

Baldness is a feature associated with mutations in the voltage-gated calcium channel Cav1.2 underlying Timothy syndrome (TS). Yucel et al. find that Cav1.2TS acts as a dominant negative to delay anagen. L-type channel blockers can overcome the TS phenotype. Cav1.2 normally regulates the production of the BMP inhibitor follistatin-like1 (Fstl1), derepressing stem cell quiescence. These findings show how these channels can regulate stem cells.
Keywords: hair follicle stem cells, bulge, calcium channel, VGCC
Abstract
The signals regulating stem cell activation during tissue regeneration remain poorly understood. We investigated the baldness associated with mutations in the voltage-gated calcium channel (VGCC) Cav1.2 underlying Timothy syndrome (TS). While hair follicle stem cells express Cav1.2, they lack detectable voltage-dependent calcium currents. Cav1.2TS acts in a dominant-negative manner to markedly delay anagen, while L-type channel blockers act through Cav1.2 to induce anagen and overcome the TS phenotype. Cav1.2 regulates production of the bulge-derived BMP inhibitor follistatin-like1 (Fstl1), derepressing stem cell quiescence. Our findings show how channels act in nonexcitable tissues to regulate stem cells and may lead to novel therapeutics for tissue regeneration.
Multipotent adult stem cells like those in the hair follicle possess the capacity for programmed organ replacement and carry the promise of induced organ repair in response to injury or damage (Li and Clevers 2010; Bermingham-McDonogh and Reh 2011). In many tissues, multipotent stem cells are found within specific tissue niches of support cells. These niches contain specific extrinsic and intrinsic cues and act to provide regulatory signals that help proliferation or differentiation (Fuchs and Segre 2000). While the relationship between stem cells and niches are known, the controls that regulate the interactions between the cells are an area of active investigation. The murine hair follicle is a great system to study adult stem cells because of its cycle dependency and relatively short switches between growth and destruction phases. Hair follicles have two parts: a permanent part that consists of sebaceous glands and the stem cell-containing or bulge region and the lower or dynamic part that goes through genetically controlled cycles of active growth (anagen), destruction (catagen), and resting phases (telogen) (Hardy 1992).
While excitable cells respond to electric signals mediated by voltage-gated calcium channels (VGCCs) (Halling et al. 2006; Striessnig et al. 2010; Bidaud and Lory 2011), the role of VGCCs in nonexcitable cells such as adult stem cells remains poorly understood. VGCCs employ three well-characterized states in regulating calcium in response to strong membrane depolarization (Catterall 2000): a nonconducting closed state, a conducting open state that allows calcium entry, and a closed inactivated state that follows the open state (Supplemental Fig. S1A). VGCCs enter the inactivated state following prolonged depolarization or elevations of intracellular calcium or by binding to channel antagonists such as the dihydropyridine or phenylalkylamine class of L-type channel inhibitors (Catterall 2000). The glycine-to-arginine mutation at position 406 that causes Timothy syndrome (TS) dramatically reduces L-type channel inactivation (Splawski et al. 2004) and therefore provides an opportunity to investigate the role of channel inactivation in various cellular processes.
Here, we investigate the baldness associated with the dominant L-type VGCC Cav1.2 mutation underlying TS (Online Mendelian Inheritance in Man [OMIM] 601005) (Splawski et al. 2004; Bidaud and Lory 2011). We found that hair follicle bulge stem cells, but not differentiating cells, express Cav1.2 but lack significant voltage-dependent calcium currents, as determined by bulge cell patch clamping and calcium imaging analyses. Expression of Cav1.2TS and loss of the channel in hair follicle bulge stem cells results in markedly delayed anagen, indicating that the TS phenotype acts in a dominant-negative manner. In contrast, treatment with L-type channel blockers that induce channel inactivation cause an early entry to anagen and can reverse the TS phenotype. In additional experiments, we found that Cav1.2 regulates the production of the bulge-derived BMP inhibitor follistatin-like1 (Fstl1), which acts to inhibit stem cell quiescence. We conclude that Cav1.2 provides a calcium-independent signal in hair follicle stem cells that inhibits quiescence and promotes tissue regeneration.
