

Abstract
Genetically modified Salmonella typhimurium VNP20009 (VNP) is a useful vehicle for cancer therapy and vaccine development but exhibits limited tumor targeting in vivo. We engineered a novel VNP derivative that expressed carcinoembryonic antigen (CEA)-specific single chain antibody fragments (scFv) on the cell surface to increase tumor-specific targeting. There was significant scFv cell surface display visualized by flow cytometry and confocal microscopy when cells were probed with fluorescently labeled CEA. Atomic force microscopy (AFM) measurements on whole bacteria confirmed binding of unlabelled CEA to the displayed scFv. The modified VNP strain exhibited increased localization in the upper gastrointestinal tract of CEA transgenic mice and accumulated in CEA-expressing tumors. Furthermore, treatment with a single dose of the VNP derivative inhibited growth of MC38CEA tumors and was associated with local accumulation of CD3+ T cells and CD11b+ macrophages. The display of antibody fragments on the surface of VNP represents a novel strategy for both targeting CEA-expressing tumors and increasing the immunogenicity of Salmonella-based vaccines for cancer.
Keywords: Salmonella, scFv display, cancer therapy, vaccine vector, carcinoembryonic antigen
1. Introduction
Salmonella enterica serovar typhimurium is a broad range host bacterium, which has been used as a vehicle to express antigens from other pathogens, and to deliver proteins to solid tumors [1, 2]. Attenuated Salmonella spp. were shown to be able to multiply preferentially in tumors and inhibit their growth but the mechanism behind this phenomenon remains unclear [1, 3]. VNP20009 (VNP) is a genetically modified strain of S. typhimurium YS72 with attenuated virulence due to deletion of the purI and msbB genes [3]. An excellent safety profile in rodents and dogs with preferential intratumoral replication of VNP in mice prompted Phase I clinical trials, which documented the safety of the bacterial regimen in cancer patients [4]. However, intratumoral localization of bacteria was limited to 2 out of 24 patients with metastatic melanoma and clinical benefits were not observed [4]. These results were in clear contrast to murine transplantable tumors that showed a 1000 to 10,000 fold enrichment of live bacteria in tumor tissue over other internal organs and correlated with a significant delay of tumor growth [5].
To date, the specific mechanisms of the VNP-induced anti-tumor effects are not clearly established. There is evidence supporting therapeutic activity related to metabolic disturbances accompanying infection of selected cells within the tumor microenvironment as well as mobilization of innate and acquired immunity against the bacteria leading to cross-presentation of tumor antigens [6]. In order to promote VNP tumor targeting, we engineered the inducible expression of high-affinity carcinoembryonic antigen (CEA)-specific single chain antibody fragments (scFv) on the surface of the bacteria. CEA is abundantly expressed in a large number of human carcinomas including gastrointestinal tract, pancreatic, non-small cell lung and breast cancers, thus constituting a convenient therapeutic target [7].
Escherichia coli OmpA protein is a major outer membrane protein, which is highly conserved among the Enterobacteriaceae and can serve as a carrier for the expression of foreign antigens on the surface of Gram-negative bacteria including Salmonella spp. [8, 9]. A method that takes advantage of efficient targeting of OmpA to the outer membrane and allows C-terminal fusion of passenger proteins to be displayed is the Lpp-OmpA expression system [10]. Apart from serving as a convenient carrier, OmpA is a prominent member of the pathogen-associated molecular pattern (PAMP) family and is able to directly stimulate macrophages, dendritic cells and NK cells through TLR-2 signaling [11]. This feature makes OmpA an interesting adjuvant, which might significantly improve the immunostimulatory properties of the bacterial vehicle and passenger proteins.
Since surface display of scFv has been largely limited to non-mutator E. coli strains, we tested the feasibility of this approach for the display of functional scFv on the surface of S. typhimurium VNP carrying several attenuating mutations but having intact recombination systems. Here, we report the technical details of strain construction and describe an experimental model enabling the study of several crucial features of VNP expressing surface scFv for cancer gene therapy or vaccination. Our data demonstrate that anti-CEA scFv can be successfully displayed on the surface of VNP using an inducible expression system. We confirmed functionality of the scFv displayed on the cell surface and examined the pattern of protein expression within bacteria. Inducible scFv expression on the cell surface resulted in accumulation of bacteria in the upper gastrointestinal tract of CEA transgenic mice and preferentially localized to CEA-expressing tumors. Moreover, immunization with the modified VNP led to substantial inhibition of tumor growth and more than doubled survival time in an MC38CEA tumor transplantation model. Inhibition of tumor growth correlated with VNP-induced mobilization of CD3+ T cells and macrophages at the tumor site. The data reported here represent a novel approach for the introduction of antibody fragments on the cell surface of Salmonella strains for vaccination and delivery of therapeutic genes.
2. Materials and Methods
2.1 Mice
Female C57Bl/6 mice were purchased from Charles River Laboratory (Wilmington, MA) and used at 8–12 weeks of age. Human CEA transgenic mice (H-2Kb) were obtained from Wolfgang Zimmerman (University of Freiberg, Freiberg, Germany) and were crossed with Apc1638 knockout mice as previously described [12]. All animals were housed in pathogen-free conditions with ample access to food and water at Columbia University according to approved institutional protocols.
2.2 Cell lines
Murine adenocarcinoma (MC38) was a gift from Dr. Nicholas Restifo (National Cancer Institute, Bethesda, MD). The CEA-expressing MC38 cells were produced by transducing MC38 with the full length human CEA cDNA using retroviral expression vector pLXSN (Clontech, Palo Alto, CA). All cell lines were grown in DMEM containing 10% FCS, 10 mM L-glutamine without antibiotics.
2.3 Bacterial strains and plasmids
TOP10F’ and DH5α E. coli strains were purchased from Invitrogen (Carlsbad, CA). S. typhimurium VNP20009 (VNP) was a gift from Dr. Mario Sznol (Vion Pharmaceuticals, New Haven, CT). Plasmid pUC18-T84.66scFv was kindly provided by Dr. Anna Wu (Beckman Research Institute of the City of Hope, Duarte, CA) and was used to derive the anti-CEA scFv sequence with an 18 amino acid flexible linker (GS18) between the variable light and heavy domains [13, 14].
