

Preface
Toxoplasma gondii is a common parasite of animals and humans that can cause serious opportunistic infections. However, the majority of infections are asymptomatic possibly because the organism has co-evolved with its many vertebrate hosts and has developed multiple strategies to persist asymptomatically for the lifetime of the host. Over the past two decades, infection studies in the mouse, combined with forward genetic approaches aimed at unraveling the molecular basis of infection, have revealed that T. gondii virulence is mediated, in part, by secretion of effector proteins into the host cell during invasion. Here, we review recent advances that illustrate how these virulence factors disarm innate immunity and promote survival of the parasite.
Introduction
Toxoplasma gondii is a member of the phylum Apicomplexa, a diverse group of primarily intracellular parasites that infect a wide range of hosts, and occasionally cause serious disease in humans and animals. T. gondii is one of the most widespread parasites of this group, found in nearly all warm-blooded vertebrates and a frequent cause of infections in wild, domesticated and companion animals1. Despite enjoying a broad host range, cats are the only host where sexual development is known to take place. Development of gametocytes within intestinal epithelial cells culminates in fertilization and shedding of infectious oocysts in the faeces (Figure 1)2. These spore-like particles can contaminate water and food and lead to infection in a wide range of animals (Figure 1)1. In the intermediate host, the ability to undergo asexual replication, in the form of fast-growing tachyzoites that replicate within nucleated host cells, allows the parasite to rapidly increase in number and disseminate throughout the body. Following a vigorous immune response, the parasite differentiates into semi-dormant tissue cysts that harbor slow growing bradyzoites. Predation and ingestion of tissue cysts by cats completes the cycle (Figure 1). Unlike related parasites, T. gondii is unique in being transmitted asexually via carnivorous/omnivorous feeding3, which may increase dissemination between its many vertebrate hosts (Figure 1). Humans do not typically transmit the parasite, yet they readily become infected by ingesting oocysts in water or tissue cysts in undercooked meat 4,5. The clinical signs in humans range from mild flu-like symptoms, to severe complications in immunocompromised individuals or following transplacental transmission to the foetus6. Human infections show a range of clinical severity, likely a consequence of many factors including host and parasite genotypes, and such associations have been noted in ocular disease7, congenital infection 8,9, and in patients with AIDS10. Although most infections do not lead to serious complications, toxoplasmosis is the third leading cause of food borne infections requiring hospitalization in the USA11.
Figure 1. The complex life cycle of T. gondii.

Cats are the definitive host where sexual replication takes place. Following replication within enterocytes of the gut (a process known as merogony), male and female gametes are formed within the host cell, as described previously 2. Fusion of gametes leads to the formation of diploid oocysts that are shed in cat faeces and undergo meiosis in the environment to yield eight haploid progeny. Oocysts are capable of surviving in the environment for long periods of time and can contaminate food and water, providing a route of infection for intermediate hosts. In the intermediate host (shown here as rodents) asexual replication occurs. Acute infection is characterized by fast replicating tachyzoites that disseminate throughout the body. Differentiation to slow-growing bradyzoites within tissue cysts leads to long-term chronic infection. Ingestion of tissue cysts via omnivorous or carnivorous feeding can lead to transmission to other intermediate hosts or to cats, which re-initiates the sexual phase of the life cycle. Many animals serve as intermediate hosts, including farm animals. Humans become infected by eating undercooked meat containing tissue cysts or by the ingestion of oocysts in contaminated water 4,5. Although most infections are mild, toxoplasmosis can cause serious symptoms in the brain and other organs (as indicated) in immunocompromised patients, as well as in the developing foetus following congenital infection.
Within its vertebrate hosts, T. gondii encounters many challenges in crossing biological barriers, avoiding immune surveillance and establishing its niche as an intracellular parasite. This journey begins in the gut where parasites invade enterocytes and replicate, but they can also cross the epithelial barrier and reach the lamina propria where they encounter resident macrophages, dendritic cells (DCs), and intra-epithelial lymphocytes12. The infection spreads rapidly to draining lymph nodes, the spleen and eventually all organs. T. gondii is equipped with an efficient system for motility that underlies its ability to invade cells and disseminate in the host (Box 1) 13,14. Once established in the host cell, T. gondii resides in a unique parasitophorous vacuole (PV) that does not fuse with the endolysosomal system15. Because tachyzoites can infect any cell type and tissue, and replication leads to cell lysis, it has a tremendous potential to cause disease. The innate immune response limits parasite growth and promotes the development of adaptive immunity, which is required for long term resistance to infection16. The immune response also induces differentiation of the parasite into its chronic semi-dormant form (bradyzoites), potentially averting a lethal showdown between the pathogen and its host. However, the immune response is unable to eradicate the tissue cysts and reactivation of the cysts can cause severe disease in patients with primary or acquired defects in T-cell mediated immunity 17.
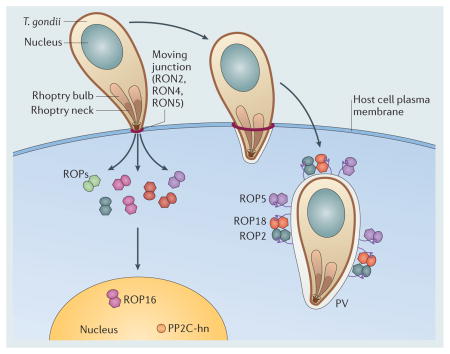
Box 1.
Host cell invasion by T. gondii involves the concerted action of protein secretion along with actin-based motility 13,14. An initial wave of secretion from micronemes is required for motility and host cell attachment 138. During invasion, the parasite invaginates the host cell plasma membrane to create a uniquely modified compartment, called the parasitophorous vacuole (PV) 15. Vacuole formation is initiated by secretion from a bulb-shaped organelle called the rhoptry, which contains a diverse array of proteins 139 that are released directly into the host cell and into the forming vacuole89. Proteins in the rhoptry neck (RONs) are initially secreted into the host cell membrane, where they help mediate formation of a moving junction that is comprised of RON2, RON4 and RON5, together with the micronemal protein AMA1140,141. Proteins in the bulb of the rhoptry (ROPs) are then released into the host cell cytosol, where they are directed to the host cell nucleus (i.e. ROP16, PP2C) or to the surface of the PV (ROP2, ROP18, ROP5) 137,142. The PV resists acidification and fusion with endosomes and lysosomes, while it recruits host mitochondria and endoplasmic reticulum, which may aid in nutrient acquisition 15. The extensive modification of the PV during invasion suggests that the parasite modulates many host cell functions as part of its intracellular lifestyle, a prediction supported by the genetic studies described in the main text. After several rounds of mitotic division daughter cells actively egress from the host cell and invade neighbouring cells.
In addition to its public health importance, T. gondii has emerged as a model organism for studying the molecular basis of pathogenesis in related apicomplexan organisms. These include the human pathogens Plasmodium and Cryptosporidium spp., as well as parasites of livestock such as Eimeria and Neospora spp. T. gondii is a convenient model system owing to its ease of use, the existence of excellent mouse models and the availability of an abundance of genetic tools 18,19. In recent years, advances in our understanding of innate immunity, combined with the development of forward genetic systems in the parasite have provided new insights into the novel mechanisms that T. gondii uses to survive in the host. The emerging picture reveals that the organism has evolved an intricate system of virulence factors that counteract the host innate immune response. The aim of this review is to highlight these recent studies and to provide a framework for future studies to delineate the molecular events that determine the outcome of infection.
Natural genetic variants of T. gondii
Despite its broad host range, comprising many orders of mammals and birds, the genus Toxoplasma contains a single species, Toxoplasma gondii, named after the African rodent (Ctenodactylus gundi) from which it was first isolated in the early 1900s20. Populations of T. gondii in North America and Europe are dominated by three clonal lineages (known as types I, II and III) 21. The three clonal lineages show evidence of being derived from a few genetic crosses among closely related parents22, and following this recent genetic bottleneck, have undergone rapid expansion3. The majority of infections in humans in these regions are due to type II strains, which likely reflects their abundance in livestock23. In marked contrast, the population structure of T. gondii in South America is more diverse, suggesting that there is a higher frequency of sexual recombination in this area23. Recent studies based on a larger collection of isolates that display wider genetic diversity reveal more than 15 lineages that fall into six major groups24. For the purposes of this review, we will focus on the more widely studied type I, II and III lineages, which exhibit very different strategies to induce and/or disrupt the host immune response and thereby assure their survival and transmission.
Rodents are natural hosts for transmission of T. gondii and hence laboratory mice provide a reasonable model to study the immunological events involved in the control of this infection 25. In laboratory mice, type I strains are uniformly lethal with an infectious dose of a single viable organism26, whereas type II have intermediate virulence that varies with mouse strain and type III parasites are considered avirulent 26. Other phenotypes that differ between the three major lineages include intrinsic growth rate27, frequency of differentiation, motility and potential to cross biological barriers28, disruption of host cell signalling (see below), induction of intestinal pathology during acute infection29 and development of CNS pathology during chronic infection in the mouse30.
The innate immune response to T. gondii
For more than 60 years T. gondii has been an important model organism for understanding how the immune system promotes resistance to intracellular pathogens 16. Most laboratory studies have used type II strains (intermediate virulence), which has facilitated the study of the immune response in mice during the acute and chronic phases of infection. These in vivo studies, combined with the analysis of mice lacking various immune effector genes, have defined important components of innate resistance (Table S1). Collectively, they have revealed that control of T. gondii requires the early production of the pro-inflammatory cytokine IL-12, which stimulates natural killer (NK) and CD4+ and CD8+ T cells to release IFN-γ 31,32,33,34 (Figure 2). Although lysis of parasite-infected cells by cytotoxic lymphocytes contributes to resistance, the primary role of these cells is in the production of IFN-γ, which upregulates antimicrobial effector mechanisms to control T. gondii35. The importance of IL-12 induction in early infection is evident from studies reporting that monocytes 36. CD8α+ DCs 37,38, plasmacytoid DCs 39,40 and neutrophils41 all contribute to the production of this activation signal (Figure 2). Selective ablation studies have implicated CD8α+ DCs 38 and inflammatory monocytes 42 as early sources of IL-12 in vivo and consistent with this, recruitment of inflammatory monocytes to the site of infection is essential to control parasite growth 43,44. In addition, myeloid differentiation primary response gene 88 (MyD88) — a central adaptor protein required for the function of multiple toll like receptors (TLRs) and several cytokines — is essential for controlling infection45 (Table S1). There has also been progress in defining the parasite molecules and host receptors involved in initial recognition, including TLR2 and TLR4 detection of glycosylphosphatidylinositol (GPI) anchored proteins 46 and TLR11 mediated detection of parasite profilin47 (Figure 2). However, deletion of individual TLRs has only modest effects on susceptibility to infection implying that multiple TLRs are involved in the recognition of T. gondii 48. Consistent this idea, triple deficiency (3d) mice, which carry a point mutation in the UNC93B1 protein and are defective in TLR3/7/9 trafficking and TLR11 signalling, are highly susceptible to infection with T. gondii 49,50.