Results and Discussion
Because VGCCs are widely expressed in nonexcitable tissues such as hair and TS individuals display a marked delay in hair growth for the first 2 yr of life (Splawski et al. 2004), we investigated how VGCCs such as Cav1.2 regulate hair growth. In the murine hair, Cav1.2 protein is not ubiquitously expressed throughout the tissue, but rather is highly expressed in the bulge stem cells as compared with interfollicular epithelium (Greco et al. 2009) and colocalizes with the outer bulge marker CD34 (Fig. 1A). As diversified channel properties arise from Cav1.2 alternate splicing (Gray et al. 2007), we compared the bulge Cav1.2 isoform with that found in excitable tissues such as neurons. RT–PCR of CACNA1C (Cav1.2) exons in FACS-sorted CD34+ α6+ bulge stem cells (B) (Nowak and Fuchs 2009) demonstrated a full-length channel isoform that also exists in P1 (postnatal day 1) mouse brains (Fig. 1B).
Figure 1.
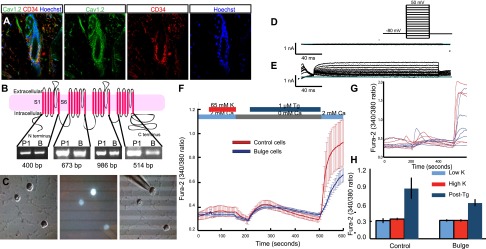
The bulge lacks voltage-gated Ca+2 currents. (A) Immunofluorescent staining of bulge stem cells with Cav1.2 antibody (green) and CD34 antibody (red). (B) Topography of the channel Cav1.2, adapted from Splawski et al. (2004) with permission from Elsevier, © 2004. There are four segments, and each segment has six membrane-spanning domains. Here are shown the RT–PCR results of the main four segments. (P1) Brain cells from P1 mice; (B) bulge stem cells. Exons 4–7 (segment 1) are 400 base pairs (bp), exons 12–15 (segment 2) are 673 bp, exons 19–29 (segment 3) are 986 bp, and exons 30–37 (segment 4) are 514 bp. (C) FACS-sorted K15-EGFP bulge cells were plated on Matrigel-treated coverslips for whole-cell patch clamping experiments. (D) Raw traces of voltage clamp recording of Ba2+ currents from a single-cell bulge cell depolarized in −10-mV steps from a holding potential of −80 mV. Two-hundred-millisecond test depolarizations ranged from −100 to +50 mV; n = 8. (E) Raw traces of voltage clamp recording of K+ currents from a single-cell bulge cell depolarized in −10-mV steps from a holding potential of −80 mV. Two-hundred-millisecond test depolarizations ranged from −100 to +40 mV; n = 8. (F) Ratiometric Ca2+ imaging was carried out using Fura-2. Cells were depolarized with 65 mM KCl to elicit voltage-gated Ca2+ entry and treated with thapsigargin (Tg) in the absence of extracellular Ca2+ to empty ER-Ca2+ stores, followed by readdition of 2 mM Ca2+ to visualize store-operated Ca2+ entry. Averaged Fura-2 responses of bulge cells (blue) and control cells (red); n = 39 for control cells; n = 11 for bulge cells. (G) Representative single-cell curves for both cell types showing cell-to-cell variability. (H) Fura-2 signals at 5 mM K (Low K), after depolarization at 65 mM K (High K), and after store depletion followed by Ca2+- addition (Post-Tg).
To determine whether these full-length isoforms carry calcium into cells, we performed electrophysiology to study FACS-isolated bulge stem cells from K15-EGFP mice (Morris et al. 2004). We used whole-cell patch clamping to measure voltage-gated currents using Ba2+ as the charge carrier in bulge cells 4–6 h after isolation (Fig. 1C). We found no detectable barium currents in response to membrane depolarization (Fig. 1D). Interestingly, most of the current observed appears to consist of a slowly inactivating delayed rectifier, consistent with store-operated channels (SOC) (Parekh and Putney 2005). To ensure that the process of isolation did not disturb the integrity of the ion channel proteins at the membrane, we confirmed the presence of outward, voltage-gated potassium currents (Fig. 1E). These results indicate that bulge Cav1.2 does not generate significant inward currents in bulge stem cells. Consistent with this result, we performed ratiometric calcium imaging in CD34+ α6+ bulge cells. Depolarization of bulge stem cells with 65 mM KCl did not elicit a voltage-dependent calcium elevation, consistent with a lack of functional VGCCs. In contrast, treatment with thapsigargin, a sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) pump inhibitor, to deplete intracellular calcium stores activated SOCs and caused a significant calcium rise (Fig. 1F–H).
