

Abstract
Androgen deprivation therapy (ADT) is the standard treatment for patients with prostate-specific antigen progression after treatment for localized prostate cancer. An alternative to continuous ADT is intermittent ADT (IADT), which allows recovery of testosterone during off-cycles to stimulate regrowth and differentiation of the regressed prostate tumor. IADT offers patients a reduction in side effects associated with ADT, improved quality of life, and reduced cost with no difference in overall survival. Our previous studies showed that IADT coupled with 5α-reductase inhibitor (5ARI), which blocks testosterone conversion to DHT could prolong survival of animals bearing androgen-sensitive prostate tumors when off-cycle duration was fixed. To further investigate this clinically relevant observation, we measured the time course of testosterone-induced regrowth of regressed LuCaP35 and LNCaP xenograft tumors in the presence or absence of a 5ARI. 5α-Reductase inhibitors suppressed the initial regrowth of regressed prostate tumors. However, tumors resumed growth and were no longer responsive to 5α-reductase inhibition several days after testosterone replacement. This finding was substantiated by bromodeoxyuridine and Ki67 staining of LuCaP35 tumors, which showed inhibition of prostate tumor cell proliferation by 5ARI on day 2, but not day 14, after testosterone replacement. 5α-Reductase inhibitors also suppressed testosterone-stimulated proliferation of LNCaP cells precultured in androgen-free media, suggesting that blocking testosterone conversion to DHT can inhibit prostate tumor cell proliferation via an intracrine mechanism. These results suggest that short off-cycle coupled with 5α-reductase inhibition could maximize suppression of prostate tumor growth and, thus, improve potential survival benefit achieved in combination with IADT.
Androgens regulate normal prostate development as well as prostate cancer cell proliferation, differentiation, and survival through the androgen receptor (AR) (1–3). Testosterone and DHT are 2 major biologically active androgens, and testosterone can be converted to the more potent ligand DHT by 5α-reductases, types 1 and 2 (4). Testosterone and DHT bind to AR with similar affinity but differ in their dissociation constants, activation potency, and ability to stabilize AR (5–8). Recent studies suggest that testosterone and DHT can differentially regulate androgen-responsive gene expression (9, 10). In testis-intact animals, blocking testosterone to DHT by 5α-reductase inhibitor (5ARI) finasteride slightly reduced the expression of androgen-responsive genes. In contrast in castrated animals, blocking testosterone to DHT enhanced the induction of androgen-responsive genes in regressed prostate by testosterone replacement (11, 12). These observations also indicate that the effect of inhibiting testosterone conversion to DHT in the intact prostate is different from that in the regressed prostate. It is likely that responses of intact prostate tumors to 5α-reductase inhibition are different from that of regressed prostate tumors, which is supported by the finding that finasteride retarded regrowth of regressed LNCaP tumors but not the growth of LNCaP tumors in testis-intact animals (13).
The standard treatment for patients with metastatic prostate cancer is androgen deprivation therapy (ADT), either by surgical or medical castration (14–17). Most patients receiving ADT develop adverse effects, including cognitive and sexual dysfunction, anemia, hot flashes, endocrine abnormalities and metabolic syndrome, cardiovascular disease, and loss of bone mineral density and muscle mass (11, 13, 18, 19). Intermittent ADT (IADT) appears to be a feasible alternative to ADT, allowing significant improvement in the quality of life (QOL) and achieving survival comparable to that observed in patients on continuous ADT (20–25). IADT consists of multiple cycles of androgen suppression, termed on-cycle, where prostate tumors undergo regression, followed by a period of testosterone recovery and tumor regrowth, or off-cycle (18, 26). Rising serum prostate-specific antigen (PSA) level is often used as an indication to restart ADT (27); however, there is currently no clearly defined guideline for timing of resuming ADT in these patients (28).
IADT creates a window of opportunity for further androgen axis manipulation during the androgen level recovery period, or off-cycle. Use of 5ARIs such as dutasteride and/or finasteride during the off-cycle could favor a testosterone-rich, DHT-poor, environment promoting tumor cell differentiation and reducing cell proliferation (29–31). We have previously investigated the impact of finasteride or dutasteride on the efficacy of IADT in animal models. Our initial studies showed that addition of finasteride to the off-cycle increased survival over IADT alone in LNCaP xenografts when the off-cycle interval was fixed (13). Subsequent studies showed that finasteride doubled the off-cycle interval in IADT, when the off-cycle was terminated based on tumor volume (28). However, prolongation of the off-cycle in the presence of finasteride did not translate into a survival benefit (28). Similarly, in a retrospective clinical study, Scholz et al (25) reported that use of finasteride during the off-cycle doubled its duration from a median of 15 months to a median of 31 months with no effect on prostate cancer progression to castration resistance, when off-cycle termination was based on serum PSA. One important and clinically relevant question is whether prostate cancer patient survival can be prolonged when off-cycle duration is fixed in a manner similar to that in the animal model.
Defining the chronology and mechanisms of finasteride or dutasteride inhibition on regrowth of regressed prostate tumors after testosterone replacement may provide new insights into the observations that finasteride inhibition prolonged survival of tumor-bearing animals when IADT off-cycle duration was fixed, but not when off-cycle duration was doubled. This may also lead to new biomarkers for switching from off-cycle to on-cycle, which will be very helpful for patients on IADT. In this study, we determined the time course of regressed LuCaP35 and LNCaP xenograft tumor regrowth in the presence or absence of 5ARI and found that 5ARI profoundly inhibited the initial phase of prostate tumor regrowth after testosterone replacement.
Materials and Methods
Animals
The BALB/c strain of athymic SCID and Hsd:Athymic Nude-Foxn1nu strain of Nude male mice were obtained from Charles River Laboratory (Montreal, Quebec, Canada) and Harlan Labs (Indianapolis, Indiana), respectively, and were kept in accordance with the National Institutes of Health guidelines under standard animal housing conditions for the care and use of experimental animals. Animal experiments were approved by the Institutional Animal Care Use Committee at the University of Pittsburgh.
