

Abstract
Optical imaging of gene expression through the use of fluorescent antisense probes targeted to the mRNA has been an area of great interest. The main obstacles to developing highly sensitive antisense fluorescent imaging agents has been the inefficient intracellular delivery of the probes and high background signal from unbound probes. Binary antisense probes have shown great promise as mRNA imaging agents because a signal can only occur if both probes are bound simultaneously to the mRNA target site. Selecting an accessible binding site is made difficult by RNA folding and protein binding in vivo and the need to bind two probes. Even more problematic, has been a lack of methods for efficient cytoplasmic delivery of the probes that would be suitable for eventual applications in vivo applications in animals. Herein we report the imaging of iNOS mRNA expression in live mouse macrophage cells with PNA·DNA binary FRET probes delivered by a cationic shell crosslinked knedel-like nanoparticle (cSCK). We first demonstrate that FRET can be observed on in vitro transcribed mRNA with both the PNA probes and the PNA•DNA hybrid probes. We then demonstrate that the FRET signal can be observed in live cells when the hybrid probes are transfected with the cSCK, and that the strength of the FRET signal is sequence specific and depends on the mRNA expression level.
INTRODUCTION
Förster resonance energy transfer (FRET) is an energy transfer mechanism that can occur between two nearby fluorescent molecules.1 The energy transfer takes place through non-radiative dipole-dipole coupling interaction, in which the energy of one molecule (the donor) at electronic excited state is transferred to the other molecule (the acceptor), which results in emission from the acceptor. The efficiency of energy transfer is given by 1/(1 + (R/R0)6), where R is the distance between the donor and acceptor and Ro is the Forster radius for the donor and acceptor 2 and is most efficient from 10 to 100 Å. Because of the strict distance requirement, FRET has found extensive use in the study of the interactions of biomolecules, such as nucleic acid hybridization,2–4 protein-protein interactions,5, 6 and protein-nucleic acid interactions.7, 8One emerging application of FRET is to monitor mRNA expression in cells using fluorescently labeled nucleic acid probes. Compared with other methods that can detect mRNA, such as RT-PCR or DNA arrays, which require the lysis of the cells or tissue, nucleic acid hybridization probes have been designed that can monitor the mRNA in live cells in real time.9–12 Because most nucleic acids and analogs are membrane impermeable, additional agents, auxiliaries, or physical methods are required to facilitate their entry into cells, and once inside often become trapped in the cell. To reduce background signal from unbound probe, the probes have been designed to light up only when bound to the mRNA.
There are two general classes of light up hybridization probes that make use of FRET for detecting mRNA in cells, unimolecular and bimolecular.9–12 Examples of unimolecular probes are hairpin molecular beacons, quenched strand displacement probes, and dual FIT probes. These probes rely on hybridization between the probe and the mRNA to change the fluorescence properties of the probe. This is accomplished by distancing or separating the fluorophore from a quencher, in the case of molecular beacons and quenched strand displacement probes, or by increasing the fluorescent quantum yield of the donor upon duplex formation as in the case of dual FIT probes.13–16 Examples of bimolecular probes are binary FRET probes and fluorogenic reaction probes. FRET probes rely on fluorescent energy transfer between a donor probe and acceptor probe when bound to adjacent sites on the target mRNA.17–19 Fluorogenic reaction probes rely on either a catalyst on one probe activating a second fluorogenic probe, or a stoichiometric reaction between two probes catalyzed by the RNA that results fluorescence activation of one probe.20–26 These latter types of fluorescence activating probes have the advantage of signal amplification, and can potentially be used to trigger the release of drugs.20, 21 Therefore such bimolecular reaction probes could have great potential for in vivo diagnostic and therapeutic applications.
A major problem in designing optimal biomolecular probes, as well as unimolecular probes, for in vivo applications is that mRNA is folded and bound by proteins making many sites on the mRNA inaccessible to the probes.27 This accessibility problem is further compounded by the fact that binary probes require a longer stretch of accessible mRNA to accommodate two probes instead of one required for unimolecular probes. In most, if not all, previously reported studies of binary and unimolecular probes, the sequences of antisense probes were chosen based on mRNA folding predictions, or on prior success with antisense agents that were chosen in a similar way, or by hit or miss experimentation. These methods of selection are not precise and may not result in the best target sites for binary probes. Furthermore, in no case as far as we know, have the fluorescence activation properties of the probes been examined on the folded mRNA target itself, except for a recent fluorogenic strand displacement probe system that we reported.28
Another major obstacle for the design of an optimal bimolecular probe system, is an efficient method for intracellular delivery. Most studies of mRNA imaging probes in cells have relied on a variety methods for intracellular delivery that would not be suitable for in vivo studies in animals. For example, mRNAs have been detected using DNA and 2’-O-methyl RNA FRET pairs, but the probes had to be directly injected into the cells.17, 19, 29 Other methods that have been successfully used for intracellular delivery of binary FRET probes include electroporation 30 and the use of reversible pore-forming agents such as Streptolysin-O (SLO).18, 23 SLO has also been used for intracellular delivery of unimolecular fluorogenic FIT probes.15 While there have been many reports of enhancing the cell permeability of antisense agents through the attachment of cell penetrating peptides, these conjugates are generally excreted rapidly in animals due to their small size.31, 32 The advantage of using nanoparticles for probe delivery is that their size and surface composition can be readily modified to optimize nucleic acid binding, targeting, cell penetration, and endosomal release.33–35 There have been some recent reports of using nanoparticles as fluorogenic antisense probes, such as nanoflares, which consist of a gold nanoparticle bound to an ODN that is hybridized to a fluorescence probe that is quenched by the gold.36–38 Entry of this nanoparticle is likely mediated by surface bound serum proteins. Gold nanoparticles have also be used to deliver a hairpin molecular beacon39 and aptamer targeted silica coated beads to deliver a quenched strand displacement probe via a reducible disulfide linker.40 Most recently, we have shown that hybrid PNA•DNA quenched strand displacement probes can be efficiently delivered into live cells by cationic shell-crosslinked knedel-like nanoparticles (cSCKs).28
In this paper, we report on the in vitro and in vivo validation of an mRNA target site and nanoparticle delivery system for the testing and optimization of binary fluorogenic probes. To this end we have designed and synthesized PNA•DNA binary FRET probed that can be efficiently delivered into mouse macrophage cells by a cSCK nanoparticle and can detect the expression of inducible nitric oxide synthase (iNOS) mRNA, a marker of inflammation 41 (Fig. 1). Because iNOS mRNA is highly inducible, it can be used to evaluate the ability of the binary probe to image mRNA expression level. The iNOS mRNA target site was chosen from a library of antisense accessible sites on native iNOS mRNA that had been mapped by a modified RT-ROL assay (reverse transcriptase-random oligonucleotide library assay)42and validated to bind ODNs and PNAs in vitro in its folded state.43 In this paper we first demonstrate the ability of the PNA•DNA binary FRET probe to detect DNA and folded iNOS mRNA in vitro. We then demonstrate the ability of cSCK nanoparticles to efficiently deliver PNA•DNA hybrid FRET probes into living RAW 264.7 cells and to image the change in iNOS mRNA expression induced by lipopolysaccharide (LPS) and γ-interferon (γ-IFN) by quantifying the FRET efficiency.