2.4 Plasmid construction
To remove the leader sequence of the scFv, a 564bp segment of the scFv was PCR amplified using the primers: 5′-CTAGTCTAGACTAGACATTGTACTGACCCAATC-3′ and 5′-TTTACTATTACCATTCGCAG-3′ cloned into XbaI/StuI digested. pGEM-T84.66scFv. The 853bp scFv gene was then were removed from plasmids by digestion with XbaI/HindIII and blunted with DNA polymerase I Klenow fragment (Promega, Madison, WI).
pMoPac2 (Figure 1A) containing lpp-ompA gene fusion under the control of Plac promoter was based on pMoPac1 [15]. The lpp-ompA fragment from pTX101 [10] was PCR amplified using AHX84 (5′-TATATACATATGAAAGCTACTAAACTGGTA-3′) and AHX85 (5′-GGCCATGGCCGGCTGGGCCGCGTTGTCCGGACGAGTGCC-3′) digested with NdeI/SfiI and ligated to NdeI/SfiI digested pMoPac1 plus a SfiI digested tet cassette. To obtain the lpp-ompA-scFv(GS18) fusion (Figure 1B), the tet cassette was removed from pMoPac2 by digestion with SfiI, the vector blunted with T4 DNA polymerase (Promega, Madison, WI) and ligated to the scFv gene. All constructs were sequenced through the Lpp-OmpA and scFv regions.
Figure 1. Schematic diagram of pMoPac2 plasmid and anti-CEA scFv display derivative.
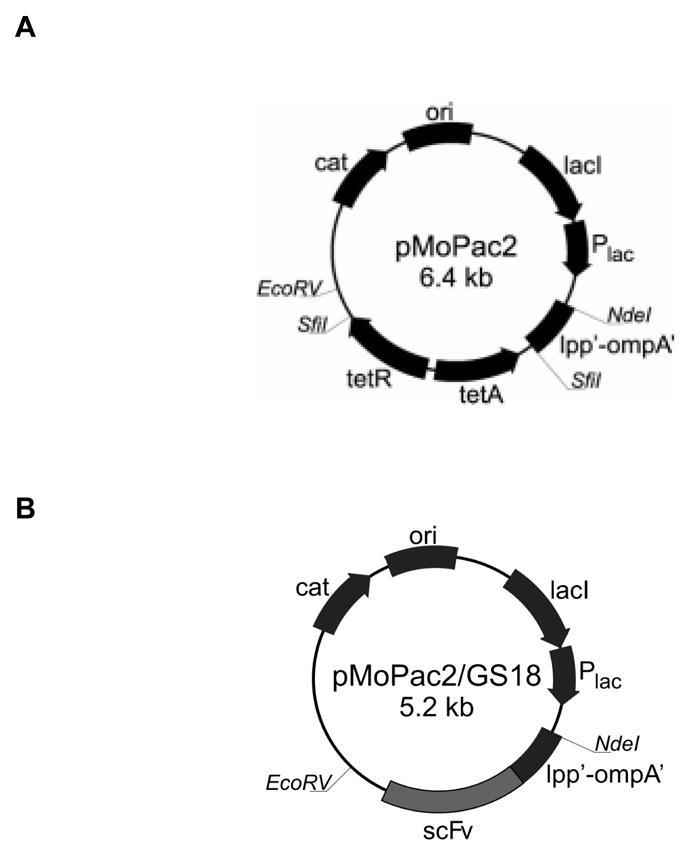
(A) pMoPac2 was used as a backbone for generating plasmids expressing scFv. (B) pMoPac2GS18 was generated by in frame cloning of a the anti-CEA scFv. cat, chloramphenicol acetyltransferase; ori, origin of replication; lacI, lac repressor, Plac, lac promoter; tetA,tetB, tetracycline resistance genes; lpp’-ompA’, fusion of major lipoprotein signal sequence fragment and portion of outer membrane protein A; scFv, single chain antibody.
2.5 Transformation of bacteria and expression of anti-CEA antibody fragments
VNP were routinely propagated in Msb medium as described elsewhere [16]. Competent E. coli were transformed with the appropriate plasmids using the calcium chloride method [17] and Salmonella were transformed through electroporation under standard conditions (2.5kV, 25μF, 200Ω) For expression, overnight cultures were subcultured 1:100 into TB and grown at 30°C to early log phase (OD600 = 0.2) at which point isopropyl-β-D-thiogalactoside (IPTG) (Sigma) was added at the specified concentrations and the bacteria were transferred to a shaking incubator set at 25°C. After 6 – 12 h bacteria were analyzed for the expression of CEA-specific antibody fragments and used in biological assays. The number of viable bacteria (CFU) was determined by plating serial dilutions of the culture onto TB agar plates with Cm.
Bacterial lysates were obtained from 12 hour cultures of VNP20009-MoPac2GS18 and proteins resolved on a 15% Laemmli SDS–PAGE gel and transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). The membranes were washed and incubated with rabbit anti-mouse polyclonal IgG (Zymed) at a dilution of 1:1000 followed by HRP-conjugated goat anti-rabbit IgG (Amersham) at a dilution of 1:10,000 and blots were developed with enhanced chemiluminescence detection reagents (PerkinElmer Life Sciences, Boston, MA) and X-OMAT-5 film (Eastman Kodak, Rochester, NY).
2.6 Bacterial staining and FACS analysis
FITC-F(ab′)2-goat anti-mouse IgG, FITC-F(ab′)2-goat anti-rabbit IgG were from Zymed (San Francisco, CA). CEA-FITC was prepared by labeling CEA (Fitzgerald, Concord, MA) with FluoroTag FITC Conjugation Kit (Sigma) or Alexa-Fluor 488 Protein Labeling Kit (Molecular Probes, Eugene, OR) according to manufacturer’s directions. Ovalbumin-FITC and CMTMR were from Molecular Probes. Rabbit polyclonal anti-Salmonella (Ab13634) were from Abcam Inc. (Cambridge, MA). 7-AAD (7-amino-actinomycin D) was from Beckman Coulter (Marseille, France). The staining was performed at 4°C on 96-well V-bottom plates using PBS with 0.2% w/v BSA for dilution of reagents and washing. Briefly, 5 μL aliquots of bacterial cultures were combined with 150 μL of PBS. Plates were centrifuged at 500 × g for 6 minutes and bacterial pellets were resuspended with 50μL of dyes at 40μg/mL FITC-F(ab′)2-goat anti-mouse IgG), 1mg/mL CEA-FITC, 0.001% w/v 7-AAD and incubated for 20 min. Acquisition and analysis were performed with a BD Biosciences FACSCalibur (San Jose, CA) equipped with CellQuest software.
