

Abstract
Background
Anandamide and 2-arachidonoylglycerol are neuromodulatory lipids interacting with cannabinoid receptors, whose availability is regulated by the balance between ‘on demand’ generation and enzymatic degradation [by fatty acid amide hydrolase (FAAH)/monoacylglycerol lipase]. Given the reported effects of anandamide on dopamine transmission, we investigated the influence of endocannabinoids and URB597, a well-known FAAH inhibitor, on the expression of tyrosine hydroxylase (TH), the rate-limiting enzyme in dopamine synthesis.
Experimental Approach
We investigated TH expression in N1E115 neuroblastoma using a reporter gene assay, as well as mRNA and protein quantifications. FAAH inhibition was confirmed by measuring radiolabelled substrate hydrolysis and endogenous endocannabinoids.
Key Results
Anandamide decreased TH promoter activity in N1E115 cells through CB1 receptor activation. Unexpectedly, URB597 reduced TH expression (pEC50 = 8.7 ± 0.2) through FAAH-independent mechanisms. Indeed, four structurally unrelated inhibitors of FAAH had no influence on TH expression, although all the inhibitors increased endocannabinoid levels. At variance with the endocannabinoid responses, the use of selective antagonists indicated that the URB597-mediated decrease in TH expression was not directed by the CB1 receptor, but rather by abnormal-cannabidiol-sensitive receptors and PPARs. Further supporting the physiological relevance of these in vitro data, URB597 administration resulted in reduced TH mRNA levels in mice brain.
Conclusions
While confirming the implication of endocannabinoids on the modulation of TH, we provide strong evidence for additional physiologically relevant off-target effects of URB597. In light of the numerous preclinical studies involving URB597, particularly in anxiety and depression, the existence of non-CB1 and non-FAAH mediated influences of URB597 on key enzymes of the catecholaminergic transmission system should be taken into account when interpreting the data.
Linked Articles
This article is part of a themed section on Cannabinoids. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2013.169.issue-4 & http://dx.doi.org/10.1111/bph.2012.167.issue-8
Keywords: endocannabinoid, carbamate, off-target, dopamine, tyrosine hydroxylase promoter, CB1-independent mechanism, Abn-CBD, cannabidiol, MAPK, PF750
Introduction
Endocannabinoids are bioactive lipids that are emerging as important modulators of synaptic transmission. The modulation of catecholaminergic (both dopaminergic and noradrenergic) neuron activity by the endocannabinoid system is thought to be involved in numerous behavioural functions, including addiction and rewarding processes (Maldonado et al., 2006), motor and cognitive functions (Hao et al., 2000; Marinelli et al., 2003), as well as emotional processing (Gobbi et al., 2005; Rubino et al., 2008) and sleep pattern (Murillo-Rodríguez et al., 2007). Further supporting the crosstalk between endocannabinoid and dopamine systems, anandamide (AEA), the first identified endocannabinoid (Devane et al., 1992), was reported to cause a reduction in dopamine levels and/or release in the nigrostriatal pathway (Romero et al., 1995b; Cadogan et al., 1997; de Lago et al., 2004). In contrast, in prefrontal cortex (Romero et al., 1995a), hypothalamus, hippocampus (Hao et al., 2000) and nucleus accumbens (Solinas et al., 2006), enhanced dopamine concentrations have been associated with AEA administration.
These somehow conflicting in vivo data probably reflect intricate mechanisms involved in the regulation of dopaminergic neurotransmission. Indeed, it is generally assumed that the modifications of catecholaminergic neuronal circuits mediated by CB1 cannabinoid receptors involve transient depression of excitatory or inhibitory synaptic transmission (Cadogan et al., 1997; Gerdeman and Lovinger, 2001; Huang et al., 2001; Kreitzer and Malenka, 2007). However, other results indicate that the endocannabinoid-mediated dopamine release is not elicited by a disinhibitory mechanism (Cachope et al., 2007). Besides, the cannabinoid receptor ligand Δ9-tetrahydrocannabinol was previously reported to modulate the activity of tyrosine hydroxylase (TH), the main enzyme involved in catecholamine biosynthesis (Bonnin et al., 1993; González et al., 2005). Similarly, studies have also found a cannabinoid-mediated regulation of TH gene expression in a neuroblastoma cell line (Bosier et al., 2007), as well as in vivo after chronic treatment with the synthetic agonist WIN 55 212-2 (Page et al., 2007), suggesting that cannabinoid receptors and endocannabinoids may also control critical neuronal functions through a delayed and more persistent control of catecholamine brain level/transmission.
To strengthen this hypothesis, it would be interesting to demonstrate a similar regulation of TH expression by the cannabinoid receptor's endogenous ligands, that is, the endocannabinoids. Because in biological systems, endocannabinoid effects are tightly regulated by their degradation, predominantly by fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) (Muccioli, 2010), an alternative way to investigate their functions is by inhibiting these hydrolysing enzymes. Among drugs eliciting such an inhibition, URB597 is a prime candidate as it elevates AEA levels in the rat and mouse brain (Kathuria et al., 2003; Tarzia et al., 2003) and magnifies its action through a selective blockade of FAAH activity (Fegley et al., 2005).
Given the reported effects of AEA on TH activity (Romero et al., 1995b), we herein investigated the influence of endocannabinoids and URB597 on TH gene expression. Corroborating our previous study (Bosier et al., 2007), the endocannabinoids AEA and 2-arachidonoylglycerol (2-AG) decreased TH promoter activity through CB1 cannabinoid receptor activation. Unexpectedly, URB597 per se regulated TH expression through CB1- and FAAH-independent mechanisms. Thus, this study provides evidence for a physiologically relevant off-target effect of URB597.
Methods
Materials
URB597 (carbamic acid, N-cyclohexyl-, 3′-(aminocarbonyl)[1,1′-biphenyl]-3-yl ester), CAY10402 (1-oxazolo[4,5-b]-pyridin-2-yl-6-phenyl-1-hexanone), CAY10499 ([4-(5-methoxy-2-oxo-1,3,4-oxadiazol-3(2H)-yl)-2-methylphenyl]-carbamic acid, phenylmethyl ester), PF750 (N-phenyl-4-(quinolin-2-ylmethyl)piperidine-1-carboxamide), cannabidiol (CBD), O-1918 (1,3-dimethoxy-5-methyl-2-[(1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-benzene) and abnormal-cannabidiol (Abn-CBD) were purchased from Cayman Chemicals (Ann Arbor, MI, USA). HU 210, troglitazone (TGZ), as well as the PPARγ and the mixed PPARγ/PPARα antagonists T0070907 (2-chloro-5-nitro-N-4-pyridinyl-benzamide) and GW9662 (2-chloro-5-nitrobenzanilide) were obtained from Tocris Cookson (Bristol, UK). The CB1 and the CB2 cannabinoid receptor inverse agonists/antagonists SR 141716A and SR 144528 were generous gifts from Dr Barth and Dr Mossé, respectively, from Sanofi-Synthélabo Research (Montpellier, France). The radiolabelled substrates [3H]-2-OG ([3H]-2-oleoylglycerol, 60 Ci·mmol−1), [3H]-AEA (60 Ci·mmol−1) and [3H]-PEA ([3H]-N-palmitoylethanolamine, 20 Ci·mmol−1) were all bought from the American Radiolabeled Chemicals (St. Louis, MO, USA). Deuterated and non-deuterated N-acylethanolamines [AEA, PEA, SEA (N-stearoylethanolamine) and OEA (N-oleoylethanolamine)] were synthesized in our laboratory from acyl chlorides and deuterared or non-deuterated ethanolamine as described by Walter et al. (2002). 2-AG and d-2-AG were obtained from Cayman Chemicals. The molecular target names used herein are in accordance with Alexander et al. (2011).
Cell culture
All cell culture media and supplements were obtained from Invitrogen (Merelbeke, Belgium). Mouse neuroblastoma N1E115 cells were grown in DMEM/NUT mix F-12 medium supplemented with 10% fetal calf serum, 100 IU·mL−1 penicillin, 100 μg·mL−1 streptomycin and 2 mM L-glutamine. All drug treatments were conducted in the same culture medium. Cells were cultured at 37°C in an atmosphere of humidified air and 5% CO2.
Gene reporter assay
N1E115 cells were plated at a density of 105 cells per well in 24-well plates and co-transfected with pTH250-Luc and pRL 138 plasmids using the phosphate co-precipitation method as previously described (Bosier et al., 2007). For drug treatments, transfected cells were washed with PBS (137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4 and 1.47 mM KH2PO4; pH 7.4) and incubated with URB597, CAY10402, CAY10499, MAFP, PF750, AEA, 2-AG or PEA at the indicated concentrations for 5 h in fresh medium. When relevant, SR 141716A, SR 144528, O-1918 or CBD were added 5 min prior to the agonists. When testing the influence of PPAR antagonists and MAP kinase kinase (MEK) inhibitor, these were added 1 h beforehand. At the end of the 5 h incubation period, cells were lysed by the addition of 100 μL of passive lysis buffer supplied in the Dual Luciferase Reporter Assay System (Promega, Leiden, the Netherlands). Firefly luciferase reporter activity was normalized for the Renilla luciferase activity. Respective measurements of light emissions were determined according to the manufacturer's instructions with a TD20/20 luminometer (Turner Design, Sunnyvale, CA, USA).
