

Abstract
Background and purpose
JZL184 is a selective inhibitor of monoacylglycerol lipase (MAGL), the enzyme that preferentially catabolizes the endocannabinoid 2-arachidonoyl glycerol (2-AG). Here, we have studied the effects of JZL184 on inflammatory cytokines in the brain and plasma following an acute immune challenge and the underlying receptor and molecular mechanisms involved.
Experimental approach
JZL184 and/or the CB1 receptor antagonist, AM251 or the CB2receptor antagonist, AM630 were administered to rats 30 min before lipopolysaccharide (LPS). 2 h later cytokine expression and levels, MAGL activity, 2-AG, arachidonic acid and prostaglandin levels were measured in the frontal cortex, plasma and spleen.
Key results
JZL184 attenuated LPS-induced increases in IL-1β, IL-6, TNF-α and IL-10 but not the expression of the inhibitor of NFkB (IκBα) in rat frontal cortex. AM251 attenuated JZL184-induced decreases in frontal cortical IL-1β expression. Although arachidonic acid levels in the frontal cortex were reduced in JZL184-treated rats, MAGL activity, 2-AG, PGE2 and PGD2 were unchanged. In comparison, MAGL activity was inhibited and 2-AG levels enhanced in the spleen following JZL184. In plasma, LPS-induced increases in TNF-α and IL-10 levels were attenuated by JZL184, an effect partially blocked by AM251. In addition, AM630 blocked LPS-induced increases in plasma IL-1β in the presence, but not absence, of JZL184.
Conclusion and implications
Inhibition of peripheral MAGL in rats by JZL184 suppressed LPS-induced circulating cytokines that in turn may modulate central cytokine expression. The data provide further evidence for the endocannabinoid system as a therapeutic target in treatment of central and peripheral inflammatory disorders.
Linked Articles
This article is part of a themed section on Cannabinoids. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2013.169.issue-4 & http://dx.doi.org/10.1111/bph.2012.167.issue-8
Keywords: monoacylglycerol lipase, JZL184, endocannabinoid, 2-AG, cytokines, IL-1β, TNF-α, IL-6, IL-10, NF-κB, frontal cortex, brain, plasma
Introduction
Neuroinflammation is a key component of various neurological diseases including Alzheimer's disease, Parkinson's disease, multiple sclerosis and psychiatric disorders such as depression. As such, the need to develop a greater understanding of the neurobiological mechanisms mediating neuroinflammation is critical at a fundamental physiological level and for the development of novel, more efficacious treatments. Accumulating evidence indicates that the endogenous cannabinoid (endocannabinoid) system plays a significant role in modulating the immune system, representing an important therapeutic target in the treatment of both central and peripheral inflammatory disorders (see Ullrich et al., 2007; Nagarkatti et al., 2009; Stella, 2009; Finn, 2010; Jean-Gilles et al., 2010; Molina-Holgado and Molina-Holgado, 2010).
The endocannabinoid system is comprised of the two cannabinoid G protein-coupled receptors (CB1 and CB2; receptor nomenclature follows Alexander et al. (2011) the endocannabinoids anandamide (N-arachidonoylethanolamide) and 2-arachidonoyl glycerol (2-AG) and the enzymes responsible for their synthesis and degradation. 2-AG, the most abundant endocannabinoid in the CNS and full agonist at both CB1 and CB2 receptors, is synthesized primarily via hydrolysis of cell membrane phospholipid precursors by diacylglycerol lipase-α (Mechoulam et al., 1995; Gao et al., 2010). Monoacylglycerol lipase (MAGL) is the enzyme primarily responsible for the metabolism of 2-AG to arachidonic acid and glycerol (Dinh et al., 2002), accounting for up to 85% of 2-AG hydrolysis in the brain (Blankman et al., 2007). Other enzymes involved in 2-AG hydrolysis include the serine hydrolyses ABHD6 and ABHD12, fatty acid amide hydrolase (FAAH), cyclooxygenase-2 (COX-2) and carboxylesterases (Di Marzo et al., 1998; Blankman et al., 2007; Xie et al., 2010; Savinainen et al., 2012). 2-AG levels are enhanced in animal models of ischaemia, Alzheimer's disease, Parkinson's disease and multiple sclerosis (Baker et al., 2001; Ferrer et al., 2003; Panikashvili et al., 2001; van der Stelt et al., 2005; 2006), and it is possible that this endocannabinoid may play a protective role in these conditions, all of which have a neuroinflammatory/neuroimmune component. Indeed, evidence from in vitro studies indicates that 2-AG suppresses immune function by reducing inflammatory cytokines such as IL-6, IL-2 and TNF-α and mediators such as nitric oxide and prostaglandins (Gallily et al., 2000; Chang et al., 2001; Facchinetti et al., 2003; Rockwell et al., 2006; Raman et al., 2011). In addition to being a substrate for COX-2, 2-AG also inhibited the increase in COX-2 expression in neurons and T cells, but not endothelial cells or macrophages, in response to inflammatory stimuli (Chang et al., 2001; Mestre et al., 2006; Zhang and Chen 2008; Raman et al., 2011). Recent studies have demonstrated that enhancing 2-AG tone protects hippocampal neurons in culture from excitotoxic- and inflammation-induced degeneration and apoptosis, an effect mediated by CB1 receptor-induced suppression of extracellular signal-regulated kinases, NF-κB phosphorlyation and COX-2 expression (Zhang and Chen 2008; Chen et al., 2011). Although, in vitro data suggest potent anti-inflammatory and neuroprotective effects of 2-AG, there has been a paucity of studies in vivo, primarily due to the lack of pharmacological agents that selectively target 2-AG metabolism. Panikashvili et al. demonstrated that 2-AG activation of CB1 receptors reduced infarct volume following closed head injury via inhibition of NF-κB signalling (Panikashvili et al., 2001; 2005). Subsequent studies from these researchers revealed that 2-AG reduced the expression of the pro-inflammatory cytokines TNF-α, IL-1β and IL-6, reactive oxygen species and blood–brain barrier permeability in this model of traumatic brain injury (McCarron et al., 2003; Panikashvili et al., 2006). Recent studies have also shown a neuroprotective effect of 2-AG in acute and chronic mouse models of multiple sclerosis, an effect accompanied by modulation of macrophage activity (Lourbopoulos et al., 2011). URB602, a MAGL inhibitor, has been shown to elicit anti-inflammatory effects in several animal models (Comelli et al., 2007; Guindon et al., 2011). However, the low potency (Hohmann et al., 2005) and lack of selectivity of URB602 for MAGL over FAAH (Vandevoorde et al., 2007) has limited the usefulness of this compound. The recent development of a novel potent MAGL inhibitor 4-nitrophenyl-4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) (Long et al., 2009a,b) has been a major advance, enabling detailed studies on the role of 2-AG in a number of physiological and pathophysiological processes including tumour cell growth, anxiety, nausea and pain (Kinsey et al., 2009; 2010; Sciolino et al., 2011; Sticht et al., 2011; Ye et al., 2011). In terms of inflammatory processes, JZL184 selectively increased gastric levels of 2-AG in mice and inhibited NSAID-induced gastric haemorrhage and associated increases in expression of the pro-inflammatory cytokines IL-1, IL-6 and TNF-α (Kinsey et al., 2011). Similarly, in a mouse model of colitis, JZL184-induced increases in 2-AG were associated with reduced macroscopic and histological colon alterations and pro-inflammatory cytokine expression, effects mediated by CB1 and CB2 receptors (Alhouayek et al., 2011). In the brain, increases in pro-inflammatory cytokine expression and PGE2 levels following an immune challenge were inhibited following genetic or pharmacological MAGL inhibition, an effect not mediated by cannabinoid receptors but possibly via modulation of the arachidonic acid cascade (Nomura et al., 2011). Similar anti-inflammatory and neuroprotective effects were observed following MAGL inhibition in a mouse model of Parkinson's disease (Nomura et al., 2011).