2.7 FACS analysis of leukocytes isolated from peripheral blood, spleen and tumor tissue
PBL and splenocytes were obtained from naïve or tumor bearing mice according to standard procedures. Tumor infiltrates were prepared by digesting of tumor tissue with the mixture of collagnenase I and IV (Sigma) at 1mg/mL, and hyaluronidase V at 0.2mg/mL in DPBS supplemented with glucose (1 mg/mL). Fcγ–receptors were blocked with FcγIII/IIR monoclonal antibodies (2.4G2) and cells were stained with FITC-anti-CD4, PE-anti-CD8, APC-anti-CD3, PE-anti-CD11b (all from BD PharMingen, San Diego, CA) and analyzed on a FACSCalibur Flow Cytometer using Cell-Quest software.
2.8 Confocal microscopy
One mL aliquots of overnight bacterial cultures in TB were diluted with 5 mL ice cold PBS containing protease inhibitors (Roche, Mannheim, Germany) and DNase (Qiagen) and spun down at 1600 x g at 4°C. Bacterial pellets were resuspended in 0.5mL of ice cold PBS and 25 – 50 μL samples were used for staining with 40μg/mL FITC-F(ab′)2-goat anti-mouse IgG (20 min on ice) or 1mg/mL CEA-Alexa-Fluor (20 min on ice). After staining bacteria were washed twice with 5 mL of ice cold PBS and after the final spin pellets were resuspended in 10 μL 4% paraformaldehyde (PAF) in PBS. 1 μL of bacterial suspension was mixed with 4 μL of deionized water on a glass coverslip and air dried for 20 min. Coverslips were washed 5 times in a large volume of deionized water, dried, and examined with laser scanning microscope Carl Zeiss LSM 510 Meta using a 100x/1.3 oil immersion objective. Images were digitally collected and processed with Adobe Photoshop software.
2.9 Atomic force microscopy (AFM)
1 × 1 cm plastic slides were cut out from the bottom of cell culture dishes (Costar) and coated with rabbit anti-Salmonella polyclonal antibodies. After 12 h incubation at 4°C slides were washed thoroughly with PBS and bacteria collected from culture at mid-log phase were allowed to adhere for 45 minutes. Weakly bound bacteria were rinsed out with DPBS and strongly adherent bacteria were analyzed with AFM as described elsewhere [18, 19]. Briefly, standard V-shaped silicon nitride (Si3N4) cantilevers with nominal spring constant of 0.01 N/m were obtained from Microlevers, Veeco (Dourdan, France). Each new tip was cleaned by immersing in acetone for 5 min, then washed in deionized water and irradiated with UV light for 20 min. Next, tips were soaked in 10% 3-aminopropyltriethoxysilane (APTES) (Sigma) water solution for 2 h, washed with deionized water, and coated with PEG water solution at 1 mg/ml followed by coating with CEA or control proteins at 50 μg/ml in DPBS for 12h. The loosely attached proteins were then removed by extensive washing with DPBS. The protein-covered tips were used immediately for making measurements. All measurements were performed in DPBS (pH 7.4) by using commercial liquid cell (Veeco) and a thermomicroscope CP (Veeco) equipped with multimode head and 100μm scanner. All measurements were performed in the AFM Laboratory at the Department of Experimental Physics, Jagiellonian University.
2.10 In vivo adhesion assay
4 – 6 months old Apc/CEA mice and their wild type littermates were fasted overnight and lightly anesthetized before being fed by oral gavage with 1 × 108 bacteria in 0.3 mL of PBS. After 1 hour, two mice per group were sacrificed, the gastrointestinal tract was isolated and luminal surfaces were washed with 5 mL of ice cold PBS. A series of 1 cm lengths of duodenum and jejunum at about 50 mg each were cut into small pieces and suspended in 0.5 mL of PBS. After sheering through 18G needles serial dilutions of the tissue suspension were plated onto TB agar with chloramphenicol at 30 μg/mL and bacterial colonies were counted after overnight culture at 30°C. All numbers were normalized to 1 g of tissue.
2.11 Tissue preparations and immunocytochemistry
Tumors excised from mice were snap frozen and cut into 10 μm-thick cryosections. Indirect double and triple immunofluorescence staining was applied to detect CD3/CD68/SMA and CEA/CD11b. Combinations of secondary antisera and streptavidin conjugated to Cy3, Cy2, AMCA were used to visualize red, green and blue fluorescence, respectively. Sections were examined with Olympus BX50 epifluorescence microscope and images were recorded with Olympus Camedia 5050 digital camera and processed with AnalySIS FIVE software.
2.12 Tumor studies
VNP, VNP/GS18 (VNP transformed with MoPac2-lpp-ompA-scFv CEA-specific) or VNP/GS18IND (VNP with IPTG-induced expression of Lpp-OmpA-scFv) at 2 × 106 cells (calculated by OD600nm) per dose were injected i.v. in 100 μL of PBS in the tail vain of C57Bl/6 mice (5 mice per group) bearing 7 days old MC38CEA tumor. Tumor sizes were evaluated by caliper every two days and the tumor area was determined by multiplying measurements of two perpendicular diameters. All experiments were repeated at least twice.
2.13 Statistical analysis
Statistical analysis was performed using the Student’s t-test with P<0.05 considered significant.
3. Results
3.1 Construction and expression of Lpp-OmpA-anti-CEA scFv fusion protein
The CEA-specific murine scFv (GS18) coding sequence was derived from pUC18-T84.66scFv [13] and cloned into pMoPac2 to create the cell surface display vector pMoPac2GS18 as described in Materials and Methods (Figure 1). The plasmid was electroporated into VNP S. typhimurium to create VNP/GS18. The inducible expression of the Lpp-OmpA-scFv fusion protein was evaluated by Western blotting probed with anti-mouse IgG polyclonal serum. Although a basal level of expression was detected as bands at the expected molecular weight (42kDa), consisting of the 15 kDa Lpp-OmpA and the 27 kDa scFv, there was a dose-dependent increase in expression following treatment with IPTG (Figure 2A). When examined by flow cytometry there was also an IPTG-induced dose-dependent increase in CEA-scFv expression as probed with FITC-F(ab′)2-goat anti-mouse (Figure 2B). There was more than a 100-fold overexpression of scFv on the surface of VNP/GS18 following IPTG induction (VNP/GS18IND).
