

Abstract
Several structural and cellular changes, including marked glial anomalies, have been observed in association with major depressive disorder. Here we review these cellular alterations and highlight the importance of glial cell pathology, especially astroglial dysfunction, in the pathophysiology of neuropsychiatric disorders with a particular interest in major depressive disorder. The functional role of astrocytes in glutamate uptake and glutamate/glutamine cycling is discussed as is the deleterious effects of chronic stress on glial cell function. Lastly, we discuss the effect of antidepressants on glial cell function and the possibility of targeting glial cells in the quest to develop novel therapeutics.
Keywords: depression, stress, glia, astrocytes, prefrontal cortex, antidepressant, EAAT
Mounting evidence now suggests that structural as well as functional changes are present in the brains of many individuals suffering with mood disorders. Numerous studies have demonstrated significant reductions in cerebral blood flow, metabolism and volume of limbic brain structures including hippocampus, prefrontal cortex and amygdala in MDD patients(1). Other studies have further identified specific cytoarchitectural abnormalities, especially reductions in glial cell number and density in individuals afflicted with mood disorders (2). Could this evidence of glial cell pathology afford us insight into some of the pathophysiological processes contributing to mood and other psychiatric disorders, and ultimately suggest new targets for drug development?
Introduction to glial cells
Since their discovery more than 150 years ago (3), glial cells have largely been considered relatively unimportant in brain physiology, serving primarily as the “glue” of the brain. However, relatively recent discoveries elucidating glial cells critical role in a host of physiological processes implicate glial cell pathology as a potential contributor to many different neuropsychiatric disorders.
Historically, glial cells have been classified into three major subgroupings, astrocytes, oligodendrocytes and microglia. Astrocyte and oligodendrocyte lineage cells are derived from neural stem cells, whereas microglia originates from the immune system. Each of these classes of glia is now known to serve a broad range of physiological roles ranging from regulation of brain energy supplies and amino acid neurotransmitter metabolism to driving aspects of synaptic remodeling. Disrupted function of any of these cell types is likely to alter normal brain function and possibly contribute to the development of neuropsychiatric disorders (4).
Oligodendrocytes ensheath long axons in the CNS with myelin and provide trophic support, allowing for rapid impulse propagation and the maintenance of normal axon transport and long-term survival. Recent findings suggest these cells may also play critical roles in the pathogenesis of several neuropsychiatric disorders (5). Microglia, the resident tissue macrophages in the CNS, provide immunomodulatory functions in response to injury or disease (6). More recently, it has also been appreciated that the microglia serve additional roles related to neural plasticity, including removal of apoptotic cells and inappropriate neural connections, and sculpting and modifying both the developing and mature CNS (7). Other recent work has implicated microglial dysfunction in a variety of neuropsychiatric disorders (8).
Astrocytes are the most abundant form of glial cells and are commonly further divided into protoplasmic and fibrous subtypes based on their presence in the grey or white brain matter respectively. However, it is now clear that there is a much greater level of heterogeneity included in this general class of astrocytes (9). The broader class of astrocytes are known to serve a large number of CNS functions, including regulating regional blood flow and energy metabolism, immune defense, amino acid neurotransmitter clearance, neurotrophin production, regulation of D-serine and glycine that serve as co-agonist in NMDA receptor excitation, stabilization and stripping of synaptic connections and adult neurogenesis (10, 11)(see figure 1). More recently, the unique complexity of higher primate and human astrocytes has been appreciated (12) and there is increasing evidence to suggest that disruption of astroglial development or function is a potential contributor to a range of neuropsychiatric disorders (13).
Figure 1. Neuron-Glial interactions within the tripartite synapse.

Glutamate is released from vesicles within presynaptic neurons on excitation. Once released the glutamate can activate a variety of ionotropic and metabotropic receptors on postsynaptic and presynaptic neurons as well as glial cells. Some additional glutamate is released into the extracellular space through the cystine/glutamate transporter (xc-) on glial cells. Glutamate is cleared from the extracellular space via high-affinity excitatory amino acid transporters (EAATs), which are located primarily on neighboring glial cells (EAAT1-2) and, to some extent, on neurons (EAAT 3). In glial cells, glutamate is converted into glutamine by glutamine (Gln) synthetase. Glutamine is then transported back into the glutamatergic neuron, where it is hydrolyzed into glutamate by glutaminase. Glial cells also provide metabolic and energy support to neurons through a supply of lactate. Additionally, Serine racemase, the D-serine-synthesizing enzyme, is expressed by astrocytes. On release, D-Serine serves as a co-agonist at NMDA receptors. Astroyctes serve additional critical physiological roles through the synthesis and release of several neurotrophic factors including glial derived neurotrophic factor and brain derived neurotrophic factor.
Evidence of Glial Pathology Associated with Mood Disorders
Human data
Following up on initial neuroimaging studies showing that the volume of the subgenual part of Brodmann’s area 24 (BA24) is reduced in familial forms of major depressive disorder (MDD) and bipolar disorder (BD), Ongur et al. (14) used unbiased stereological techniques to demonstrate that the numbers of glia were reduced in both MDD and BD. The most prominent reductions were evident in subgroups of subjects with familial MDD or BD who exhibited marked (24% and 41%, respectively) reductions in the number of glial cells compared to control subjects. Similar reductions in BA24 glial cell density were also reported by Cotter et al. (15) and most recently by Gittens and Harrison (16). Early observations of prominent glial density reductions in the orbitofrontal region and both supra- and infragranular layers of dorsolateral prefrontal cortex in depressed subjects were also reported by Rajkowska et al. (17). Later studies continued to show reductions in glial cell number and density in the dorsolateral frontal cortical regions (18–20), as well as the subgenual cingulate cortex grey matter (21) and the amygdala (22, 23). While several studies provide evidence that the reduced glial cell numbers and density are not unique to mood disorders, but also common to schizophrenia, other studies have failed to find differences between mood disorder and control cases. Table S1 provides a more comprehensive outline of the postmortem studies examining glial cell pathology in several brain regions associated with mood disorders (see Supplement).
