

Abstract
Background
The Hedgehog (HH) signaling pathway is critical for embryonic development and adult homeostasis. Recent studies have identified regulatory roles for this pathway in certain cancers with mutations in the HH pathway genes. The extent to which mutations of the HH pathway genes are involved in the pathogenesis of malignant mesothelioma (MMe) is unknown.
Methodology/Principal Findings
Real-time PCR analysis of HH pathway genes PTCH1, GLI1 and GLI2 were performed on 7 human MMe cell lines. Exon sequencing of 13 HH pathway genes was also performed in cell lines and human MMe tumors. In silico programs were used to predict the likelihood that an amino-acid substitution would have a functional effect. GLI1, GLI2 and PTCH1 were highly expressed in MMe cells, indicative of active HH signaling. PTCH1, SMO and SUFU mutations were found in 2 of 11 MMe cell lines examined. A non-synonymous missense SUFU mutation (p.T411M) was identified in LO68 cells. In silico characterization of the SUFU mutant suggested that the p.T411M mutation might alter protein function. However, we were unable to demonstrate any functional effect of this mutation on Gli activity. Deletion of exons of the PTCH1 gene was found in JU77 cells, resulting in loss of one of two extracellular loops implicated in HH ligand binding and the intracellular C-terminal domain. A 3-bp insertion (69_70insCTG) in SMO, predicting an additional leucine residue in the signal peptide segment of SMO protein was also identified in LO68 cells and a MMe tumour.
Conclusions/Significance
We identified the first novel mutations in PTCH1, SUFU and SMO associated with MMe. Although HH pathway mutations are relatively rare in MMe, these data suggest a possible role for dysfunctional HH pathway in the pathogenesis of a subgroup of MMe and help rationalize the exploration of HH pathway inhibitors for MMe therapy.
Introduction
Malignant mesothelioma (MMe) is an aggressive, incurable cancer that arises from mesothelial cells that line the serosal cavities of the pleura, peritoneum, pericardium and tunica vaginalis testis. Occupational asbestos exposure is the main risk factor for MMe, accounting for 80% of the cases in men and 40% of the cases in women [1]. The global incidence of MMe has been on the rise since the 1960s and is projected to increase until at least 2050 due to the widespread use of asbestos during the past decades [2]–[5]. MMe is highly refractory to current treatment modalities including chemotherapy, radiotherapy and surgery, with a short median survival time of less than 12 months after first diagnosis [6]. There is no doubt that successful treatment will require a paradigm shift in how MMe is viewed as a disease. Current research is focusing on the molecular mechanisms underlying MMe development and growth to identify new targets for therapeutic intervention [7].
The Hedgehog (HH) signaling pathway regulates critical aspects of embryonic development as well as adult tissue homeostasis and stem cell maintenance [8]–[10]. Recently, the HH pathway has been implicated as a major contributor to the growth and maintenance of a variety of human cancers [11]. HH acts as a ligand for the Patched (PTCH) receptor proteins, PTCH1 and PTCH2 [12]. In the absence of the HH ligand (Sonic HH (SHH), Desert HH (DHH) and Indian HH (IHH)), PTCH interaction with Smoothened (SMO) inhibits SMO function [13]. Upon ligand binding, PTCH-mediated repression of SMO is relieved and SMO transduces the signal to a SUFU-GLI complex residing in the cytoplasm, resulting in the release and activation of GLI transcription factors [14]. SUFU is the main repressor of the mammalian HH signaling pathway by sequestering GLI transcription factors in the cytoplasm and nucleus [15]. This repressor is negatively regulated by serine/threonine kinase 36 (STK36), which in turn promotes activation and nuclear accumulation of GLI [16]. In addition to SUFU, the transmembrane HH-interacting protein (HHIP) was identified as another negative regulator of the HH pathway that acts by sequestering all three HH homologs with similar affinity to that of PTCH1 protein [17]. Recently, KIF7 was identified by sequence comparison as a GLI-interacting protein [18]. KIF7 physically binds to GLI, regulating their stability and degradation and controlled GLI-mediated transcription [19]. There are 3 GLI proteins in vertebrates; GLI1, GLI2 and GLI3, which are capable of either transcriptional activation or repression of cell type-specific HH pathway target genes [20]. A schematic diagram depicting the Hedgehog signaling pathway is shown in Figure 1.
Figure 1. Hedgehog signaling pathway.