Figure 2. Innate immune responses to Toxoplasma gondii during infection.

a) Early in infection, the first cells to respond are dendritic cells (DCs) and monocytes/macrophages. Interaction of T. gondii profilin with TLR11 on DCs is important for the production of IL-12. In addition to stimulating IL-12 production, macrophages also induce TNF-α, a cofactor in antimicrobial activity, in response to the detection of GPI anchored proteins via TLR2 and TLR4. b) The immune response results in the production of IFN-γ from NK cells through the innate response and eventually from CD4+ and CD8+ T cells as the adaptive response ensues. IL-10 and IL-27 are key to modulating these pathways and prevent the overproduction of TH1 cytokines. c) Production of IFN-γ during the innate and adaptive phases is responsible for activating cells to control parasite infection. IFN-γ propagates a signal through a surface receptor (IFN-γR) to activate STAT1, a nuclear transcription factor that controls the expression of many genes. In response to STAT1 activity, nitric oxide (NO) and reactive oxygen species (ROS) are upregulated in monocytes/macrophages, both of which contribute to the control of intracellular parasites. Both haematopoetic and nonhaematopoetic cells also upregulate two families of defence proteins called immunity related GTPases (IRGs) and guanylate binding proteins (GBPs), which are recruited to pathogen-containing vacuoles and are involved in parasite clearance. The function of IRGs and GBPs depends on the autophagy protein Atg5.
IFN-γ is the major mediator of resistance to T. gondii and is crucial for the activation of a variety of antimicrobial activities in haematopoetic and non-haematopoetic cells that limit parasite replication 51,52 (Figure 2, Table S1). For example, IFN-γ alters cell metabolism, which can lead to tryptophan in fibroblasts 53 and iron starvation in enterocytes 54. This cytokine also stimulates professional phagocytes to produce reactive oxygen and nitrogen intermediates, which can lead to parasite damage and impede growth in macrophages 55,56. Studies using inducible nitric oxide synthase (iNOS) deficient mice have demonstrated the existence of an IFN-γ dependent, iNOS independent mechanism of resistance to T. gondii that operates during the acute phase of infection57. This pathway relies on immunity related GTPases (IRGs) that are induced by IFN-γ and contribute to clearance of T. gondii in multiple cell types (Figure 2, Table S1)58,59 Although the precise mechanisms are not understood, recruitment of IRGs to the parasite-containing vacuole leads to vesiculation, vacuole rupture and digestion of the parasite within the cytosol 60. A related family of GTPases known as the p67 guanylate binding proteins (GBPs) were recently shown to contibute to control of toxoplasmosis in the mouse 61. This family of proteins is more widely conserved among vertebrates 62 and they are also implicated in the control of Listeria spp. and mycobacteria 63, consistent with a more general role in host resistance to infection 64.
Although innate immune responses to T. gondii have been examined in detail, how these processes lead to the stimulation of adaptive immunity, including the ability of DCs to access antigens for priming of CD4+ and CD8+ T cells, are less well understood. Within the PV, the parasite is shielded from the major pathways for antigen processing and presentation, as discussed recently65. Moreover, infection of host cells is associated with reduced expression of major histocompatibility complex (MHC) molecules66. Despite these mechanisms of avoidance, infection with type II strains of T. gondii leads to the activation and expansion of DCs and a strong CD8+ T cell response, while infection with virulent type I strains induces a weaker response67. Several prominent endogenous antigens that are presented on class I MHC molecules include the dense granule proteins GRA6 68 and GRA4, and the rhoptry protein ROP769. These antigens are polymorphic between strains, suggesting that they may be involved in strain-dependent evasion mechanisms that also influence adaptive immunity. Other studies have highlighted the importance of highly immunogenic surface antigens (SAGs) and SAG-related surface antigen (SRS) in stimulating the adaptive immune response70.
Alterations in host signalling and immune evasion
Many innate immune effectors are under the control of transcription factors that enhance or regulate the overall immune response to invading microorganisms. In turn, successful pathogens have developed strategies to undermine important host cell immune pathways. Infection of mammalian cells with T. gondii induces many changes in host cell gene transcription, including those genes involved in energy metabolism, immune responses and signalling 71. Here, we will focus specifically on how the parasite alters the immune response, as this is crucial for our understanding of how the infection is controlled, and as seen below, the host immune response is a major target of T. gondii virulence factors.
Infection with T. gondii inhibits host cell signalling pathways involved in protective immunity, for example by blocking the transcription factors signal transducer and activator of transcription 1 (STAT1) 72 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) 73,74,75. Fibroblasts that are already infected with any of the three major natural strain types of T. gondii (i.e. strain types I, II and III) are unresponsive to the antimicrobial effects of IFN-γ and this appears to be due to defective STAT1 signalling 76. In T. gondii-infected bone marrow derived macrophages, this occurs due to modifications in chromatin structure that disrupt STAT1 binding to nuclear promoters 77. Infection with T. gondii also upregulates anti-inflammatory pathways including those involving the suppressor of cytokine signalling protein (SOCS) 1 78,79, SOCS3 80 and STAT3 81, potentially compromising host mechanisms of parasite control.
Although IFN-γ promotes antimicrobial activities in many cell types, in macrophages it is not typically sufficient for the control of T. gondii as a second signal is required to fully activate killing. The best-characterized second signals that promote control of T. gondii in vitro are provided by TNF-α or signals through CD40 82,83,84, both of which utilize the NF-κB signalling pathway. The NF-κB family of transcription factors have an important role in the immune response to T. gondii, which is expected considering their evolutionarily conserved role in the regulation of many aspects of innate and adaptive immune functions 85,86. Notably, the activation of NFκB by type II strains is associated with higher levels of IL-12 production 36, an effect that contributes to early control of infection.
Mapping and identification of virulence loci
Based on the pioneering studies of Elmer Pfefferkorn, who first demonstrated the potential for classical genetics in T. gondii 87, a number of pair-wise genetic crosses were undertaken to map the genetic loci responsible for major phenotypic differences between the clonal lineages (Box 2). These forward genetic studies have been extremely effective in identifying the underlying genetic basis for some of the most striking phenotypic differences between lineages I, II and III, notably differences in acute virulence and induction of host cell signaling 88.
Box 2.
Classical genetic crosses between the three main clonal strains have been useful for defining virulence determinants of T. gondii. Crosses were conducted by tagging strains with drug resistance markers and co-feeding cats with a mixture of cysts obtained from mice (which were infected with two different parasite strains) to generate recombinant oocysts (see Figure 1) 143. Three pair-wise genetic crosses were conducted (see table) between the different strains and single cell F1 progeny isolated from the oocysts were used to generate genetic linkage maps 144 and to identify phenotypic traits (as described in more detail previously 23,143). Analysis of these genetic crosses revealed that the genome of T. gondii, is approximately 65 x106 bp in size and is organized into 14 linear chromosomes that range in size from 2->10 Mbp 144. Recombination rates are relatively low, with a centimorgan (1% recombination rate) corresponding to roughly 100 Kbp144. These parameters predict that it should be relatively easy to map a given trait to a region of one chromosome, but more challenging to fine map and identify the gene of interest. Additional criteria that are useful in narrowing down the candidate list include differential expression levels, high levels of allelic polymorphism and the allocation of genes to specific functional pathways, such as those encoding surface or secretory proteins, which might be in contact with the host and hence more likely to contribute to pathogenesis. Genetic crosses between the three lineages have been used to analyze the segregation of several traits including acute virulence90,91,94,95, growth90, migration potential90 and effects on host cell transcription 93. Analysis of complex traits has been facilitated by the use of quantitative trait locus (QTL) mapping, which provides a statistical means of evaluating the contribution of multiple loci to a given phenotype and for examining interactions between them145. QTL analysis of growth differences between type I (fast growth) and type III (slow growth) strains of T. gondii revealed that 4 or more major loci control this trait90, which has thus far been refractory to the identification of individual genes due to the small contribution they each make. Likewise, differences in migration potential90, which may contribute to virulence146, map to a broad region of chromosome VIIa, but has not yet been localized to a specific gene(s). Genes that have been identified by mapping and their known targets are summarized in the table.
Summary of genetic crosses
| Genetic Cross | Phenotypes examined | QTLs | Genes identified | Targets |
|---|---|---|---|---|
| I × II | Acute virulence | XII | ROP5 | Activates ROP18 Binds Irga6 |
| II × III | Acute virulence Altered host gene expression IL-12 induction |
VIIa, VIIb, XII VIIb X |
ROP18, ROP5 ROP16 GRA15 |
IRGs STAT3/6 NFkB |
| I × III | Acute virulence Growth Migration |
VIIa, Ia VIIa, XI, XII, Ia VIIa |
ROP18 | IRGs, ATF6b |
Identification of rhoptry kinases as mediators of acute virulence
During invasion of host cells, secretory organelles known as rhoptries discharge their contents into the host cell (Box 1), making these primary candidates for modulating host signalling89. This prediction was confirmed when ROP18 was identified as a major factor that contributed to strain-specific differences in virulence90, 91. ROP18 is an active serine/threonine (S/T) protein kinase that is secreted into the host cell where it decorates the surface of the parasitophorous vacuole membrane (PVM) (Box 1). It is a polymorphic protein and its expression levels vary from high in type I and II strains to a low level in type III strains. Transgenic expression of ROP18 from type I or type II strains in the type III background greatly enhanced virulence confirming that this locus was responsible for the observed differences in virulence between strains 90,91. This gain of virulence required an active enzyme as transfection with an allele containing an inactive kinase allele failed to enhance virulence 90. The low expression of ROP18 in type III strains is attributed to the presence of a unique ~ 2kb region upstream of the coding sequence that is absent in type I/II strains92. A comparison of this region with the same region in the related parasite Neospora caninum suggests that it underwent a rearrangement in the ancestral progenitor to type I and II strains92, leading to upregulation of expression of ROP18 in these two lineages and enhanced virulence. Both virulent (encoded by types I and II) and avirulent (encoded by type III) alleles are stable in the population and show evidence of long term stability 92, implying that the three lineages are adapted for distinct niches.