Figure 2. Expression of anti-CEA scFv in VNP20009 transformed with pMoPac2GS18 (VNP/GS18).
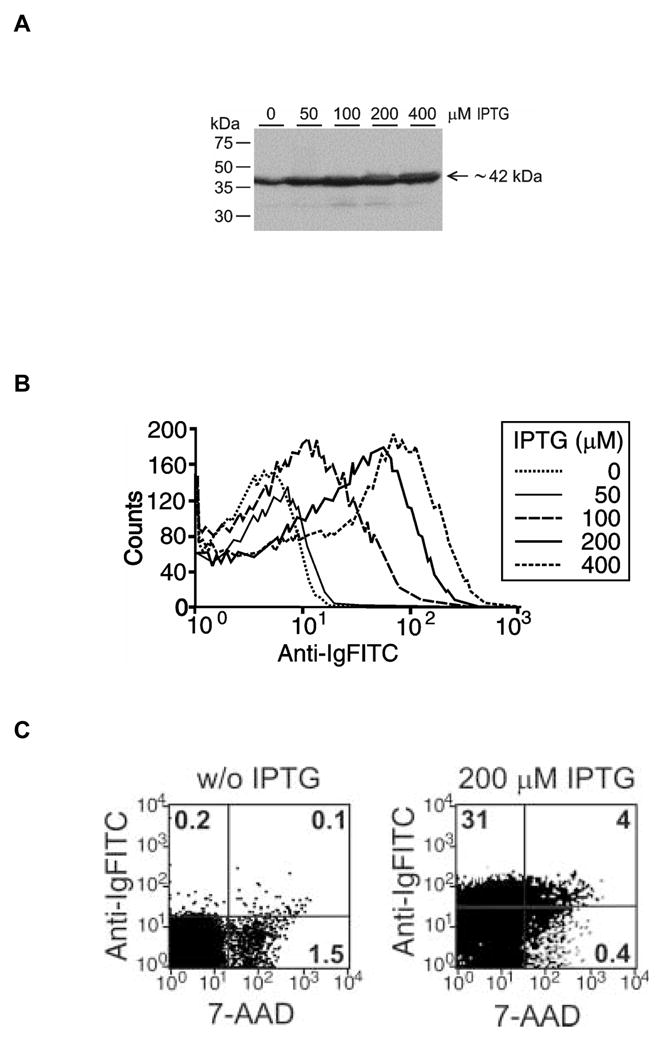
(A) Western blot analysis of Lpp-OmpA-scFv levels in whole cell lysates after induction with different IPTG concentrations. The expected 42 kDa fusion protein is identified using anti-Ig antibody as described in Materials and Methods. (B) Flow cytometric analysis of the surface expression of anti-CEA scFv using anti-Ig FITC after induction with increasing doses of IPTG. (C) Uninduced and induced (IND) cells double stained with anti-Ig FITC and 7-AAD. Data are representative of one of two (Western blot) or four (flow cytometry) experiments performed. Numbers in quadrants show percentage of the total population of cells stained with indicated reagents.
Double staining of VNP/GS18 or VNP/GS18IND with FITC-F(ab′)2-goat anti-mouse IgG (anti-Ig FITC) and 7-AAD provided a reliable method to calculate the percentage of dead bacteria expressing scFv on the surface compared to the whole population (Figure 2C). 7-AAD is a nucleic acid staining dye that can only penetrate dead or dying cells where the inner membrane has been compromised, whereas the anti-Ig FITC is too large to penetrate even the outer membrane of healthy cells. As shown in Figure 2C over 90% of cells remained undamaged when displaying this scFv.
3.2 Flow cytometry and confocal microscopy shows surface displayed Lpp-OmpA-scFv binds to CEA
The ability of the scFv domain of the fusion protein to bind CEA was determined by FACS analysis. Less than 0.5% of VNP/GS18 bound CEA-FITC without IPTG induction whereas upon IPTG induction the number of bacteria that bound CEA-FITC increased to 30% as evaluated in the whole ungated population (Figure 3A). When bacteria were gated on high FSC-H, the number of VNP/GS18IND that stained with CEA-FITC exceeded 50% representing a 100 fold increase over VNP/GS18. These results are consistent with those obtained using the anti-Ig FITC staining in Fig. 2B and 2C) suggesting that the majority of displayed scFvs are able to bind to CEA. The specificity of CEA-FITC binding was confirmed through competition with unlabeled CEA, which inhibited binding of CEA-FITC by 50% at an equimolar concentration. Under these conditions 1mg/mL of BSA, used as a negative control, did not inhibit CEA-FITC binding to VNP/GS18IND. Additionally, lack of staining with the irrelevant antigen OVA-FITC protein further confirmed the specificity of CEA binding (data not shown). A bipolar distribution of the scFv was identified by confocal microscopic analysis of VNP/GS18IND stained with CEA-Alexa-Fluor (Figure 3B).
Figure 3. Expression of functional anti-CEA scFv on the surface of VNP/GS18IND.

(A) FACS analysis of CEA-FITC binding to VNP/18GSIND. Bacteria were incubated with medium alone (control, dotted line) or with FITC-conjugated CEA (solid line), or pretreated for 15 minutes with unlabeled CEA and incubated with FITC-conjugated CEA (dashed line). Data are representative of three independent experiments. (B) Confocal images of VNP/GS18IND stained with CEA-Alexa-Fluor (left panel), CMTMR (central panel). The right panel represents the overlay of both stainings. Data are representative of two experiments.