Additional studies have provided information suggesting selective pathologies in subpopulations of glial cells. A few analyses have found evidence of significant oligodendrocyte pathology to mood disorders. A recent review by Edgar and Sibille specifically discusses the potential role impaired oligodendrocyte structure and function may have on neural circuitry, and how this potentially contributes to mood regulation in human psychiatric disorders (24). Evidence of microgliosis has also been observed in the dorsolateral PFC (DLPFC), anteriocingulate cortex, and thalamus of suicide patients (25), suggesting that there may be abnormalities of microglial function associated with MDD. A recent review thoroughly discusses these findings and the potential role of activated microglia in the pathophysiology of mood and other neuropsychiatric disorders (26).
Other studies suggest astrocyte pathology is a prominent feature of mood disorders, and this will serve as the primary focus for the remainder of this review. Glial fibrillary acidic protein (GFAP) is an intermediate filament protein that is expressed by several cell types in the body. In the CNS, it has traditionally been considered a marker of astrocytes that can be induced by degeneration, damage or aging. However, it is now known that the expression of GFAP is in fact much more complicated, with various isoforms of GFAP expressed in different cell types and unique subsets of astrocytes (9). With these caveats in mind, several studies have identified reduced levels of GFAP expression in postmortem brain tissue from regions of the hippocampus (27), prefrontal cortex (28, 29), PFC (30), anterior cingulate (16), amygdala (31) and even cerebellum (32), suggesting specific astrocyte involvement in the disorder. The fact that the reduced DLPFC levels of GFAP were most prominent in MDD subjects less than 60 years old suggests the astrocyte pathology may be an early contributor to the underlying pathophysiology of mood disorders (33).
Gene expression studies using postmortem tissue provide additional evidence that astrocytic pathology is associated with mood disorders. Reduced expression of several components critical to the function and regulation of astrocytes have been reported in studies of depressed subjects and completed suicides. Microarray analysis of anterior cingulate cortex (area 24) and left DLPFC (areas 9 and 46) from individuals diagnosed with MDD demonstrated significant down-regulation of SLC1A2, SLC1A3, and GLUL, the genes coding for the two excitatory amino acid transporters (EAATs) that are predominantly expressed in astrocytes and the key enzyme responsible for converting glutamate to glutamine in the astrocyte (34). In a follow up study, the group later demonstrated significant reductions in the mRNA expression of several astrocyte specific transcripts, including those coding for GFAP, S100 calcium binding protein, fibroblast growth factor receptor 3, gap junction proteins (connexins) Cx30 and Cx43 and aquaporin-4 in the locus coeruleus of MDD subjects (35). Similar findings of downregulated CX30 and CX43 expression, as well as reduced expression of the astrocyte-specific tropomyosin-related kinase-B receptor (TrkB.1) isoform were recently reported in the orbitofrontal cortex and the dorsolateral prefrontal cortex of suicide completers (36, 37).
In sum, this growing series of postmortem studies suggest that glial cell pathology, especially astrocyte pathology, is associated with mood disorders. Although not without contradiction, the evidence for glial cell pathology appears strongest for the prefrontal cortex of mood disorder patients (Table S1 in the Supplement). Somewhat less consistent results are seen in the ACC, dorsolateral PFC and amygdala and little support for significant glial pathology in the hippocampus is provided by these postmortem reports. Some of this inconsistency could be related to differences in the age of the subpopulations studied and the methods of cell counting employed. The literature also suggests that the pathology is not unique to MDD, but is possibly shared by BD and SCH. However, it is important to note that there are major limitations to these small postmortem studies, including differences in medication exposure between sample populations, potential lifelong differences in substance and alcohol exposure, and varying causes of death. Further limiting the ability to draw strong conclusions from the existing reports, multiple studies were conducted on tissue samples from the same cases, thereby limiting the generalizability of the data. Unfortunately, the differences in methods employed in the various studies make comparisons between studies and meta-analyses combining studies extremely difficult.
Animal model data
A number of studies employing animal models of depression support the hypothesis that astrocyte pathology is associated with mood disorders. Decreased number and somal volume of hippocampal astroglia were observed after 5 weeks of psychosocial stress in the adult tree shrew (38). Similar findings of reduced astrocyte numbers and density were also recently reported in the rodent chronic stress (39–41) and maternal deprivation models of depression (42). As the stress-induced decreases in astrocyte GFAP expression appear to be correlated with hippocampal volume reductions, questions can be raised regarding a potential causal relationship between glial pathology and neuronal atrophy, or whether these effects are due to the same mechanism and occur in parallel.
In the PFC, GFAP expressing cells were reduced following 5 weeks of chronic stress (43, 44). Similar GFAP reductions were seen in the genetically stress susceptible Wistar Kayoto rat (45) and in maternal deprivation models (42). Significant decreases in the expression of Cx43 and the diffusion of gap junction channel-permeable dye, as well as abnormal gap junctional ultrastructure were also observed in the rat PFC following exposure to CUS (46). Providing direct evidence that astrocyte damage in the PFC could be related to the behavioral deficits associated with rodent models of depression, Banasr demonstrated that local infusion of the astrocyte specific toxin, L-alpha-aminoadipic acid (L-AAA), in the PFC region of rats was sufficient to induce depressive-like behaviors similar to those seen after prolonged periods of stress (43). A more recent study provided additional evidence by demonstrating that pharmacological blockade of gap junctions in the PFC also induces depressive-like behaviors (46). Other studies are now extending the findings by showing astrocytes in regions beyond the hippocampus and PFC could be affected by chronic stress. Indeed, GFAP protein levels in the periaqueductural gray matter were found reduced after chronic restraint stress (47).
Considering the central role of astrocytes in mediating amino acid neurotransmitter clearance and metabolism (10) (figure 1), a relationship between the astrocyte pathology observed in association with mood disorders, and the emerging evidence of abnormalities within the glutamatergic neurotransmitter systems of mood disorder subjects has been postulated (48). In fact, reduced expression (34, 35) and content (49) of glial specific EAATs and glutamate synthetase (34, 35) in the brains of MDD patients suggests glutamate clearance and metabolism are likely to be impaired in some regions of the depressed brain. Evidence of reduced glutamate transporter expression and activity, especially GLT1 (rodent homologue of EAAT2), has been reported in the hippocampus of a mouse model of depression using chronic corticosterone exposure (50). GLT1 expression was also decreased in the hippocampus as well as the other two cortical regions (occipital and retrosplenial cingulate) that were examined in animals bred for the learned helplessness trait (51). The relatively strong correlation between the GLT1 expression levels with the failure to escape rates in the animals suggests a possible causative relationship. In a series of related studies, spinal cord GLT1 expression has also been shown to be decreased in stress-induced animals models of visceral hyperalgesia (52, 53). This emerging evidence showing various regions within the CNS to undergo stress-related changes in GLT1 expression, make it tempting to hypothesize that changes in glial mediated glutamate clearance could contribute to pathophysiology underlying several of the common symptom complexes commonly associated with mood, anxiety and other stress-related disorders. Supporting this hypothesis, work demonstrating that impaired glial glutamate transport can result in depressive-like behaviors is starting to emerge. Indeed, animals subjected to low doses of L-AAA, known to impair glutamate transport and glutamine synthetase activity, exhibited anhedonia-like deficits when exposed to acute stress (43). Similarly, central or cortical administration of selective inhibitors of GLT1 transporters induced anhedonia in a rodent model of intracranial self-stimulation (54, 55). Administration of DHK in the amygdala reduced social interaction (56), suggesting that glial glutamate transport impairment is involved in the expression and the development of anxiety and depressive-like symptoms (figure 2).