Genetic crosses between strains II and III also led to the identification of a protein that is responsible for the ability of these strains to differentially affect host gene transcription (Box 2) 93. This rhoptry kinase, ROP16, is unusual in that it is targeted to the host nucleus 93. Further analysis of differences in host transcription induced by type II (intermediate virulence) and type III strains (avirulent), identified a third locus encoding a polymorphic pseudokinase called ROP5 on chromosome XII (Box 2)94. Remarkably, this same ROP5 locus was the sole quantitative trait locus (QTL) identified in a genetic cross between type I (high virulence) and II (intermediate virulence) strains (Box 2)95. In both cases, genetic disruption of the ROP5 locus in a type I strain led to severe attenuation, which was restored with complementation94,95. The ROP5 locus contains a tandem repeat of 6–10 polymorphic genes that differ between strain types, and yet all are predicted to encode catalytically inactive pseudokinases94,95. Comparison of the three pair-wise crosses predicts that ROP5 and ROP18 interact and together they explain nearly 90% of the differences in acute virulence between these three lineages.
The diversity of ROP kinases in T. gondii
Protein kinases are expanded in most eukaryotic lineages, where they control a number of important signalling pathways and are defined by a series of conserved domains that fold into common N- and C-terminal lobes 96. They contain a nucleotide binding pocket characterized by a number of conserved motifs including a catalytic triad that transfers γ-phosphate from ATP to an acceptor hydroxyl group on serine, threonine or tyrosine 96. Pseudokinases encode intact proteins that typically lack the ability to catalyze phosphate transfer, but they can act as scaffolds to regulate enzymatic processes97. The realization that rhoptries contain a family of related polymorphic serine / threonine protein kinases and pseudokinases, led to a bioinformatic search that identified 44 other ROP kinase members, about half of them predicted to be active, while the remainder are predicted to be pseudokinases98. The ROP family is highly diverse in T. gondii and related apicomplexans, including N. caninum, and the abundance of non-synonymous substitutions in these proteins, often accompanied by gene duplication, indicates that they are under strong selective pressure98. Most of the predicted ROP kinase family members have not been studied, but several have been validated as bone fide rhoptry proteins that are secreted into the host cell during invasion98. In addition ROP38 from type III parasites is implicated in downregulating host MAPK signalling, as expression of this allele in a type I background suppressed this pathway 98. However the precise role of this kinase in pathogenesis is still uncertain. For the remainder of the review we will focus on the function of the ROP kinases whose functions have been elucidated.
ROP18 and ROP5 belong to a subset of the ROP kinase family typified by the pseudokinase ROP2, the first member of this family predicted to contain a kinase domain99. This prediction was borne out when the crystal structures of the kinase domain of ROP2 100,101 and ROP8101 were solved, which in turn facilitated the generation of a homology model of ROP18101 (Figure 3A). By aligning the sequence of ROP18 to known S/T kinases, all of the conserved residues known to be important in ATP binding and catalysis were identified101. It was also shown that ROP18 has a novel mechanism of activation that relies on phosphorylation of an N-terminal helical domain that wraps around the N-lobe and inhibits kinase activity when it is unphosphorylated101 In contrast to ROP18, the pseudokinases ROP2 and ROP8 have a highly degenerate nucleotide-binding pocket and are not predicted to bind or hydrolyze ATP100,101. Similarly, ROP5 is predicted to be a pseudokinase and although it is able to bind ATP, it is unlikely to carry out phosphate transfer due to an alteration in the central residue of the catalytic triad that is Arg or His instead of Asp102. Remarkably, ROP2 kinase family members are highly conserved among T. gondii strains, however, amino acid differences between strains are clustered in the substrate-binding loop101. This implies that even though they are not catalytically active, they may be capable of binding specific host substrates. ROP2 family members also have an N-terminal extension that contains three amphipathic alpha helical regions that are rich in arginine on one face and hydrophobic on the other103. This low complexity region is predicted to be involved in mediating membrane interactions and has been shown to be necessary and sufficient for targeting to the PVM 103,104 (Figure 3B,C). As recently shown, targeting of ROP18 to the PVM determines access by the kinase to host substrates and is essential for virulence105.
Figure 3. Structure and function of ROP2 family members.

a) Model of the kinase domain of ROP18 based on homology to the X-ray crystal structure of the pseudokinase ROP8 (pdb 3BYV). The two N-terminal helices (green) are important for stabilization of the kinase domain (red) and are also involved in auto-activation of ROP18. The N- and C-lobes of the kinase domain are labelled, while the N-terminal extension involved in membrane tethering, is absent from the structure. Provided by Ray Hui, Structural Genomics Consortium, Toronoto. b) Domain structure of ROP2 family members showing the signal peptide and prodomain, both of which are processed prior to secretion (arrows). The N-terminal half of the mature protein (green) contains a series of three amphipathic alpha helical regions with low sequence complexity (grey boxes) that mediate membrane targeting. The C-terminal region contains a S/T kinase domain (red). Localization of full-length ROP18 on the membrane of the parasite containing vacuole is mediated by the N-terminal amphipathic region (green). Deletion of this region disrupts vacuole association of the kinase domain (red), as shown for the truncated version of ROP18.
Host targets affected by T. gondii secretory proteins
Although multiple host signalling pathways are altered by T. gondii (Table S2), we have only recently begun to appreciate the specific parasite proteins that mediate these effects. We will focus on three parasite “effectors” that are known to target the innate immune response of the host.
Alteration of host cell transcription by ROP16
The role of ROP16 in altering host gene transcription was initially identified by analyzing the differences in host gene expression induced by different parasite strains. These studies led to a focus on genes involved in IL-4 and IL-6 responses and implicated changes in the activity of the transcription factors STAT3 and STAT6 93. Although all three strains initially induce STAT3 and STAT6 activity, only the type I or III strains (which share the same ROP16 variant, ROP16I/III) sustain this response (Figure 4, Table 1)93. Subsequent studies revealed that ROP16I/III directly phosphorylates Tyr705 in STAT3106 and Tyr641 in STAT6107, residues that are required for activation of these transcription factors. Studies using a type I strain carrying a knockout of the ROP16 allele (Δrop16) indicate that initial stimulation of STAT3 is independent of ROP16, whereas early activation of STAT6 requires ROP16 108. Comparison of the two ROP16 variants (i.e. the protein from type II strains, ROP16II and the protein common to types I and III, ROP16I/III) revealed that a single amino acid substitution at position 503 is responsible for the marked difference in sustained activation of STATs106. Prolonged STAT3/6 activation by ROP16I/III down-regulates the induction of IL-12, thus limiting the protective TH1 cytokine responses 93, which might lead to less inflammation and reduced pathology but also enhanced parasite survival (Figure 4). Consistent with this model, transgenic expression of a type I ROP16 allele in type II strains inhibits IL-12 production 93, while the Δrop16 type I strain shows increased IL-12 production 108. Additionally, the altered cytokine response of the Δrop16 type I strain provided further evidence that ROP16 controls the ability of T. gondii to block STAT3-dependent production of IL-12 and TNF-α 108.
Figure 4. The role of T. gondii virulence factors in modulating immune signalling in the host.

ROP16I/III (pink) phosphorylates STAT3 and STAT6 resulting in prolonged activation of these two transcription factors and subsequent upregulation of IL-4 while antagonizing induction of IL-12. The dense granule protein GRA15II (green) activates TRAF6, which in turn activates IKK, leading to phosphorylation of IκB and the release of NFκB following proteasomal degradation. NFκB migrates to the nucleus and drives the production of IL-12. ROP18I (red) is localized on the cytoplasmic side of the parasite containing vacuole (PV). ROP18I has been shown to phosphorylate immunity related GTPases (IRGs), thus blocking their accumulation on the vacuole and protecting the parasite from destruction. ROP18I also phosphorylates the host transcription factor ATF6β, which is involved in the unfolded protein response and may also be important for efficient antigen presentation by DCs. Genetic evidence suggests that ROP5I/III (purple), a pseudokinase important for virulence, regulates the functions of ROP18I. Allele types are shown in subscript (i.e. ROPI denotes the allele in type I strains).
Table 1.
Strain-specific virulence factors and their effects on the mouse immune response
| Strain | ROP18 | ROP5 | ROP16 | GRA15 | Effects on host | Phenotype |
|---|---|---|---|---|---|---|
| I | Type I Virulence enhancing |
Type I/III Virulence enhancing |
Type I/III Prolonged activation of STAT3/6 |
Type I/III Non-activating |
Prolonged STAT3/6 activation Reduced IL-12 production Avoidance of parasite clearance |
High virulence |
| II | Type II Virulence enhancing |
Type II Avirulent |
Type II Non-activating |
Type II Activates NFkB |
Non-sustained STAT3/6 activation High levels of IL- 12 Enhanced parasite clearance |
Intermediate virulence |
| III | Type III Hypomorphic |
Type I/III Virulence enhancing |
Type I/III Prolonged activation of STAT3/6 |
Type I/III Non-activating |
Prolonged STAT3/6 activation Reduced IL-12 production Enhanced parasite clearance |
Low virulence |
Despite having profound effects on host gene expression, ROP16 has only a moderate role in acute virulence in mice. Thus, although the Δrop16 type I strain shows slightly enhanced parasite replication in vivo compared with wild type, deletion of this allele does not reduce host mortality following ip challenge 108. ROP16 may also have a role in reducing inflammation, and thus have important roles in other infection models since transgenic expression of ROP16I/III in a type II strain was shown to reduce pathology caused by oral infection of mice109. The enhanced growth that was observed in the absence of ROP16 may be explained by the ability of this protein to activate STAT6, which leads to the induction of arginase, an enzyme associated with alternatively activated macrophages108. It is possible that arginase could have two opposing roles in T. gondii growth: enhancing parasite growth by providing polyamines, but limiting access to the essential amino acid arginine and impairing growth 110. Induction of arginase is also expected to antagonize the ability of macrophages to kill intracellular parasites since arginine also serves as a substrate for iNOS; consistent with this, elimination of arginase in macrophages promotes resistance to T. gondii 111. Thus, the balance of these two opposing roles for arginine may determine whether the pathogen persists or is eliminated in vivo.