Previous studies have shown that the presence of the RGK, small GTPase proteins, GEM, REM, and SOC activators inhibits calcium influx through L-type VGCCs (Balijepalli et al. 2004; Park et al. 2010). We found high expression of STIM1, STIM2, REM2, and GEM in the bulge (Supplemental Fig. S1B), suggesting that these proteins may act to prevent calcium influx through Cav1.2 in the bulge stem cells. Taken together, we conclude that a full-length Cav1.2 is highly enriched in bulge stem cells but does not act as a functional VGCC.
Since Cav1.2 failed to act like a classical VGCC in freshly isolated bulge stem cells, we examined its function in vivo. TS patients are born bald and display a severe delay in the onset of the first postnatal hair growth (Splawski et al. 2004). To model this dominant disease, we conditionally expressed a Cav1.2 channel carrying the TS mutation (Supplemental Fig. S2A) under the control of the Rosa26/lox–stop–lox system (Pasca et al. 2011). Cav1.2TS fx/fx; K15CrePR mice that are treated with RU486 from day 21 to day 28 (Fig. 2A) to express the mutant protein specifically in bulge stem cells demonstrate late entry to anagen by 5–8 d (n = 6) (Fig. 2B, right), reflecting the phenotype observed in TS patients. Skin sections confirmed the telogen hair follicles in Cav1.2TS mice (Fig. 2D) compared with anagen hair follicles in the control mice (Fig. 2C). We characterized the bulge to ensure that Cav1.2TS specifically disrupts stem cell timing and not the bulge structure. To examine whether the bulge stem cells are quiescent, we fluorescently stained Cav1.2TS skin with Nfatc1 (Fig. 2E). Cav1.2TS skin showed nuclear Nfatc1 staining in the bulge stem cells, indicating quiescence (Horsley et al. 2008). Skin from control mice showed cytoplasmic Nfatc1 staining (Fig. 2E). BrdU pulse-chase experiments were performed, and almost no BrdU incorporation was observed in the Cav1.2TS mutant bulge, although BrdU incorporation was observed in the bulge and hair germ of control hair follicles (Fig. 2F). Similarly, Ki67 staining demonstrated that control tissue is in anagen, but Cav1.2TS mutant skin is still in telogen (Fig. 2G; Supplemental Fig. S2F). Together, these results suggest that overexpression of a Cav1.2TS mutant channel in the bulge stem cells of the skin inhibits anagen and extends quiescence without affecting hair follicle or epidermal differentiation (Supplemental Fig. S3).
Figure 2.

Persistent telogen in Cav1.2TS mice and in pore mutants. (A) RU486 treatment strategy and C57BL6 animals' hair cycling times. (A) Anagen; (T) telogen; (C) catagen. (B) Cav1.2TS mice are still in telogen (right), while control mice are already in anagen (left). Pink skin represents telogen, and dark skin indicates anagen. Hematoxylin and eosin staining of control wild-type (WT) skin (C) and Cav1.2TS skin (D). Bar, 100 μm. (E) Immunofluorescence staining of Nfatc1 of control and Cav1.2TS bulge cells. Bar, 20 μm. (F) BrdU staining of control and Cav1.2TS bulge cells. Bar, 20 μm. (G) Ki67 staining of control and Cav1.2TS skin. Bar 100 μm. (H) Cav1.2Δ14–15 mutant mice (right) have extended telogen compared with control mice (left). Hematoxylin and eosin staining of control skin (I) and Cav1.2Δ14–15 mutant skin (J). Bar, 100 μm.
Previous studies of the Cav1.2TS mutant channel suggest that it acts dominantly in excitable cells through a loss of voltage-dependent inactivation and prolonged influx of calcium (Splawski et al. 2004). We reasoned that we might detect voltage-gated calcium currents in Cav1.2TS -expressing bulge stem cells. However, calcium imaging analysis indicated that even in Cav1.2TS mutant bulge stem cells, there is no detectable calcium current in response to polarization with KCl (Supplemental Fig. S1C).