Xenograft tumor implantation
LuCaP35 xenografts were maintained in athymic SCID mice through serial transplantation as described (32). LNCaP cells were obtained from American Type Culture Collection (Manassas, Virginia). To generate xenografts, LuCaP35 tumor bits were implanted in the flank region of SCID or Nude mice. LNCaP cells were maintained in RPMI, supplemented with 10% fetal bovine serum (FBS), glutamine, penicillin, and streptomycin. LNCaP cells underwent 4 to 8 passages in culture before mouse inoculation. Approximately 106 LNCaP cells suspended in 250 μL media were mixed with 250 μL Matrigel (Becton Dickinson Labware, Bedford, Massachusetts) and then inoculated sc in the flank region of 6- to 8-week-old male athymic SCID/Nude mice using a 25-gauge needle.
Construction of testosterone, dutasteride, and finasteride pellets and letrozole injections
Pellets were prepared as previously described (28). Briefly, approximately 7.5 mg testosterone (Sigma Chemical Co, St Louis, Missouri) was tightly packed into a silicone tube with an inner and outer diameter of 1.58 and 3.18 mm, respectively (Helix Medical, Carpenteria, California). Dutasteride (gift from GlaxoSmithKline Pharmaceuticals, Research Triangle Park, North Carolina) and finasteride (gift from Merck, Whitehouse Station, New Jersey) pellets were made similarly, ∼8 mg dutasteride or finasteride was packed into silicone tubing with an inner and outer diameter of 1.47 and 1.96 mm, respectively. Pellet ends were plugged with wooden sticks and sealed with a silicone adhesive (Dow Corning, Midland, Michigan). After overnight air-drying, pellets were sterilized with 70% ethanol for 10 minutes and stored in a light-free environment. Letrozole (S1235), purchased from Selleck Chemicals (Houston, Texas), was dissolved at 2.5 mg/ml in saline containing 0.3% hydroxypropyl cellulose. Letrozole (10 mg/kg body weight) or vehicle control was delivered to mice via daily sc injection (33).
Treatment protocol and measurement of tumor growth
The experimental design is outlined in Figure 1. Tumors were measured weekly and volume was calculated by the modified ellipsoid formula: length × width2 × 0.52 (34). For the castration group, transscrotal castration was performed under isoflurane anesthesia with proper aseptic and antiseptic technique as previously (28). Mice were initially randomized into 2 groups, testis-intact and castration, when xenograft tumors reached a volume of 200 mm3. Mice were treated using 3 different protocols: a, b, and c (Figure 1). In protocol a, mice were randomized 14 days after castration into 3 groups, consisting of 1) castration only (C), 2) castration plus testosterone replacement (C+T), and 3) castration plus testosterone replacement plus dutasteride (C+T+D). The C group was followed without any intervention. Tumors were collected at days 2, 4, and 14. At least 4 animals per group were generated. In protocol b, mice were again randomized and divided into 3 groups, consisting of C, C+T, and C+T+D, and subjected to 2 cycles of castration and testosterone replacement in the presence or absence of dutasteride. After castration, mice were followed for 10 days before implantation of testosterone and/or dutasteride. Pellets were removed 4 days after the implantation to initiate the second cycle of castration. Testosterone and/or dutasteride (C+T+D) pellets were again implanted 10 days after castration, and mice were followed for 2 days. At least 7 animals per group were generated, and tumors were collected for immunohistochemistry (IHC) 2 days after pellet implantation in the second cycle. In protocol C, mice were castrated and randomized into 5 groups, consisting of 1) C, 2) C+T, 3) C+T+D, 4) castration plus testosterone plus letrozole (C+T+L), and 5) castration plus testosterone plus dutasteride plus letrozole (C+T+D+L). Testosterone and dutasteride were delivered by pellet implantation, whereas letrozole was given via daily sc injection at 10 mg/kg body weight. Tumors for IHC were collected 2 days after the implantation and/or injection.
Figure 1.

Experimental protocols. Tumor-bearing SCID/Nude mice were castrated and subjected to 3 different protocols, a, b, and c. Animals in protocol A were followed for 14 days before randomization into 3 subgroups: implantation of testosterone (C+T), testosterone and dutasteride (C+T+D), and no intervention (C). Tumor volume was measured daily for the initial 7 days of treatment and on alternate days thereafter. Tumors were collected at days 2, 4, and 14 for IHC and qPCR (n = 4 for each group). In protocol B, mice were followed for 10 days before randomization and implantation (n = 7 for each group). Testosterone and dutasteride pellets were removed 4 days after implantation. Ten days after pellet removal, testosterone and/or dutasteride pellets were again implanted. Tumors were then collected for IHC 2 days after the pellet implantation. In protocol C, mice were followed for 10 days after castration and randomized into 5 groups, consisting of C (n = 5), C+T (n = 7), C+T+D (n = 6), C+T+L (n = 8), and C+T+D+L (n = 7). Tumors were collected at day 2 for IHC.
Response to castration was assessed as a statistically significant decrease in tumor volume compared with animals in group C+T within the first 14 days after castration. Tumors that continued to grow within the 14-day period after castration were considered castration-resistant and were excluded from the study. All pellets were implanted sc in the flank contralateral to the tumor-bearing flank. Tumor volume was measured daily. Tumors were harvested at 2, 4, 14, and 26 days of treatment, and portions were formalin-fixed or flash-frozen in liquid nitrogen for further analyses.