Induction of IL-12 production by GRA15
Infection of macrophages with type II parasites results in production of higher levels of IL-12, compared to infection with the other two lineages36. This effect is partly modulated by ROP16 since the allele expressed in type II strains is incapable of sustained phosphorylation of STAT3, as discussed above. However, this is unlikely to be the sole factor responsible for the increased levels of IL-12 during infection with type II strains. Indeed, genetic mapping identified a locus on chromosome X encoding a dense granule protein named GRA15 that is responsible for induction of IL-12 in type II strains and expression of the type II variant (GRA15II) in a type I strain enhances IL-12 production 112. The molecular mechanism of GRA15 function is uncertain as it has no homology to any other proteins in the database, nor does it contain any conserved domains. Localization studies revealed that GRA15 is secreted into the host cell together with ROP proteins in small cytoplasmic inclusions known as evacuoles112. GRA15 also localizes to the PVM 112, although its topology at this interface has not been established. Deletion of GRA15 in the type II strain ME49 prevents nuclear translocation of NFκB but the mechanism by which GRA15 activates NFκB is incompletely understood. However, it acts independently of MyD88, the IL-1 receptor family and TIR-domain-containing adapter-inducing interferon-β (TRIF)112. Since MyD88 and TRIF co-ordinate the main signalling pathways for TLR ligands it seems unlikely that GRA15 is recognized by a TLR. Nonetheless, induction of NFκB by GRA15 depends on TNF receptor associated factor 6 (TRAF6), which is an adaptor that functions with IL-1R-associated kinase 1 (IRAK1) to mediate signalling downstream of MyD88. Engagement of TRAF6 activates IKK, followed by phoshorylation of IκB, proteosome degradation, and translocation of NFκB to the nucleus 112 (Figure 4). GRA15 appears to be sufficient to induce this pathway as its expression in HeLa cells results in nuclear translocation of NFκB 112. Although GRA15II can directly drive NFκB activation and IL-12 production, this is likely to be only one contributing factor as other studies have shown that following infection by type II strains, production of IL-12 is largely Myd88-dependent in macrophages 113 and dendritic cells 45.
The polymorphic nature of ROP16 and GRA15 in different lineages of T. gondii leads to polarizing responses in cells infected with T. gondii 109,112. Infection of macrophages with parasites expressing ROP16I/III leads to activation of STAT6 and the development of alternatively activated macrophages, while those infected with strains expressing ROP16II show the classical activated phenotype 108,109. GRA15II activates NFκB resulting in high levels of IL-12 production, further enhancing the classical activation pathway in cells infected by type II parasites. Classically activated macrophages typically express chemokines and cytokines that activate cells with antimicrobial activity while alternatively activated macrophages secrete anti-inflammatory molecules that can down-regulate Th1 responses114. Hence, different variants of the effector proteins drive antagonizing responses in infected macrophages, which can have broad effects on antimicrobial effector pathways, inflammation and parasite control.
Modulation of acute virulence by ROP18 and ROP5
As noted above, differences in virulence between strains of T. gondii are associated with the S/T kinase ROP18 and the related pseudokinase ROP5. Understanding the mechanistic basis for this effect has been aided by recent observation that together this pair of parasite effectors targets host immunity by disrupting IRGs. Induced by IFN-γ, IRGs are crucial for the control of toxoplasmosis in mice 59,60. These enzymes are expanded in the mouse system but are less common in other vertebrates (for example humans have only two IRGs.)115. Although the PVM lacks most host cell proteins15, IRGs are recruited to the PVM surrounding type II and III strains where they cause vesiculation and promote parasite destruction, 116,117 a process that is also dependent on the autophagy protein Atg5 although the nature of its involvement is unclear118. Unlike strain types II and III, highly virulent type I strains avoid this fate116,119.
In IFN-γ activated cells, several IRGs including Irga6, Irgb6, and Irgb10 are phosphorylated by ROP18 and this is associated with reduced IRG recruitment to the PVM {Fentress, 2010 #6020;Steinfeldt, 2010 #6396}. Transgenic expression of the active form of ROP18 from a type I strain in a type III parasite inhibited IRG recruitment to the PVM, resulting in persistence of the parasite in activated macrophages120. RNAi suppression of Irgb6 reversed the enhanced clearance of parasites lacking ROP18 120. The interplay between ROP18 and the IRGs was revealed by mass spectrometry, which showed that ROP18 phosphorylates key threonine residues in switch region I of Irga6 and Irgb6 {Fentress, 2010 #6020;Steinfeldt, 2010 #6396}. Phosphorylation of Irga6 prevents GTP hydrolysis and oligomerization of this protein {Steinfeldt, 2010 #6396}, steps that are thought to be important for loading of IRGs onto the PVM121. These findings indicate that ROP18 contributes to virulence by prolonging survival of T. gondii in IFN-γ activated macrophages. Moreover, the role of ROP18 in blocking IRG function is not limited to professional phagocytes as similar studies have shown it is important for the avoidance of IRG recruitment to the PVM in activated fibroblasts 122.
More recently, the transcription factor ATF6β, which resides in the host ER and is important in the unfolded protein response (Figure 4), was identified as a ROP18 target123. In co-transfected cells, phosphorylation of ATF6β by ROP18 led to proteosomal degradation of ATF6β and a consequent reduction in ATF6β mediated gene expression when the unfolded protein response was induced123. The interaction of ROP18 with ATF6β in co-transfected cells was mapped to the N-terminus of the kinase and the C-terminus of ATF6β123. Since the C-terminus of ATF6β is in the lumen of the host ER, while the N-terminus of ROP18 is tethered to the PVM, this interaction suggests a precise topological rearrangement of one or both of these proteins within infected cells. A role for ATF6β in vivo was shown by the increased susceptibility of ATF6β−/− mice to challenge with an attenuated type I Δrop18 strain. This deficiency was associated with decreased induction of IFN-γ by CD8+ T cells incubated with DCs from ATF6β−/− mice123, suggesting that ATF6β might have a previously unrecognized role in antigen presentation.
Although the phenotype of Δrop18 type I parasites in IFN-γ activated macrophages in vitro is highly attenuated, this mutant is only partially attenuated in vivo120. In contrast, deletion of the pseudokinase ROP5 in the type I background significantly decreases virulence by more than a million fold94,95. However, because the ROP5 locus encodes a tandem array of predicted pseudokinases, it was initially unclear why this catalytically inactive protein would have such a dramatic effect on virulence. Insights into this question were provided by genetic studies that suggested that ROP5 and ROP18 interact. Firstly, it has been established that ROP5 binds to Irga6 and affects its ability to form GTP-dependent oligomers 124,125 which would disrupt the loading of Irga6 onto T. gondii containing vacuoles. In two separate studies, it was shown that ROP5 enhances ROP18I-mediated phosphorylation of Irga6 124 and Irgb6 126. Since ROP5 does not bind well to Irgb6, the latter result is likely due to enhanced ROP18 catalytic activity in the presence of ROP5, which has been confirmed by in vitro studies 126. This indicates that ROP5 allosterically activates ROP18, as has been recently described for several human kinases97,127. Collectively, these data suggest a model in which ROP5 contributes to acute virulence in the mouse by activating other ROP kinases, as well as disrupting IRG oligomerization, which allows type I strains to evade antimicrobial effector mechanisms.
Overview of strain-dependent mouse virulence in T. gondii
Genetic mapping studies have revealed that the marked phenotypic differences between strains of T. gondii in mice are mediated by combinations of different alleles at only a few major loci. The acutely virulent type I strains have a highly active ROP18 that functions cooperatively with ROP5 to phosphorylate host IRGs, inhibit IRG loading on the PVM and subsequently block parasite clearance in IFN-γ activated cells (Figure 4, Table 1). ROP18 also targets ATF6β and may thus compromise antigen presentation by DCs. Type I strains also express STAT3/6 activating alleles of ROP16 that can dampen TH1 cytokine responses and an allele of GRA15 that fails to induce the pro-inflammatory cytokine IL-12 (Figure 4, Table 1). The combined activities of these effectors could allow the parasite to delay induction of protective immunity systemically, while escaping innate mechanisms of control within infected cells, ultimately resulting in expansion to very high tissue burdens. Somewhat paradoxically, mice eventually succumb to infection despite producing very high levels of type I cytokines128,129. It is likely that the initial impairment of TH1 responses (via ROP16, ROP18 and possibly other factors), allows parasite expansion and results in a high parasite burden that causes tissue damage and subsequent activation of danger signals, triggering a secondary response that leads to cytokine-mediated pathology. Although the molecular basis is not fully understood, this strong acute virulence trait is also shared by numerous South American lineages 92. Although this phenotype leads to rapid death of laboratory mice, it may be an adaptation to survive in hosts such as wild deer mice 130, rats 131 or chickens 132, all of which appear innately resistant. In contrast to the extreme virulence of type I strains, type II strains induce protective immunity and exhibit intermediate virulence. Despite having a virulence enhancing ROP18 allele, they express a variant of ROP5 that is considered “avirulent” (Figure 4, Table 1). Consequently, they are less able to avoid IRG clearance within IFN-γ activated cells (Figure 4, Table 1). Although the GRA15 variant expressed by type II strains induces the production of IL-12, the inability of this strain to prolong STAT3/6 activation (due to the particular ROP16 variant it encodes) results in early IL-12 production leading to a type 1 cytokine response and effective control of acute infection (Figure 4, Table 1). Such a strategy is consistent with the high propensity of type II strains to differentiate into bradyzoites and their associated prevalence in chronic infections in animals. Finally, type III strains appear to lack an effective mechanism for blocking IRGs because they under-express ROP18 by ~ 100 fold compared to types I or II (Figure 4, Table 1). As such, they are readily cleared from IFN-γ activated murine macrophages. Type III strains also induce prolonged phosphorylation of STAT3/6, while not strongly activating NFκB and hence avoiding induction of IL-12 (Figure 4, Table 1). This strategy may be an adaptation to survive in hosts that lack the IRG pathway or where overproduction of cytokines causes detrimental pathology to the host. Although we do not know what specific hosts each of these phenotypes are adapted for, they are each common in natural isolates and likely reflect successful long-term evolutionary strategies 92.
Future directions
Recent advances have revealed that similar to other pathogens that inject virulence factors into host cells133,134,135, T. gondii injects rhoptry proteins not only into the cells it infects but also into bystander cells 136. Using this so called “kiss and spit” model137, the parasite can modify the cell it resides in, as well as potentially influencing the behavior of adjacent cells 136. An assessment of the many characterized mutants with advanced imaging techniques should allow further molecular dissection of the interactions between the parasite and its host, in order to reveal the strategies that are central to dissemination and pathogenesis of T. gondii. The questions that have received the most focus in the last decade have revolved around understanding differences in virulence between parasite strains and have provided important new perspectives on pathogenesis in the mouse model. Extending these studies to other host species will be crucial to evaluate the contribution of virulence factors to transmission dynamics and disease in natural hosts. Beyond the traits described here, a large number of other differences in host responses have been noted in previous studies, although most of these have not been examined for potential parasite strain-specific or host differences (Table S2). Thus far, most studies have focused only on the dominant strains in North America and Europe. Extension of these approaches to more diverse lineages from South America and other regions24 is likely to reveal additional layers of complexity in parasite virulence. Moreover, the current strategy for mapping of genetic differences is unlikely to uncover conserved mechanisms used by all strains of T. gondii to evade the immune response. Additional forward genetics screens will be needed to identify the molecular basis of such virulence activities. As additional more powerful experimental approaches are developed, we can look forward to learning more about the role of parasite secretory proteins in modulating host functions.