HH ligands (SHH, DHH or IHH) bind to the PTCH receptors (PTCH1 and PTCH2) and relieve the inhibition of SMO. HHIP, a negative inhibitor of HH signaling, can compete with PTCH receptors to bind HH ligand, resulting in the attenuation of HH signaling. SMO then transduces signals through the cytoplasmic SUFU-GLI complex, resulting in the activation and nuclear translocation of the downstream GLI transcription factors (GLI1-3). STK36 further promotes activation and nuclear accumulation of GLI by antagonizing SUFU. KIF7 regulates GLI-mediated transcription both positively and negatively through physical interaction with GLI and regulating the stability and degradation of GLI proteins.
Mutations in PTCH1 have been found in familial basal cell carcinoma (BCC) and medulloblastoma (MB) [21], [22]. It was later discovered that sporadic BCCs harbored somatic PTCH1 mutations [23]. PTCH1 mutations have also been found in other sporadic tumors, such as MB [24], skin trichoepitheliomas [25], esophageal squamous cell carcinomas [26], skin squamous cell carcinomas [27], meningiomas [24], breast carcinomas [24] and bladder carcinomas [28], as well as in odontogenic keratocysts [24], [27], [29]–[31]. In addition to primary tumors, missense mutations in PTCH1 have been identified in several oral squamous cell carcinoma and colon carcinoma cell lines [24], [32].
Clearly mutations in PTCH1 do not account for all cases of familial as well as sporadic BCCs and MBs. Indeed, mutations in several genes in the HH pathway have been found in familial and sporadic BCCs and MBs. In a subset of patients with sporadic BCC, mutations in SMO have been described [33]–[37] while PTCH2 mutations were detected in some cases of sporadic BCCs and MBs [38]. Unlike PTCH1, PTCH2 is overexpressed in both familial and sporadic BCCs [39] and MBs [40], suggesting that PTCH2 is a direct gene target of HH signaling and that PTCH2 may be negatively regulated by PTCH1 [39]. Loss of PTCH2 has been reported to contribute to enhanced tumorigenesis in PTCH1 haploinsufficient mice [41].
Mutations in SUFU were detected in sporadic BCCs and seemed to predispose to MB [33], [42]. Mutations in SHH have also been identified in a number of cancer types, including BCC [43], MB [43], breast carcinoma [43], glioblastoma [44] and lung cancer [45]. Oro and coworkers detected an identical somatic mutation in the putative zinc hydrolase site of SHH that caused a histidine to tyrosine substitution at codon 133 of SHH. Notably, this mutation occurred in a sporadic BCC, a MB and a breast carcinoma, hinting at the possible involvement of this nonsynonymous substitution in driving malignancies in these tumor types [43].
To our knowledge, mutations in HH pathway genes have not yet been reported in MMe. In this study, we report identification of a novel multi-exonic deletion in PTCH1 and an insertion mutation in SMO in human MMe, identified via direct sequencing of genes encoding HH signaling pathway proteins in a panel of 7 MMe cell lines. We also identified a point mutation close to the Gli-binding domain of SUFU that led to the replacement of threonine for methionine at amino acid position 411. However, Gli1 luciferase reporter assays revealed that this mutation failed to disrupt the inhibitory function of SUFU. We also genotyped a collection of primary tumors for the presence of these mutations and found one tumor sample that carried the SMO insertion mutation. In addition, we also detected a number of previously reported single nucleotide polymorphisms (SNPs) in the course of our screen. Six of these sequence variants were predicted to have substantial impact upon the function of the encoded protein.
Materials and Methods
Cell Lines and Culture Conditions
The human MMe cell lines MSTO-211H (CRL-2081), NCI-H28 (CRL-5820), NCI-H226 (CRL-5826), NCI-H2052 (CRL-5915) and NCI-H2452 (CRL-5946) as well as the mouse embryonic fibroblasts C3H/10T1/2 (CCL-226) were obtained from the American Type Culture Collection (ATCC). The 5 MMe cell lines JU77, LO68, NO36, ONE58 and STY51 were kind gifts from Professor Bruce W. Robinson, University of Western Australia, Australia [46] and REN cells were provided by Professor Steve Albelda, University of Pennsylvania, USA. These cell lines were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 4 mM L-glutamine, 100 units/ml penicillin and 100 µg/ml streptomycin (all from Invitrogen) in a humidified incubator with 5% CO2 at 37oC. In addition, normal pericardial mesothelial cells were obtained from patients undergoing thoracic surgery as described previously [47].
