DiMasi JA. Pharmaceutical innovation in the 21st century: new drug approvals in the first decade, 2000-2009. Clin Pharmacol Ther 2011;89:183–188 [DOI] [PubMed] [Google Scholar]
- 3.Roy ASA. Manhattan Institute for Policy Research, Project FDA Report, No. 5. Stifling new cures: the true cost of lengthy clinical drug trials [article online], 2012. Available from http://www.manhattan-Institute.org/html/fda_05.htm Accessed 28 December 2012
- 4.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for industry. Premarketing risk assessment [Internet], 2005. Available from http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm126958.pdf Accessed 28 December 2012
- 5.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for industry. Good pharmacovigilance practices and pharmacoepidemiologic assessment [Internet], 2005. Available from http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126834.pdf Accessed 28 December 2012
- 6.Nathan DM, Cleary PA, Backlund JY, et al. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005;353:2643–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skyler JS, Bergenstal R, Bonow RO, et al. American Diabetes Association. American College of Cardiology Foundation. American Heart Association Intensive glycemic control and the prevention of cardiovascular events: implications of the ACCORD, ADVANCE, and VA diabetes trials: a position statement of the American Diabetes Association and a scientific statement of the American College of Cardiology Foundation and the American Heart Association. Diabetes Care 2009;32:187–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008;359:1577–1589 [DOI] [PubMed] [Google Scholar]
- 9.Stratton IM, Adler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000;321:405–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Pablos-Velasco P, Parhofer KG, Bradley C, et al. Current level of glycaemic control and its associated factors in patients with type 2 diabetes across Europe: data from the PANORAMA study. Clin Endocrinol (Oxf). In press [DOI] [PubMed] [Google Scholar]
- 11.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009;58:773–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for industry diabetes mellitus: developing drugs and therapeutic biologics for treatment and prevention [Internet], 2008. Available from http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071624.pdf Accessed 7 January 2013
- 13.Bergenstal RM, Bailey CJ, Kendall DM. Type 2 diabetes: assessing the relative risks and benefits of glucose-lowering medications. Am J Med 2010;123:374.e379–374.e318 [DOI] [PubMed]
- 14.Baliga V, Sapsford R. Review article: diabetes mellitus and heart failure—an overview of epidemiology and management. Diab Vasc Dis Res 2009;6:164–171 [DOI] [PubMed] [Google Scholar]
- 15.Foley RN, Murray AM, Li S, et al. Chronic kidney disease and the risk for cardiovascular disease, renal replacement, and death in the United States Medicare population, 1998 to 1999. J Am Soc Nephrol 2005;16:489–495 [DOI] [PubMed] [Google Scholar]
- 16.Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998;339:229–234 [DOI] [PubMed] [Google Scholar]
- 17.Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009;32:834–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trautner C, Icks A, Haastert B, Plum F, Berger M. Incidence of blindness in relation to diabetes. A population-based study. Diabetes Care 1997;20:1147–1153 [DOI] [PubMed] [Google Scholar]
- 19.Lewis JD, Ferrara A, Peng T, et al. Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes Care 2011;34:916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerstein HC, Miller ME, Byington RP, et al. Action to Control Cardiovascular Risk in Diabetes Study Group Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008;358:2545–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.UK Prospective Diabetes Study (UKPDS) Group Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998;352:837–853 [PubMed] [Google Scholar]
- 22.Hampton T. Diabetes drugs tied to fractures in women. JAMA 2007;297:1645. [DOI] [PubMed] [Google Scholar]
- 23.Meier C, Kraenzlin ME, Bodmer M, Jick SS, Jick H, Meier CR. Use of thiazolidinediones and fracture risk. Arch Intern Med 2008;168:820–825 [DOI] [PubMed] [Google Scholar]
- 24.Lago RM, Singh PP, Nesto RW. Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidinediones: a meta-analysis of randomised clinical trials. Lancet 2007;370:1129–1136 [DOI] [PubMed] [Google Scholar]
- 25.Patel A, MacMahon S, Chalmers J, et al. ADVANCE Collaborative Group Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008;358:2560–2572 [DOI] [PubMed] [Google Scholar]