The specificity of CEA binding by CEA-specific scFv expressed on the bacterial surface, as well as the Lpp-OmpA-scFv distribution pattern within the bacterial wall, was further confirmed by atomic force microscopy (AFM) analysis (Figure 4A and B). From the shape of force-distance curves it can be determined if the bond between CEA protein on the tip of the AFM instrument and the scFv on the bacterial surface was created [20]. For events corresponding to the formation of a specific CEA-scFv bond a characteristic shift of a break point appears. For a non-specific interaction (e.g. direct tip-surface bond) the break point is close to the contact point (point on the curve corresponding to a moment when the bond is created) and the characteristic shift is not visible. 80% of all curves collected from measurements performed on VNP/GS18IND showed a characteristic shape confirming the specificity of CEA-scFv (Table 1). Detailed analysis of CEA binding to different regions of VNP/GS18IND (poles vs. central portion of the bacterium) confirmed that the most active clusters of scFv are expressed at the polar region (supplemental Figure 1).
Figure 4. The pattern of anti-CEA-scFv expression in VNP/GS18IND as probed with atomic force microscopy.

(A) The image of a single bacteria in AFM with descrete regions denoted as P (pole), I (intermediate region) and M (middle region). (B) Histogram (obtained from 400 force curves) of rupture forces recorded with a CEA-coated tip. Green bars represent rupture forces for a pole (P) region, orange bars – (I) and red bars – (M). The force-distance relationships have a slightly non-linear character due to polymer extension. Data represent a single measurement performed on at least two bacterial cells representative of two independent experiments.
Table 1.
Average values of rupture force and adhesion probability based on statistical analysis from AFM measurements*
| VNP modification | Tip coating | Rupture force [pN] | Adhesion probability |
|---|---|---|---|
| GS18 | CEA | 93 ± 3 | 0.35 |
| GS18IND | CEA | 123 ± 5 | 0.52 |
| GS18IND | OVA | 97 ± 5 | 0.30 |
Each data point is an average of 400 force-distance curves collected; CEA, carcinoembryonic antigen; OVA, ovalbumin.
3.3 VNP/GS18IND binds to CEA in vivo
Bacteria transferred in vivo from in vitro culture conditions undergo extensive genetic and phenotypic reprogramming allowing adjustment to a new environment [21]. Adhesion is a key initial step during bacterial colonization of the intestinal mucosa involving participation of several bacterial regulons [22]. To determine the influence of Lpp-ompA-scFv fusion protein expression on the bacterial adhesiveness in vivo, we monitored the numbers of VNP, VNP/GS18 and VNP/GS18IND bacteria one or two hours after oral gavage to wild type C57Bl/6 or CEA transgenic mice. To ensure the isolates were our administered strains, randomly chosen colonies were stained with anti-Salmonella antibody or anti-mouse IgG-FITC, and examined by flow cytometry as described in Materials and Methods. The VNP/GS18IND localized predominantly to the duodenum in CEA transgenic mice (Figure 5A) while the VNP and VNP/GS18 localized to the distal small intestine close to the cecum (Figure 5B).
Figure 5. VNP expressing anti-CEA scFv accumulate in CEA expressing intestinal cells and tumors in vivo.


Wild type (white bars) and CEA transgenic (black bars) mice were administered 108 cfu VNP, VNP/GS18, and VNP/GS18IND bacteria by oral gavage. The bars indicate the numbers of bacteria (cfu per gram of tissue) recovered 1 hour post intragastric inoculation from the upper small intestine (A) and lower small intestine (B) as described in Materials and Methods. (C) Mice were injected with 5 × 105 MC38 cells in the left flank and 5 × 105 MC38CEA cells in the right flank. Seven days later they received 106 cfu VNP/GS18IND. Tumors, liver and spleen were harvested for bacterial counts seven days later. Data are averages of two independent experiments. (A) *P < 0.05 vs wild mice; (C) *P < 0.01 vs MC38 tumor
In order to test the hypothesis that VNP expressing CEA-specific scFv would localize to CEA-expressing tumors we injected 107 cfu of theVNP/GS18IND bacteria into the tail vein of mice bearing seven day old MC38 colon carcinomas on the left flank and CEA-transduced MC38 carcinomas (MC38CEA) on the right flank. After seven days there were significantly increased numbers of bacteria within the MC38CEA tumors over MC38 tumors (p<0.01) with few colonies isolated from spleen and liver obtained from inoculated mice (Figure 5C).
3.4 Treatment with VNP/GS18IND induces local accumulation of immune cells and inhibits tumor growth
Mice bearing established MC38CEA tumors were treated with VNP, VNP/GS18 or VNP/GS18IND using a single 2 × 106 cfu dose of bacteria by i.v. injection. In contrast to treatment with VNP (control) bacteria, treatment with VNP/GS18 significantly reduced tumor growth and VNP/GS18IND resulted in complete inhibition of tumor growth in six out of ten vaccinated mice (Figure 6A) and all mice vaccinated with VNP/GS18IND survived for the duration of the experimental protocol (60 days). Tumor regression observed in mice was accompanied by transient splenomegaly (Figure 6B).
Figure 6. Effects of the modified VNP on MC38CEA tumor growth.

(A) Seven-day established MC38CEA tumors were injected i.v. with 2 × 106 cfu VNP (▲), VNP/GS18 (▼) or VNP/GS18IND (◆) or PBS (CTRL■). Tumor growth was measured every 2 – 3 days by measurement the longest perpendicular diameters and data is presented as tumor area (mm2). For VNP/GS18 and VNP/GS18IND P<0.001 vs control. Data represent average values from two independent experiments on total 10 mice per group. (B) Representative photo of tumor (upper panel) and spleen (lower panel) from mice treated with PBS (CTR) or VNP/GS18IND (VAC).
There was an increase in CD3+ T cells and CD11b+ macrophages in tumors of mice vaccinated with VNP/GS18IND (21.5% ± 1.5 vs. 10.6% ± 1.4 in control mice; P<0.001; Figure 7A). Whereas numerous necrotic foci containing large numbers of CD11b+ cells were detected in tumors from control and vaccinated mice, massive accumulation of macrophages was identified at the tumor-host tissue border in vaccinated mice (Figure 7B). Lymphocytes accumulated mainly at the border of tumors in control mice (Figure 7C) but were dispersed throughout the tumor isolated from VNP/GS18IND vaccinated mice (Figure 7D).
Figure 7. Immunocytochemical staining of MC38CEA tumors.