Ethics Statement
Use of the archival tissue blocks in this retrospective study was approved by the Sydney Local Health District Human Research Ethics Committee (Concord Hospital), as part of a larger study to identify prognostic factors in malignant pleural mesothelioma. Consent for the use of these samples was waived by the ethics committee, consistent with the Human Tissue Act (1983) and the NHMRC National Statement on Ethical Conduct in Human Research (Commonwealth of Australia, 2007).
Patients and Tumors
The MPM tumour samples used in this study (Table 1) were part of a series collected from patients who underwent extrapleural pneumonectomy at Royal Prince Alfred or Strathfield Private Hospitals (Sydney, Australia) between 1994 and 2009 [48]. The tumour content in the formalin-fixed paraffin embedded (FFPE) blocks was marked on whole sections to enable enrichment of tumour content via subsequent laser-capture microdissection.
Table 1. Demographic and clinical characteristics of patients with Mme.
| Sample ID | Subtype | Gender | Age | Survival (months) |
| 24 | Epithelioid | M | 53 | 9.56 |
| 31 | Biphasic | M | 60 | 5.42 |
| 32 | Epithelioid | M | 58 | 41.4 |
| 48 | Epithelioid | F | 48 | 44.16 |
| 50 | Epithelioid | M | 54 | 72.9 |
| 51 | Biphasic | M | 51 | 3.42 |
| 52 | Epithelioid | M | 61 | 8.28 |
| 59 | Epithelioid | M | 52 | 13.73 |
| 65 | Epithelioid | M | 64 | 6.93 |
| 72 | Epithelioid | M | 47 | 1.94 |
| 74 | Epithelioid | M | 57 | 8.34 |
| 77 | Biphasic | M | 56 | 2.56 |
| 78 | Epithelioid | F | 59 | 14.52 |
| 79 | Epithelioid | M | 56 | 10.32 |
RNA Isolation, cDNA Synthesis and Quantitative Real-time PCR Analysis (qRT-PCR) of Gene Expression
Total RNA was extracted from cell lines using an RNeasy Mini kit (Qiagen). Two micrograms of total RNA was reverse-transcribed into cDNA using random primers (Invitrogen) and Omniscript RT kit (Qiagen). TaqMan gene expression assays (Applied Biosystems) were used for quantifying the mRNA expression levels of GLI1, GLI2 and PTCH1. PGK1 was included as the endogenous control. Real-time PCR was carried out on the StepOne Plus Real-Time PCR System (Applied Biosystems) in duplicates. Fold change in gene expression relative to PGK1 was calculated using the formula as follows:
DNA Isolation, PCR Amplification and DNA Sequence Analysis
Genomic DNA was isolated from cells using the PureLink Genomic DNA kit (Invitrogen) and from FFPE samples using the FFPE DNA minikit (Qiagen), each according to the manufacturer’s instructions. PCR primers that amplify the exons and flanking intronic sequences of 13 HH pathway genes (Table 2) were either obtained from published literature [49], [50] or designed using GenBank sequences and the Vector NTI 11.0 software. The primer sequences are listed in Table S1. PCR amplification of HH pathway genes was performed as described previously with slight modifications [51]. PCR was carried out on a iCycler thermal cycler (BioRad) in a 20 µl volume containing 10 ng genomic DNA, 1x GoTaq Flexi buffer (Promega), 1.5 mM MgCl2 (Promega), 0.2 mM dNTPs (Invitrogen), 0.5 µM primers (Geneworks Pty Ltd), 6% DMSO (Sigma Aldrich), 1.25 U GoTaq Flexi DNA polymerase (Promega). The optimized PCR conditions for each primer pair are listed in Table S2. PCR products were visualized on 1% agarose gels before they were sequenced at the Australian Genome Research Facility (AGRF), Perth, Western Australia, using a Big Dye Terminator v3.1 cycle sequencing kit (Applied Biosystems) and analyzed on a 3730xl DNA Analyzer (Applied Biosystems). Base calling and quality assessment were carried out using the Sequence Scanner v1.0 software (Applied Biosystems) while sequence assembly was carried out using the Vector NTI v11.0 software (Invitrogen). All sequence variants found were confirmed by an independent PCR and sequencing reaction to exclude PCR artifacts. Seven mesothelioma cell lines (MSTO-211H, NCI-H28, JU77, LO68, NO36, ONE58 and STY51) were originally exon sequenced with a further 4 cell lines (NCI-H226, NCI-H2052, NCI-H2452 and REN) genotyped for identified mutations.