- 26.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 2007;356:2457–2471 [DOI] [PubMed] [Google Scholar]
- 27.Bailey CJ, Turner RC. Metformin. N Engl J Med 1996;334:574–579 [DOI] [PubMed] [Google Scholar]
- 28.Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 2009;25:1019–1027 [DOI] [PubMed] [Google Scholar]
- 29.Ben Salem C, Fathallah N, Hmouda H, Bouraoui K. Drug-induced hypoglycaemia: an update. Drug Saf 2011;34:21–45 [DOI] [PubMed] [Google Scholar]
- 30.Zammitt NN, Frier BM. Hypoglycemia in type 2 diabetes: pathophysiology, frequency, and effects of different treatment modalities. Diabetes Care 2005;28:2948–2961 [DOI] [PubMed] [Google Scholar]
- 31.Bailey CJ. The challenge of managing coexistent type 2 diabetes and obesity. BMJ 2011;342:d1996 [DOI] [PubMed]
- 32.Nesto RW, Bell D, Bonow RO, et al. American Heart Association. American Diabetes Association Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. October 7, 2003. Circulation 2003;108:2941–2948 [DOI] [PubMed] [Google Scholar]
- 33.Inzucchi SE, Bergenstal RM, Buse JB, et al. American Diabetes Association (ADA) European Association for the Study of Diabetes (EASD) Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012;35:1364–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malone M. Orlistat Drugs 1998;56:232–241 [PubMed] [Google Scholar]
- 35.McNeely W, Benfield P. Orlistat. Drugs 1998;56:241–249; discussion 250 [DOI] [PubMed] [Google Scholar]
- 36.Kilo C, Williamson JR. The controversial American University Group Diabetes Study—a look at sulfonylurea and biguanide therapy. Horm Metab Res Suppl 1985;15(Suppl. 15):102–104 [PubMed] [Google Scholar]
- 37.Boyne MS, Saudek CD. Effect of insulin therapy on macrovascular risk factors in type 2 diabetes. Diabetes Care 1999;22(Suppl. 3):C45–C53 [PubMed] [Google Scholar]
- 38.European Medicines Agency. European Medicines Agency recommends suspension of Avandia, Avandamet and Avaglim [Internet], 2010. Available from http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2010/09/news_detail_001119.jsp Accessed 7 January 2013
- 39.Rosiglitazone (Avandia) [package insert]]. Research Triangle Park, NC, GlaxoSmithKline, 2008. Available from http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021071s034lbl.pdf Accessed 7 January 2013
- 40.FDA document. Advisory committee meeting for NDA 21071 Avandia (rosiglitazone maleate) tablet [Internet], 2010. Available from http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM218493.pdf Accessed 7 January 2013
- 41.Loke YK, Kwok CS, Singh S. Comparative cardiovascular effects of thiazolidinediones: systematic review and meta-analysis of observational studies. BMJ 2011;342:d1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Psaty BM, Furberg CD. Rosiglitazone and cardiovascular risk. N Engl J Med 2007;356:2522–2524 [DOI] [PubMed] [Google Scholar]
- 43.Bailey CJ. The rise and fall of troglitazone. Diabet Med 2000;17:414–415 [DOI] [PubMed] [Google Scholar]
- 44.U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for industry. Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes [Internet], 2008. Available from http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/%20Guidances/UCM071627.pdf Accessed 7 January 2013
- 45.Bailey CJ, Day C, Campbell IW, Schindler C. Glycaemic control and cardiovascular outcome trials in type 2 diabetes. Br J Diabetes Vasc Dis 2012;12:161–164 [Google Scholar]
- 46.Preiss D, Sattar N, McMurray JJ. A systematic review of event rates in clinical trials in diabetes mellitus: the importance of quantifying baseline cardiovascular disease history and proteinuria and implications for clinical trial design. Am Heart J 2011;161:210–219.e1 [DOI] [PubMed] [Google Scholar]
- 47.Nissen SE, Wolski K. Rosiglitazone revisited: an updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch Intern Med 2010;170:1191–1201 [DOI] [PubMed] [Google Scholar]
- 48.Dormandy JA, Charbonnel B, Eckland DJ, et al. PROactive investigators Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet 2005;366:1279–1289 [DOI] [PubMed] [Google Scholar]
- 49.Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med 2008;358:580–591 [DOI] [PubMed] [Google Scholar]
- 50.Chatterjee DJ, Khutoryansky N, Zdravkovic M, Sprenger CR, Litwin JS. Absence of QTc prolongation in a thorough QT study with subcutaneous liraglutide, a once-daily human GLP-1 analog for treatment of type 2 diabetes. J Clin Pharmacol 2009;49:1353–1362 [DOI] [PubMed] [Google Scholar]