Supplementary Material
Acknowledgments
Work in the authors’ laboratories is supported by the National Institutes of Health (L.D.S., C.A.H.) and the State of Pennsylvania (C.A.H.). We regret not being able to cite all of the appropriate primary literature due to space limitations.
Glossary
- Apicomplexans
Protozoans of the phylum Apicomplexa known for their apical complex consisting of a specialized microtubule organizing center, called the conoid, and secretory organelles involved in host cell invasion. Toxoplasma spp. belong to the tissue-cyst forming coccidian group of the Apicomplexa
- Tachyzoite
The rapidly replicating intracellular form (from the greek tachys, meaning fast) of many tissue cyst-forming coccidians such as T. gondii
- Bradyzoite
A slow growing form of the parasite (from the greek bradys, meaning slow) residing within long lived tissue cysts that are associated with chronic infection. One of the parasitic stages that is specialized for transmission if ingested
- TLR
toll-like receptors, pattern recognition receptors that mediate immune responses by detecting pathogen associated molecular patterns
- iNOS
Inducible nitric oxide synthase (NOS2 gene) A soluble enzyme that produces nitric oxide (NO) from L-arginine. It is upregulated by IFN-γ and TNF-α, and constitutes a major antimicrobial activity of macrophages
- IRG
Immunity related GTPases: A family of 45–47 kDa GTPases that are strongly upregulated by IFN-γ and contribute to resistance to intracellular pathogens. IRGs are ubiquitous in the mouse, but are much more restricted in other vertebrates and largely absent in humans
- GBP
Guanylate binding proteins: A family of 65 kDa GTPses that are upregulated by IFN-γ and contribute to resistance to intracellular pathogens. Widely distributed in vertebrates indicating that they may play an important general role in resistance to intracellular pathogens
- MHC
Major histocompatibility complex: A family of cell surface molecules that allow recognition of epitopes from foreign or self antigens through presentation to T cells
- STAT
Signal Transducer and Activator of Transcription Cellular transcription factors: A family of transcription factors that are activated by Janus kinases (JAK) and regulate gene expression by binding to nuclear promoters. Have a role in development and the immune system
- SOCS
Suppressor of cytokine signalling: A family of intracellular proteins that regulate cytokine signalling by either direct inhibition of receptors or increased degradation of signalling proteins
- TNF-α
Tumor necrosis factor alpha: a cytokine involved in inflammation, tumor suppression and host defence
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells: A complex that controls DNA transcription in response to various signalling inputs. Plays a key role in regulating innate and adaptive immune responses
- QTL
Quantitative trait locus, Genetic region (locus) that contributes to a quantitative trait, meaning one that varies by degree. Such traits are typically polygenic, being contributed to by multiple QTLs
- Pseudokinase
Protein containing a conserved protein kinase fold but lacking key residues in the nucleotide binding pocket that result in inability to transfer phosphate to a donor substrate. Although not catalytically active, they serve regulatory or scaffolding roles
- Arginase
Enzyme in the urea cycle that mediates consumption of arginine, and thus has an antagonistic role to nitric oxide synthase as they rely on the same substrate
- Alternatively activated macrophages
Refers to one of to polar phenotype that macrophages can adopt following stimulation by cytokines. Classically activate macrophages exhibit a TH1 phenotype, promoting inflammation, while alternatively activated macrophages have a Th2 phenotype dampening inflammation and promoting repair. Arginase is typically expressed by alternatively activated macrophages, while nitric oxide synthase is produced by classically activated macrophages
- MAPK
Mitogen-activated protein (MAP) kinases: Serine/threonine kinases involved in cellular signalling in response to a diverse array of stimuli
- TRIF
TIR-domain-containing adapter-inducing interferon-β; Adaptor protein for signalling from TLRs that contain TIR domains
- TRAF6
TNF receptor associated factor 6: Protein adaptor involved in signalling through TNF, IL-1, and TLRs
- IKK
IκB kinase: a kinase comprised of three subunits α, β and γ, also known as NEMO
- IκB
Inhibitory subunit of NFKB: becomes phosphorylated by IKK, ubiquitination and degradation by the proteosome, allowing active NFκB to translocate to the nucleus
- Unfolded protein response
Cellular stress response triggered by unfolded proteins accumulating in the ER. Leads to stalled translation and upregulation of chaperones for protein folding
- Micronemes
Secretory organelles that are discharged in resposne to elevated cytoplasmic calcium. Microneme proteins (MICs) mediate substrate and host cell adhesion
- Centrimorgan
(cM) A genetic unit used for establishing genetic linkage based on recombination frequency in the progeny of a genetic cross. 1 cM is equal to the distance over which 1% recombination occurs. Named in honour of Thomas Hunt Morgan
- Oocyst
Diploid stage of parasite development that this the product of of the fusion T. gondii gametes. Oocysts are shed in cat faeces and they contaminate the environment, giving rise to infection by accidental ingestion
- GPI anchors
Glycosylphosphatidylinositol linked proteins share a common glycolipid anchor that is covalently attached to the C-terminus of variety of surface proteins as a post-translational modification
Biographies
Christopher A. Hunter
Chris Hunter is a graduate of the University of Glasgow where he specialized in Parasitology for his Ph.D. and as a post-doctoral fellow. He was also a fellow with Jack Remington at Stanford University where he focused on the immune response to Toxoplasma gondii. He joined the faculty at the University of Pennsylvania in 1994 where he is now Professor and Chair of the Department of Pathobiolology and work in his laboratory concerns studies on immune regulation.
L. David Sibley
L. David Sibley did his undergraduate studies at Oberlin College, Ohio, USA and received his Ph.D. from Louisiana State University. He was a postdoctoral fellow in the laboratory of John Boothroyd at Stanford University and subsequently joined the faculty at Washington University in St. Louis were he currently holds the Alan A and Edith L Wolff Professorship in Molecular Microbiology. His laboratory studies unique cellular adaptations and virulence mechanisms of the parasite Toxoplasma gondii.
References
- 1.Dubey JP. Toxoplasmosis of animals and humans. CRC Press; 2010. [Google Scholar]
- 2.Dubey JP, Frenkel JF. Cyst-induced toxoplasmosis in cats. Journal of Protozoology. 1972;19:155–177. doi: 10.1111/j.1550-7408.1972.tb03431.x. [DOI] [PubMed] [Google Scholar]
- 3.Su C, et al. Recent expansion of Toxoplasma through enhanced oral transmission. Science. 2003;299:414–416. doi: 10.1126/science.1078035. [DOI] [PubMed] [Google Scholar]
- 4.Jones JL, Dubey JP. Foodborne Toxoplasmosis. Clin Infect Dis. 2012 doi: 10.1093/cid/cis508. cis508 [pii] 10.1093/cid/cis508. [DOI] [PubMed] [Google Scholar]
- 5.Moura L, et al. Waterborne toxoplasmosis, Brazil, from field to gene. Emerg Infect Dis. 2006;12:326–329. doi: 10.3201/eid1202.041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joynson DH, Wreghitt TJ. Toxoplasmosis: A comprehensive clinical guide. Cambridge University Press; 2001. [Google Scholar]
- 7.Boothroyd JC, Grigg ME. Population biology of Toxoplasma gondii and its relevance to human infection: do different strains cause different disease? Current Opinion in Microbiology. 2002;5:438–442. doi: 10.1016/s1369-5274(02)00349-1. [DOI] [PubMed] [Google Scholar]
- 8.Jamieson SE, et al. Genetic and epigenetic factors at COL2A1 and ABCA4 influence clinical outcome in congenital toxoplasmosis. PLoS ONE. 2008;3:e2285. doi: 10.1371/journal.pone.0002285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLeod R, et al. Prematurity and severity are associated with Toxoplasma gondii alleles (NCCCTS, 1981–2009) Clin Infect Dis. 2012;54:1595–1605. doi: 10.1093/cid/cis258. cis258 [pii] 10.1093/cid/cis258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki Y, et al. Evidence for genetic regulation of susceptibility to toxoplasmic encephalitis in AIDS patients. Journal of Infectious Diseases. 1996;173:265–268. doi: 10.1093/infdis/173.1.265. [DOI] [PubMed] [Google Scholar]
- 11.Mead PS, et al. Food-related illness and death in the United States. Emerg Infect Dis. 1999;5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buzoni-Gatel D, Schulthess J, Menard LC, Kasper LH. Mucosal defenses against orally acquired protozoan parasites, emphasis on Toxoplasma gondii infections. Cell Microbiol. 2006;8:535–544. doi: 10.1111/j.1462-5822.2006.00692.x. [DOI] [PubMed] [Google Scholar]
- 13.Daher W, Soldati-Favre D. Mechanisms controlling glideosome function in apicomplexans. Curr Opin Microbiol. 2009;12:408–414. doi: 10.1016/j.mib.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 14.Sibley LD. How apicomplexan parasites move in and out of cells. Curr Opin Biotechnol. 2010;21:592–598. doi: 10.1016/j.copbio.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sibley LD. Invasion and intracellular survival by protozoan parasites. Immunological Reviews. 2011;240:72–91. doi: 10.1111/j.1600-065X.2010.00990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tait ED, Hunter CA. Advances in understanding immunity to Toxoplasma gondii. Mem Inst Oswaldo Cruz. 2009;104:201–210. doi: 10.1590/s0074-02762009000200013. S0074-02762009000200013 [pii] [DOI] [PubMed] [Google Scholar]
- 17.Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 18.Ajioka JW, Soldati D. Toxoplasma: molecular and cellular biology. Horizon Biosciences; 2007. [Google Scholar]
- 19.Kim K, Weiss LM. Toxoplasma gondii: the model apicomplexan. International Journal of Parasitology. 2004;34:423–432. doi: 10.1016/j.ijpara.2003.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicolle C, Manceaux LH. Sur une infection a corp de Leishman (ou organismes voisins) du gondi. Comptes Rendus de l Academie des Sciences Serie III, Sciences de la Vie. 1908;147:763–766. [Google Scholar]
- 21.Howe DK, Sibley LD. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J Infect Dis. 1995;172:1561–1566. doi: 10.1093/infdis/172.6.1561. [DOI] [PubMed] [Google Scholar]