Since the Cav1.2TS mutation enhances the probability of the open state (Supplemental Fig. S1A), we examined the effect of channel loss using the loss-of-function pore mutant Cav1.2Δ14–15. This mutation removes exons 14 and 15 (Supplemental Fig. S2B; Jeon et al. 2010) and represents the closed state (Supplemental Fig. S1A). Cav1.2Δ14–15 fx/−; K15CrePR mice were treated with RU486 from day 21 to day 28 to eliminate functional Cav1.2 in quiescent bulge stem cells (Fig. 2A); we confirmed removal of the pore region of the channel by genotyping FACS-sorted bulge stem cells (Supplemental Fig. S2C). Surprisingly, Cav1.2Δ14–15 mutant mice also entered into anagen 6–8 d later than their control littermates (n = 15) (Fig. 2H), similar to our observations in the Cav1.2TS mice. Hematoxylin and eosin staining of the skin sections showed telogen hair follicles in loss-of-function Cav1.2Δ14–15 mutant mice compared with anagen hair follicles in the control mice (Fig. 2I,J). Nfatc1 staining is nuclear in Cav1.2Δ14–15 mutant bulge cells (Supplemental Fig. S2D), indicating quiescence, and Ki67 staining also demonstrates that the Cav1.2Δ14–15 mutation inhibits proliferation (Supplemental Fig. S2E,G). Based on these results, we conclude that the dominant Cav1.2TS mutation phenocopies the Cav1.2Δ14–15 loss-of-function mutation in prolonging stem cell quiescence and indicates that Cav1.2TS acts paradoxically in a dominant-negative, rather than a constitutively active, manner in regulating hair stem cell activation.
Because the closed and open channel states demonstrated similar phenotypes in delaying anagen (Supplemental Fig. S1A), we hypothesized that inducing the inactivated state of the channel by the addition of the L-type calcium channel blockers would have the opposite effect and stimulate anagen. Indeed, treatments of L-type channel blocker verapamil between day 21 and day 28 caused a precocious entry to anagen 5–7 d prior to vehicle-treated animals (n = 8) (Fig. 3A,B). Verapamil-treated tissue showed anagen hair follicles (Fig. 3D) compared with telogen hair follicles in the control mice (Fig. 3C) in hematoxylin and eosin-stained samples and increased Ki67 staining (Fig. 3E; Supplemental Fig. S4A). Application of other structurally unrelated L-type channel blockers such as nifedipine and diltiazem showed similar results (Supplemental Fig. S4B–D). Additionally, other classes of channel blockers (Supplemental Fig. S4D) and the calcium channel pore blocker cadmium (Supplemental Fig. S8) had no effect on hair cycling, demonstrating the specificity of the response.
Figure 3.

L-type channel blockers act oppositely to pore mutants. (A) Verapamil treatment strategy. (B) Verapamil-treated mice go into anagen earlier (right) than control littermates (left). (VER) verapamil; (VEH) vehicle. Hematoxylin and eosin staining of control (C) and treated (D) skin. Bar, 100 μm. (E) Ki67 staining of control and verapamil-treated skin. Bar, 100 μm. (F) Ki67 staining of control and verapamil-treated Cav1.2Δ14–15 mutant skin. Bar, 100 μm. (G) Verapamil-treated Cav1.2Δ14–15 mutant mice (right) go into anagen later than their control littermates (left). Hematoxylin and eosin staining of control skin (H) and verapamil-treated Cav1.2Δ14–15 mutant skin (I). Bar, 100 μm.
To confirm that Cav1.2 inhibitors are acting on bulge stem cells, we treated Cav1.2Δ14–15 mutant mice that lack the channel in the bulge with verapamil. In contrast to wild-type animals, drug-treated Cav1.2Δ14–15 mutant mice phenocopy the Cav1.2Δ14–15 loss-of-function mutant mice and go into anagen 4 d later (n = 5) (Fig. 3G, right). Hematoxylin and eosin staining of skin sections from treated mice showed anagen hair follicles compared with control skin with telogen hair follicles, and Ki67 staining confirmed this result (Fig. 3F,H,I; Supplemental Fig. S5E). Furthermore, Nfatc1 staining showed cytoplasmic staining in control bulge cells as compared with nuclear staining in drug-treated Cav1.2Δ14–15 mutant bulge stem cells just like in untreated Cav1.2Δ14–15 mutant bulge stem cells (Supplemental Figs. S2D, S4E). These data provide strong support that L-type calcium channel blockers are acting on bulge stem cells to regulate hair cycling.