Figure 2.
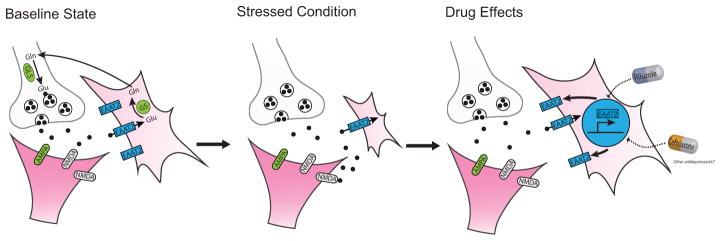
Stress and depression are associated with altered glutamate release and uptake that maybe be targeted by antidepressant development. Preclinical and clinical studies demonstrated atrophy and loss of astrocytes related to depression and animal models of depression. Astroglial anomalies include reduced EAAT2 (GLT1), glutamate metabolism and glial function. Some of these changes could be reversed by treatment with drugs that enhance EAAT2 expression, trafficking or activity such as riluzole or ceftriaxone. Identifying other drugs able to block or reverse stress-induced glial alterations could open a new avenue for antidepressant development.
At the synaptic level, the impaired ability to effectively clear extracellular glutamate is likely to result in an altered ratio of synaptic to extrasynaptic glutamate content, and an altered ratio of synaptic and extrasynaptic NMDAR activation. Several lines of evidence highlight the potentially detrimental effects of altering the ratio of synaptic and extrasynaptic NMDAR activation. Excessive activation of extrasynaptic to synaptic NMDA receptors has been linked to various forms of excitotoxicity, and has been implemented in the pathophysiology of multiple neurodegenerative disorders (57). Considering synaptic and extrasynaptic NMDAR activation have differing effects on hippocampal LTP and LTD formation (58), it is also quite possible the impaired regulation of glutamate uptake and the resulting shift in the ratio of synaptic to extrasynaptic activation could have dramatic effects on synaptic plasticity. As the transporters are generally highly efficient in clearing glutamate from the extracellular space (59), any effects of altered EAAT function could alter the balance of extrasynaptic to synaptic activation and shaping synaptic currents. Studies specifically relating GLT1 expression with LTD induction appear to demonstrate this case (60). Considering that individual astrocytes serve large numbers of synapses with minimal overlap in the synapses served by neighbouring astrocytes, the failure of a single astrocyte could impair glutamate removal at thousands of synapses (61), having large effects on glutamate neurotransmission.
In general the rodent models provide strong evidence of stress-related effects on astrocyte function. The parallel between the rodent and human postmortem findings is intriguing, but the findings must be considered in light of the fact that many of the rodent studies focused on the hippocampus, one of brain regions where the human findings are less than consistent (Table S1 in the Supplement). However, the increasing number of studies demonstrating reduced astrocyte number and function in brain regions similar to those most consistently altered in the brains of depressed patients, and studies showing targeted impairment of PFC and amygdala astrocyte function to be associated with anxious and depression-like phenotypes, suggest the findings are relevant to human disease states.
Antidepressant effects on glial cells
Data also suggests that glial cell function could be a target of existing antidepressant medications (figure 2). Observations of fluoxetine-sensitive 5-HT uptake were first reported in rat cortical astrocytes in situ nearly two decades ago (62). More recently, fluoxetine was shown to promote gliogenesis during neural differentiation in mouse embryonic stem cells (63). Studies demonstrating fluoxetine treatment prevented the stress-induced decrease in hippocampal GFAP positive astrocytes in tree shrews, suggest glioprotective effects may be relevant to the drug’s antidepressant properties (38). Although a separate study failed to find significant increases in rat PFC GFAP expression in rodents, it did show a large increase in CX43 protein expression following chronic treatment with fluoxetine (64). Another recent study reported similar findings, showing fluoxetine, as well as duloxetine and mifepristone, to reverse CUS-induced ultrasturctural alterations of astrocyte gap junctions including CX43 decreases, in prelimbic cortex (46).
Further suggesting classic antidepressants mechanism of action may involve effects on glia function, paroxetine induced a prominent up-regulation of hippocampal GFAP expression by microarray and in situ analyses (65), and both fluoxetine and paroxetine increased the expression of specific astrocyte-derived neurotrophic factors and glucose metabolism in cultured astrocytes (66, 67). Moreover, mice lacking aquaporin-4 show reduced GLT1 expression and a blunted response to fluoxetine treatment (68, 69). More directly related to glutamate uptake, treatment with fluoxetine also significantly induced GLT1 expression in hippocampus and cortex of rats, while the tricyclic desimpramine and the monoamine oxidase inhibitor tranylcypromine applications showed trend level effects (70). Chronic treatment with another tricyclic antidepressant, clomipramine, reversed the stress-induced effects on glial cell GFAP expression and GDNF (Glial-Derived Neurotrophic Factor) levels while also reversing the behavioral deficits of CUS (71, 72). The notable changes in astroglial structural plasticity in response to stress and the effects of antidepressant treatments further support the notion that astroglial changes may contribute to the pathophysiology of affective disorders as well as to the cellular actions of antidepressants.