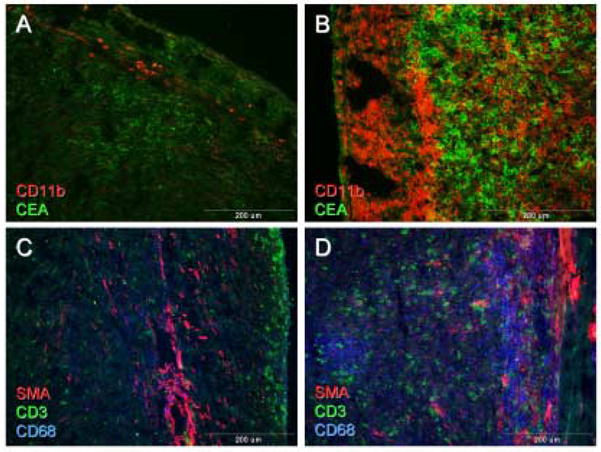
Tumors isolated from control (A, C), or VNP/GS18IND vaccinated mice (B, D) were double stained with anti-CD11b (red) and anti-CEA (green) (A, B) or triple stained with anti-CD3 (green), anti-CD68 (blue) and SMA (red) (C, D).
4. Discussion
Intracellular bacteria including Salmonella spp. have been considered as potential vaccine vectors and gene delivery vehicles for cancer therapy due to their ability to carry plasmids and localize within tumor tissue in murine models [1, 2]. Phenotypic changes accompanying several auxotrophic mutations were initially thought to be responsible for preferential intra-tumoral localization of VNP and AroA− SL7207 Salmonella strains [1], however other strains, such as SL3261, have not shown this effect [23]. To date, the mechanism of bacterial preference for localization within solid tumors in rodent models remains elusive [24]. This is important since clinical trials in humans have failed to document consistent tumor localization with Salmonella bacterial vectors [4].
Modest improvements in therapeutic responses with S. typhimurium vectors have recently been reported using a type III protein secretion system to deliver tumor-associated antigens to established tumors [25]. In this report we facilitated tumor colonization by targeting a specific tumor antigen with an engineered bacteria displaying antigen-specific scFv on its surface. We showed that CEA-specific scFv fragments as a part of a Lpp-OmpA-scFv fusion protein expressed on the VNP surface retained antigen binding potential and targeted CEA-expressing tissue in vivo. Since expression of eukaryotic proteins in bacteria can lead to aborted cell replication we utilized an IPTG-inducible expression system so that scFv could be displayed only on live bacteria immediately prior to injection. However, even the use of the inducible expression system can not completely abrogate the stress imposed on bacteria by accumulation of transgenic proteins. The deposition of such proteins and plasmid loss, under the lack of the antibiotic selection in vivo, were likely reasons for the relatively small recovery of bacteria from tumor tissue after inoculation of VNP/GS18IND in tumor-bearing mice (Figure 5C). Elevated levels of the soluble CEA often accompany human carcinomas and occurs in the Apc/CEA transgenic mice [26]. This circulating CEA could potentially suppress targeting of tumor tissue by VNP/scFv. Nevertheless, we isolated at least twice as many bacteria from CEA-expressing tumors as CEA-negative tumors which indicated efficient tumor targeting. The high level of bacterial accumulation in CEA-negative tumors is explained by the natural tendency for Salmonella to localize in murine tumors and this may mask the full potential of this vector in hosts that do not exhibit such tropism
We further evaluated the effects of oral administration of VNP/GS18IND in vivo using CEA transgenic mice, which express CEA in a similar pattern to human gastrointestinal tracts [12]. The oral administration of VNP/GS18IND bacteria resulted in accumulation of greater numbers of bacteria and in a more proximal location in the upper GI tract compared to control Salmonella, which eventually adhered to cells in the distal small intestine (Fig. 5). This has several implications for therapeutic delivery of the bacterial vector. First, the expression of the anti-CEA scFv improved adherence of the bacteria in the upper GI tract allowing for rapid absorption of bacterium into the upper GI system and portal circulation. Second, this observation suggests that the VNP/GS18IND may be useful for targeting CEA-expressing tumors of the foregut. Finally, the data suggests that the bacteria will be able to localize in CEA-expressing tumors under stringent physiological conditions.
In addition to increasing vector targeting to tumor tissue, the expression of anti-CEA scFv may also act as a vaccine and induce both local and systemic innate and adaptive immune responses. Overexpression of OmpA, which is able to induce maturation of DC as well as to directly activate NK cells through the Toll-like receptor 2 (TLR2) [11, 27], likely serves an important adjuvant role. In our preliminary studies shown here, we observed a significant reduction in tumor growth following VNP/GS18IND administration (Fig. 6), which was associated with accumulation of CD3+ T cells and CD11b+ macrophages (Fig. 7). The presence of macrophages supports the induction of a vigorous innate immune response. While similar effects were observed after treatment of mice with VNP/GS18, and to lesser extent with VNP (data not shown), the anti-tumor effects were much weaker (Fig. 6A), suggesting that the functional quality of the infiltrating immune cells and/or the induction of adaptive immunity are responsible for tumor regression. This possibility is consistent with previous studies using anti-idiotypic CEA monoclonal antibodies, which have demonstrated induction of CEA-specific T cell immunity and protection against CEA-expressing tumors [28]. Further studies to define the immune responses in this model are currently in progress.
Collectively, our data support the use of an inducible Lpp-OmpA-scFv system in S. typhimurium as a novel method for targeting tumors and inducing local anti-tumor immunity. This approach may also be useful for vaccination purposes and further studies will focus on the role of innate and adaptive immune responses to this vector. While we selected CEA as a target for this technology, the system can also be used to target other antigens and engineered to deliver various genes or gene products to further augment host immunity or deliver therapeutic agents to tumors.