Gene expression analysis
Quantitative real-time RT-PCR (qPCR) was used to determine gene expression. Total RNA was extracted using Trizol (Invitrogen, Carlsbad, California). Approximately 2 μg RNA was reverse transcribed with random primers using the high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, California). Exon-exon junction-spanning primers and TaqMan probes were designed using Primer 3 software (Totowa, New Jersey) and synthesized by Integrated DNA Technologies (Coralville, Iowa). Primers used were calreticulin, forward 5′-GGATCGAATCCAAACACAAGTC-3′ and reverse 5′-TGGCTTGTCTGCAAACCTTTAT-3′; ELL-associated factor 2 (EAF2)/U19, forward 5′-CCAGGACTCCCAATCTTGTAAA-3′ and reverse 5′-TAGCTTCTGCCTTCAGTTCTCTT-3′; ELL2, forward 5′-TGACTGCATCCAGCAAACAT-3′ and reverse 5′-TCGTTTGTTGCACACACTGTAA-3′; and PSA, forward 5′-GTCCCGGTTGTCTTCCTCA-3′ and reverse 5′-CACAATCCGAGACAGGATGAG-3′. Ex Taq 2× premix (Takara Bio Inc, Otsu, Shiga, Japan) was used for real-time PCR with 0.25μM forward and reverse primers each. Reactions were run in triplicate on a Bio-Rad IQ5 (Bio-Rad Laboratories, Hercules, California) and repeated on an ABI Step-One Plus (Applied Biosystems). 6-Carboxyl-X-rhodamine was used as passive reference dye. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as endogenous control, primer forward 5′-CATGTTCGTCATGGGTGTGA-3′ and reverse 5′-GGTGCTAAGCAGTTGGTGGT-3′. The specificity of the primer-probe combinations for their cDNA targets was confirmed by lack of amplification of human genomic DNA, mouse genomic DNA, or mouse cDNA. qPCR data were exported into MS Excel, and the expression of transcripts relative to GAPDH was calculated by the ΔCP method: relative expression = 2−ΔCP, where ΔCP is the difference between the crossing-point thresholds of target gene versus GAPDH, as described previously (35).
Immunohistochemistry
Tumor tissue samples were fixed immediately after dissection in 10% formalin overnight, dehydrated in ethanol, cleared in xylene, and embedded in paraffin blocks, as described previously (35). The 6-μm–thick sections were prepared, mounted on positively charged slides (Fisher Biotech, Pittsburgh, Pennsylvania), and air-dried overnight. After dewaxing and rehydration, samples were subjected to a heat-mediated antigen retrieval procedure using a domestic microwave oven at a maximum power of 750 W. Slides were stained for Ki-67 (1:50, sc-15402; Santa Cruz Biotechnology, Santa Cruz, California).
Hematoxylin and eosin staining was performed using the Sakura slide stainer (Sakura Finetek USA, Inc, Torrance, California). Ki-67–positive cell density was determined by analysis of sections from at least 4 different xenografts from each group. Proliferative index was determined from at least 20 fields imaged at ×40 magnification with no overlap, Ki-67–positive cells were counted to determine the average number of proliferating cells for each section. Sections were imaged using a Zeiss Axioplan2 microscope and Axiovision release 4.5 imaging software. Composite images were constructed with Photoshop CS (Adobe Systems, San Jose, California).
Cell proliferation assays
LNCaP cells were seeded at 1000 cells per well in 4-chambered slides. After allowing cells to attach for 24 hours, cells were cultured in phenol red-free RPMI medium containing 10% charcoal-stripped FBS (cFBS) for 2 days. Cells were then replaced with fresh cFBS medium containing 1nM testosterone or DHT with or without 5nM dutasteride or finasteride every 6 hours for 3 days. Bromodeoxyuridine (BrdU) labeling media was added 12 hours before staining. Cells were then washed with PBS, and BrdU incorporation was assayed according to manufacturer's instruction (Invitrogen BrdU staining kit; catalog item 93-3943). All samples were tested in triplicate, and statistical significance was determined by Student's t test using the SigmaStat program (Jandel Scientific, San Rafael, California). Slides were imaged using a Zeiss Axioplan2 microscope and Axiovision release 4.5 imaging software. Extent of BrdU staining was determined according to the presence or absence of nuclear specific staining when compared with the negative controls. BrdU-positive cell density was determined by analysis of 5 independent experiments. Assessment of proliferative index was determined from at least 10 fields imaged at ×40 magnification for each treatment group. Cell counting was performed by 3 different observers in a blinded fashion.
Statistical analysis
GraphPad Prism version 5.0 (GraphPad Software, Inc, La Jolla, California) and MS Excel 2003 (Microsoft, Redmond, Washington) were used for statistical analysis and graphical composition. Data are expressed as the mean ± SEM, and statistical significance was determined by 1-way ANOVA or Student's t test as appropriate. For Ki-67 and BrdU staining, 1-way ANOVA was calculated by Bartlett's test for equal variances using Tukey's multiple-comparison test and further verified by Bonferroni's multiple comparison test. A P value < .05 was considered statistically significant.
Results
LuCaP35 xenograft tumors were sensitive to androgen manipulation
LuCaP35 is an androgen-sensitive and PSA-producing prostate cancer xenograft model (32). LuCaP35 xenografts express the wild-type AR and respond to androgen deprivation similarly to that observed in prostate cancer patients. Thus, LuCaP35 was used in the present study. LuCaP35 xenograft tumors were established as was previously described for LNCaP tumors based on their growth response to castration (28). After castration, LuCaP35 xenograft tumor volume (C) was significantly decreased compared with intact control animals by week 2, verifying the androgen sensitivity of LuCaP35 xenograft tumors (Figure 2A). Testosterone replacement (C+T) stimulated tumor volume increase at a rate similar to that of the intact group. PSA mRNA expression levels in the tumors were also androgen sensitive (Figure 2B). After castration, PSA gene expression in castrated animals was reduced significantly by 3.3-fold compared with intact animals (P < .01); upon testosterone replacement, PSA expression increased 7.5-fold compared with that of castrated animals (P < .01) (Figure 2B).
Figure 2.

Response of LuCaP35 xenograft tumors to androgen manipulation. Panel A, Effect of castration on tumor volume in castrated (C), castrated plus testosterone replacement (C+T) and testes-intact control animals. Values expressed as percentage of original volume (200 mm3). Panel B, Effect of androgen replacement on PSA mRNA expression level relative to GAPDH. Values are presented as mean ± SEM. *, P < .05; **, P < .01. Number of animals in each group is shown in parentheses.