Targeting Astroglial Function in the Development of Novel Antidepressant Treatments
Together, these findings suggest astrocytes, especially astrocytic glutamate uptake and metabolism, may serve as a viable target for mood and anxiety disorder drug development. Riluzole, a benzothiazole drug that is currently approved by the USFDA for the treatment of amyotrophic lateral sclerosis and shown to have neuroprotective actions in a broad range of conditions (73), has been shown to facilitate glial mediated glutamate clearance. Several studies have now demonstrated the ability of riluzole to increase GLT1 (EAAT2) expression and the clearance extracellular glutamate in several regions of the CNS (74, 75). The increase in GLT1 expression and uptake has been shown to be involved in the riluzole’s cytoprotective effects, and may be mediated through a mechanism involving Heat Shock Factor 1 activation of GLT1 expression (76). However, other effects of riluzole on glial cell function, such as modulation of microglia activity and activation of neurotrophic growth factors located in glial cells, as well as the previously reported effects on pre and post-synaptic neurons, (i.e. inhibition of certain voltage-gated sodium channels and actions on glutamate receptors), could also contribute to the drug’s mechanism of action (77).
In addition to producing antidepressant-like effects in rodent models, riluzole attenuated and/or reversed the GLT1 reducing effects of chronic unpredictable stress and chronic elevated corticosteroid exposure (44)(50). Riluzole also protected against the CUS-induced reductions in GFAP expression in the PFC of rats (44), suggesting that the glioprotective effects and the enhanced capability of maintaining effective glutamate clearance under these conditions could be related to the antidepressant-like properties of the drug. In different rodent models, riluzole was also shown to have rapid anxiolytic effects (78), and to reverse the hyperemotionality and decrease the extracellular glutamate levels seen in bulbectomized animals (79).
Open label human studies and case series have repeatedly found riluzole to have antidepressant and anxiolytic activity in a broad range of psychiatric disorders including BD, MDD, general anxiety and obsessive compulsive disorders (see review (80)). These reports, outlined in Table S1 in the Supplement, consistently found riluzole, at doses between 100–200mg/day, to decrease depression and anxiety severity when used as monotherapy or augmentation therapy. However, two randomized placebo controlled studies exploring riluzole’s ability to extend and enhance the transient antidepressant effects of ketamine did not find the drug to be statistically more effective than placebo in maintaining or enhancing the antidepressant response to a single ketamine infusion (81, 82). Although the studies were neither intended nor designed to specifically examine riluzole’s antidepressant efficacy directly, the larger of the two studies performed a post hoc analysis on the subgroup of subjects that did not meet the antidepressant response criteria ketamine, comparing riluzole to placebo. This analysis failed to demonstrate a significant antidepressant effect related to riluzole. However, the authors provide several factors such as the extreme refractoriness of these subjects, the limited sample size and the concomitant exposure to ketamine that may have impacted the results. The first larger placebo controlled trial specifically designed to investigate riluzole’s antidepressant efficacy is currently underway (clinicaltrials.gov identifying number; NCT01204918).
The hypothesis that enhanced glutamate uptake and metabolism contributes to riluzole’s mechanism of antidepressant action is further supported by a human imaging study showing rapid increases in the glutamine:glutamate (Gln/Glu) ratio following riluzole treatment of bipolar depressed patients (83). The study specifically reported that the mean Gln/Glu ratio increased markedly in the anterior cingulate cortex and parietal-occipital cortex two days after starting riluzole treatment, but was largely diminished by the end of the sixth week of treatment. The rilzuole’s transient impact on the Gln/Glu ratio was postulated to reflect an increased flux through the glutamate/glutamine cycle, resulting in an early shift in the relative levels of glutamine and glutamate that returns to baseline as the cycle reaches a new steady-state. Two rodent studies employing 13C-magnetic resonance spectroscopy methodologies support this claim, demonstrating that chronic riluzole treatment enhances glucose oxidative metabolism and glutamate/glutamaine cycling (84) and blocks or reverses the effects of stress on glial cell metabolism and glutamate/glutamine cycling (44). Additional work is now being done to determine the role of the glutamate transporters, glutamate uptake and changes in glutamate/glutamine cycling in the generation of the antidepressant-like response in rodent models. As riluzole has a broad range of effects on the brain, human studies showing a strong association or causative effect of riluzole’s functional target engagement with glial cells would ultimately be required to definitively demonstrate this as the primary mechanism of the drugs antidepressant action.
Ceftriaxone and the β-lactam antibiotic class in general, have also been shown to increase GLT1 (EAAT2) expression and function (85). The increased expression and/or function of GLT1 has been shown to delay or attenuate several pathological consequences of rodent models of amyotrophic lateral sclerosis, Huntington’s chorea and stroke (85–87). Ceftriaxone’s effects on glutamate clearance have also been linked to the drugs reported ability to reduce alcohol intake (88) and prevent relapse to cocaine seeking (89) in rodent models. This is relevant knowing that both cortical administration of either the gliotoxin L-AAA or of a gap junction cortical blocker induce alcohol intake (90) within a similar drug range used for inducing depressive-like behaviors (43), and suggests that states associated with altered excitatory amino acid transporter could be related to a variety of different neuropsychiatric disorder. With regards to potential antidepressant effects, sub-chronic administration of ceftriaxone reduced immobility on the forced swim and the tail suspension tests in mice (91). Ceftriaxone was also shown to reverse the reduced GLT1 expression in aquaporin-4 knockout mice as well as the impaired LTP and fear memory reported in these mice (69). However, the relative risk associated with sub-chronic ceftriaxone treatment has limited the ability to test this drug in clinical trials to date.
The rapid antidepressant response associated NMDA receptor antagonists may at first appear contradictory to the model of impaired glial cell glutamate clearance presented above. However, it is possible that the rapid increase in glutamate release and stimulation of post-synaptic AMPA receptors following NMDAR antagonist treatment, now believed to be critical for the antidepressant effects of the drugs (92, 93) (see Krystal et al. in this issue), is capable of transiently compensating for the decreased synaptic glutamate release and impaired transmission efficiency associated with impaired glutamate clearance. As presynaptic group II metabotropic glutamate receptors provide feedback inhibition on glutamate release when activated by extrasynaptic glutamate, impaired astrocytic clearance can result in decreased synaptic glutamate release. Consistent with this effect, decreased rates of glutamate cycling and glial cell metabolism has been demonstrated in the infralimbic and prelimibic regions in the rodent CUS model (44). Additional evidence suggesting that the transient increase in glutamate release is critical to the mechanism of the NMDAR antagonists’ antidepressant action comes from studies showing group II metabotropic glutamate receptor antagonists to also generate antidepressant effects with many of the same cellular and physiological effects of the NMDAR antagonists in rodent models (94, 95). Another postmortem finding showing a robustly elevated expression of the mGlu2/3 protein in the PFC of depressed subjects relative to non-depressed controls suggests that the expression and the function of the presynaptic receptor may be up-regulated as a result of the chronically elevated levels of extracellular glutamate associated with the disorder (96).