Table 2. Genes sequenced in this study.
| Gene Symbol | Gene name | NCBI Gene ID |
| GLI1 | GLI family zinc finger 1 | 2735 |
| GLI2 | GLI family zinc finger 2 | 2736 |
| GLI3 | GLI family zinc finger 3 | 2737 |
| IHH | indian hedgehog | 3549 |
| PTCH1 | patched 1 | 5727 |
| SHH | sonic hedgehog | 6469 |
| SMO | smoothened, frizzled family receptor | 6608 |
| PTCH2 | patched 2 | 8643 |
| STK36 | serine/threonine kinase 36 | 27148 |
| DHH | desert hedgehog | 50846 |
| SUFU | suppressor of fused homolog (Drosophila) | 51684 |
| HHIP | hedgehog interacting protein | 64399 |
| KIF7 | kinesin family member 7 | 374654 |
PCR Assay for Detection of PTCH1 Exon Deletions
To detect exon deletions of the PTCH1 gene in MMe cell lines, we designed a PCR assay using exonic primers directed against exons 18, 19, 20, 21, 22, and 23 of PTCH1. The PCR condition (Protocol 1) and primer sequences are listed in Tables S2 and S3, respectively. PCR products were visualized on 4% agarose gels.
In silico Characterization of Polymorphisms in Exons
The web-based programs SIFT [52] and PolyPhen2 [53] were employed to predict the potential effect of non-synonymous amino acid substitutions resulting from the genetic alterations. The default settings were used for all parameters of each program.
Gli Luciferase Reporter Assay
C3H/10T1/2 cells were plated in triplicates on 24-well plates 24 h before transfection. Cells were cotransfected with 25 ng pRL-TK (Promega), 0.1 µg 12XGLI1 luciferase reporter construct (a kind gift from Professor Rune Toftgård, Karolinska Institutet), 0.15 µg wild-type GLI1 construct (Origene) and 0.15 µg of the appropriate SUFU construct (wild-type SUFU or SUFU (p.T411M) (Blue Heron Bio) using FuGene 6 transfection reagent (Roche), with a 3∶1 ratio (v/w) of FuGene 6 to DNA. Cells were harvested using the Dual-Glo Luciferase assay system (Promega), 48 h after transfection according to the manufacturer’s instruction. Luciferase activity was measured using a Wallac 1420 VICTOR2 multilabel plate reader (Perkin Elmer). All reporter assays were normalized to Renilla luciferase activity. All transfections were repeated in at least two independent experiments, which gave reproducible results.
Statistical Analysis
Statistical calculations were performed using Graphpad Prism 4.03 software (Graphpad Software, Inc.). Student’s t-test was used to determine the significance of luciferase reporter assays, and p<0.05 was considered significant.
Results
The Canonical HH Signaling Pathway is Active in Human MMe Cell Lines
We used qRT-PCR to demonstrate the mRNA expression of transcription factors GLI1 and GLI2, activators of the canonical HH signaling pathway and a downstream HH target gene PTCH1, in a panel of 7 human MMe cell lines (Figure 2). In contrast, normal mesothelial cells expressed extremely low levels of PTCH1 and GLI2 mRNA while GLI1 was undetectable (Figure 2). These data suggest that the components necessary for active HH signaling are expressed at significantly higher levels in all MMe cell lines analyzed, compared with controls. This is consistent with recent findings of GLI1 and HH interacting protein (HHIP) being elevated 2–6-fold in MMe tissues using qRT-PCR analysis, relative to benign pleural tissue [54]. In a similar study, the mRNA and protein levels of GLI1, GLI2 and the signal transducer SMO were found to be overexpressed in 46 MMe tissues, as analyzed by qRT-PCR and immunohistochemistry [55].
Figure 2. Relative expression (expressed in ΔCT) of the HH pathway genes.

For each gene, the relative expression of mRNA was normalized to an endogenous PGK1 control. Values represent the mean ± S.E.M. of three independent experiments each performed in duplicates. A, GLI1, B, GLI2 and C, PTCH1 in 7 MMe cell lines and 2 primary cultures of normal mesothelial cells.