In vivo study
The traditional outbred NMRI (Naval Medical Research Institute) mice (30 g) were from our in-house facility and housed in a controlled environment (12-h daylight cycle). Animals were acclimatized for 1 week with ad libitum access to food and water before starting the experiment. Furthermore, 24 h after i.p. administration of URB597 3 mg·kg−1 [in 2% ethanol, 2% dimethyl sulfoxide (DMSO), 1% Tween 80 saline solution] or vehicle alone, the different brain regions were rapidly dissected and frozen in liquid nitrogen for subsequent TH mRNA expression analysis. All experiments were approved by the local ethics committee and housing conditions were as specified by the Belgian Law of 14 November 1993 on the protection of laboratory animals (LA 1230314). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Reverse transcriptase-PCR (RT-PCR) and quantitative PCR (qPCR)
Total RNA was isolated using TriPure isolation reagent (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions. Genomic DNA contaminations were eliminated using DNase I Amplification Grade (Invitrogen) for 1 h at 37°C and then DNase I was heat-inactivated (15 min, 65°C). For qPCR experiments, RT and PCR were performed in a single step using the access RT-PCR system (Promega) and the primers mentioned in the Supporting Information Tables S1 and S2. For quantification of TH expression, qPCR was performed. cDNA was generated using the RT system (Promega). qPCR amplifications were carried out using the ABI Prism 5700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) in a final volume of 25 μL containing the cDNA template, 0.3 μM of the primers (forward, 5′-AGTTCTCCCAGGACATTGGACTT-3′; reverse, 5′-ACACAGCCCAAACTCCACAGT-3′) and the MESA qPCR® mastermix for SYBR assay (Eurogentec, Liège, Belgium). TH expression was normalized to the relative amplification of GAPDH. Quantification was performed using the 2-ΔcT method.
Cell homogenates and Western blotting
At the end of incubation with drugs, cells were pelleted and re-suspended in lysis buffer [50 mM HEPES, 50 mM KF, 1 mM Na3PO4, 1 mM EDTA, 1 mM EGTA, 0.5% 2-mercaptoethanol, 5 mM β-glycerolphosphate, 5 mM Na4P2O7, 1 mM phenylmethylsulphonyl fluoride, 100 μM Na3VO4, 1% Triton X-100, protease inhibitor complete mini EDTA (Roche, Vilvoorde, Belgium) and phosphatase inhibitor cocktail (Sigma-Aldrich, Bornem, Belgium)]. In addition, 20 μg of proteins was then diluted in loading buffer [50 mM Tris–HCl (pH 6.8), 100 mM DTT, 2% SDS, 10% glycerol, 0.1% bromophenol blue] and incubated for 5 min at 100°C. Samples were separated on a 10 or 12% (for TH and ERK1/2, respectively) SDS–polyacrylamide gel (1 h 30 migration at 120 V) and transferred (1 h, 400 mA) to nitrocellulose membranes for immunodetection. After 1 h of blocking in 5% non-fat powdered milk in TTBS (20 mM Tris, 150 mM NaCl and 0.05% Tween 20; pH 7.6), blots were probed at 4°C overnight in a 1:2500 dilution of rabbit anti-TH (Chemicon, Hampshire, UK) or a 1:1000 dilution of rabbit anti-phospho-specific ERK1/2 (Cell Signaling Technology, Bioké, Leiden, the Netherlands), antibodies. Blots were then washed thoroughly, incubated 1 h with HRP conjugated anti-rabbit (Chemicon) secondary antibody and revealed with SuperSignal West Pico Chemiluminescent Substrate System (Pierce, Erembodegem, Belgium). The consistency of the loading was validated by stripping and by re-probing the blots with an anti-actin antiserum (1:5000) (Abcam, Cambridge, UK) or antibodies that recognize both the phosphorylated and the non-phosphorylated forms of ERK1/2 (1:1000) (Cell Signaling Technology). Relative amounts of proteins were quantified by scanning densitometry using the software ImageMaster (Pharmacia Biotech Benelux, Roosendaal, the Netherlands).
Measurement of [3H]-2-OG, [3H]-AEA and [3H]-PEA hydrolysis in homogenates and intact N1E115 cells
Cells were grown up to 75–80% confluence, recovered by trypsinization, rinsed with PBS buffer and homogenized in Tris–HCl buffer prior to total protein quantification. Homogenates (0–100 μg of proteins for the hydrolytic activity and 30 μg of proteins for the IC50 determination, in 165 μL of Tris–HCl, pH 7.4) were added on ice into glass tubes containing 10 μL of either drugs or vehicle (DMSO). Hydrolysis was initiated by adding 25 μL of Tris–HCl (pH 7.4, 0.1% fatty acid free-BSA) containing [3H]-2-OG (2 nM, 55 000 dpm), [3H]-AEA (3.5 nM, 100 000 dpm) or [3H]-PEA (3.5 nM, 80 000 dpm), then tubes were incubated in a shaking water bath for 10 min at 37°C. The reactions were arrested by adding 400 μL of ice-cold MeOH–CHCl3 (1:1), followed by vigorous mixing.
[3H]-AEA hydrolysis in intact cells was measured as described by Jacobsson and Fowler (2001), with some modifications. Cells were grown for 16 h in 24-well plates. On the day of the experiment, cells were rinsed with fresh media prior to drug or vehicle addition (in 150 μL of media). After 15 min of incubation at 37°C, [3H]-AEA (50 μL, 85 000 dpm, 0.2 μM final concentration) was added to the wells. After 10 min of incubation, ice-cold MeOH (400 μL) was added and the cells were thoroughly scraped. The resulting homogenate (600 μL) was added to glass tubes containing CHCl3 (300 μL) and vigorously mixed.
After centrifugation (5 min, 1100 g, 4°C), the aqueous phase was recovered and the radiolabelled product of hydrolysis quantified by liquid scintillation. In all experiments, tubes containing buffer only, or wells with no cells (but containing media), were used to determine the chemical hydrolysis of the radiosubstrates (blank).
HPLC-MS quantification of endocannabinoids and N-acylethanolamines
Endocannabinoids (AEA and 2-AG) and N-acylethanolamines (NAEs, i.e. PEA, SEA and OEA) levels were quantified as previously described with some modifications (Muccioli et al., 2007; Muccioli and Stella, 2008). Cells (17.5 × 106 per flask) were incubated for 2 h at 37°C with medium containing drug (0.1 μM URB597, 10 μM CAY10402, 10 μM CAY10499; 10 μM MAFP; 10 μM PF750) or vehicle (0.1% DMSO). Cellular lipids were extracted using CHCl3–MeOH–H20 (10:5:2.5, v/v/v) in the presence of 200 pmol of d4-AEA, d5-2-AG, d4-PEA, d4-SEA and d4-OEA, and purified by solid-phase extraction using silica and ethyl acetate–acetone (1:1). The resulting fraction was analysed by HPLC-MS using a LTQ Orbitrap mass spectrometer (ThermoFisher Scientific, Erembodegem-Aalst, Belgium) coupled to an Accela HPLC system (ThermoFisher Scientific) as previously described (Alhouayek et al., 2011). Endocannabinoids and NAEs were quantified by isotope dilution using their respective deuterated standards (showing identical retention times). The calibration curves were generated as described (Muccioli and Stella, 2008) and, with the exception of AEA, the data were normalized to vehicle-treated cells. Note that for AEA, the data were not normalized to the vehicle-treated cells since AEA was below our detection limit in this condition. Thus, AEA data are reported as the ratio between AEA and d4-AEA signals.
Data analysis
Unless otherwise stated, data presented in the text and figures were expressed as mean percentages ± SEM of the corresponding values obtained with cells treated with vehicle alone (DMSO diluted in culture medium). GraphPad Prism 5.03 (GraphPad Software Inc., San Diego, CA, USA) was used to analyse the data, to generate dose–response curves and to perform statistics. Student's t-tests were used for the comparison between the two groups. When comparing more than two groups, one-way ANOVA was used. Post-hoc comparisons relative to control were carried out using Dunnett's post-hoc analysis, while the comparisons between all the groups were performed using Tukey's post-hoc test. Two-way ANOVA was used to compare results from experiments with two independent variables (typically the treatment and the presence of an antagonist). Except if otherwise stated, post-hoc comparison was carried out in the case of significant main factor effect and positive interaction (P < 0.05) by Bonferroni analysis.
Results
URB597 regulates TH expression in vitro and in vivo
To test the effect of endogenous cannabinoids on TH transcription, N1E115 cells were transfected with a TH promoter-controlled firefly luciferase reporter gene construct (pTH250-Luc) (Bosier et al., 2007; 2009). When assayed alone, neither endocannabinoids (AEA and 2-AG, 1 μM) nor PEA (1 μM) had an effect on TH promoter-controlled luciferase activity (Figure 1A). However, in the presence of the FAAH inhibitor URB597 (0.1 μM), AEA markedly reduced TH promoter activity, suggesting significant endocannabinoid degradation in N1E115 cells (AEA + URB597 vs. AEA). Furthermore, URB597 per se produced a significant reduction of TH promoter-directed luciferase activity (21.3 ± 2.5% decrease at 0.1 μM). The effect of URB597 was concentration-dependent, with a pEC50 value of 8.7 ± 0.2 (Figure 1B).
Figure 1.