To date, in vivo studies examining 2-AG effects on inflammation have been primarily carried out in mice. A large proportion of animal models are developed in the rat, and therefore investigation of effects across rodent species is paramount. Although JZL184 has reduced affinity for rat MAGL compared with the murine enzyme (Long et al., 2009b), systemic administration of JZL184 exhibited anxiolytic-like effects at relatively low doses in rats (Sciolino et al., 2011). However, only one study to date has reported that systemic administration of JZL184 increased 2-AG levels in the rat brain (Oleson et al., 2012). The present study demonstrates that JZL184 reduced lipopolysaccharide (LPS)-induced cytokine expression/levels both in the rat frontal cortex and plasma, effects partially attenuated by CB1 receptor antagonism. MAGL activity was inhibited, and 2-AG levels were enhanced in the spleen, but not in frontal cortex, following JZL184 administration, indicating that different mechanisms may underlie the central and the peripheral anti-inflammatory effects of JZL184.
Methods
Animals
All animal care and experimental protocols were in accordance with the guidelines of the Animal Care and Research Ethics Committee, National University of Ireland, Galway under licence from the Irish Department of Health and Children and in compliance with the European Communities Council directive 86/609. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 128 animals were used in the experiments described here.
Experiments were carried out on male Sprague Dawley rats (weight 220–260 g; Harlan, UK), housed singly in plastic bottomed cages (45 × 25 × 20 cm) containing wood shavings as bedding. The animals were maintained at a constant temperature (21 ± 2°C) under standard lighting conditions (12:12 h light–dark, lights on from 08:00 to 20:00 h). All experiments were carried out during the light phase between 08:30 h and 15:00 h. Food and water were available ad libitum. Animals were habituated to handling and received an intraperitoneal (i.p.) injection of sterile saline (0.89% NaCl) for 3–4 days before experimentation in order to minimize the influence of the injection procedure on biological endpoints.
Experimental design
Experiment 1: Effects of JZL184 on LPS-induced cytokine expression, 2-AG and arachidonic acid levels in the rat frontal cortex and plasma, and receptor mechanisms mediating these effects.
Rats were randomly assigned to one of seven groups. Vehicle–Vehicle–Saline, Vehicle–Vehicle–LPS, AM251–Vehicle–LPS, AM630–Vehicle–LPS, Vehicle–JZL184–LPS, AM251–JZL184–LPS, AM630–JZL184–LPS (n = 6–10 per group). The CB1 receptor antagonist 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide (AM251) (1 mg kg−1, Cayman Chemicals, Tallin, Estonia), CB2 receptor antagonist [6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)-methanone (AM630) (1 mg kg−1, Cayman Chemicals) or vehicle (ethanol : Cremophor : saline; 1:1:18) were administered i.p. in an injection volume of 1 mL kg−1. The doses of antagonists were chosen based on previous studies demonstrating their ability to block the effects of cannabinoid agonists in vivo (Jayamanne et al., 2006; Gonzalez et al., 2011). Immediately following the administration of antagonists, rats received either JZL184 (10 mg kg−1 in an injection volume of 2 mL kg−1, generously received from Prof Benjamin Cravatt, Scripps Institute, La Jolla, CA, USA) or vehicle (ethanol : Cremophor : saline; 1:1:18) followed 30 min later by an i.p. injection of LPS (100 μg kg−1 B0111:B4 Sigma Aldrich, Dublin, Ireland) or saline vehicle (sterile 0.89% NaCl). The dose and time of JZL184 were determined from previous studies demonstrating that a minimum of 8 mg kg−1 is required to induce behavioural effects in rats (Sciolino et al., 2011), that 10 mg kg−1 enhanced 2-AG levels in the ventral tegmental area of the rat brain (Oleson et al., 2012) and that levels of endogenous 2-AG are enhanced 30 min post-administration (Long et al., 2009a,b). The dose and time of LPS administration were chosen on the basis of previous work within our laboratory demonstrating enhanced cytokine levels in the periphery and brain 2 h post-LPS administration (Roche et al., 2006; 2008; Kerr et al., 2012). Animals were killed by decapitation 2 h post-LPS/saline, trunk blood collected into chilled lithium heparin collection tubes, centrifuged at 4°C for 15 min at 4000× g, plasma was then removed and stored at −80°C until cytokine determination. Spleen and frontal cortex were excised, snap frozen on dry ice and stored at −80°C until assayed for MAGL activity, 2-AG, arachidonic acid and prostaglandin levels and cytokine expression.
Experiment 2: Effect of JZL184 on 2-AG levels in the rat frontal cortex over time
Rats were randomly assigned to one of nine treatment groups: Vehicle–Saline, Vehicle–LPS (10 min), JZL184–LPS (10 min), Vehicle–LPS (30 min), JZL184–LPS (30 min), Vehicle–LPS (60 min), JZL184–LPS (60 min), Vehicle–LPS (90 min) and JZL184–LPS (90 min) (n = 8–12 per group). Rats were administered JZL184 (10 mg kg−1 i.p. Cayman Chemicals) or vehicle (ethanol : cremophor : saline; 1:1:18) followed 30 min later by an i.p. injection of LPS (100 μg kg−1) or saline vehicle. Rats were killed 10, 30, 60 or 90 min after LPS (or saline), the brain excised, the frontal cortex dissected out and stored at −80°C until assayed for 2-AG concentration.
Analysis of inflammatory mediators using quantitative real-time polymerase chain reaction (PCR)
RNA was extracted from cortical tissue using NucleoSpin RNA II total RNA isolation kit (Macherey-Nagel, Düren, Germany). Genomic DNA contamination was removed with the addition of DNase to the samples according to the manufacturer's instructions. RNA was reverse transcribed into cDNA using a High Capacity cDNA Archive kit (Applied Biosystems, Paisley, UK). Taqman gene expression assays (Applied Biosystems) containing forward and reverse primers and a FAM-labelled MGB Taqman probe were used to quantify the gene of interest, and real-time PCR was performed using an ABI Prism 7500 instrument (Applied Biosystems), as previously described (Kerr et al., 2012). Assay IDs for the genes examined were as follows: IL-1β (Rn00580432_m1), TNF-α (Rn99999017_m1), IL-6 (Rn00561420_m1) and IL-10 (Rn00563409_m1). In order to determine if the effects of JZL184 on cytokine expression were mediated by modulation of the NF-κB pathway, the expression of the inhibitor of NF-κB,,IκBα (Rn01473658_g1), an indirect measure of NF-κB activity (Read et al., 1994), was also assessed. PCR was performed using Taqman Universal PCR Master Mix (Applied Biosystems), and samples were run in duplicate. The cycling conditions were 90°C for 10 min and 40 cycles of 90°C for 15 min followed by 60°C for 1 min. β-actin was used as an endogenous control to normalize gene expression data. Relative gene expression was calculated using the ΔΔCT method.
Determination of plasma cytokine protein levels
Plasma TNF-α, IL-1β, IL-6 and IL-10 concentrations were determined using specific rat enzyme-linked immunosorbent assays (ELISAs) performed using antibodies and standards obtained from R&D Systems, Abingdon, UK (TNF-α and IL-10) or Peprotech, London, UK (IL-1β and IL-6) as previously described (Roche et al., 2006; 2008; Kerr et al., 2012). ELISAs were carried out according to manufacturer's instructions, and cytokine levels were expressed as pg mL−1 plasma.