We next tested whether channel ligands that induce inactivation could overcome the dominant-negative activity of the Cav1.2TS channel. In excitable tissues, channel blockers act dominantly to induce the open channel into the inactivated state (Yazawa et al. 2011). Since Cav1.2TS acts as a dominant negative, we reasoned that channel blockers would relieve inhibition and induce early anagen. Application of verapamil to the bulge cells expressing Cav1.2TS causes mice to enter anagen early (n = 2) (Supplemental Fig. S5A), as confirmed by hematoxylin and eosin staining and Ki67 quantitation (Supplemental Fig. S5B–E). We conclude that channel blockers can revert dominant-negative activity of the Cav1.2TS channel.
Intense investigation over the past decade has elucidated the major signaling regulators controlling the hair cycle. These include anagen-stimulating pathways such as Wnt and TGF-β pathways (Blanpain and Fuchs 2009; Woo and Oro 2011; Oshimori and Fuchs 2012) as well as the BMP and Nfatc1 pathways that are critical for maintaining quiescence (Kobielak et al. 2007; Horsley et al. 2008). Staining for the quiescence enforcer Nfatc1 (Horsley et al. 2008) in treated versus untreated skin demonstrated similar levels of nuclear Nfatc1 (Supplemental Fig. S5F). Nfatc1 is a transcription factor that works downstream from calcium signaling. As verapamil treatment did not change the localization of the nuclear Nfatc1 even though the tissue is anagen, Cav1.2 cannot be upstream of the Nfatc1 pathway, and verapamil's effects are not mediated through repression of nuclear Nfatc1. In contrast, we detected significant alterations in the BMP pathway in verapamil-treated versus untreated skin samples. Nuclear phospho-Smad (pSmad) 1,5,8 staining marks BMP signaling and quiescence in early telogen (Kobielak et al. 2003; Kandyba et al. 2013). Verapamil-treated bulge stem cells express significantly less nuclear pSmad 1,5,8 in early telogen, indicating that drug treatment alters BMP-mediated quiescence (Supplemental Fig. S5G).
To understand the mechanism behind how Cav1.2 blocks BMP-mediated quiescence, we performed microarray analysis on FACS-sorted verapamil- amd control-treated and also Cav1.2Δ14–15 mutant and control bulge stem cells (Fig. 4A; data not shown). We identified factors that are oppositely regulated in mutant and drug-treated bulge cells. Out of this analysis, one factor clearly emerged: Fstl1 (Fig. 4A). Fstl1 is a secreted glycoprotein that inhibits BMP signaling in lung and ureter development (Sylva et al. 2011; Xu et al. 2012). Previous studies have identified other BMP and Wnt inhibitors and activators that affect hair cycling, such as Noggin and Bambi or the TGF-β-regulated Tmeff1; however, their expression is not significantly affected or up-regulated by alterations in Cav1.2 (Fig. 4B). Fstl1 mRNA is up-regulated twofold in verapamil-treated bulge stem cells compared with control bulge stem cells and is down-regulated more than fivefold in Cav1.2Δ14–15 mutant bulge stem cells compared with control bulge cells (Fig. 4C,D). Immunofluorescence staining also indicates that Fstl1 protein levels are significantly elevated in the bulge and secondary germ of verapamil-treated skin and are reduced in Cav1.2Δ14–15 mutant and verapamil-treated Cav1.2Δ14–15 mutant bulge stem cells (Fig. 4E; data not shown), confirming the dependence of Fstl1 expression on Cav1.2 and its ligand. To understand the effect of Fstl1 on hair cycling, we subcutaneously injected two different doses of Fstl1 protein-soaked beads into day 21–22 mice. Fstl1-treated mice went into anagen 4–7 d earlier than the bovine serum albumin (BSA)-injected control animals (n = 8) (Fig. 4F–I), with a dose-dependent increase in the size of the affected area (Fig. 4F). Injection of beads containing follistatin (Fst), a highly related family member expressed in the bulge, failed to induce anagen (Supplemental Fig. S6A–C). FACS analysis of bulge stem cells treated with Fstl1, but not Fst, showed decreased pSmad 1,5,8 staining, confirming that Fstl1 acts to inhibit BMP signaling in the bulge (Supplemental Fig. S7).