- 51.Darpö B, Philip S, Macconell L, et al Exenatide at therapeutic and supratherapeutic concentrations does not prolong the QTc interval in healthy subjects. Br J Clin Pharmacol 2013;75:979–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr Relat Cancer 2009;16:1103–1123 [DOI] [PubMed] [Google Scholar]
- 53.Engel SS, Williams-Herman DE, Golm GT, et al. Sitagliptin: review of preclinical and clinical data regarding incidence of pancreatitis. Int J Clin Pract 2010;64:984–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drucker DJ, Sherman SI, Gorelick FS, Bergenstal RM, Sherwin RS, Buse JB. Incretin-based therapies for the treatment of type 2 diabetes: evaluation of the risks and benefits. Diabetes Care 2010;33:428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Butler PC, Dry S, Elashoff R. GLP-1-based therapy for diabetes: what you do not know can hurt you. Diabetes Care 2010;33:453–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahmad SR, Swann J. Exenatide and rare adverse events. N Engl J Med 2008;358:1970–1971; discussion 1971–1972 [PubMed] [Google Scholar]
- 57.U.S. Food and Drug Administration. Sitagliptin (marketed as Januvia and Janumet)— acute pancreatitis [Internet], 2009. Available from http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm183800.htm Accessed 7 January 2013
- 58.European database of suspected adverse drug reaction reports [Internet], 2012. Available from http://www.adrreports.eu/EN/index.html Accessed 7 January 2013
- 59.Lowenfels AB, Maisonneuve P, Cavallini Get al. Pancreatitis and the risk of cancer. N Engl J Med 1993;328:1433–1437 [DOI] [PubMed] [Google Scholar]
- 60.Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 2011;141:150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nyborg NCB, Mølck AM, Madsen LW, Knudsen LB. The human GLP-1 analog liraglutide and the pancreas: evidence for the absence of structural pancreatic changes in three species. Diabetes 2012;61:1243–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huxley R, Ansary-Moghaddam A, Berrington de González A, Barzi F, Woodward M. Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer 2005;92:2076–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drucker DJ, Sherman SI, Bergenstal RM, Buse JB. The safety of incretin-based therapies—review of the scientific evidence. J Clin Endocrinol Metab 2011;96:2027–2031 [DOI] [PubMed] [Google Scholar]
- 64.Monami M, Dicembrini I, Martelli D, Mannucci E. Safety of dipeptidyl peptidase-4 inhibitors: a meta-analysis of randomized clinical trials. Curr Med Res Opin 2011;27(Suppl. 3):57–64 [DOI] [PubMed] [Google Scholar]
- 65.Bjerre Knudsen L, Madsen LW, Andersen S, et al. Glucagon-like peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation [correction of: Moerch, Ulrik added. In: Endocrinology 2012;153:1000]. Endocrinology 2010;151:1473–1486 [DOI] [PubMed] [Google Scholar]
- 66.Hegedüs L, Moses AC, Zdravkovic M, Le Thi T, Daniels GH. GLP-1 and calcitonin concentration in humans: lack of evidence of calcitonin release from sequential screening in over 5000 subjects with type 2 diabetes or nondiabetic obese subjects treated with the human GLP-1 analog, liraglutide. J Clin Endocrinol Metab 2011;96:853–860 [DOI] [PubMed] [Google Scholar]
- 67.Parks M, Rosebraugh C. Weighing risks and benefits of liraglutide—the FDA’s review of a new antidiabetic therapy. N Engl J Med 2010;362:774–777 [DOI] [PubMed] [Google Scholar]
- 68.Suzuki S, Arnold LL, Pennington KL, et al. Effects of pioglitazone, a peroxisome proliferator-activated receptor gamma agonist, on the urine and urothelium of the rat. Toxicol Sci 2010;113:349–357 [DOI] [PubMed] [Google Scholar]
- 69.Spanheimer R, Erdmann E, Song E, et al. Pioglitazone and bladder malignancy during observational follow-up of PROactive: 6-year update (Abstract). Diabetologia 2012;55(Suppl. 1):787 [Google Scholar]
- 70.Colmers IN, Bowker SL, Majumdar SR, Johnson JA. Use of thiazolidinediones and the risk of bladder cancer among people with type 2 diabetes: a meta-analysis. CMAJ 2012;184:E675–E683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu Z, Shen Z, Lu Y, Zhong S, Xu C. Increased risk of bladder cancer with pioglitazone therapy in patients with diabetes: a meta-analysis. Diabetes Res Clin Pract 2012;98:159–163 [DOI] [PubMed] [Google Scholar]
- 72.Ferrara A, Lewis JD, Quesenberry CP, Jr, et al. Cohort study of pioglitazone and cancer incidence in patients with diabetes. Diabetes Care 2011;34:923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Summary of product characteristics. Actos tablets [Internet], 2012. Available from http://www.medicines.org.uk/emc/medicine/4236/SPC/Actos+Tablets Accessed 7 January 2013.