Fig. 1.
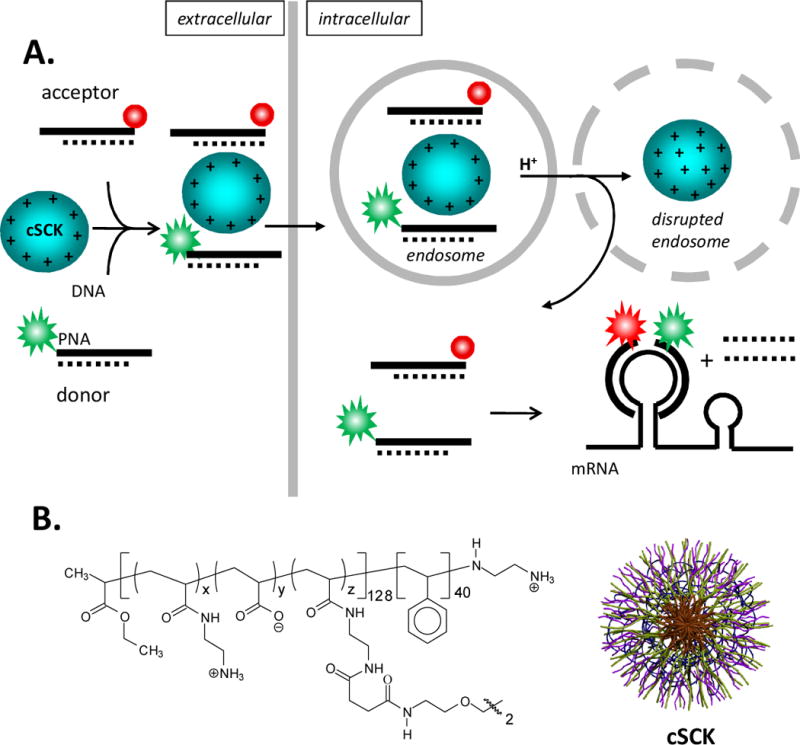
Schematic representation of cSKC-mediated delivery of FRET PNA probes for imaging mRNA expression in living cells. A) Pathway for the intracellular delivery and activation of PNA FRET probes. In the first step donor and acceptor PNAs are hybridized to partially complementary DNAs to impart negative charge for electrostatic binding to the cationic cSCK nanoparticle which can then be endocytosed by the cell. The cSCK then disrupts the endosome upon endosomal acidification through the pH sponge effect and releases the PNA•ODN FRET probes into the cytoplasm. The section of single stranded PNA (the toe-hold) can then associate with a complementary section of mRNA, and if the mRNA is completely complementary, facilitate the displacement of the DNA strand by branch migration. Upon irradiation of the donor, fluorescence from the donor will be observed (indicated by green), but emission from the acceptor, the FRET signal, (indicated by red) will only be observed when both donor and acceptor probes are adjacent to each other on the target mRNA. B) The cSCK is formed from a block copolymer that is then micellized and subsequently crosslinked to give the general structure and shape shown, where x ≈ 122, y ≈ 0 and z ≈ 6.
RESULTS AND DISCUSSION
Design and synthesis of the binary PNA FRET probes
Because traditional DNA probes are susceptible to enzymatic degradation and can trigger RNase H degradation of the mRNA target, we chose peptide nucleic acid (PNA) for constructing the binary FRET probes. PNA is a DNA analogue with N-aminoethylglycine-based polyamide backbone that is resistant to enzymatic degradation and will not induce RNase H. 44, 45PNA hybridizes to DNA or mRNA with higher binding affinity than DNA, and has been used in many applications involving nucleic acid hybridization in vitro and in vivo. PNA has been used for the construction of unimolecular fluorogenic FIT probes,14, 15, 46, 47 and quenched strand displacement probes28 for imaging mRNA in living cells. Binary PNA FRET probes have been used to monitor mRNA splicing both in solution and in fixed cells.48, 49
We chose fluorescein and Cy5 as the donor and acceptor pair based on previous work showing minimal excitation of Cy5upon excitation of the fluorescein17, 23 and on our previous in vitro work with this system.50 The sequences of the PNAs for our study were chosen to target iNOS mRNA from positions 236 to 272 (our numbering), which we have previously determined to be accessible to antisense agents in native folded iNOS mRNA by an RT-ROL(reverse transcriptase-random oligonucleotide library) assay that we modified.42 This mRNA was chosen as a target because it is a marker of inflammation 41and because its level increases approximately 100-fold to about 76,000 copies per cell in response to LPS/γIFN.51 A competition binding assay conducted on in vitro transcribed iNOS mRNA with two PNA probes targeting this site (PNA-240, TGTCCTTTTCCTCTTTCA and PNA-261, GTTTTCTTCACGTTGTTG) determined their Kds to be 10043and 68 pM (Figure S1 & S2), respectively. The target site from 236 to 272 is also optimal for PNA targeting because it is purine rich, which means that the antisense PNAs will be pyrimidine rich and hence less prone to form secondary structure and have solubility problems. The target site that we identified experimentally would not have been readily picked out on the basis of mRNA secondary structure calculations (Figures S3 & S4).
Another key consideration in designing the FRET probes was the length. Probes need to be long enough to be specific for the target mRNA, but not so long that they will bind with mismatched sequences under physiological conditions. Because of their higher affinity to the target mRNA than the DNA probes, PNA probes are usually shorter, with around 13 to 15 base pairs.48, 49We therefore chose15-mer PNA sequences for the FRET probes that were based on the PNA-240 and PNA-261 sequences described above that showed high affinity for the iNOS mRNA. Preliminary studies examining PNA FRET pairs that were separated by 2 or 6 nucleotides indicated that the one separated by 2 nucleotides (PNA-Cy5 and FAM-PNA) gave a slightly higher FRET and was therefore used for all further studies (Fig. 2). The specificity of the PNAs for iNOS mRNA was verified by the Basic Local Alignment Search Tool (BLAST). From this search, PNA-Cy5appeared to be more specific for iNOS mRNA (Table S1) than the FAM-PNA (Table S2). Since the FRET signal can only occur upon simultaneous hybridization of both probes to adjacent sites on the target mRNA, one only needs that the pair together to be highly specific as was found (Table S3). The PNAs were synthesized with a lysine group on their 3’ or 5’ end for conjugation to the donor and acceptor dyes through the ɛ-amino group. The PNAs were purified by HPLC and characterized by MALDI-TOF mass spectrometry, UV-vis and fluorescence spectroscopy.
Fig. 2.