MAGL activity assay
MAGL activity assay was conducted as previously described (Cable et al., 2011). In brief, frontal cortical or spleen tissue was weighed (∼20 mg), homogenized in 1 mL of TE buffer (50 mM Tris, 1 mM EDTA, pH 7.4) and centrifuged at 14 000× g for 15 min. The pellet was resuspended in 1 mL of TE buffer, centrifuged and resuspended in a final volume of TE buffer so as to give a 1 in 5000 or 1 in 500 dilution of the initial wet cortical or spleen tissue weights respectively. Ninety microlitres of sample aliquots or blanks were pre-incubated with 5 μL of Hanks/HEPES buffer (116 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2.2H2O, 25 mM HEPES, 0.8 mM MgSO4, 1 mM NaH2PO4.2H2O) pH 7.4, containing 1 mg mL−1 defatted albumin for 30 min at 37°C. After pre-incubation, 5 μL of substrate (500 μL 2 mM 2-OG containing 3.75μCi 2-oleoyl-[3H]-glycerol; American Radiolabelled Chemicals, Herts, UK) was added with mixing to give a final [3H]-2-OG concentration of 100 μM. The reaction was allowed to proceed for 15 min at 37°C, following which 300 μL of stop solution (8% charcoal in 0.5 M HCl) was added with mixing. Samples were allowed to stand for a further 20 min and then centrifuged at 14 000× g for 5 min to pellet the charcoal before removal of a 200 μL aliquot of the clear supernatant containing liberated [3H]glycerol for liquid scintillation counting. Homogenates were assayed in triplicate. Data were expressed as nmol min−1 g−1 tissue.
Quantitation of endocannabinoids and N-acylethanolamine concentrations using liquid chromatography – tandem mass spectrometry (LC-MS/MS)
Quantitation of endocannabinoids and N-acylethanolamines was essentially as described previously (Ford et al., 2011; Olango et al., 2011; Kerr et al., 2012). In brief, samples were homogenized in 400 μL 100% acetonitrile containing deuterated internal standards [0.014 nmol anandamide-d8, 0.48 nmol 2-AG-d8, 0.016 nmol N-palmitoylethanolamide (PEA)-d4, 0.015 nmol N-oleoylethanolamide (OEA)-d2]. Lyophilized samples were re-suspended in 40 μL 65% acetonitrile and separated on a Zorbax® C18 column (150 × 0.5 mm internal diameter; Agilent Technologies, Cork, Ireland) by reversed-phase gradient elution initially with a mobile phase of 65% acetonitrile and 0.1% formic acid, which was ramped linearly up to 100% acetonitrile and 0.1% formic acid over 10 min and held at this for a further to 20 min. Under these conditions, anandamide, 2-AG, PEA and OEA eluted at the following retention times: 11.4 min, 12.9 min, 14.4 min and 15.0 min respectively. Analyte detection was carried out in electrospray-positive ionization and multiple reaction monitoring (MRM) mode on an Agilent 1100 HPLC system coupled to a triple quadrupole 6460 mass spectrometer (Agilent Technologies Ltd, Cork, Ireland). Quantification of each analyte was performed by ratiometric analysis and expressed as nmol or pmol g−1 of tissue. The limit of quantification was 1.3 pmol g−1, 12.1 pmol g−1, 1.5 pmol g−1, 1.4 pmol g−1 for anandamide, 2-AG, PEA and OEA respectively.
Qualitative detection of JZL184 using LC-MS/MS
The system and protocol employed was similar to that described for the detection of endocannabinoid and N-acylethanolamine levels with the following modifications. Briefly, samples were prepared as for endocannabinoid determination and resuspended in 100% acetonitrile. Separation occurred by reversed-phase gradient elution initially with a mobile phase of 25% acetonitrile and 0.1% formic acid that was ramped linearly up to 100% acetonitrile and 0.1% formic acid over 10 min and held at this for a further 10 min before being returned to 25% acetonitrile. Under these conditions, JZL184 was eluted at 14 min. JZL184 detection was carried out using electrospray-positive ionization and MRM mode where the parent–daughter transition of 503.1 > 199.1 was monitored with a collision energy of 25 V.
Quantitation of PGE2 and PGD2 using LC-MS/MS
Spleen or cortical samples were homogenized in 400 μL of acetonitrile containing tetra deuterated labelled internal standards (1.7pmoles PGE2 and PGD2). Lyophilized samples were resuspended in 40 μL 25% acetonitrile and 4 μL injected onto a Zorbax SB C18 column (150 × 0.5 mm internal diameter). The mobile phases consisted of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile) maintained at a flow rate of 12 μL per min. Reversed-phase gradient elution began initially at 25% B and over 20 min was ramped linearly up to 50% B and at 25 min was ramped to 100% B and held at this for a further 5 min. Under these conditions, PGE2 and PGD2 eluted at 19.8 and 21 min respectively. Analyte detection was carried out in electrospray-negative ionization and MRM mode on an Agilent 1100 HPLC system coupled to a triple quadrupole 6460 mass spectrometer (Agilent Technologies Ltd). Quantification of each analyte was performed by ratiometric analysis and expressed as pmol g−1 tissue.
HPLC-UV determination of arachidonic acid concentration
Determination of arachidonic acid was carried out by HPLC as described (Lang et al., 1996). In brief, cortical or spleen samples (60–100 mg) were homogenized in 1 mL of mobile phase [8.5% phosphoric acid/acetonitrile (10:90 v/v)] containing 50 ng biphenyl per 20 μL as internal standard. Samples were centrifuged at 14 000× g for 15 min at 4°C and 20 μL of the supernatant or standard [200 ng arachidonic acid (Sigma, Dublin, Ireland) per 20 μL] was then injected onto the Shimadzu HPLC system (Mason Technology, Dublin, Ireland). Separation was carried out at 30°C on a Synergie 4 μm reverse phase column (Phenomenex, Cheshire, UK) at a flow rate of 1 mL per min and detected on an SPD-10A UV-vis detector at 204 nm (Mason Technology, Dublin, Ireland). Quantification of arachidonic acid levels was based on the ratiometric analysis of sample and standard peak heights at 204 nm and expressed as nmol g−1 tissue.
Data analysis
SPSS (IBM, New York, USA) was used to analyse all data. Results are expressed as group means ± SEM. Data were analysed using unpaired t-test or two-way ANOVA (following log transformation) with the factors of antagonist/vehicle and JZL184/vehicle treatment. Post hoc analysis was performed using Duncan's test when appropriate. Data were considered significant when P < 0.05.
Results
JZL184 attenuates LPS-induced increases in cytokine expression in the frontal cortex
LPS increased IL-1β (23-fold), IL-6 (21-fold), TNF-α (3.5-fold), IL-10 (17-fold) and IκBα (6.2-fold) expression when compared with saline-treated controls (Vehicle–Vehicle–Saline vs. Vehicle–Vehicle–LPS; Figure 1A–D). Systemic administration of the MAGL inhibitor JZL184 significantly attenuated the LPS-induced increase in IL-1β (JZL effect: F1,36 = 42.962, P < 0.001), IL-6 (F1,35 = 4.124, P = 0.050), TNF-α (F1,37 = 46.070, P < 0.001) and IL-10 (F1,37 = 10.977, P = 0.002) but not IκBα, expression. Administration of the CB1 receptor antagonist AM251 partially blocked the JZL184-induced attenuation of IL-1β mRNA expression following LPS administration (Antagonist × JZL184 interaction effect: F2,36 = 6.452, P = 0.004) (Figure 1A). Although there was no main effect of antagonist treatment on IL-6 expression, a strong trend for AM251-induced blockade of the action of JZL184 on the expression of this cytokine was observed. AM251 alone significantly attenuated the LPS-induced increase in IL-1β expression. Pharmacological blockade of the CB2 receptor with AM630 did not alter LPS-induced cytokine expression alone, nor did it alter the JZL-induced attenuation of LPS-induced cytokine expression.