Dutasteride inhibited initial regrowth of regressed LuCaP35 xenograft tumors
We used the LuCaP35 xenograft tumor model to define the time course of testosterone-induced regrowth of regressed prostate tumors in the absence or presence of dutasteride. Castrated SCID/Nude mice bearing LuCaP35 xenografts were treated with testosterone (C+T) or testosterone and dutasteride (C+T+D) for 14 days. Dutasteride inhibited testosterone-induced tumor regrowth of LuCaP35 xenograft tumor during the initial 4 days of treatment (Figure 3A). LuCaP35 xenograft tumor volume at day 2 was decreased by 11% in the C+T+D group in comparison with the time of initial pellet implant, whereas the C+T group tumor volume increased by 20% of the original size at day 2 with a total difference between the tumor volume in both groups of 31% (Figure 3A) (P < .01). LuCaP35 tumor volume in the C+T+D group decreased steadily on days 3 and 4, to a maximum decrease in volume of 49% on day 4 (P < .05). On day 6, tumor volume in C+T+D began to increase at a similar rate as that of C+T-implanted control mice (Figure 3A), suggesting that LuCaP35 tumors were no longer responsive to dutasteride treatment.
Figure 3.

Response of LuCaP35 and LNCaP xenograft tumors to 5ARIs- dutasteride and finasteride. Panel A, Effect of dutasteride on LuCaP35 tumor volume. Tumor volume in castrated animals with testosterone replacement (C + T) (n = 5), and animals treated with testosterone replacement along with dutasteride (C + T + D) (n = 5) at indicated time points. Panel B, Effect of finasteride on LNCaP tumor volume. Tumor volume in castrated animals with testosterone replacement (C + T) (n = 6) and in castrated animals with testosterone replacement along with finasteride (C + T + F) (n = 8) at indicated time points. Tumor volume was determined as the percentage of tumor volume at time of implantation of T, D, and/or F pellets. *, P < .05; **, P < .01.
Finasteride inhibited initial regrowth of regressed LNCaP xenograft tumors
The finding of dutasteride inhibition of initial regrowth of regressed LuCaP35 xenograft tumor encouraged us to examine whether a similar phenomenon would occur using the LNCaP xenograft tumor, another widely used androgen-sensitive prostate tumor model (36–38). Analysis of the tumor volume data showed that combination of testosterone with finasteride inhibited regrowth of regressed LNCaP tumors over the initial 4 days of treatment (Figure 3B). Tumor volume in C+T+F-treated animals was 23% less than that of C+T animals (P < .01). As in the LuCaP35 xenograft tumors treated with dutasteride, LNCaP tumor volume in C+T+F-treated mice began to increase at a similar rate as the C+T-treated animals by day 7, suggesting that 5α-reductase inhibition of tumor regrowth was most effective in the initial 4 days of treatment in LNCaP tumor models.
Dutasteride decreased cellular proliferation during initial regrowth of LuCaP35 and LNCaP xenograft tumors
To evaluate whether dutasteride inhibition of initial regrowth in LuCaP35 xenografts (C+T+D) was mediated by a reduction in cellular proliferation, we studied proliferative markers BrdU and Ki-67 to detect dividing cells in the xenograft tumors. The number of BrdU-positive cells in LuCaP35 tumors treated with dutasteride (C+T+D) was decreased by 12-fold compared with C+T animals on day 2 of treatment (Figure 4, A–C) (P < .0001). The number of BrdU-positive cells in the C+T+D group was not statistically different from the castration group (C), suggesting that dutasteride was capable of repressing initial testosterone-mediated growth. However, at day 14, the number of BrdU-positive cells in the C+T+D group was not statistically different from that of the testosterone-treated group (C+T). These results further suggest that dutasteride inhibited the initial regrowth of regressed xenograft tumors only. Our findings were confirmed by Ki-67 staining of cells, an additional marker of cellular proliferation (Figure 4, D–F). Tumors in testosterone-treated mice (C+T) had an 11-fold increase in Ki-67 index compared with castrated control animals after 2 days of pellet implant. At day 14, the Ki-67 index was 3-fold higher in C+T+D than in C+T (P < .0001). Similar to the BrdU index, the Ki-67 index in C+T+D-treated xenografts at day 14 was not significantly different from that of control animals, again suggesting that inhibition of cell proliferation was effective only during the initial 4 days of treatment. The lack of correlation between BrdU and Ki-67 index at day 14 in C+T tumors compared with C+T+D tumors could be due in part to variability in the rate of diffusion of BrdU after ip injection (39).
Figure 4.

Effects of dutasteride on cellular proliferation in LuCaP35 and LNCaP tumors. Panel A, BrdU labeling in transverse sections of LuCaP35 xenograft tumors from C, C+T, and C+T+D mice 2 and 14 days after testosterone implantation. Panels B and C, Quantification of BrdU label-retaining cells in LuCaP35 tumors at days 2 and 14 after castration. Panel D, Ki-67 immunostaining in transverse sections of LuCaP35 xenograft tumors from C, C+T, and C+T+D mice 2 and 14 days after testosterone implantation. Panels E and F, Quantification of Ki-67–positive cells in LuCaP35 tumors at days 2 and 14 after castration. Data represent the average of 4 mice per group, which is indicated in parentheses. Panel G, Ki-67 immunostaining in transverse sections of LNCaP xenograft tumors from C, C+T, and C+T+D mice collected at day 2 of the second cycle of testosterone/dutasteride implantation. Panel H, Quantification of Ki-67–positive cells in LNCaP tumors at day 2 of the second cycle after testosterone replacement. Number of animals in each group is shown in parentheses. Scale bars, 50 μm. ***, P < .0001.
Similar results were observed when animals bearing LNCaP tumors were subjected to 2 rounds of intermittent therapy followed by tumor growth analysis. The Ki-67 index in the testosterone replacement (C+T) cohort was 4-fold higher when compared with the C+T+D cohort 2 days after androgen replacement in the second cycle (Figure 4, G and H).
Letrozole administration does not affect the antiproliferative effect of dutasteride during initial regrowth of LNCaP xenograft tumors
Testosterone could be converted to estrogens via aromatase. Blockade of testosterone to DHT conversion by dutasteride could elevate the level of estradiol, which could affect prostate tumor growth. To address this possibility, mice bearing LNCaP tumors were treated with testosterone (C+T) or C+T+D in the presence or absence of letrozole (Figure 5, A and B). As expected, the C+T group showed a 7-fold increase in Ki-67 index in comparison with the C group or C+T+D group (P < .0001). No difference in Ki-67 index was found between the C+T and C+T+L group or between group C+T+D and C+T+D+L, suggesting that the antiproliferative effect of 5ARIs during short-term exposure was unlikely to be mediated through testosterone metabolism to estradiol.