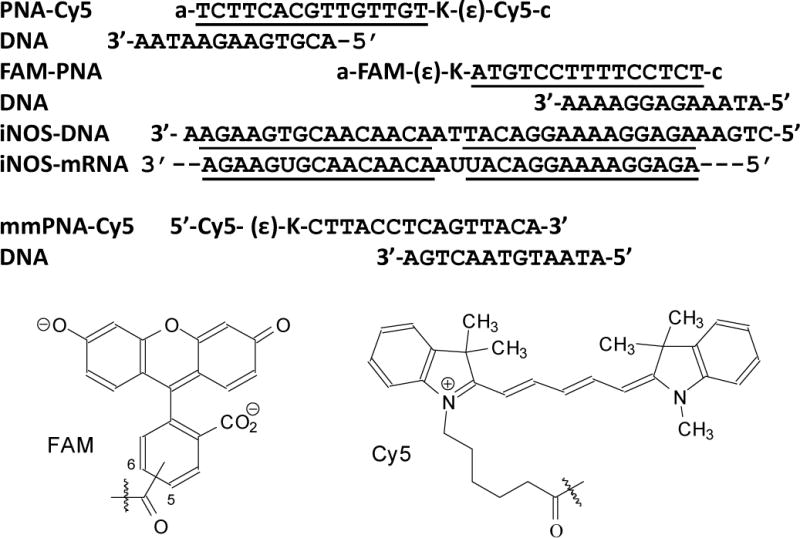
PNA•DNA FRET probes used in this study. The sequences of the PNAs are shown with their orientation indicated by “a” for the amino terminus and “c” for the carboxy terminus, and the DNAs with the 5’- and 3’-ends as indicated. The sections of complementarity are underlined. The donor and acceptor fluorophores shown below are connected through the indicated carboxy group to the ɛ-amino terminus of the lysines on the PNA.
FRET study with a DNA template
When 0.5 μM FAM-PNA was excited by 488 nm light in the presence of ca. 5-fold excess of the PNA-Cy5 in the absence of the iNOS-DNA template, the fluorescence from the FAM was virtually the same as that of the FAM-PNA hybridized to DNA in the absence of PNA-Cy5 (Figure 3). Most importantly, very little fluorescence emission for the Cy5 was observed at 670 nm which could be largely accounted for by direct excitation of the Cy5 (2.4 μM), as deduced from the emission observed from PNA-Cy5 alone or hybridized to iNOS-DNA (0.6 μM). These results indicated that there no significant intermolecular FRET was taking place at the concentrations of probes used. When 0.5 μM of FAM-PNA was incubated with 0.3 μM of PNA-Cy5 in the presence of 0.6 μM of iNOS-DNA, a ca.40% reduction in the fluorescence of FAM and an increase in the fluorescence of Cy5 to 6.4 % that of the FAM signal that was observed upon irradiation of the FAM-PNA•iNOS-DNA at 488 nm. When the amount of PNA-Cy5 was further increased to 0.6 μM, the fluorescence of the FAM decreased by 55%, and the fluorescence of the Cy5 increased to 8.6 %.
Fig. 3.
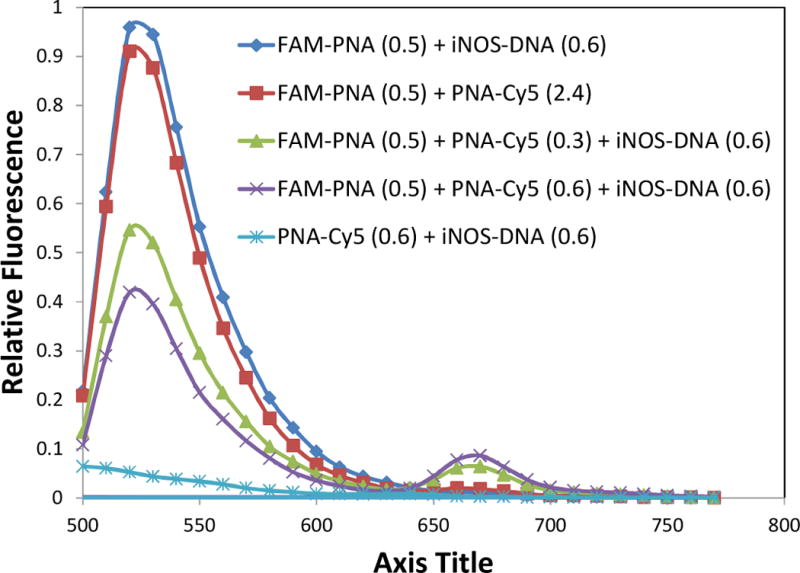
Fluorescence spectra of FRET probes in the presence and absence of a complementary DNA template. PNA-Cy5 and FAM-PNA were incubated at the concentrations indicated (μM) in 100 mM Tris 5 mM MgCl2, pH 7.1. in the presence or absence of a complementary DNA and excited at 488 nm. FAM-PNA •DNA and PNA-Cy5•DNA were used as controls for the individual PNAs bound to DNA.
FRET study with in vitro transcribed mRNA in solution
To see if the FRET probes would be suitable for imaging iNOS mRNA in cells, their ability to detect iNOS mRNA was first tested in vitro (Figure 4A). After mixing FAM-PNA and PNA-Cy5 with an equal amount of in vitro transcribed iNOS mRNA at 0.2 μM concentration in Opti-MEM® medium a significant FRET signal of 4.7 %was detected at 670 nm that was significantly greater than that from the PNA-Cy5 alone bound to the mRNA. The FRET signal was somewhat less than observed with the DNA template, possibly due to incomplete binding, or to a different geometry of the probes on the mRNA template compared to the DNA template. The specificity of the FRET was demonstrated by the lack of a significant FRET signal (slightly less than observed for the PNA-Cy5 alone with the mRNA) when FAM-PNA was incubated with a mismatched Cy5 bearing sequence (mmPNA-Cy5). Heating the mRNA to 65°C and then cooling further increased the FRET signal to 6.9 %, presumably because transiently unfolding the mRNA may have made the target site more accessible and trapped by the PNA. It is important to note that native mRNA in vivo may have a different secondary structure(s)and may be further bound by proteins that may make it more or less accessible than in vitro.
Fig. 4.