In conclusion, there is now mounting evidence to suggest glial cell pathology contributes to pathophysiology and possible pathogenesis of several neuropsychiatric disorders including mood and anxiety disorders. This new appreciation of the glial’s potential contributions to neuropsychiatric disorders has presented several novel targets for the development of therapeutic drugs in a range of neuropsychiatric disorders. Recent preclinical studies provide early evidence suggesting drugs, such as ceftriaxone and riluzole, that increase glutamate uptake via GLT1, possess antidepressant and anxiolytic activity. Moreover, this model predicts that drugs capable of modulating the group II metabotropic receptor mediated inhibitory effects on glutamate release, such as mGluR2 antagonist, or transiently stimulating glutamate release, such as the NMDAR antagonists could have rapid antidepressant-like effects. However, considering the hypothesized underlying impairment in glutamate clearance, the potential long-term adverse effects of these approaches should be examined carefully in this patient population.
Future preclinical studies should help us understand the mechanisms underlying the development of the glial cell pathology that are associated with mood and other neuropsychiatric disorders. However, proof of clinical efficacy for drugs with more selective actions targeting glial function will be required to ultimately demonstrate the true relevance of these glial abnormalities to treatment of mood and other neuropsychiatric disorders.
Supplementary Material
Acknowledgments
This work was supported by NIMH sponsored R01 MH081211 and R01 MH08055, The State of Connecticut DMHAS through the Connecticut Mental Health Center, and NARSAD. We would like to thank Dr. Ben Kelmendi for his assistance with the figure artwork.
Footnotes
Financial Disclosure statement:
Dr. Sanacora has received consulting fees form Abbott Laboratories, AstraZeneca, Avanier Pharmaceuticals, Bristol-Myers Squibb, Evotec, Eli Lilly & Co., Hoffman La-Roche, Novartis (all < $10,000year), and Noven Pharmaceuticals (>$10,000) over the last 24 months. He has also received additional grant support from AstraZeneca, Bristol-Myers Squibb, Hoffman La-Roche, and Merck & Co., and Servier. In addition he is a co-inventor on filed patent application by Yale University (PCTWO06108055A1) with Drs. J. Krystal and V. Coric.
Dr. Banasr reports no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Price JL, Drevets WC. Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn Sci. 2012;16:61–71. doi: 10.1016/j.tics.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 2.Miguel-Hidalgo JJ, Rajkowska G. Morphological brain changes in depression: can antidepressants reverse them? CNS Drugs. 2002;16:361–372. doi: 10.2165/00023210-200216060-00001. [DOI] [PubMed] [Google Scholar]
- 3.Virchow RLK. Die Cellularpathologie in ihrer Begründung auf physiologische und pathologische Gewebelehre. Berlin: A. Hirschwald; 1858. [PubMed] [Google Scholar]
- 4.Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010;11:275–283. doi: 10.1038/nrn2797. [DOI] [PubMed] [Google Scholar]
- 6.David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12:388–399. doi: 10.1038/nrn3053. [DOI] [PubMed] [Google Scholar]
- 7.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blank T, Prinz M. Microglia as modulators of cognition and neuropsychiatric disorders. Glia. 2012 doi: 10.1002/glia.22372. [DOI] [PubMed] [Google Scholar]
- 9.Oberheim NA, Goldman SA, Nedergaard M. Heterogeneity of astrocytic form and function. Methods Mol Biol. 2012;814:23–45. doi: 10.1007/978-1-61779-452-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ransom BR, Ransom CB. Astrocytes: multitalented stars of the central nervous system. Methods Mol Biol. 2012;814:3–7. doi: 10.1007/978-1-61779-452-0_1. [DOI] [PubMed] [Google Scholar]
- 11.Tsai HH, Li H, Fuentealba LC, Molofsky AV, Taveira-Marques R, Zhuang H, et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science. 2012;337:358–362. doi: 10.1126/science.1222381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, et al. Uniquely hominid features of adult human astrocytes. J Neurosci. 2009;29:3276–3287. doi: 10.1523/JNEUROSCI.4707-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, et al. Astrocytes and disease: a neurodevelopmental perspective. Genes & development. 2012;26:891–907. doi: 10.1101/gad.188326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Archives of General Psychiatry. 2001;58:545–553. doi: 10.1001/archpsyc.58.6.545. [DOI] [PubMed] [Google Scholar]
- 16.Gittins RA, Harrison PJ. A morphometric study of glia and neurons in the anterior cingulate cortex in mood disorder. J Affect Disord. 2011;133:328–332. doi: 10.1016/j.jad.2011.03.042. [DOI] [PubMed] [Google Scholar]
- 17.Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression.[comment] Biological Psychiatry. 1999;45:1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 18.Rajkowska G, Halaris A, Selemon LD. Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder.[comment] Biological Psychiatry. 2001;49:741–752. doi: 10.1016/s0006-3223(01)01080-0. [DOI] [PubMed] [Google Scholar]
- 19.Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophrenia Research. 2004;67:269–275. doi: 10.1016/S0920-9964(03)00181-6. [DOI] [PubMed] [Google Scholar]
- 20.Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cerebral Cortex. 2002;12:386–394. doi: 10.1093/cercor/12.4.386. [DOI] [PubMed] [Google Scholar]
- 21.Williams MR, Hampton T, Pearce RK, Hirsch SR, Ansorge O, Thom M, et al. Astrocyte decrease in the subgenual cingulate and callosal genu in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2012 doi: 10.1007/s00406-012-0328-5. [DOI] [PubMed] [Google Scholar]
- 22.Hamidi M, Drevets WC, Price JL. Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biological Psychiatry. 2004;55:563–569. doi: 10.1016/j.biopsych.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Bowley MP, Drevets WC, Ongur D, Price JL. Low glial numbers in the amygdala in major depressive disorder. Biological Psychiatry. 2002;52:404–412. doi: 10.1016/s0006-3223(02)01404-x. [DOI] [PubMed] [Google Scholar]