Figure 1.
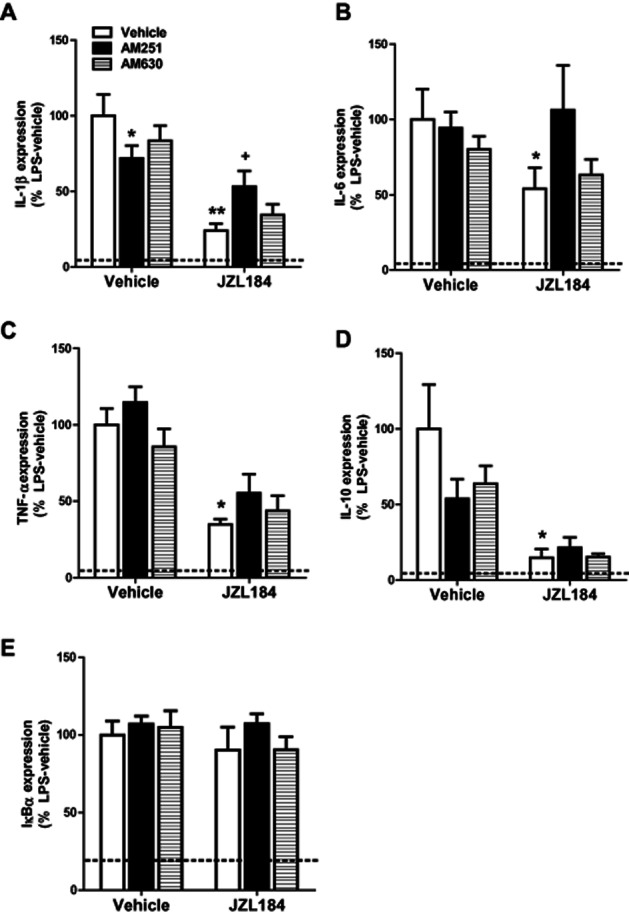
JZL184 attenuates LPS-induced increases in cytokine expression in the rat frontal cortex. JZL184 (10 mg kg−1 i.p.) significantly attenuated LPS-induced increases in IL-1β (A), IL-6 (B), TNF-α (C) IL-10 (D) mRNA expression in the rat frontal cortex. AM251 alone attenuated the LPS-induced increase in IL-1β mRNA expression while also partially preventing the JZL184-induced attenuation of IL-1β following LPS administration (A). Effect of LPS and JZL184 on IκBα (E). Data expressed as means ± SEM (n = 6–10 per group). Dotted line represents Vehicle–Vehicle–Saline. **P < 0.01; *P < 0.05 versus Vehicle–Vehicle–LPS. +P < 0.05 versus Vehicle–JZL184–LPS.
JZL184 attenuates LPS-induced increases in TNF-α and IL-10 levels in plasma, an effect partially attenuated by CB1 receptor antagonism
LPS increased IL-1β (287-fold), IL-6 (5.9-fold), TNF-α (1300-fold) and IL-10 (169-fold) levels in the plasma when compared with saline-treated controls (Vehicle–Vehicle–Saline vs. Vehicle–Vehicle–LPS; Figure 2A–D). JZL184 significantly attenuated the LPS-induced increases in TNF-α (JZL effect: F1,31 = 40.334, P < 0.001) and IL-10 (F1,30 = 7.337, P = 0.011) but not IL-1β nor IL-6, levels (Figure 2C–D). AM251 and AM630 partially attenuated the JZL184-induced attenuation of TNF-α (Antagonist x JZL184 interaction effect: F2,31 = 4.216 P = 0.024) following LPS administration. Furthermore, the JZL184-induced attenuation of the increase in IL-10 levels following LPS was blocked by AM251 (Antagonist × JZL184 interaction effect: F2,30 = 8.888, P = 0.001; Figure 2D). AM251 alone attenuated the LPS-induced increase in IL-10 plasma levels (Figure 2D). AM630 significantly attenuated LPS-induced increase in IL-1β cytokine levels in the presence of JZL184 (Antagonist × JZL184 interaction effect: F2,32 = 4.614, P = 0.017; Figure 2A).
Figure 2.
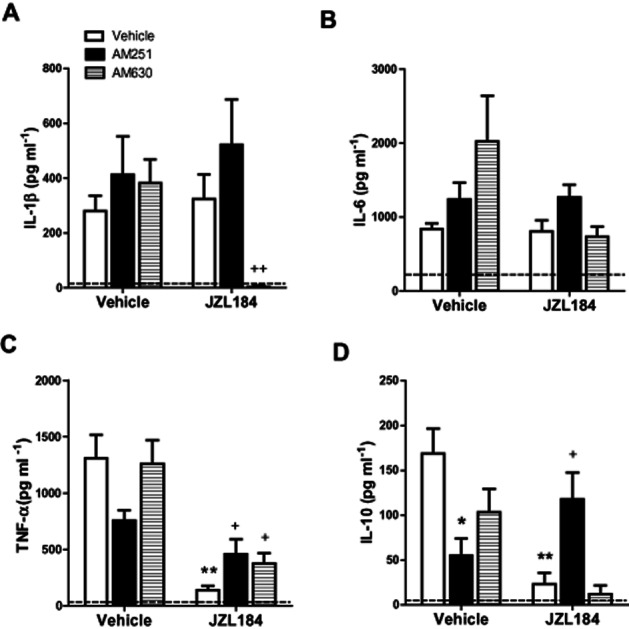
JZL184 attenuates LPS-induced increases in TNF-α and IL-10 levels in the plasma, an effect partially mediated by CB1 receptors. LPS significantly increased IL-1β (A), IL-6 (B), TNF-α (C) and IL-10 (D) plasma levels expression when compared with saline-treated controls (dotted line). JZL184 (10 mg kg−1 i.p.) attenuated the LPS-induced increase in TNF-α (C) and IL-10 (D), effects partially attenuated by CB1 receptor antagonism with AM251. AM630 also partially reversed the JZL184-induced attenuation of TNF-α levels following LPS administration (C). In the presence of JZL184, AM630 completely blocked the LPS-induced increase in IL-1β (A). AM251 alone attenuated the LPS-induced increase in IL-10 levels (D). Data expressed as means ± SEM (n = 6–10 per group). Dotted line represents Vehicle–Vehicle–Saline. *P < 0.05; **P < 0.01 versus Vehicle–Vehicle–LPS. +P < 0.05;++P < 0.01 versus Vehicle–JZL184–LPS.
Systemic administration of JZL184 inhibits MAGL activity and increases 2-AG levels in the rat spleen but not in the frontal cortex
Systemic administration of JZL184 resulted in a significant inhibition of MAGL activity (P = 0.002) and an associated increase in 2-AG levels (P = 0.023) in the spleen, but not in the frontal cortex, of LPS-treated rats (Figure 3A,B). There was no effect of JZL184 on the levels of anandamide, OEA or PEA in either the frontal cortex or in the spleen (Figure 3C).
Figure 3.