- 22.Boyle JP, et al. Just one cross appears capable of dramatically altering the population biology of a eukaryotic pathogen like Toxoplasma gondii. Proc Natl Acad Sci (USA) 2006;103:10514–10519. doi: 10.1073/pnas.0510319103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sibley LD, Ajioka JW. Population structure of Toxoplasma gondii: Clonal expansion driven by infrequent recombination and selective sweeps. Ann Rev Microbiol. 2008;62:329–351. doi: 10.1146/annurev.micro.62.081307.162925. [DOI] [PubMed] [Google Scholar]
- 24.Su CL, et al. Globally diverse Toxoplasma gondii isolates comprise six major clades originating from a small number of distinct ancestral lineages. Proc Natl Acad Sci (USA) 2012;109:5844–5849. doi: 10.1073/pnas.1203190109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munoz M, Liesenfeld O, Heimesaat MM. Immunology ofToxoplasma gondii. Immunol Rev. 2011;240:269–285. doi: 10.1111/j.1600-065X.2010.00992.x. 10.1111/j.1600-065X.2010.00992.x. [DOI] [PubMed] [Google Scholar]
- 26.Sibley LD, Boothroyd JC. Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature (Lond) 1992;359:82–85. doi: 10.1038/359082a0. [DOI] [PubMed] [Google Scholar]
- 27.Radke JR, et al. Defining the cell cycle for the tachyzoite stage of Toxoplasma gondii. Molec Biochem Parasitol. 2001;115:165–175. doi: 10.1016/s0166-6851(01)00284-5. [DOI] [PubMed] [Google Scholar]
- 28.Barragan A, Sibley LD. Transepithelial migration of Toxoplasma gondii is linked to parasite motility and virulence. J Exp Med. 2002;195:1625–1633. doi: 10.1084/jem.20020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liesenfeld O. Oral infection of C57BL/6 mice with Toxoplasma gondii: a new model of inflammatory bowel disease? J Infect Dis. 2002;185:S96–101. doi: 10.1086/338006. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki Y, Joh K. Effect of the strain of Toxoplasma gondii on the development of toxoplasmic encephalitis in mice treated with antibody to interferon-gamma. Parasitology Research. 1994;80:125–130. doi: 10.1007/BF00933779. [DOI] [PubMed] [Google Scholar]
- 31.Khan IA, Matsuura T, Kasper LH. Interleukin-12 enhances murine survival against acute toxoplasmosis. Infect Immun. 1994;62:1639–1642. doi: 10.1128/iai.62.5.1639-1642.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson LL. SCID mouse models of acute and relapsing chronic Toxoplasma gondii infections. Infect Immun. 1992;60:3719–3724. doi: 10.1128/iai.60.9.3719-3724.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gazzinelli RT, Hieny S, Wynn TA, Wolf S, Sher A. Interleukin 12 is required for the T-lymphocyte-independent induction of interferon gamma by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc Natl Acad Sci U S A. 1993;90:6115–6119. doi: 10.1073/pnas.90.13.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunter CA, Subauste CS, Van Cleave VH, Remington JS. Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice: regulation by interleukin-10, interleukin-12, and tumor necrosis factor alpha. Infect Immun. 1994;62:2818–2824. doi: 10.1128/iai.62.7.2818-2824.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denkers EY, et al. Perforin-mediated cytolysis plays a limited role in host resistance to Toxoplasma gondii. J Immunol. 1997;159:1903–1908. [PubMed] [Google Scholar]
- 36.Robben PM, et al. Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J Immunol. 2004;172:3686–3694. doi: 10.4049/jimmunol.172.6.3686. [DOI] [PubMed] [Google Scholar]
- 37.Reis e Sousa C, et al. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J Exp Med. 1997;186:1819–1829. doi: 10.1084/jem.186.11.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mashayekhi M, et al. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity. 2011;35:249–259. doi: 10.1016/j.immuni.2011.08.008. S1074-7613(11)00313-X [pii] 10.1016/j.immuni.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bierly AL, Shufesky WJ, Sukhumavasi W, Morelli AE, Denkers EY. Dendritic cells expressing plasmacytoid marker PDCA-1 are Trojan horses during Toxoplasma gondii infection. J Immunol. 2008;181:8485–8491. doi: 10.4049/jimmunol.181.12.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pepper M, et al. Plasmacytoid dendritic cells are activated by Toxoplasma gondii to present antigen and produce cytokines. J Immunol. 2008;180:6229–6236. doi: 10.4049/jimmunol.180.9.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bliss SK, Butcher BA, Denkers EY. Rapid recruitment of neutrophils containing prestored IL-12 during microbial infection. J Immunol. 2000;165:4515–4521. doi: 10.4049/jimmunol.165.8.4515. [DOI] [PubMed] [Google Scholar]
- 42.Goldszmid RS, et al. NK Cell-Derived Interferon-gamma Orchestrates Cellular Dynamics and the Differentiation of Monocytes into Dendritic Cells at the Site of Infection. Immunity. 2012;36:1047–1059. doi: 10.1016/j.immuni.2012.03.026. S1074-7613(12)00242-7 [pii] 10.1016/j.immuni.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunay IR, et al. Gr1+ inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robben PR, LaRegina M, Kuziel WA, Sibley LD. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J Exp Med. 2005;201:1761–1769. doi: 10.1084/jem.20050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scanga CA, et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
- 46.Debierre-Grockiego F, et al. Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J Immunology. 2007;179:1129–1137. doi: 10.4049/jimmunol.179.2.1129. [DOI] [PubMed] [Google Scholar]
- 47.Yarovinsky F, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 48.Gazzinelli RT, Denkers EY. Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat Rev Immunol. 2006;6:895–906. doi: 10.1038/nri1978. nri1978 [pii] 10.1038/nri1978. [DOI] [PubMed] [Google Scholar]
- 49.Melo MB, et al. UNC93B1 mediates host resistance to infection with Toxoplasma gondii. PLoS Pathog. 2010;6:e1001071. doi: 10.1371/journal.ppat.1001071. 10.1371/journal.ppat.1001071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pifer R, Benson A, Sturge CR, Yarovinsky F. UNC93B1 is essential for TLR11 activation and IL-12-dependent host resistance to Toxoplasma gondii. J Biol Chem. 2011;286:3307–3314. doi: 10.1074/jbc.M110.171025. M110.171025 [pii] 10.1074/jbc.M110.171025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-γ: the major mediator of resistance against Toxoplasma gondii. Science. 1988;240:516–518. doi: 10.1126/science.3128869. [DOI] [PubMed] [Google Scholar]
- 52.Yap GS, Sher A. Effector cells of both nonhemopoietic and hemopoietic origin are required for interferon (IFN)-γ- and tumor necrosis factor (TNF)- α-dependent host resistance to the intracellular pathogen, Toxoplasma gondii. Journal Experimental Medicine. 1999;189:1083–1092. doi: 10.1084/jem.189.7.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pfefferkorn ER. Interferon-γ blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host to degrade tryptophan. Proc Natl Acad Sci USA. 1984;81:908–912. doi: 10.1073/pnas.81.3.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dimier IH, Bout DT. Interferon-γ-activated primary enterocytes inhibit Toxoplasma gondii replication: a role for intracellular iron. Immunology. 1998;94:488–495. doi: 10.1046/j.1365-2567.1998.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murray HW, Rubin BY, Carriero SM, Harris AM, Jaffee EA. Human mononuclear phagocyte antiprotozoal mechanisms: oxygen-dependent vs oxygen-independent activity against intracellular Toxoplasma gondii. Journal Immunology. 1985;134:1982–1988. [PubMed] [Google Scholar]
- 56.Adams LB, Hibbs JB, Taintor RR, Krahenbuhl JL. Microbiostatic effect of murine activated macrophages for Toxoplasma gondii Role for synthesis of inorganic nitrogen oxides from L-Arginine. Journal Immunology. 1990;144:2725–2729. [PubMed] [Google Scholar]
- 57.Scharton-Kersten TM, Yap G, Magram J, Sher A. Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. Journal of Experimental Medicine. 1997;185:1261–1273. doi: 10.1084/jem.185.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taylor GA, Feng CG, Sher A. p47 GTPases: regulators of immunity to intracellular pathogens. Nat Rev Immunol. 2004;4:100–109. doi: 10.1038/nri1270. [DOI] [PubMed] [Google Scholar]
- 59.Taylor GA, Feng CG, Sher A. Control of IFN-gamma-mediated host resistance to intracellular pathogens by immunity-related GTPases (p47 GTPases) Microb Infect. 2007;9:1644–1651. doi: 10.1016/j.micinf.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 60.Howard JC, Hunn JP, Steinfeldt T. The IRG protein-based resistance mechanism in mice and its relation to virulence in Toxoplasma gondii. Curr Opin Microbiol. 2011;14:414–421. doi: 10.1016/j.mib.2011.07.002. S1369-5274(11)00086-5 [pii] 10.1016/j.mib.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 61.Yamamoto M, et al. A cluster of Interferon-gamma-inducible p65 GTPases plays a critical role in host defense againstToxoplasma gondii. Immunity. 2012;37 doi: 10.1016/j.immuni.2012.06.009. org/10.1016/j.immuni.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 62.Shenoy AR, et al. Emerging themes in IFN-gamma-induced macrophage immunity by the p47 and p65 GTPase families. Immunobiol. 2008;212:771–784. doi: 10.1016/j.imbio.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim BH, et al. A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science. 2011;332:717–721. doi: 10.1126/science.1201711. 332/6030/717 [pii] 10.1126/science.1201711. [DOI] [PubMed] [Google Scholar]
- 64.MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol. 2012;12:367–382. doi: 10.1038/nri3210. 10.1038/nri3210. nri3210 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blanchard N, Shastri N. Topological journey of parasite-derived antigens for presentation by MHC class I molecules. Trends Immunol. 2010;31:414–421. doi: 10.1016/j.it.2010.08.004. S1471–4906(10)00120-1 [pii] 10.1016/j.it.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 66.Luder CGK, Lang T, Beuerle B, Gross U. Down-regulation of MHC class II molecules and inability to up-regulate class I molecules in murine macrophages after infection with Toxoplasma gondii. Clinical and Experimental Immunology. 1998;112:308–316. doi: 10.1046/j.1365-2249.1998.00594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tait ED, et al. Virulence of Toxoplasma gondii is associated with distinct dendritic cell responses and reduced numbers of activated CD8+ T cells. J Immunol. 2010;185:1502–1512. doi: 10.4049/jimmunol.0903450. jimmunol.0903450 [pii] 10.4049/jimmunol.0903450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blanchard N, et al. Immunodominant, protective response to the parasite Toxoplasma gondii requires antigen processing in the endoplasmic reticulum. Nat Immunol. 2008;9:937–944. doi: 10.1038/ni.1629. ni.1629 [pii] 10.1038/ni.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frickel EM, et al. Parasite stage-specific recognition of endogenous Toxoplasma gondii-derived CD8+ T cell epitopes. J Infect Dis. 2008;198:1625–1633. doi: 10.1086/593019. 10.1086/593019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lekutis C, Ferguson DJ, Grigg ME, Camps M, Boothroyd JC. Surface antigens of Toxoplasma gondii: variations on a theme. Int J Parasitology. 2001;31:1285–1292. doi: 10.1016/s0020-7519(01)00261-2. [DOI] [PubMed] [Google Scholar]