- 24.Edgar N, Sibille E. A putative functional role for oligodendrocytes in mood regulation. Translational psychiatry. 2012;2:e109. doi: 10.1038/tp.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res. 2008;42:151–157. doi: 10.1016/j.jpsychires.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 26.Beumer W, Gibney SM, Drexhage RC, Pont-Lezica L, Doorduin J, Klein HC, et al. The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J Leukoc Biol. 2012;92:959–975. doi: 10.1189/jlb.0212100. [DOI] [PubMed] [Google Scholar]
- 27.Muller MB, Lucassen PJ, Yassouridis A, Hoogendijk WJ, Holsboer F, Swaab DF. Neither major depression nor glucocorticoid treatment affects the cellular integrity of the human hippocampus. Eur J Neurosci. 2001;14:1603–1612. doi: 10.1046/j.0953-816x.2001.01784.x. [DOI] [PubMed] [Google Scholar]
- 28.Webster MJ, Knable MB, Johnston-Wilson N, Nagata K, Inagaki M, Yolken RH. Immunohistochemical localization of phosphorylated glial fibrillary acidic protein in the prefrontal cortex and hippocampus from patients with schizophrenia, bipolar disorder, and depression. Brain, Behavior, & Immunity. 2001;15:388–400. doi: 10.1006/brbi.2001.0646. [DOI] [PubMed] [Google Scholar]
- 29.Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey EF, et al. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol Psychiatry. 2000;5:142–149. doi: 10.1038/sj.mp.4000696. [DOI] [PubMed] [Google Scholar]
- 30.Si X, Miguel-Hidalgo JJ, O’Dwyer G, Stockmeier CA, Rajkowska G. Age-dependent reductions in the level of glial fibrillary acidic protein in the prefrontal cortex in major depression. Neuropsychopharmacology. 2004;29:2088–2096. doi: 10.1038/sj.npp.1300525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altshuler LL, Abulseoud OA, Foland-Ross L, Bartzokis G, Chang S, Mintz J, et al. Amygdala astrocyte reduction in subjects with major depressive disorder but not bipolar disorder. Bipolar Disord. 2010;12:541–549. doi: 10.1111/j.1399-5618.2010.00838.x. [DOI] [PubMed] [Google Scholar]
- 32.Fatemi SH, Laurence JA, Araghi-Niknam M, Stary JM, Schulz SC, Lee S, et al. Glial fibrillary acidic protein is reduced in cerebellum of subjects with major depression, but not schizophrenia. Schizophrenia Research. 2004;69:317–323. doi: 10.1016/j.schres.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 33.Miguel-Hidalgo JJ, Baucom C, Dilley G, Overholser JC, Meltzer HY, Stockmeier CA, et al. Glial fibrillary acidic protein immunoreactivity in the prefrontal cortex distinguishes younger from older adults in major depressive disorder. Biological Psychiatry. 2000;48:861–873. doi: 10.1016/s0006-3223(00)00999-9. [DOI] [PubMed] [Google Scholar]
- 34.Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci U S A. 2005;102:15653–15658. doi: 10.1073/pnas.0507901102. Epub 12005 Oct 15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernard R, Kerman IA, Thompson RC, Jones EG, Bunney WE, Barchas JD, et al. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ernst C, Chen ES, Turecki G. Histone methylation and decreased expression of TrkB.T1 in orbital frontal cortex of suicide completers. Mol Psychiatry. 2009;14:830–832. doi: 10.1038/mp.2009.35. [DOI] [PubMed] [Google Scholar]
- 37.Ernst C, Nagy C, Kim S, Yang JP, Deng X, Hellstrom IC, et al. Dysfunction of astrocyte connexins 30 and 43 in dorsal lateral prefrontal cortex of suicide completers. Biol Psychiatry. 2011;70:312–319. doi: 10.1016/j.biopsych.2011.03.038. [DOI] [PubMed] [Google Scholar]
- 38.Czeh B, Simon M, Schmelting B, Hiemke C, Fuchs E. Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology. 2006;31:1616–1626. doi: 10.1038/sj.npp.1300982. [DOI] [PubMed] [Google Scholar]
- 39.Gong Y, Sun XL, Wu FF, Su CJ, Ding JH, Hu G. Female early adult depression results in detrimental impacts on the behavioral performance and brain development in offspring. CNS Neurosci Ther. 2012;18:461–470. doi: 10.1111/j.1755-5949.2012.00324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Araya-Callis C, Hiemke C, Abumaria N, Flugge G. Chronic psychosocial stress and citalopram modulate the expression of the glial proteins GFAP and NDRG2 in the hippocampus. Psychopharmacology (Berl) 2012;224:209–222. doi: 10.1007/s00213-012-2741-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye Y, Wang G, Wang H, Wang X. Brain-derived neurotrophic factor (BDNF) infusion restored astrocytic plasticity in the hippocampus of a rat model of depression. Neurosci Lett. 2011;503:15–19. doi: 10.1016/j.neulet.2011.07.055. [DOI] [PubMed] [Google Scholar]
- 42.Leventopoulos M, Ruedi-Bettschen D, Knuesel I, Feldon J, Pryce CR, Opacka-Juffry J. Long-term effects of early life deprivation on brain glia in Fischer rats. Brain Res. 2007 doi: 10.1016/j.brainres.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 43.Banasr M, Duman RS. Glial Loss in the Prefrontal Cortex Is Sufficient to Induce Depressive-like Behaviors. Biol Psychiatry. 2008 doi: 10.1016/j.biopsych.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banasr M, Chowdhury GM, Terwilliger R, Newton SS, Duman RS, Behar KL, et al. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psychiatry. 2010;15:501–511. doi: 10.1038/mp.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gosselin RD, Gibney S, O’Malley D, Dinan TG, Cryan JF. Region specific decrease in glial fibrillary acidic protein immunoreactivity in the brain of a rat model of depression. Neuroscience. 2009;159:915–925. doi: 10.1016/j.neuroscience.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 46.Sun JD, Liu Y, Yuan YH, Li J, Chen NH. Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology. 2012;37:1305–1320. doi: 10.1038/npp.2011.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imbe H, Kimura A, Donishi T, Kaneoke Y. Chronic restraint stress decreases glial fibrillary acidic protein and glutamate transporter in the periaqueductal gray matter. Neuroscience. 2012;223:209–218. doi: 10.1016/j.neuroscience.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 48.Sanacora G, Rothman DL, Mason GF, Krystal JH. Clinical studies implementing glutamate neurotransmission in mood disorders. In: Moghaddam B, Wolf ME, editors. Glutamate and Disorders of Cognition and Motivation. 2003:292–308. doi: 10.1196/annals.1300.018. [DOI] [PubMed] [Google Scholar]