PTCH1, SMO and SUFU Mutations in MMe Cell Lines
Through de novo sequencing, all the exons of 13 genes encoding components of the HH signaling pathway were screened for mutations in a panel of 7 MMe cell lines. No mutation was found in the exonic regions of SHH, DHH, IHH, PTCH1, PTCH2, HHIP, KIF7, GLI1, GLI2 and GLI3 genes. However, one non-synonymous mutation in SUFU was identified (Table 3). This missense mutation, which was found in LO68, involved a C>T transition at nucleotide 1232 in exon 10 of SUFU, which substitutes threonine for methionine at position 411 (p.T411M) (Figure 3A). This mutation was characterized by SIFT [52] and PolyPhen2 [53] and determined to be damaging. We have also cross-checked with 1000 Genomes and confirmed that this insertion is not a germline variant. In addition, as shown in Figure 3B, amino acid sequence alignment from various species revealed that Thr411 of SUFU was evolutionarily highly conserved.
Table 3. Mutations identified in MMe cell lines.
| Type of alteration | Gene | Exon | Region | Nucleotidechange | Amino acid change | SIFT | PolyPhen2 | Cell line |
| Point mutation | SUFU | 10 | Coding | 1232 C>T | T411M | Damaging | Possibly damaging | LO68 |
| Deletion | PTCH1 | 18 | Coding | – | – | Unknown | Unknown | JU77 |
| PTCH1 | 19 | Coding | – | – | Unknown | Unknown | JU77 | |
| PTCH1 | 20 | Coding | – | – | Unknown | Unknown | JU77 | |
| PTCH1 | 21 | Coding | – | – | Unknown | Unknown | JU77 | |
| PTCH1 | 22 | Coding | – | – | Unknown | Unknown | JU77 | |
| PTCH1 | 23 | Coding | – | – | Unknown | Unknown | JU77 | |
| Insertion | SMO | 1 | Coding | 69_70insCTG | 23L_24GinsL | Unknown | Unknown | LO68 |
Figure 3. Identification of a SUFU T411M mutation in LO68 cell line.

A, Electropherogram of SUFU gene. Left, the sequencing result of MSTO-211H cell line, showing the wildtype SUFU gene. Right, the sequencing result of LO68 carrying the mutant allele. B, Amino acid sequence alignment of SUFU from various species. Shown in the red box is the T411 residue that is evolutionarily conserved among these different species. Numbers indicate the position of amino acid residue with the start codon (methionine) as number 1.
Although no point mutations were identified in the exonic region, homozygous deletion of exons 18, 19, 20, 21, 22 and 23 of PTCH1 in JU77 cell line was observed and had been suspected by the absence of PCR product on agarose gel electrophoresis (Figure 4). Subsequently, this was confirmed by PCR using exonic primers and demonstrated complete absence of PCR amplicon in the cell line (Figure 5). In addition to the identification and characterization of point mutations and exon deletions, a 3 base-pair insertion of CTG between nucleotides 69 and 70 in exon 1 of the SMO gene in LO68 cell line was identified. This 69_70insCTG mutation resulted in an in-frame addition of a leucine residue after amino acid 23 of the SMO protein (p.23L_24GinsL) with unknown functional role (Figure 6). The same in-frame insertion was recently reported in two out of 39 gastric tumors [56].
Figure 4. Agarose gel electrophoretic analysis of amplified exons 18–23 of PTCH1 gene from MMe cell lines.

JU77 has a homozygous deletion of all 6 exons. Lanes are labeled according to the cell lines, “NTC” = No template control. Exons assayed and size markers (“M”), in base pairs, are shown on left side of the gel image.
Figure 5. Confirmatory PCR. JU77 showing a deletion of exons 18–23 of the PTCH1 gene.

Lanes are labeled according to the cell lines (“L” = LO68, “J” = JU77 and “−” = NTC) and exons assayed and size markers (“M”), in base pairs, shown on both sides of the gel image.
Figure 6. Identification of a SMO 23insL mutation in LO68 cell line.

A, Wild-type sequence in MSTO-211H cell line. B, 3-bp CTG insertion in LO68 cell line.