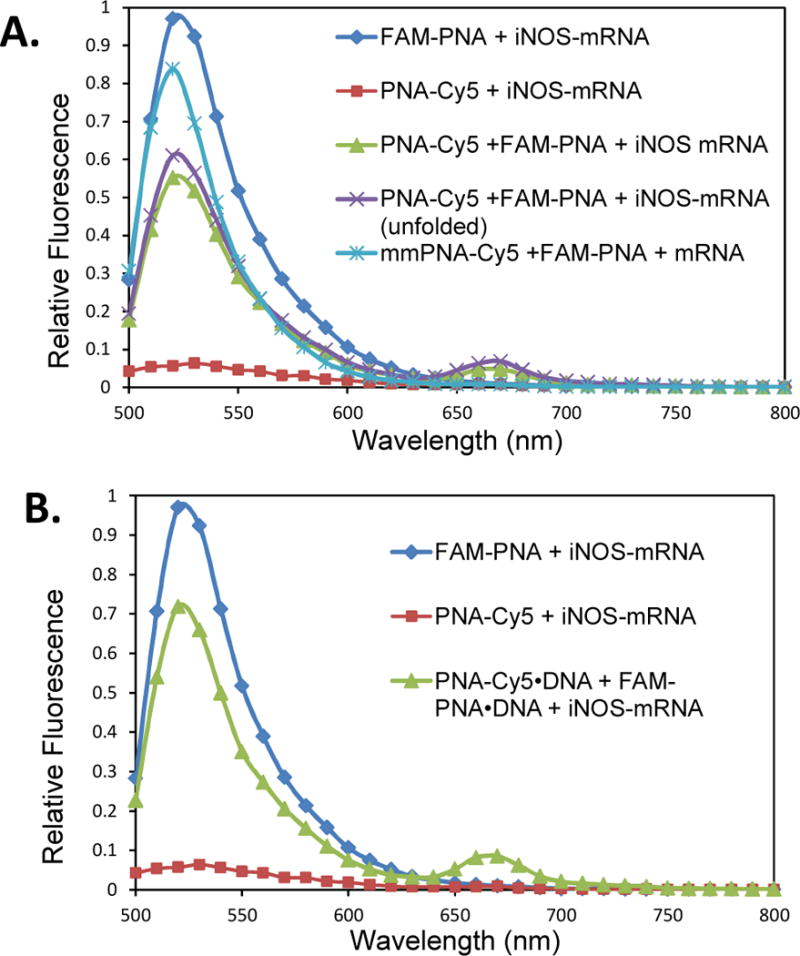
Fluorescence spectra of FRET PNA probes in the presence or absence of in vitro transcribed iNOS mRNA. A) 0.2μM FAM-PNA and PNA-Cy5 or mmPNA-Cy5 (mismatched) were incubated with 0.2 μM in vitro transcribed iNOS mRNA at 37°C for 30 min in Opti-MEM® medium and excited at 488 nm. The PNAs were also heated with iNOS mRNA to 65°C for 1 min before incubation at 37°C for 30 min. B) 0.2 μM FAM-PNA and PNA-Cy5 were annealed to their partially complementary ODNs, and incubated with the iNOS mRNA at 37 °C for 30 min. As controls, 0.2μMFAM-PNA (0.2 μM) and PNA-Cy5 (0.2 μM) were separately incubated with iNOS mRNA under the same conditions.
Because our strategy for delivering the PNA FRET probes into cells requires the PNAs to be annealed to complementary negatively charged DNAs to enable them to electrostatically bind to the nanoparticles, we also examined the FRET efficiency of the PNA•DNA hybrid probes. To enable the PNAs to bind to the mRNA rapidly, the DNAs were made to be only partially complementary so as to leave a 6 nucleotide toehold on the PNA that could initiate binding to an mRNA which is then followed by displacement of the DNA strand by branch migration.52 We have recently shown that strand displacement can take place in vitro and cells with a PNA-donor•DNA-quencher hybrid probe.51 As one can see from Figure 4B, the FRET intensity with the PNA•DNA probes is similar (8.5 %) to that observed in the absence of the complementary DNA indicating that the DNA does not interfere with probe binding.
FRET imaging of iNOS mRNA in living cells
One promising approach for the intracellular delivery of nucleic acid-based probes that would potentially be suitable for in vivo delivery in animals is to make use of cationic nanoparticles which have been widely investigated for the intracellular delivery of oligonucleotides and analogs for controlling gene expression.34, 53, 54 The positive character of these agents promotes the electrostatic association with the negatively charged nucleic acids and with cell membranes to promote cell entry via endocytosis or membrane fusion. Because the binary PNA FRET probes are uncharged, they first have to be hybridized to a negatively charged ODN to form complexes with cationic lipids or nanoparticles.55 Recently, the Wooley group has developed a cationic shell-cross-linked (cSCK) nanoparticle (Fig. 1B) that we have shown can efficiently deliver DNA plasmids, oligonucleotide phosphorothioates, and PNA•DNA hybrids into HeLa cells.56–58 These nanoparticles are composed of a hydrophobic polystyrene core linked to a hydrophilic crosslinked poly(acrylamidoethylamine) shell. Delivery of the PNA•DNA hybrids by the cSCK was more efficient and less toxic than by Lipofectamine 2000. More recently, we have used cSCKs to deliver a fluorogenic quenched PNA•DNA strand displacement probe into mouse macrophages to detect iNOS mRNA.28 We have also used the cSCK to deliver 123I-radiolabeled PNA•DNA hybrids into the lungs of live mice to detect iNOS expression following treatment with LPS/γIFN.59
For live cell imaging studies the binary FRET PNA•DNA hybrid probes were delivered with cSCK nanoparticles which had a zeta potential of about 23 mV at pH 7 and a hydrodynamic diameter of approximately 15 nm.56RAW264.7 cells were chosen because this mouse macrophage cell line is known to greatly upregulate iNOS expression in response to LPS/γ-IFN,51, 60and because alveolar macrophages are largely responsible for iNOS induction in acute lung injury (ALI)61 for which we are developing these probes. We have previously found that the optimal N/P ratio of for transfection needs to be 4 or higher when using 100 nM PNA•DNA probe, where N refers to the number of basic amines on the cSCK, which is related to the number of positive charges at a given pH, and P refers to the number of phosphates (negative charges) on the DNA backbone. The amines are located in the outer shell of the cSCK (Fig. 1B) and when protonated also serve to electrostatically interact with the cell surface and promote endocytosis. Unprotonated amines in the shell serve as a proton sponge to buffer the contents of the endosome during endosomal acidification thereby causing an excess of protons and counter ions to accumulate within the endosome and eventually cause its disruption and release of the probes.58
For the imaging studies aPNA•DNA probe concentration of 0.5 μM was found to be optimal which required 14.5 μg/mL of cSCK to achieve an N/P ratio of 8 needed to bind the probes tightly. The cytotoxicity of the cSCK at the concentration used in this experiment was not determined, but a closely related cSCK with composition PAEA160-b-PS30 cSCK-pa100 showed about 52% viability at 15 μg/mL 24 h after induction with LPS/γ-IFN as determined by the MTT assay.62 It is noteworthy that at a slightly lower concentration of 10 μg/mL, the viability was found to be about 85%, but we never carried out experiments at this concentration.
There was concern that background FRET might occur from binding of the donor and acceptor probes to the same cSCK nanoparticle, but control experiments did not show detectable FRET under similar conditions used for the in vivo experiment (0.2 μM PNA•DNA, N/P=10) (Figure S5). It also appeared that the cSCK could inhibit the FRET as indicated by a diminished FRET when the donor and acceptor probes were first prebound to iNOS DNA(Figure S5). Even so, the donor and acceptor probes were separately bound to the cSCK prior to treating the cells to minimize the possibility of FRET. For experiments in which iNOS mRNA expression was stimulated, cells were first treated with LPS and γ-IFN for 18 h before incubation with the cSCK•PNA•DNA complexes for an additional 24 h before imaging. LPS and γ-IFN were added again during the cSCK transfection step to maintain a high level of iNOS mRNA expression.