- 71.Blader I, Manger ID, Boothroyd JC. Microarray analysis reveals previously unknown changes in Toxoplasma gondii infected human cells. J Biol Chem. 2001;276:24223–24231. doi: 10.1074/jbc.M100951200. [DOI] [PubMed] [Google Scholar]
- 72.Luder CG, Walter W, Beuerle B, Maeurer MJ, Gross U. Toxoplasma gondii down-regulates MHC class II gene expression and antigen presentation by murine macrophages via interference with nuclear translocation of STAT1α. Eur J Immunol. 2001;31:1475–1484. doi: 10.1002/1521-4141(200105)31:5<1475::AID-IMMU1475>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 73.Butcher BA, Kim L, Johnson PF, Denkers EY. Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NF-κB. J Immunol. 2001;167:2193–2201. doi: 10.4049/jimmunol.167.4.2193. [DOI] [PubMed] [Google Scholar]
- 74.Shapira S, et al. Initiation and termination of NF-kappaB signaling by the intracellular protozoan parasite Toxoplasma gondii. J Cell Sci. 2005;118:3501–3508. doi: 10.1242/jcs.02428. 118/15/3501 [pii] 10.1242/jcs.02428. [DOI] [PubMed] [Google Scholar]
- 75.Shapira S, Speirs K, Gerstein A, Caamano J, Hunter CA. Suppression of NF-kappaB activation by infection with Toxoplasma gondii. Journal of Infectious Diseases. 2002;185 (Suppl 1):S66–72. doi: 10.1086/338000. [DOI] [PubMed] [Google Scholar]
- 76.Kim SK, Fouts AE, Boothroyd JC. Toxoplasma gondii dysregulates IFN-γ inducible gene expression in human fiboblasts: insights from a genome-wide transcriptional profiling. J Immunol. 2007;178:5154–5165. doi: 10.4049/jimmunol.178.8.5154. [DOI] [PubMed] [Google Scholar]
- 77.Lang C, et al. Impaired chromatin remodelling at STAT1-regulated promoters leads to global unresponsiveness of Toxoplasma gondii-infected macrophages to IFN-gamma. PLoS Pathog. 2012;8:e1002483. doi: 10.1371/journal.ppat.1002483. 10.1371/journal.ppat.1002483. PPATHOGENS-D-11-00905 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stutz A, Kessler H, Kaschel ME, Meissner M, Dalpke AH. Cell invasion and strain dependent induction of suppressor of cytokine signaling-1 byToxoplasma gondii. Immunobiology. 2012;217:28–36. doi: 10.1016/j.imbio.2011.08.008. S0171-2985(11)00188-4 [pii] 10.1016/j.imbio.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zimmermann S, Murray PJ, Heeg K, Dalpke AH. Induction of suppressor of cytokine signaling-1 by Toxoplasma gondii contributes to immune evasion in macrophages by blocking IFN-γ signaling. J Immunol. 2006;176:1840–1847. doi: 10.4049/jimmunol.176.3.1840. [DOI] [PubMed] [Google Scholar]
- 80.Whitmarsh RJ, et al. A critical role for SOCS3 in innate resistance to Toxoplasma gondii. Cell Host Microbe. 2011;10:224–236. doi: 10.1016/j.chom.2011.07.009. 10.1016/j.chom.2011.07.009. S1931-3128(11)00230-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Butcher BA, et al. Cutting edge: IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-alpha in host macrophages. J Immunology. 2005;174:3148–3152. doi: 10.4049/jimmunol.174.6.3148. [DOI] [PubMed] [Google Scholar]
- 82.Andrade RM, Wessendarp M, Gubbels JM, Striepen B, Subaste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006;116:2366–2377. doi: 10.1172/JCI28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reichmann G, et al. The CD40/CD40 ligand interaction is required for resistance in toxoplasmic encephalitis. Infection and Immunity. 2000;68:1312–1318. doi: 10.1128/iai.68.3.1312-1318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sibley LD, Adams LB, Fukutomi Y, Krahenbuhl JL. Tumor necrosis factor-α tiggers antitoxoplasmal activity of IFN-γ primed macrophages. Journal of Immunology. 1991;147:2340–2345. [PubMed] [Google Scholar]
- 85.Denkers EY, Butcher BA, Del Rio L, Kim L. Manipulations of mitogen-activated protein kinase/nuclear factor-kappaB-signalling cascades during intracellular Toxoplasma gondii infection. Immunol Rev. 2004;201:191–205. doi: 10.1111/j.0105-2896.2004.00180.x. [DOI] [PubMed] [Google Scholar]
- 86.Mason NJ, Artis D, Hunter CA. New lessons from old pathogens: what parasitic infections have taught us about the role of nuclear factor-kappaB in the regulation of immunity. Immunol Rev. 2004;201:48–56. doi: 10.1111/j.0105-2896.2004.00189.x. 10.1111/j.0105-2896.2004.00189.x. IMR189 [pii] [DOI] [PubMed] [Google Scholar]
- 87.Pfefferkorn ER, Kasper LH. Toxoplasma gondii: Genetic crosses reveal phenotypic suppression of hydroxyurea resistance by fluorodeoxyuridine resistance. Experimental Parasitology. 1983;55:207–218. doi: 10.1016/0014-4894(83)90015-2. [DOI] [PubMed] [Google Scholar]
- 88.Sibley LD, et al. Forward genetics in Toxoplasma gondii reveals a family of rhoptry kinases that mediates pathogenesis. Eukaryot Cell. 2009;8:1085–1093. doi: 10.1128/EC.00107-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hakansson S, Charron AJ, Sibley LD. Toxoplasma evacuoles: a two-step process of secretion and fusion forms the parasitophorous vacuole. Embo J. 2001;20:3132–3144. doi: 10.1093/emboj/20.12.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Taylor S, et al. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogenToxoplasma gondii. Science. 2006;314:1776–1780. doi: 10.1126/science.1133643. Forward genetic analysis of acute virulence in type I strains of Toxopalsma revealed that ROP18 is primarily reposible for the differences with avirulent type III strains. [DOI] [PubMed] [Google Scholar]
- 91.Saeij JPJ, et al. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science. 2006;314:1780–1783. doi: 10.1126/science.1133690. Forward genetic analysi of differences in mouse infectivity bewteen type II and III strains identified ROP18 along with several other loci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Khan A, Taylor S, Ajioka JW, Rosenthal BM, Sibley LD. Selection at a single locus leads to widespread expansion of Toxoplasma gondii lineages that are virulence in mice. PLoS Genetics. 2009;5:e1000404. doi: 10.1371/journal.pgen.1000404. Analyis of different populations of Toxoplasma in South America revealed that many share acute virulence of type I strians, and express virulent type I alleles of ROP18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saeij JPJ, et al. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 2007;445:324–327. doi: 10.1038/nature05395. Forward genetic anlaysis lead to discovery of ROP16, a polymorphic kinase that affects host cel gene expresison. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reese ML, Zeiner GM, Saeij JP, Boothroyd JC, Boyle JP. Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc Natl Acad Sci U S A. 2011;108:9625–9630. doi: 10.1073/pnas.1015980108. 1015980108 [pii] 10.1073/pnas.1015980108. Forward genetic analysis indicating that ROP5 contributes substaintialy to differences between intermediate type II and aviruelnt type III strians of Toxoplasma in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Behnke MS, et al. Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseuodokinases. Proc Natl Acad Sci (USA) 2011;108:9631–9636. doi: 10.1073/pnas.1015338108. Forward genetic analysis demonstrating that ROP5 modulates most of the difference in acute virulence between highly virulent type I and intermediate type II stains in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB Journal. 1995;9:576–596. [PubMed] [Google Scholar]
- 97.Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16:443–452. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 98.Peixoto L, et al. Integrative genomics approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe. 2010;8:208–218. doi: 10.1016/j.chom.2010.07.004. Bioinformatic analysis that provides a complete summary of the kinome in Toxoplasma and demonstrates the divergent and unique nature of the ROP kinase family. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.El Hajj H, et al. The ROP2 family of Toxoplasma gondii rhoptry proteins: proteomic and genomic characterization and molecular modeling. Proteomics. 2006;6:5773–5784. doi: 10.1002/pmic.200600187. [DOI] [PubMed] [Google Scholar]
- 100.Labesse G, et al. ROP2 from Toxoplasma gondii: a virulence factor with a protein-kinase fold and no enzymatic activity. Structure. 2009;17:139–146. doi: 10.1016/j.str.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 101.Qiu W, et al. Novel structural and regulatory features of rhoptry secretory kinases in Toxoplasma gondii. EMBO J. 2008;28:969–979. doi: 10.1038/emboj.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reese ML, Boothroyd JC. A conserved non-canonical motif in the pseudoactive site of the ROP5 pseudokinase domain mediates its effect on Toxoplasma virulence. J Biol Chem. 2011;286:29366–29375. doi: 10.1074/jbc.M111.253435. M111.253435 [pii] 10.1074/jbc.M111.253435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reese ML, Boothroyd JC. A helical membrane-binding domain targets the Toxoplasma ROP2 family to the parasitophorous vacuole. Traffic. 2009;10:1458–1470. doi: 10.1111/j.1600-0854.2009.00958.x. TRA958 [pii] 10.1111/j.1600-0854.2009.00958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.El Hajj H, et al. ROP18 is a rhoptry kinase controlling the intracellular proliferation of Toxoplasma gondii. PLoS Pathogens. 2007;3:e14. doi: 10.1371/journal.ppat.0030014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fentress SJ, Sibley LD. An arginine-rich domain of ROP18 is necessary for vacuole targeting and virulence inToxopalsma gondii. Cell Microbiol. 2012 doi: 10.1111/cmi.12022. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamamoto M, et al. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med. 2009;206:2747–2760. doi: 10.1084/jem.20091703. jem.20091703 [pii] 10.1084/jem.20091703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ong YC, Reese ML, Boothroyd JC. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J Biol Chem. 2010;285:28731–28740. doi: 10.1074/jbc.M110.112359. M110.112359 [pii] 10.1074/jbc.M110.112359. References 106 and 107 demonstrate that the molecular targets of ROP16 include STAT3 and STAT6, and that these transcription factors are directly phosphorylated on key tyrosine residues, resulting in their prolonged activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Butcher BA, et al. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog. 2011;7:e1002236. doi: 10.1371/journal.ppat.1002236. 10.1371/journal.ppat.100223610 -PLPA-RA-4201. [pii] Demonstrates the complexity signaling affected by ROP16, which leads to inhibition of cytokine responses and can lead to induction of parasite control. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jensen KD, et al. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe. 2011;9:472–483. doi: 10.1016/j.chom.2011.04.015. S1931-3128(11)00167-3 [pii] 10.1016/j.chom.2011.04.015. The combination of alleles at ROP16 and GRA15 were shown to determine the polarization of macrophage phenotypes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Denkers EY, Bzik DJ, Fox BA, Butcher BA. An inside job: hacking into Janus kinase/signal transducer and activator of transcription signaling cascades by the intracellular protozoan Toxoplasma gondii. Infect Immun. 2012;80:476–482. doi: 10.1128/IAI.05974-11. IAI.05974-11 [pii] 10.1128/IAI.05974-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.El Kasmi KC, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol. 2008;9:1399–1406. doi: 10.1038/ni.1671. ni.1671 [pii] 10.1038/ni.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosowski EE, et al. Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. [10.1084/jem.20100717]. J Exp Med. 2011;208:195–212. doi: 10.1084/jem.20100717. jem.20100717 [pii] Forward genetic mapping identified a dense granule protein as primarily responsible for strain dependent differences in activation of NFkB by Toxoplasma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim L, et al. Toxoplasma gondii genotype determines MyD88-dependent signaling in infected macrophages. J Immunol. 2006;177:2584–2591. doi: 10.4049/jimmunol.177.4.2584. [DOI] [PubMed] [Google Scholar]
- 114.Goerdt S, et al. Alternative versus classical activation of macrophages. Pathobiology. 1999;67:222–226. doi: 10.1159/000028096. 28096 [pii] [DOI] [PubMed] [Google Scholar]
- 115.Bekpen C, et al. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol. 2005;6:R92. doi: 10.1186/gb-2005-6-11-r92. gb-2005-6-11-r92 [pii] 10.1186/gb-2005-6-11-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Khaminets A, et al. Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell Microbiol. 2010;12:939–961. doi: 10.1111/j.1462-5822.2010.01443.x. CMI1443 [pii] 10.1111/j.1462-5822.2010.01443.x. Together with reference 119, this work demonstarated the correlation between virulent strains of Toxoplasma and avoidance of IRG recruitment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martens S, et al. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. Plos Pathogens. 2005;1:e24. doi: 10.1371/journal.ppat.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao Z, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao Y, et al. Virulent Toxoplasma gondii evade immunity-related GTPas-mediated parasite vacuole disruption within primed macrophages. J Immunol. 2009;182:3775–3781. doi: 10.4049/jimmunol.0804190. Together with reference 116, this work demonstarated the correlation between virulent strains of Toxoplasma and avoidance of IRG recruitment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fentress SJ, et al. Phosphorylation of immunity-related GTPases by a parasite secretory kinase promotes macrophage survival and virulence. Cell Host Microbe. 2010;16:484–495. doi: 10.1016/j.chom.2010.11.005. Demonstrated that ROP18 phosphorylates IRG proteins in activated macrophages, thus pretecting the parasite against clearance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Martens S, et al. Mechanisms regulating the positioning of mouse p47 resistance GTPases LRG-47 and IIGP1 on cellular membranes: retargeting to plasma membrane induced by phagocytosis. J Immunol. 2004;173:2594–2606. doi: 10.4049/jimmunol.173.4.2594. 173/4/2594 [pii] [DOI] [PubMed] [Google Scholar]
- 122.Steinfeldt T, et al. Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol. 2010;8:e1000576. doi: 10.1371/journal.pbio.1000576. The specific sites phosphorylated by ROP18 in Irga6 were shown to disrupt GTPase activity, and prevent oligomerization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yamamoto M, et al. ATF6-beta is a host cellular target of the Toxoplasma gondii virulence factor ROP18. J Exp Med. 2011;208:1533–1546. doi: 10.1084/jem.20101660. jem.20101660 [pii] 10.1084/jem.20101660. Using a yeast two hybrid screen, these authors found that ROP18 also phosphorylates ATF6β, a transcription factor involvedin the unfolded protein response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fleckenstein MC, et al. A Toxoplasma gondii Pseudokinase Inhibits Host IRG Resistance Proteins. PLoS Biol. 2012;10:e1001358. doi: 10.1371/journal.pbio.1001358. 10.1371/journal.pbio.1001358. PBIOLOGY-D-11-05197 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Niedelman W, et al. The Rhoptry Proteins ROP18 and ROP5 Mediate Toxoplasma gondii Evasion of the Murine, But Not the Human, Interferon-Gamma Response. PLoS Pathog. 2012;8:e1002784. doi: 10.1371/journal.ppat.1002784. 10.1371/journal.ppat.1002784. PPATHOGENS-D-11-02761 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Behnke MS, et al. The polymorphic pseudokinase ROP5 controls virulence in Toxoplasma gondii by regulating the active kinase ROP18. PLoS Path. 2012 doi: 10.1371/journal.ppat.1002992. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zeqiraj E, van Aalten DM. Pseudokinases-remnants of evolution or key allosteric regulators? Curr Opin Struct Biol. 2010;20:772–781. doi: 10.1016/j.sbi.2010.10.001. S0959-440X(10)00150-8 [pii] 10.1016/j.sbi.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gavrilescu LC, Denkers EY. IFN-γ overproduction and high level apoptosis are associated with high but not low virulence Toxoplasma gondii infection. J Immunol. 2001;167:902–909. doi: 10.4049/jimmunol.167.2.902. [DOI] [PubMed] [Google Scholar]
- 129.Mordue DG, Monroy F, La Regina M, Dinarello CA, Sibley LD. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J Immunol. 2001;167:4574–4584. doi: 10.4049/jimmunol.167.8.4574. [DOI] [PubMed] [Google Scholar]
- 130.Frenkel JK. Host, strain and treatment variation as factors in the pathogenesis of toxoplasmosis. American Journal of Tropical Medicine and Hygiene. 1953;2:390–415. doi: 10.4269/ajtmh.1953.2.390. [DOI] [PubMed] [Google Scholar]
- 131.Dubey JP, Frenkel JK. Toxoplasmosis of rats: a review, with considerations of their value as an animal model and their possible role in epidemiology. Veterinary Parasitology. 1998;77:1–32. doi: 10.1016/s0304-4017(97)00227-6. [DOI] [PubMed] [Google Scholar]
- 132.Dubey JP. Toxoplasma gondii infections in Chickens (Gallus domesticus): Prevalence, clinical disease, diagnosis, and public health significance. Zoonoses and Public Health. 2008;57:60–73. doi: 10.1111/j.1863-2378.2009.01274.x. [DOI] [PubMed] [Google Scholar]
- 133.Backert S, Meyer TF. Type IV secretion systems and their effectors in bacterial pathogenesis. Current Opinion in Microbiology. 2006;9:207–217. doi: 10.1016/j.mib.2006.02.008. 10.1016/j.mib.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 134.Cascales E, Christie PJ. The versatile bacterial type IV secretion systems. Nature Reviews Microbiology. 2003;1:137–149. doi: 10.1038/nrmicro753. 10.1038/nrmicro753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Erhardt M, Namba K, Hughes KT. Bacterial nanomachines: the flagellum and type III injectisome. Cold Spring Harb Perspect Biol. 2010;2:a000299. doi: 10.1101/cshperspect.a000299. cshperspect.a000299 [pii] 10.1101/cshperspect.a000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Koshy AA, et al. Toxoplasma co-opts host cells it does not invade. PLoS Path. 2012;8:e1002825. doi: 10.1371/journal.ppat.1002825. Demonstrates that Toxoplasma can inject rhoptry proteins into cells that it does not directly infect, thus potentially altering the behavior of non-infected cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Boothroyd JC, Dubremetz JF. Kiss and spit: the dual roles of Toxoplasma rhoptries. Nat Rev Microbiol. 2008;6:79–88. doi: 10.1038/nrmicro1800. [DOI] [PubMed] [Google Scholar]
- 138.Carruthers VB, Tomley FM. Microneme proteins in apicomplexans. Subcell Biochem. 2008;47:33–45. doi: 10.1007/978-0-387-78267-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bradley PJ, et al. Proteomic analysis of rhoptry organelles reveals many novel constituents for host-parasite interactions in T. gondii. J Biol Chem. 2005;280:34245–34258. doi: 10.1074/jbc.M504158200. [DOI] [PubMed] [Google Scholar]
- 140.Besteiro S, Dubremetz JF, Lebrun M. The moving junction of apicomplexan parasites: a key structure for invasion. Cell Microbiol. 2011;13:797–805. doi: 10.1111/j.1462-5822.2011.01597.x. 10.1111/j.1462-5822.2011.01597.x. [DOI] [PubMed] [Google Scholar]
- 141.Shen B, Sibley LD. The moving junction, a key portal to host cell invasion by apicomplexan parasites. Curr Opin Microbiol. 2012 doi: 10.1016/j.mib.2012.02.007. S1369-5274(12)00023-9 [pii] 10.1016/j.mib.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bradley PJ, Sibley LD. Rhoptries: an arsenal of secreted virulence factors. Curr Opin Microbiol. 2007;10:582–587. doi: 10.1016/j.mib.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sibley LD. Development of forward genetics in Toxoplasma gondii. Intl J Parasitol. 2009;39:915– 924. doi: 10.1016/j.ijpara.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Khan A, et al. Composite genome map and recombination parameters derived from three archetypal lineages ofToxoplasma gondii. Nuc Acids Res. 2005;33:2980–2992. doi: 10.1093/nar/gki604. Genetic linkage maps of Toxoplasma constructed from three, pairwise crosses between types I, II and III strains. This platform enabled the discovery of genes controlling important traits such as virulence and host gene transcription. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Barragan A, Sibley LD. Migration of Toxoplasma gondii across biological barriers. Trends Microbiol. 2003;11:426– 430. doi: 10.1016/s0966-842x(03)00205-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.