Figure 4.
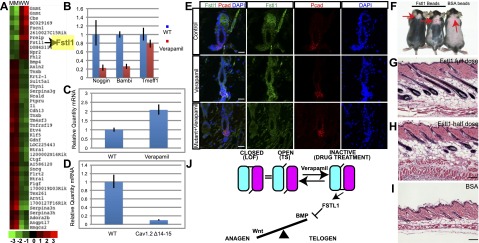
L-type channel blockers regulate anagen by inducing Fstl1. (A) A heat map of microarray results of Cav1.2Δ14–15 mutant and control bulge stem cells. The first two rows are genes from mutant bulge cells (M), and the last two rows are genes from wild-type bulge cells (W). (B) Other BMP inhibitors go down or do not change in verapamil-treated bulge stem cells analyzed by quantitative PCR (qPCR). qPCR results of Fstl1 in control and verapamil-treated bulge cells (C) and control and Cav1.2Δ14–15 mutant bulge cells (D). (E) Immunofluorescence staining of Fstl1 in control, verapamil-treated, and verapamil-treated Cav1.2Δ14–15 mutant bulge. Bar, 20 μm. (F) Injection of Fstl1 protein-soaked beads caused early entry to anagen. The mouse at the left received a full dose (4 μg) of Fstl1 protein, and the mouse in the middle received a half dose (2 μg) of Fstl1. The mouse at the right received 4 μg of BSA. Red arrows point to the injection site. (G–I) Hematoxylin and eosin staining of skin of the mice from F. Bar, 100 μm. (J) Model of Cav1.2 in murine hair cycling. During telogen, Cav1.2 is in the closed state, and BMPs are expressed. This state is represented by the Cav1.2Δ14–15 loss-of-function mutant. The open state is represented by the Cav1.2TS mutant, and this state is the same as the closed state in hair follicle stem cells. Verapamil binding will put Cav1.2 into the inactive state, which in turn will activate Fstl1, which blocks BMPs and lets anagen begin.
Through the study of the Cav1.2TS protein in nonexcitable tissues, our genetic and biochemical studies uncovered the surprising role for VGCCs in hair follicle stem cell regulation. This is the first report that shows that a calcium-independent L-type channel regulates stem cell quiescence. We could not detect significant voltage-gated currents in the bulge and could not rule out the role for smaller, transient currents that fall below our detection methods. An alternative explanation is that Cav1.2 and the calcium channel blockers act like a more traditional ligand–receptor pair. Upon binding to its receptor, verapamil induces allosteric changes in the channel that result in increased Fstl1 expression. Our efforts to understand the connections between Cav1.2 and Fstl1 induction have been thwarted by the inability of bulge stem cells to maintain their properties in long-term culture. We are currently searching for other in vitro systems to study these nonvoltage channel properties of this versatile family of proteins.
Hair follicle stem cell proliferation, like those in other tissues, requires tight regulation of competing activation and quiescence pathways. These pathways are in balance until one side wins over the other. Our study shows that Cav1.2 provides a key signal to break this symmetry and induce tissue regeneration through the novel BMP antagonist Fstl1 (Fig. 4J). Cav1.2 serves as a pacemaker in cardiac tissue, and its expression in many tissues and stem cell compartments suggests that a similar role might be played in nonexcitable tissues. As-yet-unidentified endogenous channel ligands promoting the inactivated state could affect stem cell and regenerative functions independent of calcium-gating activities. These unique ligands would have novel therapeutic applications for treating channelopathies like TS and in regenerative medicine.
Materials and methods
Mice
All mouse studies were approved by and conformed to the policies and regulations of the Institutional Animal Care and Use Committees at Stanford University. Cav1.2TS mice conditionally expressing Cav1.2G406R under the Rosa26 promoter in response to Cre recombinase were generated as previously described (Pasca et al. 2011). These mice were then crossed to K15CrePR (Ito et al. 2005) mice to be able to express the TS channel in the bulge stem cells. Cav1.2 Δ14–15 fx/− mice were a gift from Jean Pierre Kinet (Harvard University) (Jeon et al. 2010).