Stimulated cells that were incubated with the iNOS-targeted PNA•DNA binary FRET probes showed diffuse fluorescence throughout much of the cell from both the FAM and the Cy5 probes upon direct excitation of each fluorophore (Fig.5, panels A and C). Most importantly an easily discernible FRET signal from the Cy5 was observed upon excitation of the FAM at 488 nm (Fig. 5, panel B). To quantify the FRET signal, emission spectra of individual cells were obtained by collecting images at 10 nm intervals from 500 to 690 nm (Fig. 5 panels D & E). Analysis of the emission spectra showed that there was a 12.4 ± 3 % FRET signal at 660 nm (Cy5 emission) relative to 520 nm (FAM emission) indicating that the PNA•DNA FRET probes were binding together on the iNOS mRNA target. An alternate explanation for the FRET signal could have been that it arose from the proximity of the donor and acceptor probes that were bound by the same cSCK nanoparticle. Though we had ruled out this explanation from fluorescence experiments on the cSCK complexes with the probes, it is also ruled out by the much lower FRET signal of 2.4 ± 0.9 % in stimulated cells incubated with the mismatched mmPNA-Cy5 acceptor probe in place of the targeted PNA-Cy5 probe (Figure 6E). In non-stimulated cells, a 3.3 ± 0.6 %FRET signal was observed with the targeted FRET probes, which was significantly weaker than the FRET signal in the stimulated cells (Fig.7).
Fig. 5.
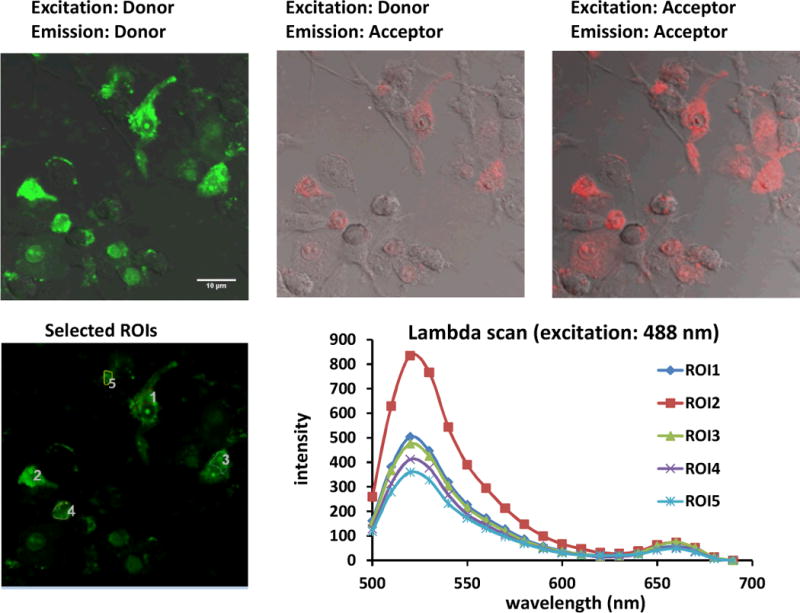
Confocal images of stimulated RAW 264.7 cells treated with iNOS PNA•DNA FRET probes. Cells were stimulated with LPS and γ-IFN and then again after 18 h at which point they were also treated with 0.5μM FAM-PNA•DNA, 0.5 μM PNA-Cy5•DNA and 14.5 μg/mLc SCK at an N/P ratio of 8. The cells were incubated for an additional 24 h before confocal microscopy. Green channel: FAM emission, red channel: Cy5. The first two panels show images resulting from excitation at 488 nm, while panel C shows and image following excitation at 633. The second panel shows the FRET emission at 660 nm resulting from excitation at 488 nm. The fourth panel shows regions of interest (ROIs) from which the emission spectra shown in the last panel were obtained.
Fig. 6.
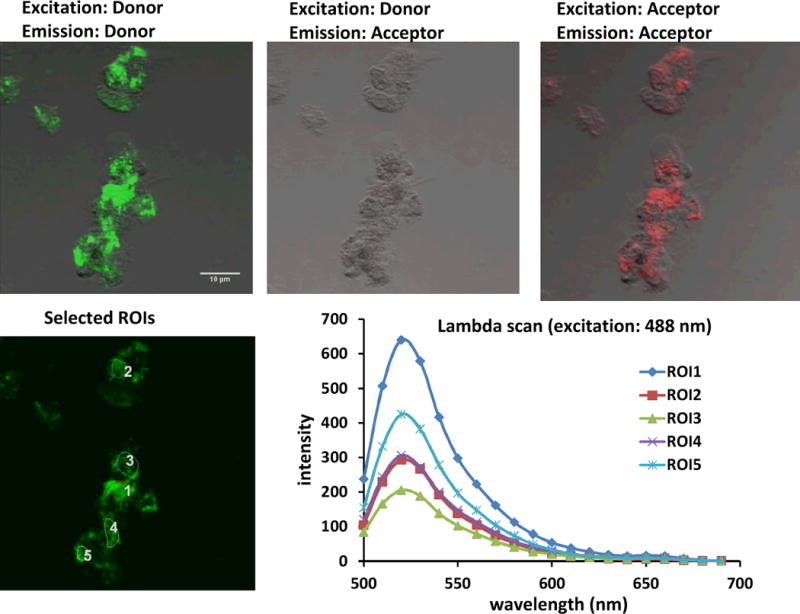
Confocal images of stimulated RAW 264.7 cells treated with matched donor FAM-PNA and mismatched acceptor mmPNA-Cy5•DNA FRET probes. The experiment was carried out in the same way as described in Figure 5, but with non-targeted mmPNA-Cy5•DNA in place of PNA-Cy5•DNA.
Fig. 7.
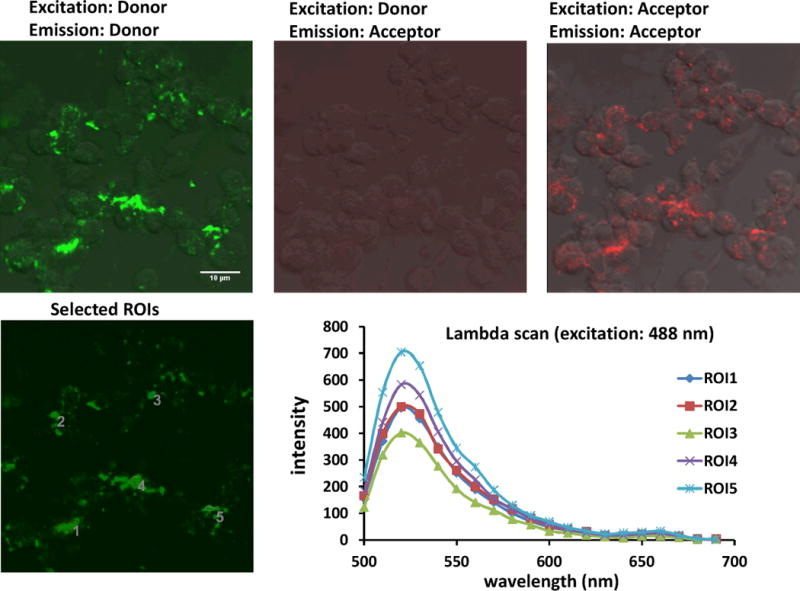
Confocal images of unstimulated cells treated with cSCK and PNA-Cy5•DNA and FAM-PNA•DNA FRET probes. The experiment was carried out as described for Fig. 5, except that unstimulated RAW 264.7 cells were used.