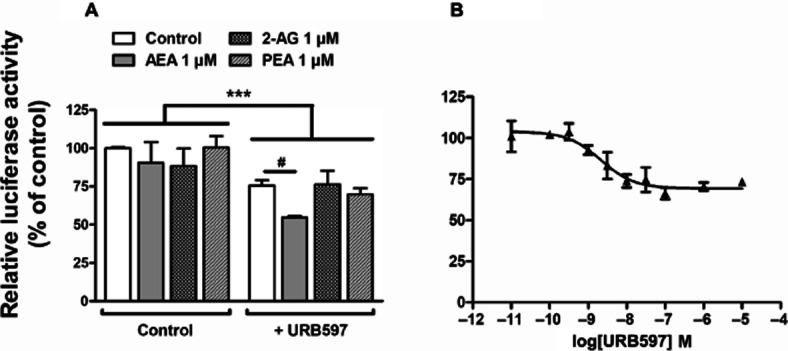
Endocannabinoids and URB597-mediated regulation of TH promoter activity. Luciferase activity was measured in N1E115 cells transiently transfected with pTH250-Luc and treated for 5 h with AEA, 2-AG, PEA or vehicle, each at 1 μM (A). The responses to these endocannabinoids were also measured in cells concomitantly treated with URB597 (0.1 μM). (B) Concentration–response modulation of luciferase activity with URB597; pEC50 value derived from non-linear analysis of concentration–response curves is indicated in the text. Results are given as the percentages of relative luciferase activity (firefly luciferase relative to Renilla luciferase) relative to control values. Data shown are means with SEM values of three to six experiments performed in triplicate. Two-way ANOVA indicates a general effect of URB597 (***P = 0.0002, f = 20.60, residual d.f. = 22). #P < 0.05 using one-way ANOVA performed inside the URB597-treated group, relative to control cells treated with URB597 alone.
To validate the results obtained with the reporter gene assay used here, we measured TH mRNA and protein contents consecutively to URB597 exposure. As shown in Figure 2A,B, TH mRNA and protein levels were reduced after 24 h of incubation with URB597. Because URB597 is also widely used in vivo as FAAH inhibitor, and to further strengthen the physiological relevance of our findings, we went on to determine whether URB597 was able to modify TH expression in vivo. We therefore measured brain TH mRNA level 24 h after URB597 (3 mg·kg−1, i.p.) administration to mice. Consistent with what we observed in vitro, mice exhibited a significant decrease of TH mRNA levels in the hippocampus and striatum after a single injection of URB597 (Figure 2C). In addition, although it failed to reach statistical significance, URB597 decreased TH mRNA expression in the cerebellum (P < 0.07 relative to controls respectively).
Figure 2.
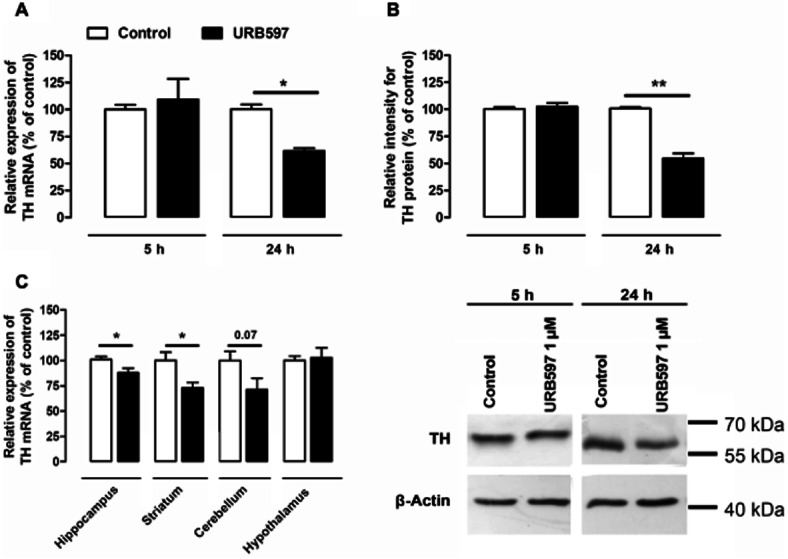
In vitro and in vivo URB597-mediated modification of TH expression. Quantifications of TH mRNA (A) and protein (B) expression were performed on neuroblastoma exposed for 5 or 24 h to 1 μM URB597. Quantitative PCR of TH mRNA was performed on total RNA extracts. The expression of TH mRNA (A) was normalized against GAPDH expression and results are given as the relative expression of treated versus control cells/animals. The densitometric analysis of the TH protein signals (60 kDa) shown in (B) was normalized against the measured signals corresponding to actin (42 kDa). A typical immunoblot is shown at the bottom right corner. Results are given as the percentages of expression relative to control cells. Data shown are means with SEM values of three experiments performed in triplicate. Two-tailed paired Student's t-test, *P < 0.05, **P < 0.01, relative to control at corresponding time (P = 0.0314, t = 5.507, residual d.f. = 2) and (P = 0.0087, t = 10.64, residual d.f. = 2) for mRNA and protein dosages respectively. In mice, TH mRNA contents were evaluated in hippocampus, striatum, cerebellum, cortex and hypothalamus tissues (C) 24 h after a single injection of URB597 (3 mg·kg−1, i.p.). Results are given as the percentages relative to control animals injected with vehicle only. Values are means with SEM of seven animals in each group. Two-tailed unpaired Student's t-test, *P < 0.05, relative to control (P = 0.0305, t = 2.452, residual d.f. = 12) (P = 0.0153, t = 2.824, residual d.f. = 12) (P = 0.0705, t = 1.985, residual d.f. = 2) in the hippocampus, the striatum and the cerebellum respectively.
URB597-mediated regulation of TH expression does not require CB1 cannabinoid receptor activation nor FAAH inhibition
We found that N1E115 cells express the mRNA of all the endocannabinoid-hydrolysing enzymes reported so far (Supporting Information Fig. S1). Using radiolabelled substrates, we detected the hydrolysing activities corresponding to these enzymes. Indeed, [3H]-AEA and [3H]-PEA were hydrolysed by cell homogenates (pH 7.4) in time- and protein-dependent manner, revealing specific activities of 0.061 ± 0.003 and 0.037 ± 0.003 pmol·μg−1·min−1 respectively. Similarly, the monoglyceride 2-OG, a substrate of MAGL, was hydrolysed by the same cell preparation with a specific activity of 0.81 ± 0.12 pmol·μg−1·min−1. In accordance with its described potent FAAH inhibition (Kathuria et al., 2003; Lichtman et al., 2004), we found that URB597 dose-dependently inhibited [3H]-AEA and [3H]-PEA hydrolysis by N1E115 cell homogenate (IC50 values of 31 ± 3.5 and 600 ± 101 nM, respectively), suggesting that this FAAH inhibitor increases endocannabinoid levels in these cells. Therefore, we investigated whether the herein reported effects of URB597 were related to enhanced endocannabinoid levels.
First, we exogenously increased AEA levels up to 10 μM and found a significant reduction in luciferase activity (Figure 3A). Consistent with our previous reports on cannabinoid-mediated control of TH promoter activity (Bosier et al., 2007), the CB1 selective inverse agonist SR 141716A completely reversed the AEA-mediated effect. However, in the presence of URB597, SR 141716A only partially reversed the effect mediated by AEA (Figure 3A). Indeed, in the presence of SR 141716A, luciferase activity is still reduced by 20%, which corresponds to the magnitude of URB597 effect (Figure 3A, dashed line). This suggests that URB597-induced reduction of TH transcription is not mediated by the CB1 cannabinoid receptor, whereas the effect of AEA alone is CB1-dependent. As an additional control, we incubated the cells in the presence of URB597 alone and with SR 141716A (Figure 3B). In these conditions, SR 141716A failed to antagonize URB597-directed regulation of TH promoter activity, although it completely reversed HU 210-mediated effects, as previously reported (Bosier et al., 2007).
Figure 3.
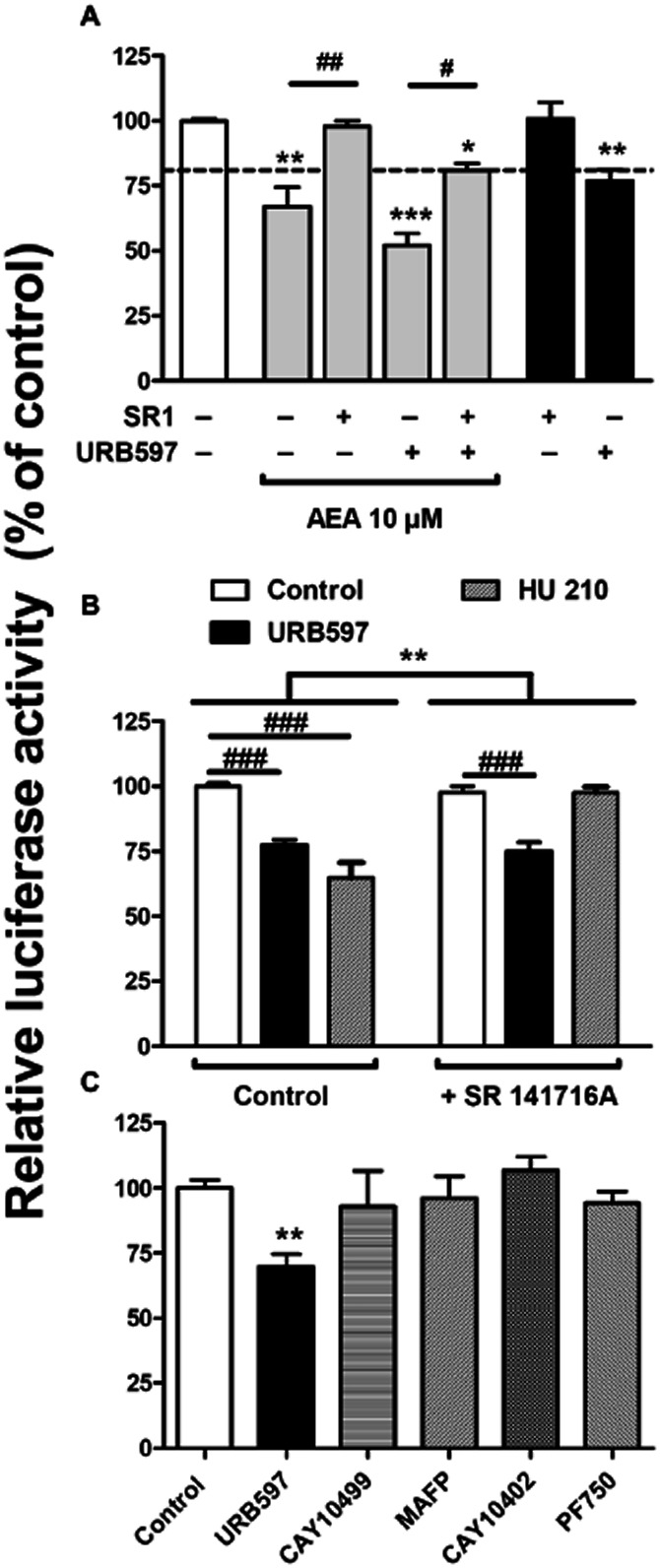
URB597-mediated regulation of TH promoter activity is CB1 cannabinoid receptor- and FAAH-independent. (A) Luciferase activity determined on cells transfected with pTH250-Luc and treated with AEA at 10 μM in the presence or the absence of SR 141716A (1 μM) and/or URB597 (0.1 μM). One-way ANOVA followed by Tukey's post-test, ***P < 0.001, **P < 0.01, *P < 0.05, relative to control; and ##P < 0.01, #P < 0.05, as indicated between treated cells. (B) illustrates the influence of SR 141716A (1 μM) on the regulation of TH promoter activity mediated by URB597 or HU 210 (both at 0.1 μM). Two-way ANOVA indicates a general effect of SR 141716A (**P = 0.024, f = 10.26, residual d.f. = 48) with a positive interaction on the treatment (P = 0.0001, f = 11.16). ###P < 0.001 as indicated between treated cells, determined with Bonferroni post-test. The responses induced by URB597 (0.1 μM) and other FAAH inhibitors (all at 10 μM) were compared to investigate the involvement of FAAH (C). One-way ANOVA indicated a global treatment effect (P = 0.0246, f = 3.396, residual d.f. = 18) with **P < 0.01 relative to control, given by Dunnett's post-test. All the results are given as the percentages of relative luciferase activity (firefly luciferase relative to Renilla liciferase) relative to control values. Data shown are means with SEM values of three to six experiments performed in triplicate.