Immunostaining
Immunofluorescence was carried out overnight on frozen sections fixed in 4% paraformaldehyde and dropped in sucrose. Alternatively, skin samples were embedded in OCT directly and then post-fixed in 4% paraformaldehyde for 2–5 min. Antibodies used were Cav1.2 (1:200; Millipore, #AB5156), Nfatc1 (1:200; Santa Cruz Biotechnology, #sc-7294), CD34 (1:750; BD Pharmingen #553731), pSmad 1,5,8 (1:50; Cell Signaling, #9511), BrdU (1:500; Abcam, #ab6326), Ki67 (1:200; Neomarkers/LabVision, clone SP6), Fstl1 (1:200; Abcam, #71548), and Pcad (1:1000; R&D Systems, #MAB761).
FACS and RNA isolation
FACS isolation (Stanford Shared FACS Facility) of stem cells was performed as previously described (Nowak and Fuchs 2009). Antibodies used were PE rat anti-human CD49f (1:200; BD Biosciences, #555736) and biotin anti-mouse CD34 antibody (1:100; eBioscience, #13-0341-81). Alternatively, K15-EGFP mice from Jaks Laboratories were used for isolation of the bulge stem cells. RNA isolation was carried out using a Mirvana kit from Ambion or RNAeasy Mini Plus kit from Qiagen. cDNA synthesis was carried out by using High-Capacity RNA-to-cDNA Master Mix from Applied Biosystems. pSmad FACS analysis was reported elsewhere (Kandyba et al. 2013)
Real-time PCR
Total RNA or cDNA (see above) were used in a Stratagene Mx3000P using SYBR green quantitative RT–PCR master mix (Agilent) with technical and biological replicates.
Microarray analysis
Illumina MouseRef-8 version 2.0 expression BeadChip was used. Microarray analysis was performed by Stanford Functional Genomics Facility. SAMS analysis was performed for statistical analysis (Tusher et al. 2001). Data were submitted to Gene Expression Omnibus (accession no. 16773750).
Patch clamping
K15EGFP bulge cells that were FACS-isolated were plated on Matrigel-treated (BD Biosciences) coverslips and incubated for 4–6 h at 37°C prior to recording. Coverslips were mounted on the stage of an inverted Nikon fluorescence microscope (Ellipse TE2000) in a standard recording chamber at room temperature. Cells were visualized under a 40×- air lens using GFP fluorescence. Whole-cell barium currents were recorded using EPC 10 amplifier (HEKA) with the following external solution: 5 mM BaCl2, 160 mM TEACl, and 10 mM HEPES (pH adjusted to 7.4 with TEAOH). The pipette solution contained 135 mM CsMeSO3, 5 mM CsCl, 0.5 mM EGTA, 1 mM MgCl2, 4 mM Mg-ATP, 0.5 mM Na-GTP, and 10 mM HEPES (pH adjusted to 7.4 with CsOH). Potassium currents were measured using the following external solution: 129 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 25 mM HEPES, and 30 mM sucrose (pH 7.4). The pipette solution contained 140 mM KCH3SO3, 2 mM MgCl2, 2 mM EGTA, 5 mM KCl, and 20 mM HEPES (pH 7.2). Recording pipettes made of borosilicate glass (Sutter Instruments, BF150-110-10) had a resistance of 4–6 MΩ when filled with intracellular solution. Leak and capacity currents were subtracted. Data were filtered at 2 kHz and digitized at 5 kHz. Data were collected and initially analyzed with Patchmaster software (HEKA).
Acknowledgments
We thank R. Lewis, J. Van Arnam, and the Oro laboratory members for critical input; Masayuki Yazawa for sharing the TS mice before publication; and Eve Kandyba and Krzysztof Kobielak for help with pSmad 1,5,8 FACS analysis. This work was supported by NIH grant AR052785 (to A.E.O.), NIH grant F32-GM090538 (to G.Y.), and the California Institute for Regenerative Medicine.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.216556.113.