Figure 5.
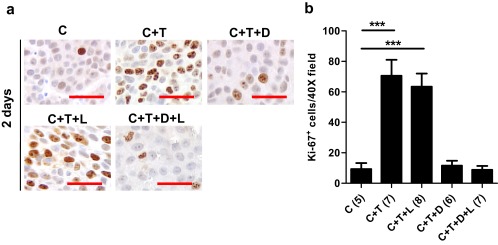
Effect of aromatase inhibitor letrozole on cellular proliferation marker Ki-67 expression in LNCaP tumor xenografts. A, Ki-67 immunostaining in transverse sections of xenograft tumors from C, C+T, C+T+D, C+T+L, and C+T+D+L mice 2 days after testosterone replacement. Panel B, Quantification of Ki-67–positive cells in LNCaP tumors at day 2 post testosterone replacement. Error bars represent SEM. Number of animals in each group is shown in parentheses. ***, P < .0001.
Dutasteride enhanced the expression of androgen-responsive genes EAF2 and calreticulin on day 4 but not day 14 after testosterone replacement in LuCaP35 xenograft tumors
Previously, we reported that dutasteride enhanced the testosterone-induced expression of androgen-responsive genes EAF2 and calreticulin during initial regrowth of castrated LNCaP xenograft tumors on day 3 after testosterone replacement (12). In the current study, the expression of 4 known androgen-responsive genes, EAF2, PSA, ELL2, and calreticulin, was determined by qPCR on days 4 and 14 of testosterone and/or dutasteride replacement in LuCaP35 tumors. Calreticulin gene expression was enhanced 2-fold (P < .05) on day 4 of treatment in in C+T+D-treated LuCaP35 xenografts (Figure 6A). Up-regulation of EAF2 and PSA in the C+T+D group compared with the C+T group was not statistically significant, and ELL2 expression remained unaffected by dutasteride treatment (Figure 6, B–D). This lack of up-regulation could be due to in part to the small number of animals analyzed or to differences in promoters and/or enhancers present in different androgen-responsive genes. In contrast, on day 14, dutasteride treatment resulted in the down-regulation of these androgen-responsive genes (Figure 7). Dutasteride (C+T+D) caused a significant decrease in calreticulin (P < .05) and EAF2 (P < .01) expression compared with testosterone alone (C+T) on day 14 after testosterone replacement (Figure 7, A and B).
Figure 6.

Effect of dutasteride on the expression of various androgen-responsive genes in LuCaP35 tumors 4 days after testosterone implantation. Data shown are qPCR analysis of androgen-responsive genes, calreticulin, EAF2, PSA, and ELL2, in C, C+T, and C+T+D LuCaP35 xenograft tumors. Data were normalized to GAPDH. Number of animals in each group is shown in parentheses. Each dot represents a single sample; the line depicts the mean. *, P < .05.
Figure 7.

Effect of dutasteride on the expression of various androgen-responsive genes in LuCaP35 tumors 14 days after testosterone implantation. Data shown are qPCR analysis of androgen-responsive genes, calreticulin, EAF2, PSA, and ELL2, in C, C+T, and C+T+D LuCaP35 xenograft tumors. Data were normalized to GAPDH. Number of animals in each group is shown in parentheses. Each dot represents a single sample; the line depicts the mean. *, P < .05.
Finasteride and dutasteride inhibited testosterone-induced proliferation of cFBS-conditioned LNCaP cells in culture
Having demonstrated suppression of testosterone-induced regrowth of prostate tumor xenografts by 5ARIs in vivo, we next tested whether 5ARIs could also suppress testosterone-induced proliferation in vitro using cultured LNCaP cells as a model. Cells grown in medium containing 10% cFBS, mimicking the castrated condition, exhibited a BrdU index of 3% (Figure 8). Treatment with testosterone and DHT increased the BrdU index to 65.4% and 93.5%, respectively. Finasteride and dutasteride significantly reduced the testosterone-induced BrdU index to 9.6% and 12.2%, respectively (P < .0001). In contrast, finasteride or dutasteride only slightly inhibited DHT induction of BrdU index (Figure 8, A and B).
Figure 8.

Effects of dutasteride or finasteride on testosterone-stimulated cellular proliferation in cultured LNCaP cells. Panel A, Representative images of BrdU immunostaining in LNCaP cells precultured in charcoal-stripped media for 48 hours and subsequently cultured for 3 days under the following conditions: dimethylsulfoxide vehicle control (C), 5nM finasteride (F), 5nM dutasteride (D), 1nM testosterone (T), 1nM testosterone plus 5nM finasteride (T+F), 1nM testosterone plus 5nM dutasteride (T+D), 1nM DHT, 1nM DHT plus 5nM finasteride (DHT+F), 1nM DHT plus 5nM dutasteride (DHT+D). Panel B, Quantification of BrdU-positive cells. Data are representative of 5 independent experiments. Error bars represent SEM. ***, P < .0001.
Discussion
Understanding the effect of 5α-reductase inhibition on the regrowth of regressed prostate tumors is clinically relevant, specifically to optimize the off-cycle duration in IADT because it involves prostate tumor regression during on-cycles and regrowth of regressed tumor during off-cycles. Finasteride administration during off-cycles in a clinical study doubled the off-cycle interval when serum PSA level was used as the trigger for switching from off-cycle to on-cycle, which improved the QOL for patients. However, finasteride administration during the off-cycles had no significant effect on the progression of prostate cancer to castration resistance (25). Our previous studies demonstrated that blocking testosterone to DHT conversion by 5α-reductase inhibition prolonged the survival of immunodeficient mice bearing androgen-sensitive LNCaP xenograft prostate tumors when the off-cycle duration was fixed (13). However, the survival benefits of 5α-reductase inhibition of IADT were lost when the switch from off-cycle to on-cycle was based on tumor volume that resulted in prolongation of off-cycle interval (40). This suggested that prolongation of the off-cycle compromised potential survival benefits of IADT in the presence of finasteride. Our finding of finasteride or dutasteride inhibition of the initial phase of regrowth of regressed xenograft prostate tumors, but not after a prolonged period of androgen replacement, is consistent with the potential negative effect of longer off-cycles. This suggests that short off-cycle intervals in IADT in the presence of finasteride or dutasteride during the off-cycle may result in significant tumor growth retardation and prolonged survival.