- 49.Miguel-Hidalgo JJ, Waltzer R, Whittom AA, Austin MC, Rajkowska G, Stockmeier CA. Glial and glutamatergic markers in depression, alcoholism, and their comorbidity. J Affect Disord. 2010;127:230–240. doi: 10.1016/j.jad.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gourley SL, Espitia JW, Sanacora G, Taylor JR. Antidepressant-like properties of oral riluzole and utility of incentive disengagement models of depression in mice. Psychopharmacology (Berl) 2012;219:805–814. doi: 10.1007/s00213-011-2403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zink M, Vollmayr B, Gebicke-Haerter PJ, Henn FA. Reduced expression of glutamate transporters vGluT1, EAAT2 and EAAT4 in learned helpless rats, an animal model of depression. Neuropharmacology. 2010;58:465–473. doi: 10.1016/j.neuropharm.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 52.Gosselin RD, O’Connor RM, Tramullas M, Julio-Pieper M, Dinan TG, Cryan JF. Riluzole normalizes early-life stress-induced visceral hypersensitivity in rats: role of spinal glutamate reuptake mechanisms. Gastroenterology. 2010;138:2418–2425. doi: 10.1053/j.gastro.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 53.Bradesi S, Golovatscka V, Ennes HS, McRoberts JA, Karagiannides I, Bakirtzi K, et al. Role of astrocytes and altered regulation of spinal glutamatergic neurotransmission in stress-induced visceral hyperalgesia in rats. Am J Physiol Gastrointest Liver Physiol. 2011;301:G580–589. doi: 10.1152/ajpgi.00182.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bechtholt-Gompf AJ, Walther HV, Adams MA, Carlezon WA, Jr, Ongur D, Cohen BM. Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology. 2010;35:2049–2059. doi: 10.1038/npp.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.John CS, Smith KL, Van’t Veer A, Gompf HS, Carlezon WA, Jr, Cohen BM, et al. Blockade of astrocytic glutamate uptake in the prefrontal cortex induces anhedonia. Neuropsychopharmacology. 2012;37:2467–2475. doi: 10.1038/npp.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee Y, Gaskins D, Anand A, Shekhar A. Glia mechanisms in mood regulation: a novel model of mood disorders. Psychopharmacology (Berl) 2007 doi: 10.1007/s00213-006-0652-4. [DOI] [PubMed] [Google Scholar]
- 57.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M, et al. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 59.Beart PM, O’Shea RD. Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150:5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Omrani A, Melone M, Bellesi M, Safiulina V, Aida T, Tanaka K, et al. Up-regulation of GLT-1 severely impairs LTD at mossy fibre--CA3 synapses. J Physiol. 2009;587:4575–4588. doi: 10.1113/jphysiol.2009.177881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng K, Scimemi A, Rusakov DA. Receptor actions of synaptically released glutamate: the role of transporters on the scale from nanometers to microns. Biophys J. 2008;95:4584–4596. doi: 10.1529/biophysj.108.129874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dave V, Kimelberg HK. Na(+)-dependent, fluoxetine-sensitive serotonin uptake by astrocytes tissue-printed from rat cerebral cortex. J Neurosci. 1994;14:4972–4986. doi: 10.1523/JNEUROSCI.14-08-04972.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kusakawa S, Nakamura K, Miyamoto Y, Sanbe A, Torii T, Yamauchi J, et al. Fluoxetine promotes gliogenesis during neural differentiation in mouse embryonic stem cells. J Neurosci Res. 2010;88:3479–3487. doi: 10.1002/jnr.22509. [DOI] [PubMed] [Google Scholar]
- 64.Fatemi SH, Folsom TD, Reutiman TJ, Pandian T, Braun NN, Haug K. Chronic psychotropic drug treatment causes differential expression of connexin 43 and GFAP in frontal cortex of rats. Schizophr Res. 2008;104:127–134. doi: 10.1016/j.schres.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 65.Sillaber I, Panhuysen M, Henniger MS, Ohl F, Kuhne C, Putz B, et al. Profiling of behavioral changes and hippocampal gene expression in mice chronically treated with the SSRI paroxetine. Psychopharmacology (Berl) 2008;200:557–572. doi: 10.1007/s00213-008-1232-6. [DOI] [PubMed] [Google Scholar]
- 66.Allaman I, Fiumelli H, Magistretti PJ, Martin JL. Fluoxetine regulates the expression of neurotrophic/growth factors and glucose metabolism in astrocytes. Psychopharmacology (Berl) 2011;216:75–84. doi: 10.1007/s00213-011-2190-y. [DOI] [PubMed] [Google Scholar]
- 67.Ubhi K, Inglis C, Mante M, Patrick C, Adame A, Spencer B, et al. Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of alpha-synucleinopathy. Exp Neurol. 2012;234:405–416. doi: 10.1016/j.expneurol.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kong H, Sha LL, Fan Y, Xiao M, Ding JH, Wu J, et al. Requirement of AQP4 for antidepressive efficiency of fluoxetine: implication in adult hippocampal neurogenesis. Neuropsychopharmacology. 2009;34:1263–1276. doi: 10.1038/npp.2008.185. [DOI] [PubMed] [Google Scholar]
- 69.Li YK, Wang F, Wang W, Luo Y, Wu PF, Xiao JL, et al. Aquaporin-4 deficiency impairs synaptic plasticity and associative fear memory in the lateral amygdala: involvement of downregulation of glutamate transporter-1 expression. Neuropsychopharmacology. 2012;37:1867–1878. doi: 10.1038/npp.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zink M, Rapp S, Donev R, Gebicke-Haerter PJ, Thome J. Fluoxetine treatment induces EAAT2 expression in rat brain. J Neural Transm. 2011;118:849–855. doi: 10.1007/s00702-010-0536-y. [DOI] [PubMed] [Google Scholar]