Second, we tested a battery of other potent FAAH inhibitors, that is, MAFP, CAY10499, CAY10402 and PF750, which are structurally unrelated to URB597 (Leung et al., 2003; Ahn et al., 2007; Muccioli et al., 2008). Unexpectedly, none of these inhibitors mimicked the URB597-mediated modulation of TH promoter activity (Figure 3C). Of note, PF750, a highly selective FAAH inhibitor, completely prevented [3H]-AEA hydrolysis in intact cells (Supporting Information Fig. S2), indicating that this inhibitor properly reached its target. Furthermore, when measuring endocannabinoid levels in the presence of these inhibitors, we found that all of them, although to various extent, were able to increase AEA, 2-AG, PEA, OEA and SEA amounts in intact N1E115 cells (Figure 4). Together, these results indicate that URB597 regulates TH transcription by a mechanism independent of FAAH inhibition and CB1 cannabinoid receptor activation.
Figure 4.

Increase in endocannabinoid and N-acylethanolamine contents in N1E115 cells induced by different FAAH inhibitors. N1E115 cell contents in AEA (A), 2-AG (B), PEA (C), OEA (D) and SEA (E) were measured by HPLC-MS using an isotope dilution method after exposure to URB597 (0.1 μM), MAFP (10 μM), CAY10499 (10 μM), CAY10402 (10 μM), PF750 (10 μM) or vehicle alone. Results are expressed as the percentages of endocannabinoid or N-acylethanolamine contents relative to control except for (A), for which the basal level of AEA was below the detection limit (N.D.). For this panel only, results ere expressed as the ratio between AEA and its internal standard d4-AEA. Data are means with SEM values of three experiments performed in triplicate. One-way ANOVA followed by Dunnett's post-test ***P < 0.001, **P < 0.01, relative to control.
Additional cannabinoid targets are involved in URB597-mediated regulation of TH promoter activity
Next, we sought to further characterize the putative targets expressed by N1E115 cells that could mediate URB597 effects on TH promoter activity. As expected (Bosier et al., 2007), a positive amplification for CB1 cannabinoid receptor was obtained using the appropriate primers and N1E115 cDNA samples, while no specific PCR signal was detected for the CB2 cannabinoid receptor. Besides, the PCR study ( Figure 5A) revealed the genetic expression of GPR55 and GPR119 receptors as well as the nuclear PPARs PPARα and PPARγ. Contrasting with this, the expected amplification product for the ligand-gated ion channel vanilloid receptor (TRPV1) was absent, whereas a positive amplification was achieved using a cDNA sample from the mouse brain as positive control (Figure 5A).
Figure 5.
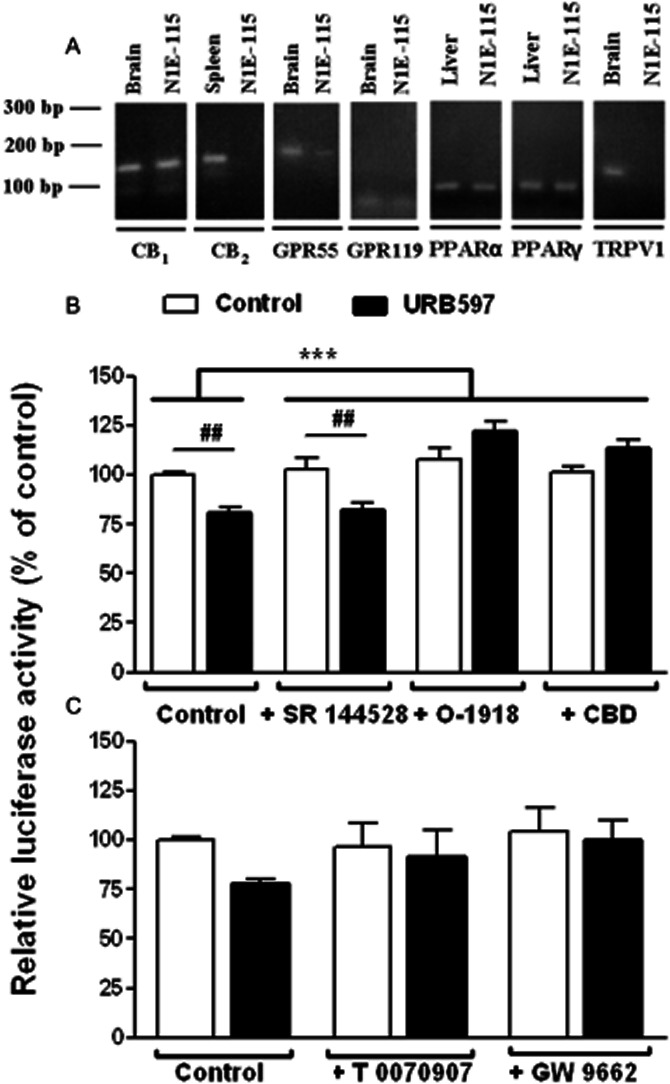
Involvement of non-CB1/non-CB2 cannabinoid receptors and PPARs in the URB597-mediated regulation of TH promoter activity. (A) RT-PCR was performed on N1E115 cell mRNA extracts with specific primers targeting the different endocannabinoid molecular targets. RT-PCR yielded the predicted amplification products for CB1, GPR55 and GPR119 receptors as well as for PPARα and PPARγ. No bands were detected for CB2 cannabinoid receptor and TRPV1 despite a correct amplification in spleen and brain tissues respectively. Negative controls were performed without any DNA template (not shown). The influence of SR 144528 at 1 μM (B), O-1918 at 30 μM (B), CBD at 10 μM (B) or PPAR antagonists (C) on luciferase activity was examined using transfected N1E115 cells carrying pTH250-Luc. Cells were concomitantly treated with the indicated antagonists and 0.1 μM URB597. The tested PPAR antagonists were T 0070907 (PPARγ antagonist, 10 μM) or GW 9662 (PPARγ/PPARα antagonist, 20 μM). Results are given as the percentages of relative luciferase activity (firefly luciferase relative to Renilla luciferase) relative to control values. Data are means with SEM values of at least three experiments performed in triplicate. Two-way ANOVA indicated a general effect of some of the antagonists (***P < 0.0001, f = 18.76, residual d.f. = 18) with a positive interaction (***P < 0.0001, f = 12.13) for (B). ##P < 0.01, as indicated for treated cells and determined by Bonferroni post-test. In (C), Bartlett's statistic revealed that variances significantly differ between these groups. Despite a strong trend (P = 0.062 for a general effect of URB597 in two-way ANOVA), statistical significance was not reached using the non-parametric Kruskal–Wallis test effect.
Based on the earlier discussion, the involvement of non-CB1/non-CB2 cannabinoid receptors in the control of TH gene expression elicited by URB597 was further investigated. Even though the CB2 cannabinoid receptor is not expressed, SR 144528 was tested as an antagonist of the SR 144528-sensitive non-CB1/non-CB2 receptor (Calignano et al., 2001). Excluding the involvement of the SR 144528-sensitive non-CB1/non-CB2 binding site, this antagonist failed to reverse the effect induced by URB597 (Figure 5B). However, O-1918 and CBD, which have been described as antagonists of both the non-CB1 endothelial cannabinoid receptor (Járai et al., 1999; Offertáler et al., 2003) and the GPR55 receptor (Ryberg et al., 2007), abrogated URB597-induced reduction in TH expression.