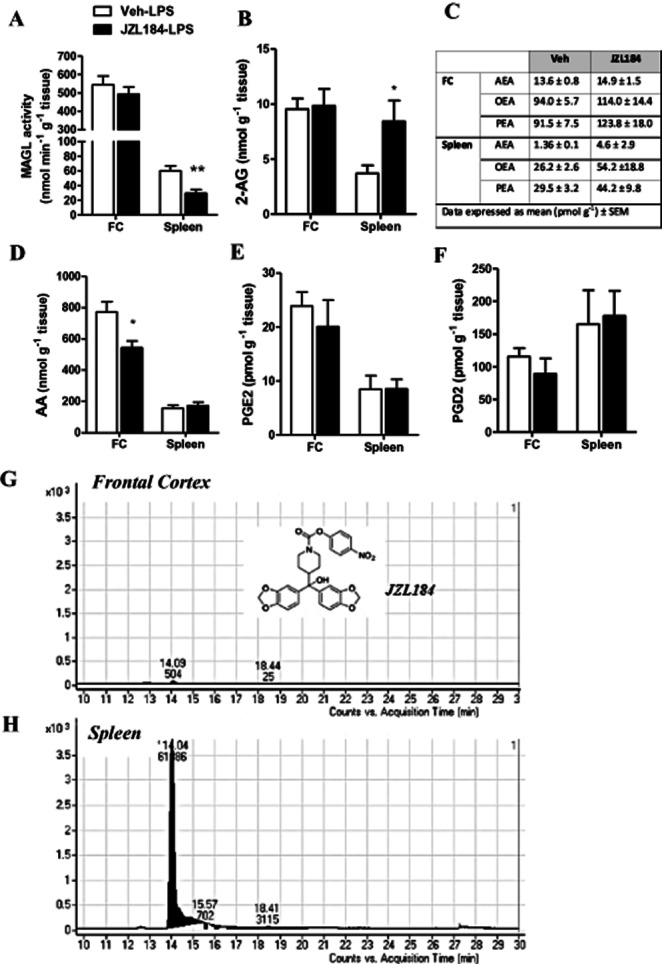
Effect of systemic administration JZL184 on MAGL activity, 2-AG, arachidonic acid and prostaglandin levels in the frontal cortex and spleen. Systemic administration of JZL184 (10 mg kg−1, i.p.) inhibited MAGL activity (A) and increased 2-AG levels (B) in the spleen but not frontal cortex. JZL184 did not alter concentrations of anandamide, OEA or PEA in either the frontal cortex or spleen (C). Arachidonic acid levels were decreased in the frontal cortex but not in the spleen of JZL184-treated rats (D). Levels of PGE2 (E) and PGD2 (F) in the frontal cortex or spleen of LPS-treated animals were not altered by prior administration of JZL184. JZL184 could be detected in the spleen (H) but not in the frontal cortex (G) following systemic administration. Structure of JZL184 presented as insert (G). Data expressed as means ± SEM (n = 6–10 per group). **P < 0.01, *P < 0.05 versus Vehicle–LPS.
JZL184 reduces arachidonic acid levels but does not alter PGE2 or PGD2 levels in the frontal cortex of LPS-treated rats
Arachidonic acid levels were reduced in the frontal cortex (P = 0.020), but not in the spleen, of JZL184–LPS-treated rats (Figure 3D). There was no effect of JZL184 on levels of PGE2 or PGD2 in the frontal cortex or in the spleen (Figure 3E,F).
The effect of JZL184 on 2-AG levels in the frontal cortex over time
As JZL184 did not alter 2-AG levels in the frontal cortex 2.5 h after administration (2 h following LPS), we sought to determine if the reduction in arachidonic acid might have resulted from increased 2-AG levels at an earlier time point than that examined in the studies described above. JZL184, administered 30 min before LPS, did not alter 2-AG levels in the frontal cortex measured at 10, 30, 60 and 90 min after LPS administration (Table 1). In addition, LPS alone did not alter 2-AG levels in the frontal cortex at any of the time points examined.
Table 1.
Effect of systemic administration of JZL184 (10 mg kg−1) on 2-AG levels in the frontal cortex over time
| Time (min) | ||||
|---|---|---|---|---|
| 10 | 30 | 60 | 90 | |
| Vehicle–LPS | 6.80 ± 0.47 | 6.49 ± 0.55 | 7.64 ± 0.72 | 7.19 ± 0.81 |
| JZL184–LPS | 6.68 ± 0.40 | 7.13 ± 0.40 | 6.24 ± 0.78 | 7.70 ± 0.57 |
Vehicle–vehicle–saline: 7.00 ± 0.43 nmol g tissue. Data expressed as mean 2-AG concentration ± SEM in nmol g−1 tissue. (n = 7–11 per group). 2-AG, 2-arachidonyl glycerol; LPS, lipopolysaccharide.
JZL184 is present in the rat spleen but not frontal cortex following systemic administration
As 2-AG levels were not enhanced in the frontal cortex at any of the time points examined, LC-MS/MS analysis was preformed to qualitatively determine if JZL184 was present in the samples following systemic administration. Analysis revealed that although JZL184 was readily detectable in the rat spleen, the MAGL inhibitor could not be detected in the frontal cortex 2.5 h after administration (2 h after LPS) (Figure 3G,H).
Discussion
The present study demonstrated that systemic administration of the MAGL inhibitor JZL184 robustly attenuated LPS-induced increases in cytokine expression in the rat frontal cortex. Although CB1 receptor antagonism attenuated the JZL184-induced decrease in IL-1β expression, this occurred in the absence of any JZL184-induced inhibition of MAGL activity or increase in 2-AG levels in the frontal cortex. Although arachidonic acid levels in this brain region were reduced in JZL184–LPS-treated rats, this was not accompanied by changes in PGE2 or PGD2 levels. In comparison, JZL184 inhibited MAGL activity and increased 2-AG levels in the spleen, and attenuated the LPS-induced increases in plasma IL-10 and TNF-α levels, effects partially attenuated by CB1 receptor antagonism. Together, these data demonstrate potent anti-inflammatory effects of JZL184 in the rat, both centrally and peripherally, although the mechanisms underlying these effects may differ.
Although JZL184 robustly and selectively enhanced 2-AG levels in the brain and peripheral organs of mice (Long et al., 2009a,b; Alhouayek et al., 2011; Kinsey et al., 2011; Nomura et al., 2011), to our knowledge only one study to date (Oleson et al., 2012) has reported an increase in 2-AG levels in the rat brain (ventral tegmental area). In comparison with this latter study, the present study demonstrated that JZL184 inhibited MAGL activity and increased 2-AG levels in the rat spleen but not frontal cortex, 2.5 h after administration. Further analysis revealed that JZL184 could be detected in the spleen, but not in frontal cortex, following systemic administration. Although the dose of JZL184 (10 mg kg−1) was comparable between the present study and that of Oleson et al. (2012), the divergent results with respect to the ability of JZL184 to inhibit MAGL activity and increase 2-AG levels in the brain may result from differences between the two studies, such as the routes of administration (i.p. vs. i.v.) or the brain regions under investigation. The lack of increase in 2-AG in the brain is also in contrast to that observed in mice at an equivalent time point (Long et al., 2009a,b). It should be noted that the affinity of JZL184 for rat MAGL is 10-fold lower than that for mouse or human MAGL (Long et al., 2009b) and, as such, higher doses of JZL184 in the present study may be required to inhibit MAGL and increase 2-AG in the rat brain. However, the present findings indicate that the dose of JZL184 used was capable of inhibiting MAGL and increasing 2-AG levels in the spleen. These data, taken together with the data demonstrating detectable levels of JZL184 in the spleen but not in the frontal cortex, suggest that the lack of efficacy of the drug in the frontal cortex relates to insufficient brain permeability rather than dose.