Hedgehog Pathway Gene Variants in MMe Cell Lines
Screening all the exons of each of the 13 HH pathway genes revealed a total of 35 SNPs using our panel of 7 MMe cell lines (Table 4). Examination of the NCBI dbSNP database revealed that all the SNPs were previously reported variations. No polymorphism was found in the exonic regions of DHH and SHH genes. Among the 35 SNPs detected, all were located in the coding regions and 16 SNPs would cause substitution of an amino acid (Table 4). The most frequent SNPs include: (i) GLI2 rs2592595, ii) GLI2 rs3738880, iii) GLI2 rs10167980, iv) GLI2 rs12711538, v) KIF7 rs3803531, vi) KIF7 rs8037349, vii) KIF7 rs8179066 and SMO rs2228617, which were identified in all 7 cell lines. In silico analysis of potential functional impact of the 16 non-synonymous SNPs were characterized by the SIFT and PolyPhen2 programs. There was an overlap between SIFT and PolyPhen2 predictions: only one SNP (KIF7 rs8179065) was predicted to be a non-deleterious substitution by both SIFT and PolyPhen2 (Table 4). The remaining five non-synonymous SNPs (PTCH1 rs357564, STK36 rs1344642 and rs1863704, KIF7 rs138354681 and GLI1 rs2228224) were predicted by PolyPhen2 as damaging but benign by SIFT (Table 4).
Table 4. SNPs identified in MMe cell lines.
| Gene | Exon | Region | Nucleotide change | Amino acid change | dbSNP ID | SIFT | PolyPhen2 | Cell line |
| GLI1 | 5 | Coding | 576 G>A | E192E | rs2228225 | Tolerated | Benign | JU77, LO68, NO36, STY51 |
| GLI1 | 11 | Coding | 2798 G>A | G933D | rs2228224 | Tolerated | Damaging | JU77, LO68, NO36, STY51 |
| GLI1 | 11 | Coding | 3298 G>C | E1100Q | rs2228226 | Tolerated | Benign | JU77, LO68, NO36, STY51 |
| GLI2 | 5 | Coding | 801 G>A | S267S | rs2592595 | Tolerated | Benign | MSTO-211H, NCI-H28, JU77, LO68, NO36, ONE58, STY51 |
| GLI2 | 13 | Coding | 3466 G>T | A1156S | rs3738880 | Tolerated | Benign | MSTO-211H, NCI-H28, JU77, LO68, NO36, ONE58, STY51 |
| GLI2 | 13 | Coding | 3916 G>A | D1306N | rs12711538 | Tolerated | Benign | MSTO-211H, NCI-H28, JU77, LO68, NO36, ONE58, STY51 |
| GLI2 | 13 | Coding | 3939 A>G | P1313P | rs10167980 | Tolerated | Benign | MSTO-211H, NCI-H28, JU77, LO68, NO36, ONE58, STY51 |
| GLI3 | 4 | Coding | 547 A>G | T183A | rs846266 | Tolerated | Benign | MSTO-211H, NCI-H28, LO68, ONE58, STY51 |
| GLI3 | 14 | Coding | 2993 C>T | P998L | rs929387 | Tolerated | Benign | JU77, NO36, ONE58 |
| HHIP | 13 | Coding | 2058 T>C | I686I | rs11727676 | Tolerated | Benign | MSTO-211H, NCI-H28 |
| IHH | 3 | Coding | 600 G>A | T200T | rs3731878 | Tolerated | Benign | STY51 |
| IHH | 3 | Coding | 753 T>C | P251P | rs3731881 | Tolerated | Benign | MSTO-211H, NCI-H28, JU77, LO68, ONE58 |
| IHH | 3 | Coding | 1128 T>C | T376T | rs394452 | Tolerated | Benign | MSTO-211H, NCI-H28, JU77, LO68, ONE58, STY51 |
| KIF7 | 1 | Coding | 154 G>A | D52N | rs8179065 | Tolerated | Benign | MSTO-211H, NCI-H28, LO68 |
| KIF7 | 1 | Coding | 195 G>C | A65A | rs8179066 | Tolerated | Benign | MSTO-211H, NCI-H28, JU77, LO68, NO36, ONE58, STY51 |
| KIF7 | 4 | Coding | 1102 A>G | T368A | rs8037349 | Tolerated | Benign | MSTO-211H, NCI-H28, JU77, LO68, NO36, ONE58, STY51 |