Moreover, the role of PPAR activation in the URB597-induced modulations of TH expression was examined using PPARγ (T 0070907) (Lee et al., 2002) and PPARγ/PPARα (GW 9662) (Seimandi et al., 2005) antagonists. While basal luciferase activity was not influenced by T 0070907 (10 μM) and GW 9662 (20 μM), our results indicated that they both prevented the regulation of TH promoter activity by URB597 (Figure 5C). This result suggests that PPAR activation is, in part, responsible for the action of URB597 on TH expression. However, note that while significant in all other experiments, in this last one, the effect of URB597 failed to reach significance. This is probably due to the difference in the variance observed in the presence of the PPAR inhibitors.
The effects of O-1918 and CBD suggest the implication of additional receptors (the non-CB1 endothelial cannabinoid receptor or the GPR55 receptor) having in common their activation by Abn-CBD. We tested this hypothesis by directly incubating N1E115 cells with Abn-CBD. Similar to URB597, Abn-CBD decreased TH promoter activity through a CBD-dependent mechanism (Figure 6A). In addition, further suggesting the involvement of common signalling pathways, the Abn-CBD-mediated reduction in luciferase activity was blocked by PPAR antagonists as well (Figure 6B). Finally, undeniably confirming the involvement of PPARs in the regulation of TH expression, a reduction of TH-controlled luciferase activity was observed after treatment with TGZ (20 μM), a PPARγ/PPARα agonist, which was completely prevented either by T 0070907 or GW 9662 (Figure 6C).
Figure 6.
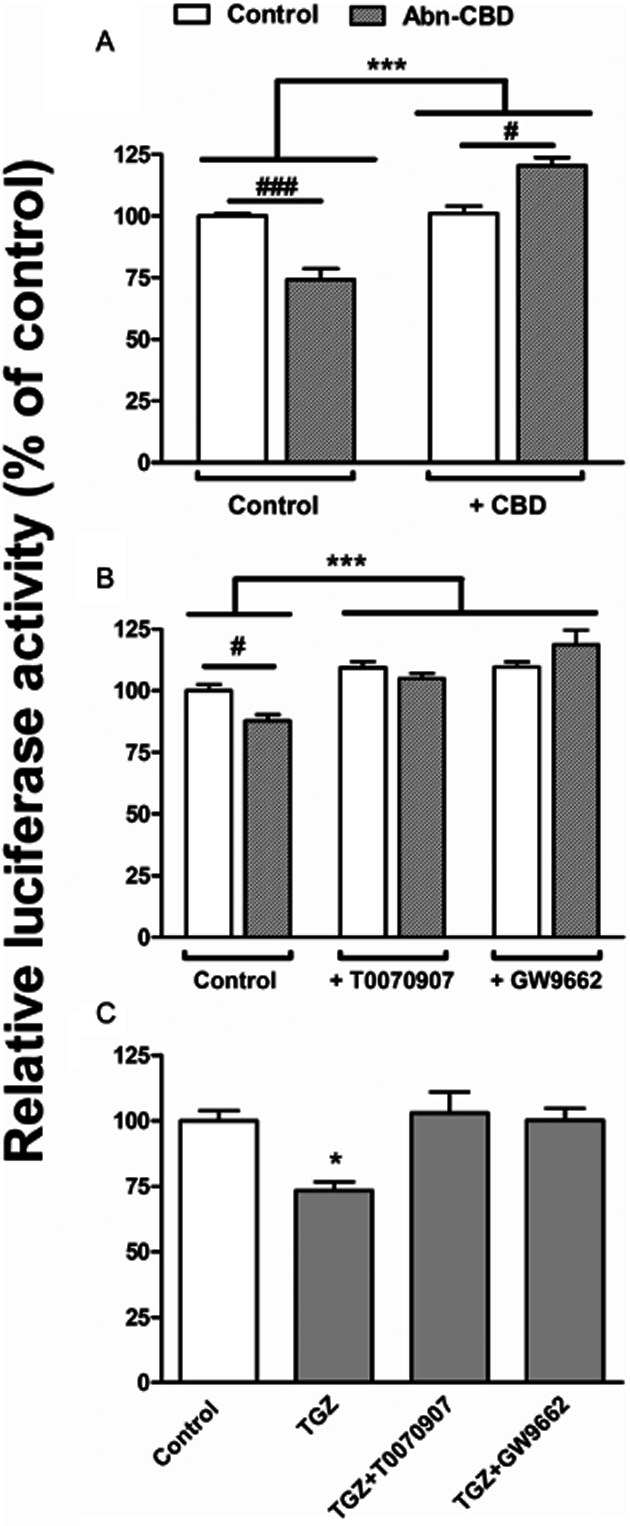
Regulation of TH promoter activity by direct activation of the non-CB1/non-CB2 cannabinoid receptors and PPARs. To confirm the involvement of the non-CB1/non-CB2 cannabinoid receptors in the URB597-mediated effect, the influence of Abn-CBD (10 μM, 5 h) was investigated on N1E115 cells transfected with pTH250-Luc. The involved signalling cascades were examined by concomitantly treating the cells either with non-CB1/non-CB2 cannabinoid receptors antagonists (CBD, 10 μM) (A) or PPAR antagonists (T 0070907 at 10 μM or GW 9662 at 20 μM) (B). (A) Two-way ANOVA indicated a general effect of CBD as antagonist (***P < 0.0001, f = 52.37, residual d.f. = 10) with a positive interaction (***P < 0.0001, f = 65.28). #P < 0.05 and ###P < 0.001, as indicated for treated cells and determined by Bonferroni post-test. (B) Two-way ANOVA indicated a general effect of PPAR antagonists (***P < 0.0001, f = 19.49, residual d.f. = 18) with a positive interaction (*P = 0.0193, f = 5.026). #P < 0.05 as indicated for treated cells and determined by Bonferroni post-test. (C) The involvement of PPARs in the regulation of TH expression was confirmed by examining the influence of troglitazone (TGZ, 20 μM, 5 h) alone and in the presence of the PPAR antagonists (T 0070907 at 10 μM or GW 9662 at 20 μM). One-way ANOVA indicated a global treatment effect (P = 0.0128, f = 6.949, residual d.f. = 8) with *P < 0.05 relative to control, given by Dunnett's post-test. All the results are given as the percentages of relative luciferase activity (firefly luciferase relative to Renilla luciferase) relative to control values. Data shown are means with SEM values of at least three experiments performed in triplicate.
Role of MAPK in URB597's effects
It is well known that MAPK activation can regulate PPAR activity (Gelman et al., 2005). Therefore, we investigated their role in URB597-mediated effects. Here, we show that decreasing p44/42 MAPK (also known as ERK1/2) activation, using the MAPK/ERK kinase 1 and 2 (MEK1/2) inhibitor U0126, results in lower TH promoter activity (Figure 7A). In addition, URB597 failed to induce a further decrease when co-incubated with U0126, suggesting that common pathways are involved. Indeed, distinct pathways would result in additive effects between U0126 and URB597. In agreement with a single pathway, URB597 controls ERK1/2 phosphorylation (Figure 7B). As for TH promoter activity (Figure 3B), SR 141716A failed to prevent URB597-mediated ERK1/2 activation.
Figure 7.
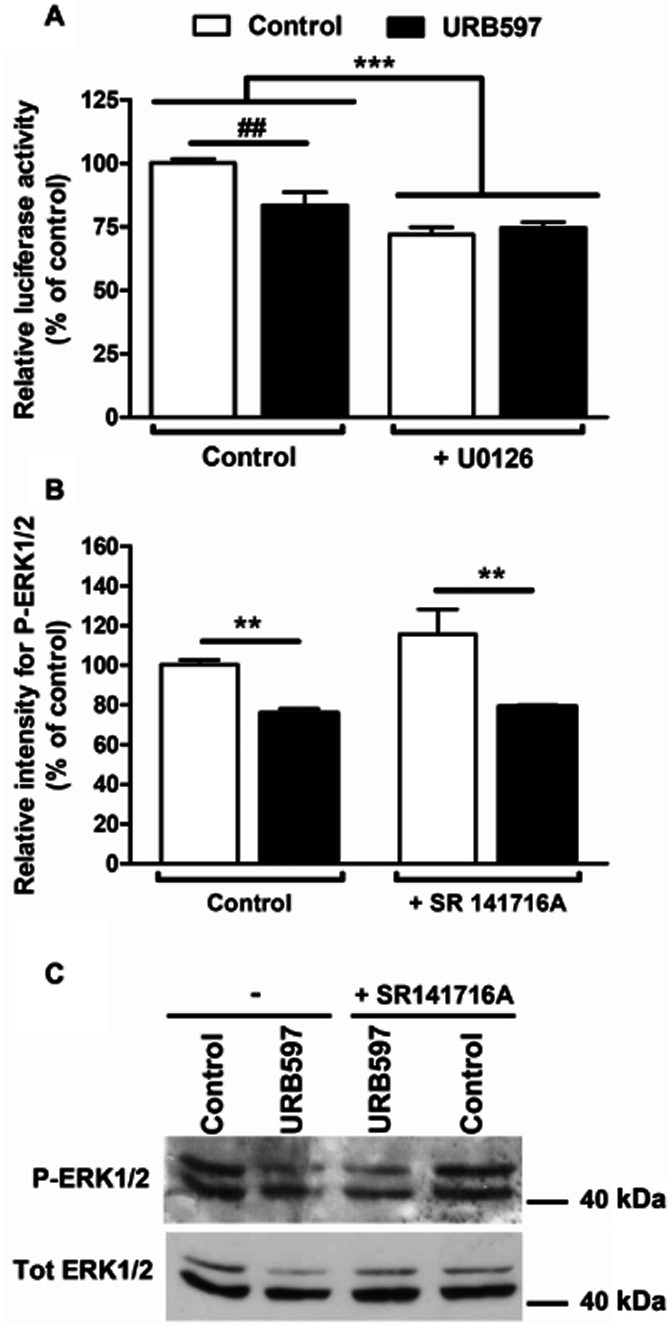
Role of MAPK in URB597 control of TH promoter activity. The influence of MAPK signalling was investigated in N1E115 cells transfected with pTH250-Luc. To examine the role of this signalling cascade, cells were treated for 1 h with MEK1/2 inhibitor U0126 (5 μM) before adding URB597 (0.1 μM) for an additional 5 h (A). Results are given as the percentages of relative luciferase activity (firefly luciferase relative to Renilla luciferase) relative to control values. Data shown are means with SEM values of three experiments performed in triplicate. One-way ANOVA indicated a general effect of the MEK1/2 inhibitor (***P = 0.0003, f = 36.33, residual d.f. = 8) with a positive interaction (*P = 0.0139, f = 9.831). ##P < 0.01 as indicated for treated cells and determined by Bonferroni post-test. (B) ERK1/2 activation was determined using immunoblot detection of phosphorylated ERK1/2 (P-ERK1/2 – 42 and 44 kDA). Cells were treated with URB597 (1 μM, 15 min) in the presence or the absence of SR 141716A (1 μM). Phosphorylated ERK1/2 values were normalized to total ERK1/2 (Tot ERK1/2). Results are expressed as percentages of relative density. A representative immunoblot is shown in (C). Data shown are means with SEM values of three experiments. One-way ANOVA indicated a general effect of URB597 (**P < 0.0016, f = 21.80, residual d.f. = 8). Neither significant effect for SR 141716A nor an interaction were observed, indicating the absence of antagonism by SR 141716A.