Our studies showed that finasteride or dutasteride significantly inhibited the regrowth of regressed LNCaP or LuCaP35 xenograft prostate tumors within the first 4 days of testosterone replacement. This was also true for the second cycle of the intermittent treatment where dutasteride exhibited an antiproliferative effect during the initial tumor regrowth. This observation suggests that the initial regrowth of regressed prostate tumors remained sensitive to dutasteride after cycles of the intermittent androgen deprivation. In addition, similar results from using 2 different 5ARIs in different models suggested that the efficacy of finasteride or dutasteride was unlikely to be mediated through off-target effects of the inhibitors.
There appears to be a switch during testosterone-induced regrowth from being susceptible to dutasteride or finasteride to being resistant to 5α-reductase inhibition, because tumor regrowth resumed 4 days after testosterone replacement in the presence of dutasteride or finasteride (Figure 3). This finding indicates that regressed prostate tumors behave differently from intact tumors in response to testosterone in the presence of 5ARIs. Androgen deprivation induces cell death as well as dedifferentiation in prostate tumors because surviving androgen-sensitive prostate cancer cells in regressed prostate tumors do not express or express very low levels of androgen-responsive genes, including PSA, which is a marker for the fully differentiated prostatic luminal epithelial phenotype (41). According to our findings, androgen-sensitive prostate cancer cells in regressed prostate tumor could be stimulated by DHT, but not testosterone, to undergo rapid proliferation (Figure 4). However, in the regressed prostate tumor cells, the expression of androgen-responsive genes could be readily induced in regressed tumors by testosterone in the presence of dutasteride, leading to differentiation (Figures 6 and 7). Expression of androgen-responsive genes such as PSA is indicative of differentiation of prostate tumor cells. It is possible that differentiated prostate tumor cells can be stimulated by testosterone to proliferate in the presence of dutasteride. The differentiation of regressed prostate cancer may take 3 to 4 days in LNCaP and LuCaP35 tumor models because the transition of testosterone-induced regrowth from being dutasteride-sensitive to insensitive occurred around 4 days after testosterone replacement (Figure 3). The hypothesis that androgen-conditioned prostate tumor cells can be stimulated by testosterone and/or DHT to undergo proliferation is supported by the observation that LNCaP xenograft tumor growth is insensitive to finasteride in testis-intact animals (28). This hypothesis is also consistent with the observation that dutasteride did not inhibit the proliferation of primary prostate tumors in patients naive to androgen deprivation (42). Further studies are needed to determine whether dutasteride can inhibit testosterone-induced proliferation during the initial regrowth of regressed prostate tumors in patients in clinical trials.
The aromatase inhibitor letrozole did not affect proliferation during testosterone-stimulated initial regrowth of regressed LNCaP xenografts in the presence or absence of dutasteride (Figure 5). This observation suggests that conversion of testosterone to estradiol, when testosterone to DHT is blocked, does not play an important role in modulating cellular proliferation in LNCaP xenografts. One potential mechanism for dutasteride or finasteride to inhibit initial regrowth of regressed prostate tumors but not androgen-conditioned prostate tumors may involve supra-induction of androgen-responsive genes in the initial regrowth phase but not after extended exposure of prostate tumors to androgens. Some androgen-responsive genes are growth-suppressive or tumor-suppressive. EAF2 and calreticulin are examples of tumor suppressors that inhibit cell growth in the prostate (43, 44). Elevated expression of these tumor-suppressive androgen-responsive genes is likely to contribute in part to the growth suppression by finasteride or dutasteride during the initial regrowth phase of regressed prostate tumors.
Finasteride or dutasteride inhibited the initial regrowth of regressed prostate tumors in both LNCaP and LuCaP35 models. Similarly, dutasteride or finasteride enhanced the induction of androgen-responsive genes in testosterone-induced initial regrowth in both models. This suggests that finasteride or dutasteride should have a similar effect on regressed prostate tumor regrowth in other models.
Finasteride and dutasteride were able to dramatically inhibit testosterone-induced proliferation but only slightly inhibited DHT-induced proliferation of LNCaP cells cultured in androgen-free media. This observation suggests that finasteride or dutasteride inhibition of testosterone-stimulated proliferation of cFBS-conditioned LNCaP cells is mediated through blocking testosterone conversion to DHT. Also, this finding suggests that the effect of 5ARI on testosterone-induced LNCaP proliferation does not require other types of cells and is likely mediated through an intracrine mechanism.
The finding of finasteride or dutasteride inhibition of initial regrowth of regressed prostate tumors provides an explanation for our previous observation that finasteride or dutasteride could prolong the survival of animals bearing LNCaP xenograft tumors on IADT when the off-cycle interval was fixed but not when the off-cycle was prolonged. Short intervals of off-cycles in the presence of finasteride or dutasteride will stimulate the expression of prostate-specific genes such as PSA, which is indicative of prostatic differentiation, although minimizing proliferation. In contrast, prolongation of the off-cycle in the presence of finasteride or dutasteride will stimulate both prostatic differentiation and proliferation, with proliferation induction occurring subsequent to differentiation. Although doubling of off-cycle intervals in the presence of finasteride or dutasteride can improve the QOL, it will allow more time for prostate tumors to grow, which could offset the potential benefits associated with tumor growth suppression in the initial phase of regrowth by finasteride or dutasteride in off-cycles.
Based on the current and previous studies, we propose that the efficacy of IADT can be improved by finasteride or dutasteride administration when short fixed off-cycle intervals are used (27). Ideally, the off-cycle should be terminated at the time when finasteride or dutasteride can no longer inhibit the regrowth of regressed tumors. Furthermore, determining the mechanisms by which finasteride or dutasteride inhibit the initial phase of regrowth of regressed prostate cancer will be important and may lead to novel approaches for improving treatment of patients with prostate cancer.