| KIF7 | 11 | Coding | 2501 A>G | Q834R | rs138354681 | Tolerated | Damaging | ONE58 |
| KIF7 | 12 | Coding | 2658 A>C | A886A | rs3803531 | Tolerated | Benign | MSTO-211H, NCI-H28, JU77, LO68, NO36, ONE58, STY51 |
| KIF7 | 13 | Coding | 2873 G>T | S958I | rs3803530 | Tolerated | Benign | |
| KIF7 | 14 | Coding | 3013 G>A | G1005R | rs12900805 | Tolerated | Benign | JU77, ONE58, STY51 |
| KIF7 | 14 | Coding | 3048 G>A | S1016S | rs9672286 | Tolerated | Benign | JU77, ONE58, STY51 |
| PTCH1 | 23 | Coding | 3944 C>T | P1315L | rs357564 | Tolerated | Damaging | MSTO-211H, NCI-H28, ONE58, STY51 |
| PTCH2 | 2 | Coding | 90 G>A | L30L | rs45573433 | Tolerated | Benign | MSTO-211H, NCI-H28 |
| PTCH2 | 8 | Coding | 1080 G>T | V360V | rs11573579 | Tolerated | Benign | LO68 |
| PTCH2 | 14 | Coding | 1821 A>G | E607E | rs2295997 | Tolerated | Benign | NO36 |
| PTCH2 | 14 | Coding | 2055 T>C | A685A | rs7525308 | Tolerated | Benign | JU77, NO36 |
| PTCH2 | 16 | Coding | 2487 C>T | D829D | rs2295996 | Tolerated | Benign | JU77 |
| SMO | 1 | Coding | 74 A>G | D25G | rs41304185 | Tolerated | Unknown | MSTO-211H, NCI-H28, LO68, NO36 |
| SMO | 2 | Coding | 384 C>T | A128A | rs45571737 | Tolerated | Benign | MSTO-211H |
| SMO | 4 | Coding | 808 G>A | V270I | rs111694017 | Tolerated | Benign | LO68 |
| SMO | 4 | Coding | 852 G>A | Q284Q | rs45445295 | Tolerated | Benign | ONE58 |
| SMO | 6 | Coding | 1164 G>C | G388G | rs2228617 | Tolerated | Benign | MSTO-211H, NCI-H28, JU77, LO68, NO36, ONE58, STY51 |
| STK36 | 13 | Coding | 1748 G>A | R583Q | rs1344642 | Tolerated | Damaging | JU77 |
| STK36 | 24 | Coding | 3008 G>A | G1003D | rs1863704 | Tolerated | Damaging | JU77 |
| SUFU | 11 | Coding | 1299 T>C | I433I | rs17114803 | Tolerated | Benign | NO36 |
PTCH1, SMO and SUFU Mutations in FFPE MMe Tumors
To validate the mutations that were identified in the cell lines, we analyzed DNA from micro-dissected FFPE tumors from 14 patients with MMe for PTCH1 exonic deletions, SMO insertion and SUFU point mutation. Sequencing results showed that one patient had a tumor harboring the CTG insertion in the SMO gene. The patient was a male Caucasian who survived for 3.4 months after being diagnosed at age 51 years with biphasic MMe. As blood was not collected from the patient, the germline vs. somatic origin of the mutation could not be determined. Lastly, we did not detect the presence of the PTCH1 exon deletions or the SUFU mutation in the patient cohort.
Functional Characterization of SUFU Mutant
To corroborate the SIFT and PolyPhen2 predictions that the p.T411M mutation affects SUFU function, we transiently transfected C3H10T1/2 cells with a Gli1 luciferase reporter construct and expression constructs for wild-type Gli1, wild-type SUFU and the SUFU p.T411M mutant. Transfection of wild-type Gli1 resulted in increased Gli1 reporter activity whereas cotransfection of wild-type SUFU inhibited Gli1-induced reporter activity. However, the SUFU mutant failed to suppress Gli1 reporter activity (Figure 7).
Figure 7. p.T411M mutation fails to disrupt the inhibitory function of SUFU.