Although pharmacological blockade of MAGL with JZL184 reduced LPS-induced cytokines in both the frontal cortex and plasma, the profile and magnitude of the cytokine changes differed between these regions. JZL184 almost completely blocked LPS-induced expression of mRNA for cytokines (IL-1β, TNF-α, IL-6 and IL-10) in the frontal cortex, similar to that previously reported in mouse brain (Alhouayek et al., 2011; Nomura et al., 2011). Further studies are required to determine if alterations in mRNA expression translate to changes in protein levels. Unlike these latter studies, this anti-inflammatory profile following JZL184 administration was not accompanied by an increase in 2-AG levels. It has been proposed that in the CNS, the inhibition of MAGL may shunt the hydrolysis of 2-AG onto other pathways such as COX-2, which would account for the lack of increase in 2-AG in the frontal cortex following JZL184. Such an effect would result in decreased arachidonic acid production via the MAGL hydrolysis of 2-AG, as observed in the current study. However, MAGL activity was not inhibited in frontal cortex following systemic administration of JZL184 and thus this is not a likely explanation. Nomura et al. have suggested that the anti-inflammatory effects of JZL184 in the brain are not directly mediated through cannabinoid receptors but rather due to reduced levels of arachidonic acid, with a consequent reduction in production of inflammatory mediators such as PGE2 (Nomura et al., 2011). Although arachidonic acid levels were reduced in the frontal cortex of JZL184-treated animals in the current study, this effect was not accompanied by alterations in PGE2 or PGD2 levels. In addition, CB1 receptor antagonism with AM251 partially attenuated the JZL184-induced decrease in frontal cortical IL-1β following LPS administration, indicating a potential role for the CB1 receptor in mediating this response. 2-AG activation of CB1 receptors has been shown to attenuate pro-inflammatory cytokine expression and protect against closed head injury via modulation of NF-κB signalling (Panikashvili et al., 2005; 2006). In addition, recent in vitro studies have demonstrated that JZL184-induced increases in 2-AG results in reduced phosphorlyation of NF-κB and COX-2 expression in hippocampal neurons via activity at CB1 receptors (Zhang and Chen, 2008; Du et al., 2011). However, the role of the CB1 receptor in mediating the decrease in LPS-induced cytokine expression in the frontal cortex following JZL184 administration in the current study was not clear in light of the absence of an increase in 2-AG or changes in IκBα expression, an indirect measure of NF-κB signalling (Read et al., 1994). Taken together, the data in the current study suggest that the anti-inflammatory effects of JZL184 in the rat frontal cortex, and their blockade by the CB1 receptor antagonist, are most likely an indirect consequence of 2-AG-induced decreases in circulating cytokine levels following JZL184. Increased levels of circulating pro-inflammatory cytokines can communicate with the brain via many routes (diffusion into brain across the blood brain barrier deficient areas, sensory signals and vagus nerve stimulation) and induce cytokine synthesis within the CNS, which leads to a state of acute neuroinflammation. Thus, modulation of peripheral cytokines can profoundly affect brain neuroinflammatory processes. This has important implications as novel treatments targeting the levels of 2-AG in peripheral tissues or organs may indirectly modulate neuroimmune function.
In contrast to effects in the brain, JZL184 reduced LPS-induced increases in plasma levels of TNF-α and IL-10, but not IL-1β or IL-6, effects accompanied by elevated 2-AG concentrations in the spleen, a major immune organ and source of circulating cytokines. To our knowledge, this is the first study to examine the effects of JZL184 on circulating cytokine levels following an acute immune challenge. The present findings correlate with recent studies demonstrating that a JZL184-induced increase in 2-AG was associated with reduced expression of several cytokines including TNF-α and IL-10 in models of gastric haemorrhage and colitis (Alhouayek et al., 2011; Kinsey et al., 2011). 2-AG-induced activation of CB1 receptors was shown to prevent NSAID-induced gastric haemorrhage (Kinsey et al., 2011). Both CB1 and CB2 receptors appear to be involved in the JZL184-induced amelioration of colon alterations in the mouse model of colitis; however, antagonism of these receptors only partially attenuated the JZL184-induced decrease in cytokine expression in the colon (Alhouayek et al., 2011). In the current study, pharmacological antagonism of the CB1 receptor with AM251 fully attenuated the JZL-induced decrease in plasma IL-10 levels, whereas antagonism of both CB1 and CB2 receptors partially blocked the decrease in TNF-α levels. Therefore, JZL184 inhibition of rat MAGL increased peripheral 2-AG levels, which probably acted via CB1/2 receptors to attenuate LPS-induced increases in cytokine (IL-10 and TNF-α) levels. Although JZL184 did not alter levels of plasma IL-1β or IL-6, effects on these cytokines at time points other than those examined in the present study cannot be ruled out.
It should be noted that a combination of CB2 receptor antagonism and JZL184 resulted in complete inhibition of the LPS-induced increase in plasma IL-1β, an effect not observed in the absence of MAGL inhibition. Although the significance of this finding is unclear, we propose that activation of other receptors by 2-AG, under circumstances where CB2 receptors are blocked, may be responsible for this effect. For example, 2-AG suppressed IL-2 production via activation of PPARγ (Rockwell et al., 2006) and a similar mechanism may account for the reduction in IL-1β levels observed here. PPARγ activation has been repeatedly shown to elicit anti-inflammatory effects, including reductions in IL-1β, and recent evidence indicates that this occurs by interfering with toll-like receptor 4 (TLR4), the LPS receptor, and its downstream signalling components (Maggi et al., 2000; Ji et al., 2011). Thus, we propose that the tonic activity of 2-AG at PPARγ is minimal; however, concomitant JZL184-induced MAGL inhibition and blockade of CB2 receptors results in increased 2-AG availability, shunting its activity away from CB2 receptors and onto PPARγ, consequently inhibiting TLR4 signalling and LPS-induced IL-1β production.
Immunosuppressive effects of cannabinoid receptor antagonists/inverse agonists have been previously reported, although the precise mechanisms by which these effects are mediated remain to be determined. Our data demonstrate that AM251 alone reduces LPS-induced IL-1β expression in the frontal cortex and IL-10 levels in the plasma. Studies from our laboratory have previously reported that AM251 reduced plasma TNF-α levels and, to a lesser degree, IL-1β and IL-6 (Roche et al., 2008) and that another CB1 receptor antagonist/inverse agonist, rimonabant, reduced IL-1β levels in the brain and IL-1β and TNF-α in the plasma (Roche et al., 2006) under similar conditions. In addition, rimonabant has recently been shown to reduce the TNF-α, IL-6 and MCP-1 expression in a mouse model of colitis (Alhouayek et al., 2011). Although in the current study, AM251 reduced IL-10 levels in rat plasma, rimonabant enhanced LPS-induced IL-10 levels in mice (Smith et al., 2000), indicating potential, species-related, differences in the effects of the two compounds. As highlighted earlier, the immunosuppressive effects of cannabinoid antagonists may be due to the unmasking of endocannabinoid actions at other receptors or direct activity at alternative targets such as GPR55 (Ryberg et al., 2007). In addition, it has also been suggested that the cannabinoid receptor antagonists may act as partial agonists at CB1 and CB2 receptors when administered alone (Smith et al., 2000; Croci et al., 2003; Roche et al., 2006; 2008). Further studies are required in order to elucidate the mechanism underlying the immunomodulatory effects of these antagonists.