References
- Balijepalli RC, Foell JD, Kamp TJ 2004. Blocking the L-type Ca2+ channel with a gem: A paradigm for a more specific Ca2+ channel blocker. Circ Res 95: 337–339 [DOI] [PubMed] [Google Scholar]
- Bermingham-McDonogh O, Reh TA 2011. Regulated reprogramming in the regeneration of sensory receptor cells. Neuron 71: 389–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidaud I, Lory P 2011. Hallmarks of the channelopathies associated with L-type calcium channels: A focus on the Timothy mutations in Cav1.2 channels. Biochimie 93: 2080–2086 [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E 2009. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat Rev Mol Cell Biol 10: 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA 2000. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16: 521–555 [DOI] [PubMed] [Google Scholar]
- Fuchs E, Segre JA 2000. Stem cells: A new lease on life. Cell 100: 143–155 [DOI] [PubMed] [Google Scholar]
- Gray AC, Raingo J, Lipscombe D 2007. Neuronal calcium channels: Splicing for optimal performance. Cell Calcium 42: 409–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco V, Chen T, Rendl M, Schober M, Pasolli HA, Stokes N, Dela Cruz-Racelis J, Fuchs E 2009. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell 4: 155–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halling DB, Aracena-Parks P, Hamilton SL 2006. Regulation of voltage-gated Ca2+ channels by calmodulin. Sci STKE 2006: er1. [DOI] [PubMed] [Google Scholar]
- Hardy MH 1992. The secret life of the hair follicle. Trends Genet 8: 55–61 [DOI] [PubMed] [Google Scholar]
- Horsley V, Aliprantis AO, Polak L, Glimcher LH, Fuchs E 2008. NFATc1 balances quiescence and proliferation of skin stem cells. Cell 132: 299–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Liu Y, Yang Z, Nguyen J, Liang F, Morris RJ, Cotsarelis G 2005. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat Med 11: 1351–1354 [DOI] [PubMed] [Google Scholar]
- Jeon D, Kim S, Chetana M, Jo D, Ruley HE, Lin SY, Rabah D, Kinet JP, Shin HS 2010. Observational fear learning involves affective pain system and Cav1.2 Ca2+ channels in ACC. Nat Neurosci 13: 482–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandyba E, Leung Y, Chen YB, Widelitz R, Chuong CM, Kobielak K 2013. Competitive balance of intrabulge BMP/Wnt signaling reveals a robust gene network ruling stem cell homeostasis and cyclic activation. Proc Natl Acad Sci 110: 1351–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobielak K, Pasolli HA, Alonso L, Polak L, Fuchs E 2003. Defining BMP functions in the hair follicle by conditional ablation of BMP receptor IA. J Cell Biol 163: 609–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobielak K, Stokes N, de la Cruz J, Polak L, Fuchs E 2007. Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proc Natl Acad Sci 104: 10063–10068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Clevers H 2010. Coexistence of quiescent and active adult stem cells in mammals. Science 327: 542–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RJ, Liu Y, Marles L, Yang Z, Trempus C, Li S, Lin JS, Sawicki JA, Cotsarelis G 2004. Capturing and profiling adult hair follicle stem cells. Nat Biotechnol 22: 411–417 [DOI] [PubMed] [Google Scholar]
- Nowak JA, Fuchs E 2009. Isolation and culture of epithelial stem cells. Methods Mol Biol 482: 215–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshimori N, Fuchs E 2012. Paracrine TGF-β signaling counterbalances BMP-mediated repression in hair follicle stem cell activation. Cell Stem Cell 10: 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Putney JW Jr 2005. Store-operated calcium channels. Physiol Rev 85: 757–810 [DOI] [PubMed] [Google Scholar]
- Park CY, Shcheglovitov A, Dolmetsch R 2010. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science 330: 101–105 [DOI] [PubMed] [Google Scholar]
- Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, et al. 2011. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med 17: 1657–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, et al. 2004. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119: 19–31 [DOI] [PubMed] [Google Scholar]
- Striessnig J, Bolz HJ, Koschak A 2010. Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflugers Arch 460: 361–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylva M, Li VS, Buffing AA, van Es JH, van den Born M, van der Velden S, Gunst Q, Koolstra JH, Moorman AF, Clevers H, et al. 2011. The BMP antagonist follistatin-like 1 is required for skeletal and lung organogenesis. PLoS ONE 6: e22616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci 98: 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo WM, Oro AE 2011. SnapShot: Hair follicle stem cells. Cell 146: 334–334.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Qi X, Gong J, Yu M, Zhang F, Sha H, Gao X 2012. Fstl1 antagonizes BMP signaling and regulates ureter development. PLoS ONE 7: e32554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, Dolmetsch RE 2011. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471: 230–234 [DOI] [PMC free article] [PubMed] [Google Scholar]