The difference in FRET signal intensity for the stimulated and non-stimulated cells was 3.7 ± 1 –fold. This difference does not, however, nearly correspond to the expected increase in iNOS expression of 100-fold determined by RT-PCR analysis of whole cell extracts.51 The difference detected by the binary FRET system is similar, however, to that of 4.1 ± 2.3 –fold and 7.0 ± 4.7 –fold determined in two separate experiments by a quenched strand displacement probe that was targeted to a different iNOS site.28 The similarity in the measured increase determined by these two different methods suggests that the small difference in induction level is either real, or that it results from the same type of experimental artifact. If real, it would further support our previous suggestion that the expression levels of iNOS mRNA being imaged by the probes are those of the cytoplasm, while those being measured by RT-PCR are those of the whole cell. Alternatively, it may be that iNOS expression may have become induced by the cSCK. It is also possible, however, that the increase is not being measured accurately, because the level of the background signal is above that that would arise from the basal mRNA level. Clearly, more studies will be needed to address these issues.
EXPERIMENTAL PROCEDURES
Materials
Anhydrous N,N-dimethylformamide (DMF), diisopropylethylamine (DIPEA), trifluoroacetic acid (TFA), meta-cresol, dichloromethane (DCM), N-methylpyrrolidone (NMP)were purchased from Sigma-Aldrich (St Louis, MO). PNA monomers were purchased from PolyOrg, Inc (Leominster, MA). Fmoc protected amino acids were purchased from EMD chemicals (Gibbstown, NJ). 2-(1H-7-Azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uranium hexafluorophosphate (HATU) was purchased from GenScript (Piscataway, NJ). (5,6)-Fluorescein-N-succinimidyl ester (FAM-NHS ester) was purchased from Sigma-Aldrich (St Louis, MO). Cy5-N-hydroxysuccinimidyl ester (Cy5-NHS ester) was synthesized by according to a literature procedure. Fmoc-PAL-PEG-PS resin for the solid phase PNA synthesis was purchased from Applied Biosystems (Carlsbad, CA). The PNAs were synthesized by solid-phase Fmoc chemistry on an Expedite 8909 DNA/PNA synthesizer on a 2 μmol scale. All the oligodeoxynucleotides were purchased from Integrated DNA Technologies (Coralville, IA). The crude PNA probes were purified by reversed phase high-performance liquid chromatography (HPLC) on a Beckman Gold System with a UV array detector and a Varian Microsorb-MV column (C-18, 5μm, 300Å pore size, 4.6 × 250 mm internal diameter and length) using a gradient of 0.1% TFA/acetonitrile in 0.1% TFA/water. The purified probes were characterized by MALDI-TOF mass spectrometry on an Applied Biosystems 4700 MALDI-TOF mass spectrometer. The concentration of the DNAs was determined from the absorbance at 260 nm on a Bausch and Lomb Spectronic 1001 spectrophotometer. PNA concentrations were determined from the absorbance at 260 nm at 70°C. The cSCK nanoparticle was prepared as previously described.56
Synthesis of the FRET probes
PNAs were synthesized on the Expedite 8909 PNA synthesizer. Fmoc-Lys (Mtt)-OH was attached to the N terminus on the donor PNA and to the C terminus on the acceptor PNA. After the PNA synthesis was completed, the column was removed from the synthesizer and the N-terminal NH2 of the PNA was capped for 10 min with 5% acetic anhydride and 6% 2,6-lutidine in DMF using a pair of syringes. Then the Mtt group on the lysine was then removed by washing the column with 1% TFA in DCM until the yellow color of the deprotection mixture faded away. The column was then washed twice with DCM followed by 5% DIPEA in DCM for 5 min. The column was dried by nitrogen gas and infused overnight with FAM-NHS ester (4 μmol, 2eq), Cy3-NHS (4 μmol, 2eq) or Cy5-NHS (4 μmol, 2eq) ester in dry DMSO with DIPEA (4 μmol, 2eq). The excess dyes were then washed off the column by DMF and DCM. The dried resin was taken out from column and cleaved by cleavage cocktail (20% m-cresol in TFA, 250 μL) for 2 to 4 h. Crude PNA products were precipitated by cold ether, dried, dissolved in 0.1% TFA water and filtered through an Xpertek syringe filter (13 mm, 0.45 μm) before HPLC purification. The purified PNA FRET probes were dried in a SC110A Thermo Savant speed vacuum system and re-dissolved in de-ionized water to make stock solutions. The concentration of the solutions was determined from the absorbance at 260 nm at 70°C. All probes were characterized by MALDI-TOF mass spectrometry: FAM-PNA: 4500.3 calcd. 4505.5 obsvd; PNA-Cy5: 4686.6 calcd., 4689.4 obsvd., mmPNA-Cy5: 4634.6 calcd., 4639.3 obsvd..
FRET studies with DNA templates in solution
PNA-Cy5 (0μM, 0.3μM and 0.6μM) was added to FAM-PNA and the iNOS-DNA target (0.6μM each) in 100 mM Tris 5 mM MgCl2 buffer. For negative controls, one sample contained 0.6 μM mmPNA-Cy5 and 0.6 μM iNOS DNA and another contained 0.5 μM FAM-PNA mixed with excess PNA-Cy5 (2.4 μM) in the absence of iNOS-DNA. All the solutions were heated at 95°C for 2 min and allowed to anneal at 37°C prior to collecting an emission spectrum with excitation at 488 nm on a Varian Eclipse Fluorimeter.
FRET probes to detect in vitro transcribed iNOS mRNA
E. coli containing the PCMV-SPORT6 vector containing the iNOS mRNA gene(American Type Culture Collection, Manassas, VA) was inoculated in LB media at 37°C for 18 h. The plasmid was extracted using the HiPure Plasmid Maxprep kit (Invitrogen) following the manufacturer’s protocol. The plasmid was then linearized with Xbal (Promega, WA) and transcribed with the MEGAtranscript® SP6 (Ambion) kit to produce an mRNA of about 3.9 K bases that was verified by gel electrophoresis on a 1% agarose gel using DEPC treated TAE buffer. The concentration of the mRNA was quantified by its absorbance at 260 nM and then dissolved in OPTI-MEM® solution to make a 0.2μM solution, together with equal molar amounts of FAM-PNA or PNA-Cy5 or both. Samples were incubated at 37°C for 30 min prior to obtaining the fluorescence emission spectra with excitation at 488 nm. For a negative control, 0.2μM each of FAM-PNA and the mismatched mmPNA-Cy5 sequence were mixed with 0.2 μM iNOS-mRNA. For the positive control, equal molar FRET probes and mRNA were mixed and heated at 65°C for 2 min before incubation at 37°C for 30 min. PNA·DNA duplex FRET probes were tested by hybridizing the 15-mer PNA FRET probes to 13-mer ODNs, which have 9 bases complementary to the PNA probes with a 4-nucleotide DNA overhang. The duplex probes (0.2 μM) were then incubated with 0.2 µM iNOS mRNA in OPTI-MEM® solution for 30 min at 37°C.