Discussion
Modulation of TH activity and expression is one way by which noradrenergic and dopaminergic signalling can be regulated. Given the variety of neurological processes or disorders related to the dopamine and/or norepinephrine transmission systems, understanding the mechanisms controlling TH gene expression is of high physiopathological relevance. The present study provides evidence for the control of TH gene expression by the FAAH inhibitor URB597 via mechanisms that are independent of FAAH and of the CB1 cannabinoid receptor. We also suggest that URB597 possibly acts through Abn-CBD-sensitive receptors and PPARs (Figure 8).
Figure 8.
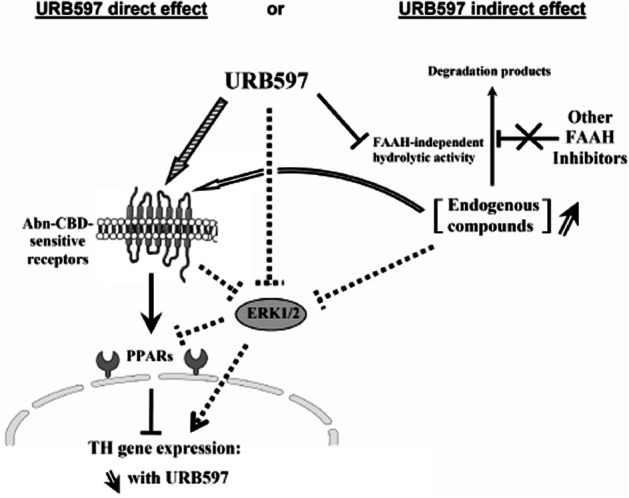
Schematic representation of the potential targets involved in the URB597-metiated regulation of TH. Besides the well-documented FAAH inhibition and endocannabinoid/NAE elevations, URB597 could either directly (striped arrow) or indirectly, putatively through inhibition of an alternative (non-FAAH) enzymatic activity (open arrow), interact with some off-targets. This results in a regulation of the Abn-CBD-sensitive receptor, which, in turn, could decrease TH gene expression by interacting with PPARs. However, our investigations also support the involvement of ERK1/2 signalling pathway. Therefore, we may hypothesize that the URB597-mediated reduction in ERK1/2 phosphorylation (through direct or indirect Abn-CBD-sensitive receptor-dependent mechanisms) also contributes to the regulation of PPARs (dotted lines). The resulting effect is a decrease in TH gene expression. Contrasting with URB597 effect, other tested FAAH inhibitors failed to regulate these targets and to modulate TH gene expression.
We previously reported that synthetic cannabinoid agonists regulate TH mRNA expression (Bosier et al., 2007). Here, we show that TH gene expression is also controlled – through activation of the CB1 cannabinoid receptor – by exogenously added AEA. However, with the exception of URB597, the different FAAH inhibitors tested here failed to induce a similar effect, while increasing endocannabinoid levels, suggesting that the endogenous concentration of AEA is not sufficient to control TH transcription. This is in agreement with the high concentrations of exogenously added AEA (10 μM) required to control TH promoter activity. In addition, probably due to its rapid degradation, when used at a moderate concentration (1 μM), exogenous AEA required a protection from FAAH hydrolytic activity to induce its effect on TH regulation. Probably reflecting the lack of URB597-mediated protection against MAGL activity (Kathuria et al., 2003), no effects were observed with 2-AG in similar conditions. However, when used at 10 μM, this endogenous agonist also triggered a reduction of TH transcription, which was abrogated by SR141716A (data not shown). Possibly having pathophysiological consequences, these results suggest that, as long as sufficient concentration is reached, the endocannabinoid system may directly interfere with the main limiting enzyme involved in dopamine synthesis.
On the other hand, we also found here that URB597 per se has pronounced effects on TH gene expression. As shown by using SR 141716A, this was not a CB1-mediated mechanism. Because URB597 was developed as a FAAH inhibitor that increases endocannabinoid levels, one would expect activation of CB1 cannabinoid receptors in neuronal cell lines to be responsible for the URB597-direct effect. However, our data are in line with recent papers that reported CB1-independent URB597-induced effects. Thus, for instance, TRPV1 (Maione et al., 2006) and PPARs (Jhaveri et al., 2008; Melis et al., 2008; Sagar et al., 2008; Mazzola et al., 2009) were described as potential targets of the FAAH-controlled mediators.
Excluding the involvement of TRPV1 as potential target in the herein reported effects, N1E115 neuroblastoma cells do not express this channel receptor (Figure 5A). However, even though PEA which is also recognized as a PPAR agonist (Lo Verme et al., 2005) failed to modulate TH expression (Figure 1), we found that preventing PPAR signalling resulted in the loss of URB597-induced modulation of TH transcription. Moreover, confirming the involvement of these nuclear receptors in the control of TH promoter activity, direct activation of PPARs with TGZ significantly regulated TH expression. While this could appear conflicting, one should note that in this assay, PEA was used at 1 μM, whereas the reported pEC50 of PEA for PPARα activation is 3 μM (Lo Verme et al., 2005). Besides, further reconciling our results with the involvement of PPARs, when tested at 10 μM PEA elicited a reduction of TH promoter activity of about 25% (data not shown). Together, this is in line with previous reports demonstrating the involvement of PPARs in URB597-mediated control of dopaminergic functions (Melis et al., 2008; Scherma et al., 2008; Luchicchi et al., 2010).
In addition, we also found that by interfering with the signalling of the Abn-CBD-sensitive receptors [i.e. GPR55 and/or the non-CB1 endothelial cannabinoid receptor (Járai et al., 1999; Ryberg et al., 2007)], CBD and O-1918 prevented URB597-mediated effects on TH promoter activity. Further corroborating this assumption, Abn-CBD per se was able to decrease TH transcription, whereas CBD prevented this effect. Of note, the later results could explain the reported increase in dopamine levels after CBD administration to rats (Murillo-Rodríguez et al., 2006). Moreover, supporting the existence of crosstalk between the Abn-CBD receptors and PPARs, the Abn-CBD-mediated reduction of TH transcription was totally prevented using PPAR antagonists. These results are consistent with the notion that besides increasing AEA levels, FAAH inhibition regulates other bioactive lipid levels such as NAEs (e.g. PEA and OEA), N-acyltaurines and N-acyldopamines, which, in turn, could activate TRPV1 or PPAR (Long et al., 2011). For instance, NAEs were reported to participate in some of the URB597 effects, either through PPARα (Jhaveri et al., 2008; Melis et al., 2008) or TRPV1 (Maione et al., 2006) activation.
However, quite more unexpectedly, we found that the effect of URB597 on TH expression does not involve FAAH inhibition. Indeed, a battery of other tested FAAH inhibitors did not regulate TH gene expression, even though all the inhibitors enhance endocannabinoid and NAE levels in N1E115 cells. The following elements rule out FAAH as the molecular target mediating the herein reported effect of URB597. (i) In addition to URB597, we used four potent FAAH inhibitors, MAFP, CAY10402 CAY10499 and PF750, which are structurally unrelated and differ by their mechanism of FAAH inhibition and their selectivity towards NAE degradation enzymes. (ii) We confirmed in our neuroblastoma model that PF750 (a highly selective FAAH inhibitor), MAFP (non-selective irreversible inhibitor that also binds cannabinoid receptors) and CAY10499 (potent FAAH and MAGL inhibitor) increase endocannabinoid and NAE levels to the same extent than URB597. Note that CAY10402 only moderately increased OEA and SEA levels. PF750, which increases NAE levels to a similar extent than URB597, completely prevented [3H]-AEA hydrolysis in intact N1E115 cells (Supporting Information Fig. S2). This suggests that the NAE increase is a consequence of FAAH inhibition. (iii) MAFP, CAY10499, CAY10402, as well as PF750, used at up to 100-fold times of URB597 concentration, failed to regulate TH promoter activity despite the inhibition of FAAH activity and the subsequent NAE increase. Together, this represents compelling evidence for a FAAH-independent action of URB597.