Acknowledgments
We are grateful to Alexandria Kenetake, Aiyuan Zhang, Katie Leschak, Krystal Roskov, Marie Acquafondata, Kelley Knizner, and Marianne Notaro for technical support.
This investigation was supported in part by National Institutes of Health Grants 1 P50 CA90386, R01 CA 120386, 5 R37 DK51193, and T32 DK007774. This project used the University of Pittsburgh Cancer Institute Cancer Center Grant-supported Animal Facility and Tissue and Research Pathology Services (TARPS) and was supported in part by Award P30CA047904. K.M.Z. is a recipient of the Mellam Family Foundation Scholarship. L.E.P. is a Tippins scholar.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ADT
- androgen deprivation therapy
- AR
- androgen receptor
- 5ARI
- 5α-reductase inhibitor
- BrdU
- bromodeoxyuridine
- C
- castration only
- cFBS
- charcoal-stripped FBS
- C+T
- castration plus testosterone replacement
- C+T+D
- castration plus testosterone replacement plus dutasteride
- C+T+D+L
- castration plus testosterone plus dutasteride plus letrozole
- C+T+L
- castration plus testosterone plus letrozole
- EAF2
- ELL-associated factor 2
- FBS
- fetal bovine serum
- IADT
- intermittent ADT
- IHC
- immunohistochemistry
- PSA
- prostate-specific antigen
- QOL
- quality of life
- qPCR
- quantitative real-time RT-PCR.
References
- 1. Coffey DS, Shimazaki J, Williams-Ashman HG. Polymerization of deoxyribonucleotides in relation to androgen-induced prostatic growth. Arch Biochem Biophys. 1968;124:184–198 [DOI] [PubMed] [Google Scholar]
- 2. Huggins Endocrine-induced regression of cancers. Cancer Research. 1967;27:1925–1930 [PubMed] [Google Scholar]
- 3. Yang Q, Fung KM, Day WV, Kropp BP, Lin HK. Androgen receptor signaling is required for androgen-sensitive human prostate cancer cell proliferation and survival. Cancer Cell Int. 2005;5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thomas LN, Douglas RC, Lazier CB, et al. Levels of 5α-reductase type 1 and type 2 are increased in localized high grade compared to low grade prostate cancer. J Urol. 2008;179:147–151 [DOI] [PubMed] [Google Scholar]
- 5. Zhou ZX, Lane MV, Kemppainen JA, French FS, Wilson EM. Specificity of ligand-dependent androgen receptor stabilization: receptor domain interactions influence ligand dissociation and receptor stability. Mol Endocrinol. 1995;9:208–218 [DOI] [PubMed] [Google Scholar]
- 6. George FW. Androgen metabolism in the prostate of the finasteride-treated, adult rat: a possible explanation for the differential action of testosterone and 5α-dihydrotestosterone during development of the male urogenital tract. Endocrinology. 1997;138:871–877 [DOI] [PubMed] [Google Scholar]
- 7. Grino PB, Griffin JE, Wilson JD. Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology. 1990;126:1165–1172 [DOI] [PubMed] [Google Scholar]
- 8. Saartok T, Dahlberg E, Gustafsson JA. Relative binding affinity of anabolic-androgenic steroids: comparison of the binding to the androgen receptors in skeletal muscle and in prostate, as well as to sex hormone-binding globulin. Endocrinology. 1984;114:2100–2106 [DOI] [PubMed] [Google Scholar]
- 9. Biancolella M, Valentini A, Minella D, et al. Effects of dutasteride on the expression of genes related to androgen metabolism and related pathway in human prostate cancer cell lines. Investigational new drugs. 2007;25:491–497 [DOI] [PubMed] [Google Scholar]
- 10. Lin TM, Chang C. Cloning and characterization of TDD5, an androgen target gene that is differentially repressed by testosterone and dihydrotestosterone. Proc Natl Acad Sci U S A. 1997;94:4988–4993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dadras SS, Cai X, Abasolo I, Wang Z. Inhibition of 5α-reductase in rat prostate reveals differential regulation of androgen-response gene expression by testosterone and dihydrotestosterone. Gene Expr. 2001;9:183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gupta S, Wang Y, Ramos-Garcia R, Shevrin D, Nelson JB, Wang Z. Inhibition of 5α-reductase enhances testosterone-induced expression of U19/Eaf2 tumor suppressor during the regrowth of LNCaP xenograft tumor in nude mice. Prostate. 2010;70:1575–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eggener SE, Stern JA, Jain PM, et al. Enhancement of intermittent androgen ablation by “off-cycle” maintenance with finasteride in LNCaP prostate cancer xenograft model. Prostate. 2006;66:495–502 [DOI] [PubMed] [Google Scholar]
- 14. Sato N, Gleave ME, Bruchovsky N, et al. Intermittent androgen suppression delays progression to androgen-independent regulation of prostate-specific antigen gene in the LNCaP prostate tumour model. J Steroid Biochem Mol Biol. 1996;58:139–146 [DOI] [PubMed] [Google Scholar]
- 15. Mohler JL, Armstrong AJ, Bahnson RR, et al. Prostate cancer, version 3.2012: featured updates to the NCCN guidelines. J Natl Compr Canc Netw. 2012;10:1081–1087 [DOI] [PubMed] [Google Scholar]
- 16. Schulman C, Irani J, Aapro M. Improving the management of patients with prostate cancer receiving long-term androgen deprivation therapy. BJU Int. 2012;109(Suppl 6):13–21 [DOI] [PubMed] [Google Scholar]
- 17. Ismail M, Ferroni M, Gomella LG. Androgen suppression strategies for prostate cancer: is there an ideal approach? Curr Urol Rep. 2011;12:188–196 [DOI] [PubMed] [Google Scholar]