Supplementary Material
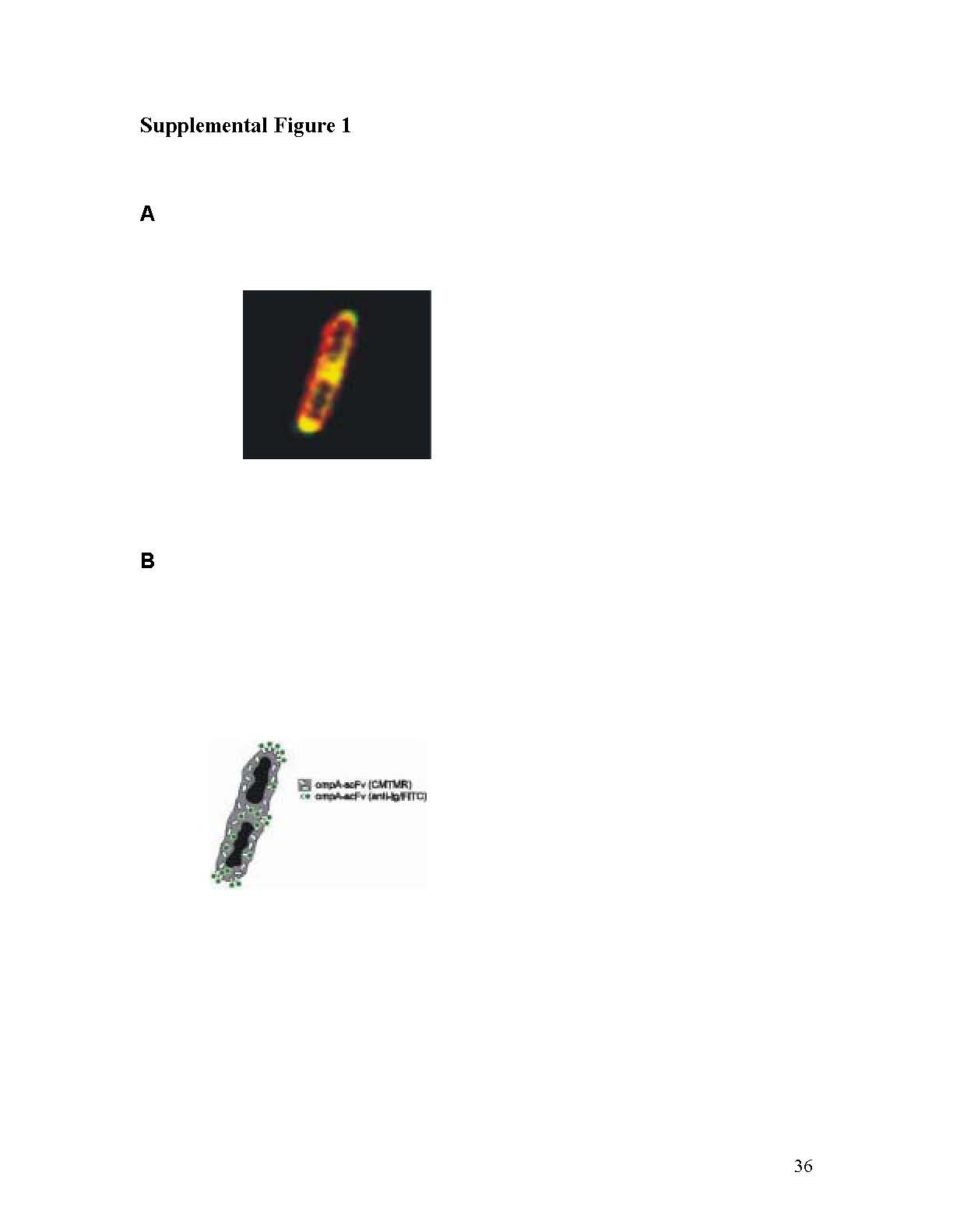
(A) Magnified confocal image of two unseparated VNPGS18IND bacteria. Periplasmic accumulation of scFv was visualized by CMTMR staining (red) and bipolar surface expression was labeled with anti-Ig-FITC (green). (B) Details of staining are shown on the schematic diagram.
{kind=link}
Acknowledgments
We thank Dorota Moroziewicz and Robert Glover for technical assistance and Greg Bereta for the help with graphics. A special acknowledgement shall go to the AFM Laboratory at Jagiellonian University, Poland, and its head Dr. Marek Szymonski for making available atomic force microscope. This work was supported by grants KO8 CA79881 from the NIH (H.L.K.), 2PO4B00230 from the Polish Ministry of Science and Education (M.B.), C06 RR12087 from the NIH, and charitable donations from the USAA Foundation and SFBR (A.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bermudes D, Zheng LM, King IC. Live bacteria as anticancer agents and tumor-selective protein delivery vectors. Curr Opin Drug Discov Devel. 2002;5(2):194–9. [PubMed] [Google Scholar]
- 2.Rosenkranz CD, Chiara D, Agorio C, Baz A, Pasetti MF, Schreiber F, et al. Towards new immunotherapies: targeting recombinant cytokines to the immune system using live attenuated Salmonella. Vaccine. 2003;21(7–8):798–801. doi: 10.1016/s0264-410x(02)00602-3. [DOI] [PubMed] [Google Scholar]
- 3.Clairmont C, Lee KC, Pike J, Ittensohn M, Low KB, Pawelek J, et al. Biodistribution and genetic stability of the novel antitumor agent VNP20009, a genetically modified strain of Salmonella typhimurium. J Infect Dis. 2000;181(6):1996–2002. doi: 10.1086/315497. [DOI] [PubMed] [Google Scholar]
- 4.Toso JF, Gill VJ, Hwu P, Marincola FM, Restifo NP, Schwartzentruber DJ, et al. Phase I study of the intravenous administration of attenuated Salmonella typhimurium to patients with metastatic melanoma. J Clin Oncol. 2002;20(1):142–52. doi: 10.1200/JCO.2002.20.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Low KB, Ittensohn M, Le T, Platt J, Sodi S, Amoss M, et al. Lipid A mutant Salmonella with suppressed virulence and TNFalpha induction retain tumor-targeting in vivo. Nat Biotechnol. 1999;17(1):37–41. doi: 10.1038/5205. [DOI] [PubMed] [Google Scholar]
- 6.Sundquist M, Rydstrom A, Wick MJ. Immunity to Salmonella from a dendritic point of view. Cell Microbiol. 2004;6(1):1–11. doi: 10.1046/j.1462-5822.2003.00336.x. [DOI] [PubMed] [Google Scholar]
- 7.Marshall J. Carcinoembryonic antigen-based vaccines. Semin Oncol. 2003;30(3 Suppl 8):30–6. doi: 10.1016/s0093-7754(03)00233-1. [DOI] [PubMed] [Google Scholar]
- 8.Schorr J, Knapp B, Hundt E, Kupper HA, Amann E. Surface expression of malarial antigens in Salmonella typhimurium: induction of serum antibody response upon oral vaccination of mice. Vaccine. 1991;9(9):675–81. doi: 10.1016/0264-410x(91)90194-b. [DOI] [PubMed] [Google Scholar]
- 9.Lee JS, Shin KS, Pan JG, Kim CJ. Surface-displayed viral antigens on Salmonella carrier vaccine. Nat Biotechnol. 2000;18(6):645–8. doi: 10.1038/76494. [DOI] [PubMed] [Google Scholar]
- 10.Francisco JA, Campbell R, Iverson BL, Georgiou G. Production and fluorescence-activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc Natl Acad Sci U S A. 1993;90(22):10444–8. doi: 10.1073/pnas.90.22.10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chalifour A, Jeannin P, Gauchat JF, Blaecke A, Malissard M, N’Guyen T, et al. Direct bacterial protein PAMP recognition by human NK cells involves TLRs and triggers alpha-defensin production. Blood. 2004;104(6):1778–83. doi: 10.1182/blood-2003-08-2820. [DOI] [PubMed] [Google Scholar]
- 12.Horig H, Wainstein A, Long L, Kahn D, Soni S, Marcus A, et al. A new mouse model for evaluating the immunotherapy of human colorectal cancer. Cancer Res. 2001;61(23):8520–6. [PubMed] [Google Scholar]
- 13.Wu AM, Chen W, Raubitschek A, Williams LE, Neumaier M, Fischer R, et al. Tumor localization of anti-CEA single-chain Fvs: improved targeting by non-covalent dimers. Immunotechnology. 1996;2(1):21–36. doi: 10.1016/1380-2933(95)00027-5. [DOI] [PubMed] [Google Scholar]