- 71.Liu Q, Li B, Zhu HY, Wang YQ, Yu J, Wu GC. Clomipramine treatment reversed the glial pathology in a chronic unpredictable stress-induced rat model of depression. Eur Neuropsychopharmacol. 2009;19:796–805. doi: 10.1016/j.euroneuro.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 72.Liu Q, Zhu HY, Li B, Wang YQ, Yu J, Wu GC. Chronic clomipramine treatment restores hippocampal expression of glial cell line-derived neurotrophic factor in a rat model of depression. J Affect Disord. 2012;141:367–372. doi: 10.1016/j.jad.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 73.Verhave PS, Jongsma MJ, Van Den Berg RM, Vanwersch RA, Smit AB, Philippens IH. Neuroprotective effects of riluzole in early phase Parkinson’s disease on clinically relevant parameters in the marmoset MPTP model. Neuropharmacology. 2012;62:1700–1707. doi: 10.1016/j.neuropharm.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 74.Yoshizumi M, Eisenach JC, Hayashida K. Riluzole and gabapentinoids activate glutamate transporters to facilitate glutamate-induced glutamate release from cultured astrocytes. Eur J Pharmacol. 2012;677:87–92. doi: 10.1016/j.ejphar.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578:171–176. doi: 10.1016/j.ejphar.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 76.Liu AY, Mathur R, Mei N, Langhammer CG, Babiarz B, Firestein BL. Neuroprotective drug riluzole amplifies the heat shock factor 1 (HSF1)- and glutamate transporter 1 (GLT1)-dependent cytoprotective mechanisms for neuronal survival. J Biol Chem. 2011;286:2785–2794. doi: 10.1074/jbc.M110.158220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pittenger C, Coric V, Banasr M, Bloch M, Krystal JH, Sanacora G. Riluzole in the treatment of mood and anxiety disorders. CNS Drugs. 2008;22:761–786. doi: 10.2165/00023210-200822090-00004. [DOI] [PubMed] [Google Scholar]
- 78.Sugiyama A, Saitoh A, Iwai T, Takahashi K, Yamada M, Sasaki-Hamada S, et al. Riluzole produces distinct anxiolytic-like effects in rats without the adverse effects associated with benzodiazepines. Neuropharmacology. 2012;62:2489–2498. doi: 10.1016/j.neuropharm.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 79.Takahashi K, Murasawa H, Yamaguchi K, Yamada M, Nakatani A, Yoshida M, et al. Riluzole rapidly attenuates hyperemotional responses in olfactory bulbectomized rats, an animal model of depression. Behav Brain Res. 2011;216:46–52. doi: 10.1016/j.bbr.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 80.Pittenger C, Kelmendi B, Wasylink S, Bloch MH, Coric V. Riluzole augmentation in treatment-refractory obsessive-compulsive disorder: a series of 13 cases, with long-term follow-up. J Clin Psychopharmacol. 2008;28:363–367. doi: 10.1097/JCP.0b013e3181727548. [DOI] [PubMed] [Google Scholar]
- 81.Ibrahim L, Diazgranados N, Franco-Chaves J, Brutsche N, Henter ID, Kronstein P, et al. Course of Improvement in Depressive Symptoms to a Single Intravenous Infusion of Ketamine vs Add-on Riluzole: Results from a 4-Week, Double-Blind, Placebo-Controlled Study. Neuropsychopharmacology. 2012;37:1526–1533. doi: 10.1038/npp.2011.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mathew SJ, Murrough JW, Aan Het Rot M, Collins KA, Reich DL, Charney DS. Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: a pilot randomized, placebo-controlled continuation trial. Int J Neuropsychopharmacol. 2009:1–12. doi: 10.1017/S1461145709000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brennan BP, Hudson JI, Jensen JE, McCarthy J, Roberts JL, Prescot AP, et al. Rapid Enhancement of Glutamatergic Neurotransmission in Bipolar Depression Following Treatment with Riluzole. Neuropsychopharmacology. 2009 doi: 10.1038/npp.2009.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chowdhury GM, Banasr M, de Graaf RA, Rothman DL, Behar KL, Sanacora G. Chronic riluzole treatment increases glucose metabolism in rat prefrontal cortex and hippocampus. J Cereb Blood Flow Metab. 2008 doi: 10.1038/jcbfm.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 86.Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, et al. Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience. 2008;153:329–337. doi: 10.1016/j.neuroscience.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thone-Reineke C, Neumann C, Namsolleck P, Schmerbach K, Krikov M, Schefe JH, et al. The beta-lactam antibiotic, ceftriaxone, dramatically improves survival, increases glutamate uptake and induces neurotrophins in stroke. J Hypertens. 2008;26:2426–2435. doi: 10.1097/HJH.0b013e328313e403. [DOI] [PubMed] [Google Scholar]
- 88.Sari Y, Sakai M, Weedman JM, Rebec GV, Bell RL. Ceftriaxone, a beta-lactam antibiotic, reduces ethanol consumption in alcohol-preferring rats. Alcohol Alcohol. 2011;46:239–246. doi: 10.1093/alcalc/agr023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Knackstedt LA, Melendez RI, Kalivas PW. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry. 2010;67:81–84. doi: 10.1016/j.biopsych.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miguel-Hidalgo J, Shoyama Y, Wanzo V. Infusion of gliotoxins or a gap junction blocker in the prelimbic cortex increases alcohol preference in Wistar rats. J Psychopharmacol. 2009;23:550–557. doi: 10.1177/0269881108091074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mineur YS, Picciotto MR, Sanacora G. Antidepressant-Like Effects of Ceftriaxone in Male C57BL/6J Mice. Biol Psychiatry. 2007;61:250–252. doi: 10.1016/j.biopsych.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 92.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res. 2011;224:107–111. doi: 10.1016/j.bbr.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 94.Dwyer JM, Lepack AE, Duman RS. mTOR activation is required for the antidepressant effects of mGluR(2)/(3) blockade. Int J Neuropsychopharmacol. 2012;15:429–434. doi: 10.1017/S1461145711001702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koike H, Iijima M, Chaki S. Involvement of the mammalian target of rapamycin signaling in the antidepressant-like effect of group II metabotropic glutamate receptor antagonists. Neuropharmacology. 2011;61:1419–1423. doi: 10.1016/j.neuropharm.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 96.Feyissa AM, Woolverton WL, Miguel-Hidalgo JJ, Wang Z, Kyle PB, Hasler G, et al. Elevated level of metabotropic glutamate receptor 2/3 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:279–283. doi: 10.1016/j.pnpbp.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.