- 74.FDA briefing document: NDA 202293 dapagliflozin tablets, 5 and 10 mg. Sponsor: Bristol-Myers Squibb. Advisory Committee meeting July 19, 2011 [Internet], 2011. Available from http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/ucm262994.pdf Accessed 7 January 2013
- 75.Santer R, Calado J. Familial renal glucosuria and SGLT2: from a Mendelian trait to a therapeutic target. Clin J Am Soc Nephrol 2010;5:133–141 [DOI] [PubMed] [Google Scholar]
- 76.Janghorbani M, Dehghani M, Salehi-Marzijarani M. Systematic review and meta-analysis of insulin therapy and risk of cancer. Horm Cancer; 2012;3:137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pollak M, Russell-Jones D. Insulin analogues and cancer risk: cause for concern or cause célèbre? Int J Clin Pract 2010;64:628–636 [DOI] [PubMed] [Google Scholar]
- 78.Gerstein HC. Does insulin therapy promote, reduce, or have a neutral effect on cancers? JAMA 2010;303:446–447 [DOI] [PubMed] [Google Scholar]
- 79.Smith U, Gale EA. Does diabetes therapy influence the risk of cancer? Diabetologia 2009;52:1699–1708 [DOI] [PubMed] [Google Scholar]
- 80.Gerstein HC, Bosch J, Dagenais GR, et al. ORIGIN Trial Investigators Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med 2012;367:319–328 [DOI] [PubMed] [Google Scholar]
- 81.Sommerfeld MR, Müller G, Tschank G, et al. In vitro metabolic and mitogenic signaling of insulin glargine and its metabolites. PLoS ONE 2010;5:e9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Giovannucci E, Harlan DM, Archer MC, et al. Diabetes and cancer: a consensus report. Diabetes Care 2010;33:1674–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Habib ZA, Havstad SL, Wells K, Divine G, Pladevall M, Williams LK. Thiazolidinedione use and the longitudinal risk of fractures in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab 2010;95:592–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Loke YK, Singh S, Furberg CD. Long-term use of thiazolidinediones and fractures in type 2 diabetes: a meta-analysis. CMAJ 2009;180:32–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Riche DM, King ST. Bone loss and fracture risk associated with thiazolidinedione therapy. Pharmacotherapy 2010;30:716–727 [DOI] [PubMed] [Google Scholar]
- 86.Mabilleau G, Chappard D, Baslé MF. Cellular and molecular effects of thiazolidinediones on bone cells: a review. Int J Biochem Mol Biol 2011;2:240–246 [PMC free article] [PubMed] [Google Scholar]
- 87.Singer FR, Eyre DR. Using biochemical markers of bone turnover in clinical practice. Cleve Clin J Med 2008;75:739–750 [DOI] [PubMed] [Google Scholar]
- 88.Lewiecki EM. Benefits and limitations of bone mineral density and bone turnover markers to monitor patients treated for osteoporosis. Curr Osteoporos Rep 2010;8:15–22 [DOI] [PubMed] [Google Scholar]