- 18. Buchan NC, Goldenberg SL. Intermittent androgen suppression for prostate cancer. Nat Rev Urol. 2010;7:552–560 [DOI] [PubMed] [Google Scholar]
- 19. Spry NA, Kristjanson L, Hooton B, et al. Adverse effects to quality of life arising from treatment can recover with intermittent androgen suppression in men with prostate cancer. Eur J Cancer. 2006;42:1083–1092 [DOI] [PubMed] [Google Scholar]
- 20. Bruchovsky N, Klotz LH, Sadar M, et al. Intermittent androgen suppression for prostate cancer: Canadian Prospective Trial and related observations. Mol Urol. 2000;4:191–199;discussion 201 [PubMed] [Google Scholar]
- 21. Crook JM, O'Callaghan CJ, Duncan G, et al. Intermittent androgen suppression for rising PSA level after radiotherapy. N Engl J Med. 2012;367:895–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crook JM, Szumacher E, Malone S, Huan S, Segal R. Intermittent androgen suppression in the management of prostate cancer. Urology. 1999;53:530–534 [DOI] [PubMed] [Google Scholar]
- 23. Gleave M, Bruchovsky N, Goldenberg SL, Rennie P. Intermittent androgen suppression for prostate cancer: rationale and clinical experience. Eur Urol. 1998;34:37–41 [DOI] [PubMed] [Google Scholar]
- 24. Klotz LH, Herr HW, Morse MJ, Whitmore WF., Jr Intermittent endocrine therapy for advanced prostate cancer. Cancer. 1986;58:2546–2550 [DOI] [PubMed] [Google Scholar]
- 25. Scholz MC, Jennrich RI, Strum SB, Johnson HJ, Guess BW, Lam RY. Intermittent use of testosterone inactivating pharmaceuticals using finasteride prolongs the time off period. J Urol. 2006;175:1673–1678 [DOI] [PubMed] [Google Scholar]
- 26. De La Taille A, Zerbib M, Conquy S, et al. Intermittent androgen suppression in patients with prostate cancer. BJU Int. 2003;91:18–22 [DOI] [PubMed] [Google Scholar]
- 27. Akakura K, Bruchovsky N, Goldenberg SL, Rennie PS, Buckley AR, Sullivan LD. Effects of intermittent androgen suppression on androgen-dependent tumors. Apoptosis and serum prostate-specific antigen. Cancer. 1993;71:2782–2790 [DOI] [PubMed] [Google Scholar]
- 28. Wang Y, Gupta S, Hua V, et al. Prolongation of off-cycle interval by finasteride is not associated with survival improvement in intermittent androgen deprivation therapy in LNCaP tumor model. Prostate. 2010;70:147–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nacusi LP, Tindall DJ. Targeting 5α-reductase for prostate cancer prevention and treatment. Nat Rev Urol. 2011;8:378–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Montorsi F, Alcaraz A, Desgrandchamps F, Hammerer P, Schröder F, Castro R. A broader role for 5ARIs in prostate disease? Existing evidence and emerging benefits. Prostate. 2009;69:895–907 [DOI] [PubMed] [Google Scholar]
- 31. Dörsam J, Altwein J. 5α-Reductase inhibitor treatment of prostatic diseases: background and practical implications. Prostate Cancer Prostatic Dis. 2009;12:130–136 [DOI] [PubMed] [Google Scholar]
- 32. Corey E, Quinn JE, Buhler KR, et al. LuCaP 35: a new model of prostate cancer progression to androgen independence. Prostate. 2003;55:239–246 [DOI] [PubMed] [Google Scholar]
- 33. Yue W, Wang J, Savinov A, Brodie A. Effect of aromatase inhibitors on growth of mammary tumors in a nude mouse model. Cancer Res. 1995;55:3073–3077 [PubMed] [Google Scholar]
- 34. Euhus DM, Hudd C, LaRegina MC, Johnson FE. Tumor measurement in the nude mouse. J Surg Oncol. 1986;31:229–234 [DOI] [PubMed] [Google Scholar]
- 35. O'Malley KJ, Langmann G, Ai J, Ramos-Garcia R, Vessella RL, Wang Z. Hsp90 inhibitor 17-AAG inhibits progression of LuCaP35 xenograft prostate tumors to castration resistance. Prostate. 2012;72:1117–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lim DJ, Liu XL, Sutkowski DM, Braun EJ, Lee C, Kozlowski JM. Growth of an androgen-sensitive human prostate cancer cell line, LNCaP, in nude mice. Prostate. 1993;22:109–118 [DOI] [PubMed] [Google Scholar]
- 37. Stephenson RA, Dinney CP, Gohji K, Ordóñez NG, Killion JJ, Fidler IJ. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst. 1992;84:951–957 [DOI] [PubMed] [Google Scholar]
- 38. Horoszewicz JS, Leong SS, Kawinski E, et al. LNCaP model of human prostatic carcinoma. Cancer Res. 1983;43:1809–1818 [PubMed] [Google Scholar]
- 39. Kee N, Sivalingam S, Boonstra R, Wojtowicz JM. The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods. 2002;115:97–105 [DOI] [PubMed] [Google Scholar]
- 40. Deslypere JP, Young M, Wilson JD, McPhaul MJ. Testosterone and 5α-dihydrotestosterone interact differently with the androgen receptor to enhance transcription of the MMTV-CAT reporter gene. Mol Cell Endocrinol. 1992;88:15–22 [DOI] [PubMed] [Google Scholar]
- 41. Isaacs JT. Prostate stem cells and benign prostatic hyperplasia. Prostate. 2008;68:1025–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gleave M, Qian J, Andreou C, et al. The effects of the dual 5α-reductase inhibitor dutasteride on localized prostate cancer: results from a 4-month pre-radical prostatectomy study. Prostate. 2006;66:1674–1685 [DOI] [PubMed] [Google Scholar]
- 43. Alur M, Nguyen MM, Eggener SE, et al. Suppressive roles of calreticulin in prostate cancer growth and metastasis. Am J Pathol. 2009;175:882–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xiao W, Zhang Q, Jiang F, Pins M, Kozlowski JM, Wang Z. Suppression of prostate tumor growth by U19, a novel testosterone-regulated apoptosis inducer. Cancer Res. 2003;63:4698–4704 [PubMed] [Google Scholar]