FRET imaging of iNOS mRNA in living cells
Mouse macrophage RAW 264.7 cells were seeded on 14 mm glass-bottom dish (MatTek, MA) at 1 × 105 cells/well and grown overnight until reaching about 70% confluence. A portion of the cells were treated with 1 μg/mL LPS and 300 ng/mL γ-IFN for 18 h to stimulate iNOS expression. FAM-PNA and PNA-Cy5 or mmPNA-Cy5 were first annealed to their complementary DNAs separately in 25 μL Opti-MEM® and then separately incubated together with cSCK nanoparticles at an N/P ratio of 8:1 and at room temperature for 20 min. The solutions were then mixed with 100 μL DMEM medium containing 10% FBS for a final volume of 150 μL containing 0.5 μM probe and 14.5 μg/ml cSCK which was then added to the cells. For stimulated samples, the DMEM was supplemented with LPS (1 μg/ml) and γ-IFN (0.3μg/ml)to maintain iNOS mRNA induction. Cells were incubated at 37 °C in a wet chamber with 5% CO2 for 24 h and then washed and visualized by confocal microscopy using an Olympus Fluoview FV1000 confocal microscope and a 20x water immersion objective. Cells were excited at 488 nm and emission wavelengths were collected in 10 nm increments from 500 to 700 nm.
Conclusion
We have validated a target site and delivery system for detecting the expression of iNOS mRNA with a binary antisense imaging agent both in vitro and in vivo. Though the sequence-specific FRET observed with the PNA-Cy5•DNA, FAM-PNA•DNA pair demonstrates that the site is accessible both in vitro and in vivo, the signal intensity is low, and is therefore unlikely to be useful for precise quantification of mRNA induction, or for in vivo use in animals. Currently, we are working on more red-shifted binary PNA FRET pairs with greater FRET efficiency, and using complementary DNAs with quenchers to eliminate background fluorescence from unbound probes. Ultimately, we plan to use this iNOS target and next generation cSCK delivery systems to further develop and validate catalytic nucleic acid triggered probe and drug activating systems in cells and eventually in animals.20, 22
Supplementary Material
Acknowledgments
This material is based upon work supported by the National Heart Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology(HHSN268201000046C)and by the Washington University NIH Mass Spectrometry Resource (Grant No. P41 RR000954).
References
- 1.Lakowicz JR. Principles of fluorescence spectroscopy. 3. Springer; New York: 2006. [Google Scholar]
- 2.Didenko VV. Biotechniques. 2001;31:1106–1116. 1118, 1120–1101. doi: 10.2144/01315rv02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, Zhou X, Ye D. Biochem Biophys Res Commun. 2008;373:457–461. doi: 10.1016/j.bbrc.2008.05.038. [DOI] [PubMed] [Google Scholar]
- 4.Juskowiak B. Anal Bioanal Chem. 2011;399:3157–3176. doi: 10.1007/s00216-010-4304-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piston DW, Kremers GJ. Trends Biochem Sci. 2007;32:407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Hoch DA, Stratton JJ, Gloss LM. J Mol Biol. 2007;371:971–988. doi: 10.1016/j.jmb.2007.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hillisch A, Lorenz M, Diekmann S. Curr Opin Struct Biol. 2001;11:201–207. doi: 10.1016/s0959-440x(00)00190-1. [DOI] [PubMed] [Google Scholar]
- 8.Buning R, van Noort J. Biochimie. 2010;92:1729–1740. doi: 10.1016/j.biochi.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Santangelo P, Nitin N, Bao G. Ann Biomed Eng. 2006;34:39–50. doi: 10.1007/s10439-005-9003-6. [DOI] [PubMed] [Google Scholar]
- 10.Tyagi S. Nat Methods. 2009;6:331–338. doi: 10.1038/nmeth.1321. [DOI] [PubMed] [Google Scholar]
- 11.Bao G, Rhee WJ, Tsourkas A. Annu Rev Biomed Eng. 2009;11:25–47. doi: 10.1146/annurev-bioeng-061008-124920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armitage BA. Curr Opin Chem Biol. 2011;15:806–812. doi: 10.1016/j.cbpa.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Socher E, Knoll A, Seitz O. Organic & Biomolecular Chemistry. 2012;10:7363–7371. doi: 10.1039/c2ob25925g. [DOI] [PubMed] [Google Scholar]
- 14.Kummer S, Knoll A, Socher E, Bethge L, Herrmann A, Seitz O. Bioconjug Chem. 2012;23:2051–2060. doi: 10.1021/bc300249f. [DOI] [PubMed] [Google Scholar]
- 15.Kummer S, Knoll A, Socher E, Bethge L, Herrmann A, Seitz O. Angew Chem Int Ed Engl. 2011;50:1931–1934. doi: 10.1002/anie.201005902. [DOI] [PubMed] [Google Scholar]
- 16.Köhler O, Jarikote DV, Seitz O. ChemBioChem. 2005;6:69–77. doi: 10.1002/cbic.200400260. [DOI] [PubMed] [Google Scholar]
- 17.Tsuji A, Koshimoto H, Sato Y, Hirano M, Sei-Iida Y, Kondo S, Ishibashi K. Biophys J. 2000;78:3260–3274. doi: 10.1016/S0006-3495(00)76862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santangelo PJ, Nix B, Tsourkas A, Bao G. Nucleic Acids Res. 2004;32:e57. doi: 10.1093/nar/gnh062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okabe K, Harada Y, Zhang J, Tadakuma H, Tani T, Funatsu T. Nucleic Acids Res. 2011;39:e20. doi: 10.1093/nar/gkq1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma Z, Taylor JS. Proc Natl Acad Sci USA. 2000;97:11159–11163. doi: 10.1073/pnas.97.21.11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Z, Taylor JS. Bioorg Med Chem. 2001;9:2501–2510. doi: 10.1016/s0968-0896(01)00245-0. [DOI] [PubMed] [Google Scholar]
- 22.Cai J, Li X, Yue X, Taylor JS. J Am Chem Soc. 2004;126:16324–16325. doi: 10.1021/ja0452626. [DOI] [PubMed] [Google Scholar]
- 23.Abe H, Kool ET. Proc Natl Acad Sci USA. 2006;103:263–268. doi: 10.1073/pnas.0509938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pianowski ZL, Winssinger N. Chemical communications. 2007:3820–3822. doi: 10.1039/b709611a. [DOI] [PubMed] [Google Scholar]