This constitutes a provocative result because the potential therapeutic effects of URB597 on depression, anxiety and pain have been attributed to the accumulation of AEA and consecutive CB1 cannabinoid receptor (Kathuria et al., 2003; Gobbi et al., 2005; Piomelli et al., 2006) or CB2 cannabinoid receptor (Jayamanne et al., 2006) activation. The cannabinoid receptor-independent effect reported in this study could be related to pharmacological responses as well. Indeed, we found that URB597 administration to mice resulted in a reduction of TH gene expression in several, but not all, brain areas. Considering that TH is the rate-limiting enzyme in the synthesis of dopamine, one could speculate that some of the URB597-reported effects result from modification of the dopaminergic or noradrenergic basal tone. Indeed, previous studies showed that URB597 increases waking (Murillo-Rodríguez et al., 2007) and counteracts the addictive properties of nicotine (Melis et al., 2008; Scherma et al., 2008) through either modulation of dopamine content or dopamine neuron activity in different brain regions.
Although generally regarded as a selective FAAH inhibitor, proteome-wide studies identified off-target enzymes for URB597 (Lichtman et al., 2004). Among those are several carboxypeptidase isoforms (Zhang et al., 2007) and possibly ABHD6 and/or ABHD12, two enzymes responsible for 2-AG metabolism (Blankman et al., 2007). Noteworthy, the 20-fold difference in URB597 potency for the inhibition of AEA and PEA hydrolysis could be due to inhibition of an alternative enzyme. However, URB597 does not inhibit NAAA, ruling out this N-acylethanolamine degradation enzyme (Sun et al., 2005; Tsuboi et al., 2007). Because we found a similar increase in NAE levels with URB597, MAFP, CAY10499 and PF750, these bioactive lipids are not responsible for URB597-mediated reduction in TH gene expression. These data raise the question of an original (non-FAAH/non-CB1/non-CB2) target controlled by URB597 and involved in the regulation of TH gene expression. For instance, the existence of off-target enzymes suggests that URB597 could inhibit the degradation of some endogenous compounds, differing from FAAH substrates, which increased levels, in turn, may control TH gene expression through PPAR and/or Abn-CBD receptors. Alternatively, URB597 could directly bind to a receptor, thus controlling TH promoter activity. Supporting this hypothesis, Niforatos et al. showed that URB597 binds and activates some members of the TRP ion channel superfamily (Niforatos et al., 2007). While this remains an open question, we have identified here a potential pathway linking URB597 to TH gene expression (see Figure 8).
Indeed, our results strongly suggest the involvement of Abn-CBD receptors. These include both GPR55 and the non-CB1 endothelial cannabinoid receptor. Both are GPCRs (Offertáler et al., 2003; Ryberg et al., 2007) known to activate different MAPK family members (Offertáler et al., 2003; Oka et al., 2007). On the other hand, MAPK including ERK1/2 have been shown to both activate and inhibit PPARs (Gelman et al., 2005). In accordance with this, our results (Figure 7) suggest that ERK1/2 could represent the molecular link between Abn-CBD-sensitive receptors and PPARs. We therefore propose the following mechanism explaining the concomitant involvement of Abn-CBD receptors and PPARs in the control of TH transcription by URB597: either directly or indirectly, URB597 regulates Abn-CBD receptors, which, in turn, control ERK1/2 phosphorylation, leading to PPAR regulation and modulation of TH gene transcription.
In conclusion, we report a URB597-mediated alteration of TH gene expression in N1E115 cells. Contrasting with its largely reported effects, URB597 acts through non-CB1- and non-FAAH-dependent pathways. Although the precise mechanism of action remains to be elucidated, we identified PPARs and Abn-CBD receptors as putative targets mediating URB597 effects, independent of FAAH inhibition. Therefore, this study provides the first pharmacologically relevant off-target for URB597. In light of the numerous preclinical studies involving URB597, particularly in anxiety and depression, the effects on the catecholaminergic transmission system suggested in the present study should be taken into account when interpreting the data.
Acknowledgments
B. B. is a FRS-FNRS Postdoctoral Researcher. We are grateful to Maité Graux, Owein Guillemot-Legris, Mireille Alhouayek and Damien Naslain for skilful technical assistance. This study was supported by grants from the Belgian National Fund for Scientific Research (FRFC No. 2.4.654.06; FRFC No. 2.4555.08; crédit au chercheur 1.B144.10) and FSR grants from the Université catholique de Louvain to D. M. L. and G. G. M.
Glossary
- Abn-CBD
abnormal-cannabidiol
- AEA
anandamide
- CBD
cannabidiol
- FAAH
fatty acid amide hydrolase
- MAGL
monoacylglycerol lipase
- MEK
MAP kinase kinase
- NAE
N-acylethanolamine
- OEA
N-oleoylethanolamine
- PEA
N-palmitoylethanolamine
- SEA
N-stearoylethanolamine
- TGZ
troglitazone
- TH
tyrosine hydroxylase
- TRPV1
ligand-gated ion channel vanilloid receptor 1
Conflict of interest
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Expression of endocannabinoid-hydrolysing enzyme mRNAs in NIE115 cells.
RT-PCR was performed on N1E115 cell mRNA extracts with specific primers targeting the different endocannabinoid-hydrolysing enzymes. RT-PCR yielded the predicted amplification product for FAAH, NAAA, MAGL, ABHD6 and ABHD12. Negative controls were performed without any DNA template (not shown).
Figure S2 Inhibition of AEA hydrolysis by URB597 and PF750 in N1E115 cells.
Measurements of [3H]-AEA hydrolysis by N1E115 intact cells incubated with URB597, PF750 (10 and 0.1 μM) or vehicle alone, during 25 min. Results are expressed as the percentages of substrate hydrolysis relative to control values. Data are means with SEM values of 3 experiments performed in triplicate. One-way ANOVA followed by Dunnett's post-test ***P < 0.001, relative to control.
Table S1 Nucleotide sequences for the primers used for the PCR amplifications of cannabinoid targets, the corresponding amplicons and the annealing temperatures.
Table S2 Nucleotide sequences for the primers used for the PCR amplifications of endocannabinoid-degrading enzymes, the corresponding amplicons and the annealing temperatures.
References
- Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, et al. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry. 2007;46:13019–13030. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhouayek M, Lambert DM, Delzenne NM, Cani PD, Muccioli GG. Increasing endogenous 2-arachidonoylglycerol levels counteracts colitis and related systemic inflammation. FASEB J. 2011;25:2711–2721. doi: 10.1096/fj.10-176602. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin A, Fernandez-Ruiz JJ, Martin M, Rodriguez de Fonseca F, Hernandez ML, Ramos JA. delta 9-Tetrahydrocannabinol affects mesolimbic dopaminergic activity in the female rat brain: interactions with estrogens. J Neural Transm Gen Sect. 1993;92:81–95. doi: 10.1007/BF01244868. [DOI] [PubMed] [Google Scholar]
- Bosier B, Tilleux S, Najimi M, Lambert DM, Hermans E. Agonist selective modulation of tyrosine hydroxylase expression by cannabinoid ligands in a murine neuroblastoma cell line. J Neurochem. 2007;102:1996–2007. doi: 10.1111/j.1471-4159.2007.04679.x. [DOI] [PubMed] [Google Scholar]
- Bosier B, Hermans E, Lambert DM. Concomitant activation of adenylyl cyclase suppresses the opposite influences of CB(1) cannabinoid receptor agonists on tyrosine hydroxylase expression. Biochemistry. 2009;77:216–227. doi: 10.1016/j.bcp.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Cachope R, Mackie K, Triller A, O'Brien J, Pereda AE. Potentiation of electrical and chemical synaptic transmission mediated by endocannabinoids. Neuron. 2007;56:1034–1047. doi: 10.1016/j.neuron.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadogan AK, Alexander SP, Boyd EA, Kendall DA. Influence of cannabinoids on electrically evoked dopamine release and cyclic AMP generation in the rat striatum. J Neurochem. 1997;69:1131–1137. doi: 10.1046/j.1471-4159.1997.69031131.x. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Piomelli D. Antinociceptive activity of the endogenous fatty acid amide, palmitylethanolamide. Eur J Pharmacol. 2001;419:191–198. doi: 10.1016/s0014-2999(01)00988-8. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther. 2005;313:352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Gelman L, Michalik L, Desvergne B, Wahli W. Kinase signalling cascades that modulate peroxisome proliferator-activated receptors. Curr Opin Cell Biol. 2005;17:216–222. doi: 10.1016/j.ceb.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Gerdeman G, Lovinger DM. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol. 2001;85:468–471. doi: 10.1152/jn.2001.85.1.468. [DOI] [PubMed] [Google Scholar]
- Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, et al. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci U S A. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González S, Mena MA, Lastres-Becker I, Serrano A, de Yébenes JG, Ramos JA, et al. Cannabinoid CB(1) receptors in the basal ganglia and motor response to activation or blockade of these receptors in parkin-null mice. Brain Res. 2005;1046:195–206. doi: 10.1016/j.brainres.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Hao S, Avraham Y, Mechoulam R, Berry EM. Low dose anandamide affects food intake, cognitive function, neurotransmitter and corticosterone levels in diet-restricted mice. Eur J Pharmacol. 2000;392:147–156. doi: 10.1016/s0014-2999(00)00059-5. [DOI] [PubMed] [Google Scholar]