- 14.Wu AM, Williams LE, Zieran L, Padma A, Sherman M, Bebb GG, et al. Anti-carcinoembryonic antigen (CEA) diabody for rapid tumor targeting and imaging. Tumor Targeting. 1999;4:47–58. [Google Scholar]
- 15.Hayhurst A, Happe S, Mabry R, Koch Z, Iverson BL, Georgiou G. Isolation and expression of recombinant antibody fragments to the biological warfare pathogen Brucella melitensis. J Immunol Methods. 2003;276(1–2):185–96. doi: 10.1016/s0022-1759(03)00100-5. [DOI] [PubMed] [Google Scholar]
- 16.Murray SR, Bermudes D, de Felipe KS, Low KB. Extragenic suppressors of growth defects in msbB Salmonella. J Bacteriol. 2001;183(19):5554–61. doi: 10.1128/JB.183.19.5554-5561.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maniatis JSEFFT. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 18.Hinterdorfer P, Baumgartner W, Gruber HJ, Schilcher K, Schindler H. Detection and localization of individual antibody-antigen recognition events by atomic force microscopy. Proc Natl Acad Sci U S A. 1996;93(8):3477–81. doi: 10.1073/pnas.93.8.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lekka M, Laidler P, Dulinska J, Labedz M, Pyka G. Probing molecular interaction between concanavalin A and mannose ligands by means of SFM. Eur Biophys J. 2004;33(7):644–50. doi: 10.1007/s00249-004-0412-5. [DOI] [PubMed] [Google Scholar]
- 20.Dupres V, Menozzi FD, Locht C, Clare BH, Abbott NL, Cuenot S, et al. Nanoscale mapping and functional analysis of individual adhesins on living bacteria. Nat Methods. 2005;2(7):515–20. doi: 10.1038/nmeth769. [DOI] [PubMed] [Google Scholar]
- 21.Bajaj V, Lucas RL, Hwang C, Lee CA. Co-ordinate regulation of Salmonella typhimurium invasion genes by environmental and regulatory factors is mediated by control of hilA expression. Mol Microbiol. 1996;22(4):703–14. doi: 10.1046/j.1365-2958.1996.d01-1718.x. [DOI] [PubMed] [Google Scholar]
- 22.Baranov V, Hammarstrom S. Carcinoembryonic antigen (CEA) and CEA-related cell adhesion molecule 1 (CEACAM1), apically expressed on human colonic M cells, are potential receptors for microbial adhesion. Histochem Cell Biol. 2004;121(2):83–9. doi: 10.1007/s00418-003-0613-5. [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg SA, Spiess PJ, Kleiner DE. Antitumor effects in mice of the intravenous injection of attenuated Salmonella typhimurium. J Immunother. 2002;25(3):218–25. doi: 10.1097/01.CJI.0000014623.45316.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forbes NS, Munn LL, Fukumura D, Jain RK. Sparse initial entrapment of systemically injected Salmonella typhimurium leads to heterogeneous accumulation within tumors. Cancer Res. 2003;63(17):5188–93. [PubMed] [Google Scholar]
- 25.Nishikawa H, Sato E, Briones G, Chen LM, Matsuo M, Nagata Y, et al. In vivo antigen delivery by a Salmonella typhimurium type III secretion system for therapeutic cancer vaccines. J Clin Invest. 2006;116(7):1946–54. doi: 10.1172/JCI28045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steele G, Jr, Zamcheck N. The use of carcinoembryonic antigen in the clinical management of patients with colorectal cancer. Cancer Detect Prev. 1985;8(3):421–7. [PubMed] [Google Scholar]
- 27.Jeannin P, Renno T, Goetsch L, Miconnet I, Aubry JP, Delneste Y, et al. OmpA targets dendritic cells, induces their maturation and delivers antigen into the MHC class I presentation pathway. Nat Immunol. 2000;1(6):502–9. doi: 10.1038/82751. [DOI] [PubMed] [Google Scholar]
- 28.Bhattacharya-Chatterjee M, Chatterjee SK, Foon KA. Anti-idiotype vaccine against cancer. Immunol Lett. 2000;74(1):51–8. doi: 10.1016/s0165-2478(00)00249-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Magnified confocal image of two unseparated VNPGS18IND bacteria. Periplasmic accumulation of scFv was visualized by CMTMR staining (red) and bipolar surface expression was labeled with anti-Ig-FITC (green). (B) Details of staining are shown on the schematic diagram.