In conclusion, the current study demonstrated that JZL184 inhibited MAGL activity and increased 2-AG levels in a primary immune organ (the spleen) and attenuated LPS-induced increases in circulating cytokine levels in the rat, effects partially mediated by CB1 receptors. In the frontal cortex, JZL184 robustly attenuated LPS-induced cytokine expression without elevating 2-AG levels or inhibiting MAGL activity, suggesting that the effects on central cytokine expression may be mediated indirectly via suppression of LPS-induced peripheral cytokine production. These results provide further evidence that MAGL inhibition may constitute a novel approach for the treatment of central and peripheral inflammatory disorders.
Acknowledgments
The authors would like to thank Benjamin Cravatt and Jonathan Long (Scripps Institute, La Jolla, CA, USA) for providing the JZL184 used in this study and Stephen Alexander (University of Nottingham, UK) for his assistance with establishing the MAGL activity assay. Funding for this project was received from the Millennium Fund, National University of Ireland, Galway and Science Foundation Ireland.
Glossary
- 2-AG
2-arachidonyl glycerol
- AM251
1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide
- AM630
[6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)-methanone
- FAAH
fatty acid amide hydrolase
- IκBα
inhibitor of NF-κB
- JZL184
4-nitrophenyl-4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate
- MAGL
monoacylglycerol lipase
- NF-κB
nuclear factor kappa B
- OEA
N-oleoylethanolamide
- PEA
N-palmitoylethanolamide
Conflict of interest
The authors declare no conflict of interest.
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhouayek M, Lambert DM, Delzenne NM, Cani PD, Muccioli GG. Increasing endogenous 2-arachidonoylglycerol levels counteracts colitis and related systemic inflammation. FASEB J. 2011;25:2711–2721. doi: 10.1096/fj.10-176602. [DOI] [PubMed] [Google Scholar]
- Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Makriyannis A, et al. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001;15:300–302. doi: 10.1096/fj.00-0399fje. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cable JC, Tan GD, Alexander SP, O'Sullivan SE. The activity of the endocannabinoid metabolising enzyme fatty acid amide hydrolase in subcutaneous adipocytes correlates with BMI in metabolically healthy humans. Lipids Health Dis. 2011;10:129. doi: 10.1186/1476-511X-10-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YH, Lee ST, Lin WW. Effects of cannabinoids on LPS-stimulated inflammatory mediator release from macrophages: involvement of eicosanoids. J Cell Biochem. 2001;81:715–723. doi: 10.1002/jcb.1103. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhang J, Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons against beta-amyloid insults. Neuroscience. 2011;178:159–168. doi: 10.1016/j.neuroscience.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comelli F, Giagnoni G, Bettoni I, Colleoni M, Costa B. The inhibition of monoacylglycerol lipase by URB602 showed an anti-inflammatory and anti-nociceptive effect in a murine model of acute inflammation. Br J Pharmacol. 2007;152:787–794. doi: 10.1038/sj.bjp.0707425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croci T, Landi M, Galzin AM, Marini P. Role of cannabinoid CB1 receptors and tumor necrosis factor-alpha in the gut and systemic anti-inflammatory activity of SR 141716 (rimonabant) in rodents. Br J Pharmacol. 2003;140:115–122. doi: 10.1038/sj.bjp.0705412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Bisogno T, Sugiura T, Melck D, De Petrocellis L. The novel endogenous cannabinoid 2-arachidonoylglycerol is inactivated by neuronal- and basophil-like cells: connections with anandamide. Biochem J. 1998;331(Pt 1):15–19. doi: 10.1042/bj3310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Chen X, Zhang J, Chen C. Inhibition of COX-2 expression by endocannabinoid 2-arachidonoylglycerol is mediated via PPAR-gamma. Br J Pharmacol. 2011;163:1533–1549. doi: 10.1111/j.1476-5381.2011.01444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchinetti F, Del Giudice E, Furegato S, Passarotto M, Leon A. Cannabinoids ablate release of TNFalpha in rat microglial cells stimulated with lypopolysaccharide. Glia. 2003;41:161–168. doi: 10.1002/glia.10177. [DOI] [PubMed] [Google Scholar]
- Ferrer B, Asbrock N, Kathuria S, Piomelli D, Giuffrida A. Effects of levodopa on endocannabinoid levels in rat basal ganglia: implications for the treatment of levodopa-induced dyskinesias. Eur J Neurosci. 2003;18:1607–1614. doi: 10.1046/j.1460-9568.2003.02896.x. [DOI] [PubMed] [Google Scholar]
- Finn DP. Endocannabinoid-mediated modulation of stress responses: physiological and pathophysiological significance. Immunobiology. 2010;215:629–646. doi: 10.1016/j.imbio.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Ford GK, Kieran S, Dolan K, Harhen B, Finn DP. A role for the ventral hippocampal endocannabinoid system in fear-conditioned analgesia and fear responding in the presence of nociceptive tone in rats. Pain. 2011;152:2495–2504. doi: 10.1016/j.pain.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Gallily R, Breuer A, Mechoulam R. 2-Arachidonylglycerol, an endogenous cannabinoid, inhibits tumor necrosis factor-alpha production in murine macrophages, and in mice. Eur J Pharmacol. 2000;406:R5–R7. doi: 10.1016/s0014-2999(00)00653-1. [DOI] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci. 2010;30:2017–2024. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Herradon E, Abalo R, Vera G, Perez-Nievas BG, Leza JC, et al. Cannabinoid/agonist WIN 55,212-2 reduces cardiac ischaemia-reperfusion injury in Zucker diabetic fatty rats: role of CB2 receptors and iNOS/eNOS. Diabetes Metab Res Rev. 2011;27:331–340. doi: 10.1002/dmrr.1176. [DOI] [PubMed] [Google Scholar]
- Guindon J, Guijarro A, Piomelli D, Hohmann AG. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br J Pharmacol. 2011;163:1464–1478. doi: 10.1111/j.1476-5381.2010.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Gilles L, Gran B, Constantinescu CS. Interaction between cytokines, cannabinoids and the nervous system. Immunobiology. 2010;215:606–610. doi: 10.1016/j.imbio.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Ji Y, Liu J, Wang Z, Li Z. PPARgamma agonist rosiglitazone ameliorates LPS-induced inflammation in vascular smooth muscle cells via the TLR4/TRIF/IRF3/IP-10 signaling pathway. Cytokine. 2011;55:409–419. doi: 10.1016/j.cyto.2011.05.020. [DOI] [PubMed] [Google Scholar]
- Kerr DM, Burke NN, Ford GK, Connor TJ, Harhen B, Egan LJ, et al. Pharmacological inhibition of endocannabinoid degradation modulates the expression of inflammatory mediators in the hypothalamus following an immunological stressor. Neuroscience. 2012;204:53–63. doi: 10.1016/j.neuroscience.2011.09.032. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O'Neal ST, Abdullah RA, Poklis JL, Boger DL, et al. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, Cravatt BF, Lichtman AH. Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti-allodynic effects in mice through distinct cannabinoid receptor mechanisms. J Pain. 2010;11:1420–1428. doi: 10.1016/j.jpain.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey SG, Nomura DK, O'Neal ST, Long JZ, Mahadevan A, Cravatt BF, et al. Inhibition of monoacylglycerol lipase attenuates nonsteroidal anti-inflammatory drug-induced gastric hemorrhages in mice. J Pharmacol Exp Ther. 2011;338:795–802. doi: 10.1124/jpet.110.175778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang W, Qin C, Hill WA, Lin S, Khanolkar AD, Makriyannis A. High-performance liquid chromatographic determination of anandamide amidase activity in rat brain microsomes. Anal Biochem. 1996;238:40–45. doi: 10.1006/abio.1996.0247. [DOI] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009a;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009b;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourbopoulos A, Grigoriadis N, Lagoudaki R, Touloumi O, Polyzoidou E, Mavromatis I, et al. Administration of 2-arachidonoylglycerol ameliorates both acute and chronic experimental autoimmune encephalomyelitis. Brain Res. 2011;1390:126–141. doi: 10.1016/j.brainres.2011.03.020. [DOI] [PubMed] [Google Scholar]