- Huang CC, Lo SW, Hsu KS. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J Physiol. 2001;532:731–748. doi: 10.1111/j.1469-7793.2001.0731e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsson SO, Fowler CJ. Characterization of palmitoylethanolamide transport in mouse Neuro-2a neuroblastoma and rat RBL-2H3 basophilic leukaemia cells: comparison with anandamide. Br J Pharmacol. 2001;132:1743–1754. doi: 10.1038/sj.bjp.0704029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Járai Z, Wagner JA, Varga K, Lake KD, Compton DR, Martin BR, et al. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc Natl Acad Sci U S A. 1999;96:14136–14141. doi: 10.1073/pnas.96.24.14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Robinson I, Garle MJ, Patel A, Sun Y, et al. Inhibition of fatty acid amide hydrolase and cyclooxygenase-2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator-activated receptor-alpha in a model of inflammatory pain. Neuropharmacology. 2008;55:85–93. doi: 10.1016/j.neuropharm.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- de Lago E, de Miguel R, Lastres-Becker I, Ramos JA, Fernandez-Ruiz J. Involvement of vanilloid-like receptors in the effects of anandamide on motor behavior and nigrostriatal dopaminergic activity: in vivo and in vitro evidence. Brain Res. 2004;1007:152–159. doi: 10.1016/j.brainres.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, et al. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277:19649–19657. doi: 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- Leung D, Hardouin C, Boger DL, Cravatt BF. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat Biotechnol. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, et al. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- Long JZ, LaCava M, Jin X, Cravatt BF. An anatomical and temporal portrait of physiological substrates for fatty acid amide hydrolase. J Lipid Res. 2011;52:337–344. doi: 10.1194/jlr.M012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchicchi A, Lecca S, Carta S, Pillolla G, Muntoni AL, Yasar S, et al. Effects of fatty acid amide hydrolase inhibition on neuronal responses to nicotine, cocaine and morphine in the nucleus accumbens shell and ventral tegmental area: involvement of PPAR-alpha nuclear receptors. Addict Biol. 2010;15:277–288. doi: 10.1111/j.1369-1600.2010.00222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione S, Bisogno T, DeNovellis V, Palazzo E, Cristino L, Valenti M, et al. Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. J Pharmacol Exp Ther. 2006;316:969–982. doi: 10.1124/jpet.105.093286. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Valverde O, Berrendero F. Involvement of the endocannabinoid system in drug addiction. Trends Neurosci. 2006;29:225–232. doi: 10.1016/j.tins.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Marinelli S, Di Marzo V, Berretta N, Matias I, Maccarrone M, Bernardi G, et al. Presynaptic facilitation of glutamatergic synapses to dopaminergic neurons of the rat substantia nigra by endogenous stimulation of vanilloid receptors. J Neurosci. 2003;23:3136–3144. doi: 10.1523/JNEUROSCI.23-08-03136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzola C, Medalie J, Scherma M, Panlilio LV, Solinas M, Tanda G, et al. Fatty acid amide hydrolase (FAAH) inhibition enhances memory acquisition through activation of PPAR-alpha nuclear receptors. Learn Mem. 2009;16:332–337. doi: 10.1101/lm.1145209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Pillolla G, Luchicchi A, Muntoni AL, Yasar S, Goldberg SR, et al. Endogenous fatty acid ethanolamides suppress nicotine-induced activation of mesolimbic dopamine neurons through nuclear receptors. J Neurosci. 2008;28:13985–13994. doi: 10.1523/JNEUROSCI.3221-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccioli GG. Endocannabinoid biosynthesis and inactivation, from simple to complex. Drug Discov Today. 2010;15:474–483. doi: 10.1016/j.drudis.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Muccioli GG, Stella N. An optimized GC-MS method detects nanomolar amounts of anandamide in mouse brain. Anal Biochem. 2008;373:220–228. doi: 10.1016/j.ab.2007.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccioli GG, Xu C, Odah E, Cudaback E, Cisneros JA, Lambert DM, et al. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J Neurosci. 2007;27:2883–2889. doi: 10.1523/JNEUROSCI.4830-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccioli GG, Labar G, Lambert DM. CAY10499, a novel monoglyceride lipase inhibitor evidenced by an expeditious MGL assay. Chembiochem. 2008;9:2704–2710. doi: 10.1002/cbic.200800428. [DOI] [PubMed] [Google Scholar]
- Murillo-Rodríguez E, Millán-Aldaco D, Palomero-Rivero M, Mechoulam R, Drucker-Colín R. Cannabidiol, a constituent of Cannabis sativa, modulates sleep in rats. FEBS Lett. 2006;580:4337–4345. doi: 10.1016/j.febslet.2006.04.102. [DOI] [PubMed] [Google Scholar]
- Murillo-Rodríguez E, Vázquez E, Millán-Aldaco D, Palomero-Rivero M, Drucker-Colin R. Effects of the fatty acid amide hydrolase inhibitor URB597 on the sleep-wake cycle, c-Fos expression and dopamine levels of the rat. Eur J Pharmacol. 2007;562:82–91. doi: 10.1016/j.ejphar.2007.01.076. [DOI] [PubMed] [Google Scholar]
- Niforatos W, Zhang XF, Lake MR, Walter KA, Neelands T, Holzman TF, et al. Activation of TRPA1 channels by the fatty acid amide hydrolase inhibitor 3′-carbamoylbiphenyl-3-yl cyclohexylcarbamate (URB597) Mol Pharmacol. 2007;71:1209–1216. doi: 10.1124/mol.106.033621. [DOI] [PubMed] [Google Scholar]
- Offertáler L, Mo FM, Bátkai S, Liu J, Begg M, Razdan RK, et al. Selective ligands and cellular effectors of a G protein-coupled endothelial cannabinoid receptor. Mol Pharmacol. 2003;63:699–705. doi: 10.1124/mol.63.3.699. [DOI] [PubMed] [Google Scholar]
- Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochemistry. 2007;362:928–934. doi: 10.1016/j.bbrc.2007.08.078. [DOI] [PubMed] [Google Scholar]
- Page ME, Oropeza VC, Sparks SE, Qian Y, Menko AS, Van Bockstaele EJ. Repeated cannabinoid administration increases indices of noradrenergic activity in rats. Pharmacol Biochem Behav. 2007;86:162–168. doi: 10.1016/j.pbb.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, et al. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597) CNS Drug Rev. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero J, de Miguel R, Garcia-Palomero E, Fernandez-Ruiz JJ, Ramos JA. Time-course of the effects of anandamide, the putative endogenous cannabinoid receptor ligand, on extrapyramidal function. Brain Res. 1995a;694:223–232. doi: 10.1016/0006-8993(95)00835-e. [DOI] [PubMed] [Google Scholar]
- Romero J, Garcia L, Cebeira M, Zadrozny D, Fernandez-Ruiz JJ, Ramos JA. The endogenous cannabinoid receptor ligand, anandamide, inhibits the motor behavior: role of nigrostriatal dopaminergic neurons. Life Sci. 1995b;56:2033–2040. doi: 10.1016/0024-3205(95)00186-a. [DOI] [PubMed] [Google Scholar]
- Rubino T, Realini N, Castiglioni C, Guidali C, Vigano D, Marras E, et al. Role in anxiety behavior of the endocannabinoid system in the prefrontal cortex. Cereb Cortex. 2008;18:1292–1301. doi: 10.1093/cercor/bhm161. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar DR, Kendall DA, Chapman V. Inhibition of fatty acid amide hydrolase produces PPAR-alpha-mediated analgesia in a rat model of inflammatory pain. Br J Pharmacol. 2008;155:1297–1306. doi: 10.1038/bjp.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherma M, Panlilio LV, Fadda P, Fattore L, Gamaleddin I, Le Foll B, et al. Inhibition of anandamide hydrolysis by cyclohexyl carbamic acid 3′-carbamoyl-3-yl ester (URB597) reverses abuse-related behavioral and neurochemical effects of nicotine in rats. J Pharmacol Exp Ther. 2008;327:482–490. doi: 10.1124/jpet.108.142224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seimandi M, Lemaire G, Pillon A, Perrin A, Carlavan I, Voegel JJ, et al. Differential responses of PPARalpha, PPARdelta, and PPARgamma reporter cell lines to selective PPAR synthetic ligands. Anal Biochem. 2005;344:8–15. doi: 10.1016/j.ab.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Solinas M, Justinova Z, Goldberg SR, Tanda G. Anandamide administration alone and after inhibition of fatty acid amide hydrolase (FAAH) increases dopamine levels in the nucleus accumbens shell in rats. J Neurochem. 2006;98:408–419. doi: 10.1111/j.1471-4159.2006.03880.x. [DOI] [PubMed] [Google Scholar]
- Sun YX, Tsuboi K, Zhao LY, Okamoto Y, Lambert DM, Ueda N. Involvement of N-acylethanolamine-hydrolyzing acid amidase in the degradation of anandamide and other N-acylethanolamines in macrophages. Biochim Biophys Acta. 2005;1736:211–220. doi: 10.1016/j.bbalip.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, et al. Design, synthesis, and structure-activity relationships of alkylcarbamic acid aryl esters, a new class of fatty acid amide hydrolase inhibitors. J Med Chem. 2003;46:2352–2360. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Zhao LY, Okamoto Y, Araki N, Ueno M, Sakamoto H, et al. Predominant expression of lysosomal N-acylethanolamine-hydrolyzing acid amidase in macrophages revealed by immunochemical studies. Biochim Biophys Acta. 2007;1771:623–632. doi: 10.1016/j.bbalip.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Moller T, Stella N. Astrocytes in culture produce anandamide and other acylethanolamides. J Biol Chem. 2002;277:20869–20876. doi: 10.1074/jbc.M110813200. [DOI] [PubMed] [Google Scholar]
- Zhang D, Saraf A, Kolasa T, Bhatia P, Zheng GZ, Patel M, et al. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology. 2007;52:1095–1105. doi: 10.1016/j.neuropharm.2006.11.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.