- 25.Franzini RM, Kool ET. J Am Chem Soc. 2009;131:16021–16023. doi: 10.1021/ja904138v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dutta S, Flottmann B, Heilemann M, Mokhir A. Chemical communications. 2012;48:9664–9666. doi: 10.1039/c2cc33827k. [DOI] [PubMed] [Google Scholar]
- 27.Rhee WJ, Santangelo PJ, Jo H, Bao G. Nucleic Acids Res. 2008;36:e30. doi: 10.1093/nar/gkn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Zhang K, Wooley KL, Taylor JS. Journal of nucleic acids. 2012;2012:962652. doi: 10.1155/2012/962652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsuji A, Sato Y, Hirano M, Suga T, Koshimoto H, Taguchi T, Ohsuka S. Biophys J. 2001;81:501–515. doi: 10.1016/S0006-3495(01)75717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen AK, Davydenko O, Behlke MA, Tsourkas A. Nucleic Acids Res. 2010;38:e148. doi: 10.1093/nar/gkq436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganguly S, Chaubey B, Tripathi S, Upadhyay A, Neti PV, Howell RW, Pandey VN. Oligonucleotides. 2008;18:277–286. doi: 10.1089/oli.2008.0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun X, Fang H, Li X, Rossin R, Welch MJ, Taylor JS. Bioconjug Chem. 2005;16:294–305. doi: 10.1021/bc049783u. [DOI] [PubMed] [Google Scholar]
- 33.Dominska M, Dykxhoorn DM. J Cell Sci. 2010;123:1183–1189. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- 34.Tan SJ, Kiatwuthinon P, Roh YH, Kahn JS, Luo D. Small. 2011;7:841–856. doi: 10.1002/smll.201001389. [DOI] [PubMed] [Google Scholar]
- 35.Elsabahy M, Wooley KL. Chem Soc Rev. 2012;41:2545–2561. doi: 10.1039/c2cs15327k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seferos DS, Giljohann DA, Hill HD, Prigodich AE, Mirkin CA. J Am Chem Soc. 2007;129:15477–15479. doi: 10.1021/ja0776529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prigodich AE, Seferos DS, Massich MD, Giljohann DA, Lane BC, Mirkin CA. ACS Nano. 2009;3:2147–2152. doi: 10.1021/nn9003814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prigodich AE, Randeria PS, Briley WE, Kim NJ, Daniel WL, Giljohann DA, Mirkin CA. Analytical Chemistry. 2012;84:2062–2066. doi: 10.1021/ac202648w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xue J, Shan L, Chen H, Li Y, Zhu H, Deng D, Qian Z, Achilefu S, Gu Y. Biosens Bioelectron. 2012 doi: 10.1016/j.bios.2012.06.062. [DOI] [PubMed] [Google Scholar]
- 40.Kim JK, Choi KJ, Lee M, Jo MH, Kim S. Biomaterials. 2012;33:207–217. doi: 10.1016/j.biomaterials.2011.09.023. [DOI] [PubMed] [Google Scholar]
- 41.Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, Murphy WJ. Proc Natl Acad Sci USA. 1993;90:9730–9734. doi: 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang H, Shen Y, Taylor JS. RNA. 2010;16:1429–1435. doi: 10.1261/rna.1940610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shrestha R, Shen Y, Pollack KA, Taylor JS, Wooley KL. Bioconjug Chem. 2012;23:574–585. doi: 10.1021/bc200629f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shakeel S, Karim S, Ali A. Journal of Chemical Technology & Biotechnology. 2006;81:892–899. [Google Scholar]
- 45.Nielsen PE. Chem Biodivers. 2010;7:786–804. doi: 10.1002/cbdv.201000005. [DOI] [PubMed] [Google Scholar]
- 46.Kam Y, Rubinstein A, Nissan A, Halle D, Yavin E. Molecular Pharmaceutics. 2012;9:685–693. doi: 10.1021/mp200505k. [DOI] [PubMed] [Google Scholar]
- 47.Torres AG, Fabani MM, Vigorito E, Williams D, Al-Obaidi N, Wojciechowski F, Hudson RH, Seitz O, Gait MJ. Nucleic Acids Res. 2012;40:2152–2167. doi: 10.1093/nar/gkr885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robertson KL, Yu L, Armitage BA, Lopez AJ, Peteanu LA. Biochemistry. 2006;45:6066–6074. doi: 10.1021/bi052050s. [DOI] [PubMed] [Google Scholar]
- 49.Blanco AM, Rausell L, Aguado B, Perez-Alonso M, Artero R. Nucleic Acids Res. 2009;37:e116. doi: 10.1093/nar/gkp551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oquare BY, Taylor JS. Bioconjug Chem. 2008;19:2196–2204. doi: 10.1021/bc800284x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhenghui Wang KZ, Wooley Karen L, Taylor John-Stephen. J Nucleic Acids. doi: 10.1155/2012/962652. in press, xxxx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang DY, Winfree E. J Am Chem Soc. 2009;131:17303–17314. doi: 10.1021/ja906987s. [DOI] [PubMed] [Google Scholar]
- 53.Tamura A, Nagasaki Y. Nanomedicine (Lond) 2010;5:1089–1102. doi: 10.2217/nnm.10.76. [DOI] [PubMed] [Google Scholar]
- 54.Jeong JH, Park TG, Kim SH. Pharm Res. 2011;28:2072–2085. doi: 10.1007/s11095-011-0412-y. [DOI] [PubMed] [Google Scholar]
- 55.Koppelhus U, Nielsen PE. Adv Drug Deliv Rev. 2003;55:267–280. doi: 10.1016/s0169-409x(02)00182-5. [DOI] [PubMed] [Google Scholar]
- 56.Zhang K, Fang H, Wang Z, Taylor JS, Wooley KL. Biomaterials. 2009;30:968–977. doi: 10.1016/j.biomaterials.2008.10.057. [DOI] [PubMed] [Google Scholar]
- 57.Zhang K, Fang H, Shen G, Taylor JS, Wooley KL. Proc Am Thorac Soc. 2009;6:450–457. doi: 10.1513/pats.200902-010AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fang H, Zhang K, Shen G, Wooley KL, Taylor JS. Mol Pharm. 2009;6:615–626. doi: 10.1021/mp800199w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen Y, Shrestha R, Ibricevic A, Gunsten SP, Welch MJ, Wooley KL, Brody SL, Taylor JSA, Liu Y. Interface Focus. 2013 doi: 10.1098/rsfs.2012.0059. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noda T, Amano F. J Biochem. 1997;121:38–46. doi: 10.1093/oxfordjournals.jbchem.a021566. [DOI] [PubMed] [Google Scholar]
- 61.Altmann C, Andres-Hernando A, McMahan RH, Ahuja N, He Z, Rivard CJ, Edelstein CL, Barthel L, Janssen WJ, Faubel S. Am J Physiol Renal Physiol. 2012;302:F421–432. doi: 10.1152/ajprenal.00559.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen Y, Fang H, Zhang K, Shrestha R, Wooley KL, Taylor JSA. Nucleic Acid Therapeutics. 2013 doi: 10.1089/nat.2012.0390. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.