- McCarron RM, Shohami E, Panikashvili D, Chen Y, Golech S, Strasser A, et al. Antioxidant properties of the vasoactive endocannabinoid, 2-arachidonoyl glycerol (2-AG) Acta Neurochir Suppl. 2003;86:271–275. doi: 10.1007/978-3-7091-0651-8_59. [DOI] [PubMed] [Google Scholar]
- Maggi LB, Jr, Sadeghi H, Weigand C, Scarim AL, Heitmeier MR, Corbett JA. Anti-inflammatory actions of 15-deoxy-delta 12,14-prostaglandin J2 and troglitazone: evidence for heat shock-dependent and -independent inhibition of cytokine-induced inducible nitric oxide synthase expression. Diabetes. 2000;49:346–355. doi: 10.2337/diabetes.49.3.346. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Mestre L, Correa F, Docagne F, Clemente D, Guaza C. The synthetic cannabinoid WIN 55,212-2 increases COX-2 expression and PGE2 release in murine brain-derived endothelial cells following Theiler's virus infection. Biochem Pharmacol. 2006;72:869–880. doi: 10.1016/j.bcp.2006.06.037. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado E, Molina-Holgado F. Mending the broken brain: neuroimmune interactions in neurogenesis. J Neurochem. 2010;114:1277–1290. doi: 10.1111/j.1471-4159.2010.06849.x. [DOI] [PubMed] [Google Scholar]
- Nagarkatti P, Pandey R, Rieder SA, Hegde VL, Nagarkatti M. Cannabinoids as novel anti-inflammatory drugs. Future Med Chem. 2009;1:1333–1349. doi: 10.4155/fmc.09.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olango WM, Roche M, Ford GK, Harhen B, Finn DP. The endocannabinoid system in the rat dorsolateral periaqueductal grey mediates fear-conditioned analgesia and controls fear expression in the presence of nocicpetive tone. Br J Pharmacol. 2011;165:2549–2560. doi: 10.1111/j.1476-5381.2011.01478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleson EB, Beckert MV, Morra JT, Lansink CS, Cachope R, Abdullah RA, et al. Endocannabinoids shape accumbal encoding of cue-motivated behavior via CB1 receptor activation in the ventral tegmentum. Neuron. 2012;73:360–373. doi: 10.1016/j.neuron.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, et al. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Panikashvili D, Mechoulam R, Beni SM, Alexandrovich A, Shohami E. CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J Cereb Blood Flow Metab. 2005;25:477–484. doi: 10.1038/sj.jcbfm.9600047. [DOI] [PubMed] [Google Scholar]
- Panikashvili D, Shein NA, Mechoulam R, Trembovler V, Kohen R, Alexandrovich A, et al. The endocannabinoid 2-AG protects the blood-brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol Dis. 2006;22:257–264. doi: 10.1016/j.nbd.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Raman P, Kaplan BL, Thompson JT, Vanden Heuvel JP, Kaminski NE. 15-Deoxy-delta12,14-prostaglandin J2-glycerol ester, a putative metabolite of 2-arachidonyl glycerol, activates peroxisome proliferator activated receptor gamma. Mol Pharmacol. 2011;80:201–209. doi: 10.1124/mol.110.070441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read MA, Whitley MZ, Williams AJ, Collins T. NF-kappa B and I kappa B alpha: an inducible regulatory system in endothelial activation. J Exp Med. 1994;179:503–512. doi: 10.1084/jem.179.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche M, Diamond M, Kelly JP, Finn DP. In vivo modulation of LPS-induced alterations in brain and peripheral cytokines and HPA axis activity by cannabinoids. J Neuroimmunol. 2006;181:57–67. doi: 10.1016/j.jneuroim.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Roche M, Kelly JP, O'Driscoll M, Finn DP. Augmentation of endogenous cannabinoid tone modulates lipopolysaccharide-induced alterations in circulating cytokine levels in rats. Immunology. 2008;125:263–271. doi: 10.1111/j.1365-2567.2008.02838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwell CE, Snider NT, Thompson JT, Vanden Heuvel JP, Kaminski NE. Interleukin-2 suppression by 2-arachidonyl glycerol is mediated through peroxisome proliferator-activated receptor gamma independently of cannabinoid receptors 1 and 2. Mol Pharmacol. 2006;70:101–111. doi: 10.1124/mol.105.019117. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinainen JR, Saario SM, Laitinen JT. The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol (Oxf) 2012;204:267–276. doi: 10.1111/j.1748-1716.2011.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciolino NR, Zhou W, Hohmann AG. Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacol Res. 2011;64:226–234. doi: 10.1016/j.phrs.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SR, Terminelli C, Denhardt G. Effects of cannabinoid receptor agonist and antagonist ligands on production of inflammatory cytokines and anti-inflammatory interleukin-10 in endotoxemic mice. J Pharmacol Exp Ther. 2000;293:136–150. [PubMed] [Google Scholar]
- Stella N. Endocannabinoid signaling in microglial cells. Neuropharmacology. 2009;56:244–253. doi: 10.1016/j.neuropharm.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stelt M, Fox SH, Hill M, Crossman AR, Petrosino S, Di Marzo V, et al. A role for endocannabinoids in the generation of parkinsonism and levodopa-induced dyskinesia in MPTP-lesioned non-human primate models of Parkinson's disease. FASEB J. 2005;19:1140–1142. doi: 10.1096/fj.04-3010fje. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Mazzola C, Esposito G, Matias I, Petrosino S, De Filippis D, et al. Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci. 2006;63:1410–1424. doi: 10.1007/s00018-006-6037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticht MA, Long JZ, Rock EM, Limebeer CL, Mechoulam R, Cravatt BF, et al. The MAGL inhibitor, JZL184, attenuates LiCl-induced vomiting in the Suncus murinus and 2AG attenuates LiCl-induced nausea-like behavior in rats. Br J Pharmacol. 2011;165:2425–2435. doi: 10.1111/j.1476-5381.2011.01407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Merker K, Timm J, Tauber S. Immune control by endocannabinoids – New mechanisms of neuroprotection? J Neuroimmunol. 2007;184:127–135. doi: 10.1016/j.jneuroim.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Vandevoorde S, Jonsson KO, Labar G, Persson E, Lambert DM, Fowler CJ. Lack of selectivity of URB602 for 2-oleoylglycerol compared to anandamide hydrolysis in vitro. Br J Pharmacol. 2007;150:186–191. doi: 10.1038/sj.bjp.0706971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Borazjani A, Hatfield MJ, Edwards CC, Potter PM, Ross MK. Inactivation of lipid glyceryl ester metabolism in human THP1 monocytes/macrophages by activated organophosphorus insecticides: role of carboxylesterases 1 and 2. Chem Res Toxicol. 2010;23:1890–1904. doi: 10.1021/tx1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Zhang B, Seviour EG, Tao KX, Liu XH, Ling Y, et al. Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett. 2011;307:6–17. doi: 10.1016/j.canlet.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Zhang J, Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons by limiting COX-2 elevation. J Biol Chem. 2008;283:22601–22611. doi: 10.1074/jbc.M800524200. [DOI] [PMC free article] [PubMed] [Google Scholar]