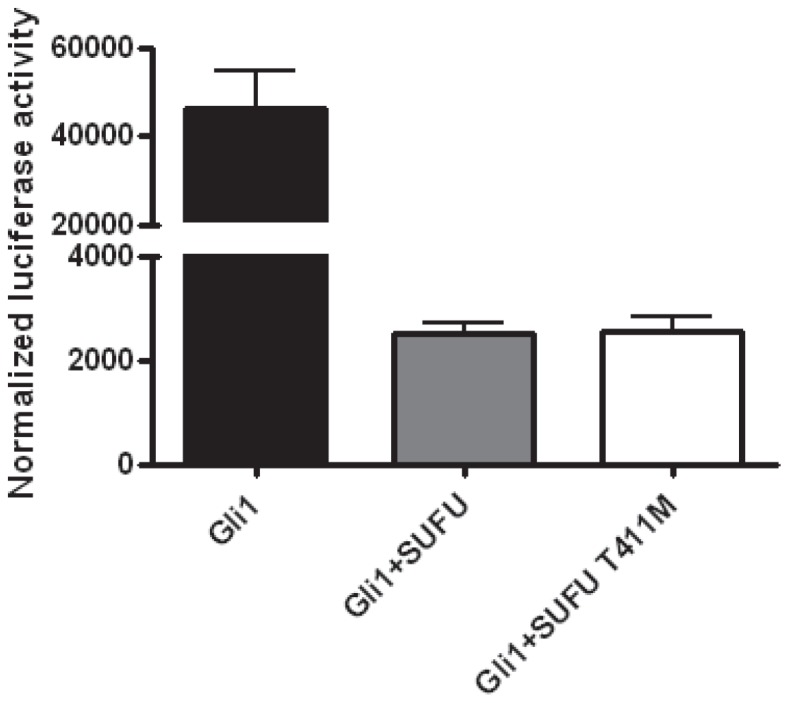
C3H10T1/2 cells were transfected with Gli-luciferase reporter construct with Renilla luciferase plasmid for 48 h before they were harvested for luciferase activity determination. Values represent the mean ± S.E.M. of a representative experiment performed in triplicates.
Discussion
We examined the expression profiles of the HH gene components in MMe cell lines and cultured primary mesothelial cells. We found that the HH pathway components GLI1, GLI2 and PTCH1 are expressed at high levels in MMe cell lines compared to normal mesothelial cells. This suggests that HH signaling is active in MMe, which corroborated findings from an earlier study [54], [55].
Aberrant activation of HH signaling in human cancers could result from genetic alterations in pathway components, including PTCH1, SMO, SUFU and GLI1 [37], [42], [57], [58]. Utilizing de novo sequencing of PCR-amplified DNA fragments, we present for the first time a mutational analysis of 13 genes encoding components of the HH signaling pathway in 7 MMe cell lines. The rationale for screening HH pathway genes for genetic alterations is based on several observations: 1) this pathway plays a critical role in development and growth, 2) high frequency of mutations in the HH pathway genes screened to date, and 3) the HH pathway components were found to be overexpressed in primary MMe tumors and cell lines [54], [55].
In the present study, only three mutations were found in 3 out of 13 genes screened and they were detected in only two cell lines (Table 3). The first mutation that we have identified is a novel multiple-exon deletion in the PTCH1 gene in JU77 cell line, whereby exons 18–23 were deleted. This mutation would result in a putative truncated PTCH1 protein, in which one of two extracellular loops and the cytoplasmic C-terminal domain are lost, or could undergo nonsense-mediated mRNA decay. However, we did not detect this multi-exonic deletion in our cohort of tumor samples from patients with MMe, but this may be due to the small sample size of the patient cohort.
The tumor suppressor gene PTCH1 was cloned in 1996 and subsequently shown to be involved in repression of the HH pathway [13], [21], [22]. As expected for a classical tumor suppressor gene, PTCH1 was found to be mutated in Gorlin syndrome and many other cancers [59]. Previously published studies on mutations in PTCH1 have found more than 300 mutations, most of which appeared to be clustered in the large extracellular and intracellular loops of the PTCH1 protein [59], [60]. Although the functional impact of the deletion of exons 18–23 in PTCH1 has yet to be elucidated, the key role of the extracellular loops in mediating HH ligand binding and the role of the cytoplasmic C-terminal domain in mediating subcellular localization and turnover of PTCH1 as well as inhibition of HH gene targets suggest that the receptor lacking exons 18–23 may lead to an aberrantly activated HH pathway [61]–[63]. Recent studies also suggest that the C-terminal fragment of PTCH1 could be localized to the nuclear region of the cell where it represses the transcriptional activity of Gli1, even though the canonical view of PTCH1 as a transmembrane protein is deeply entrenched in the literature [64]. In addition to the exonic deletion, we found a SNP (c.3944C>T) in exon 23 of PTCH1, which was also reported in cancers of the skin, bone and vulva [32], [65]–[68]. This non-synonymous SNP leads to substitution of leucine for proline at position 1315 in the C-terminal domain of the PTCH1. Modeling analysis suggested that the Leu1315 substitution might alter the secondary structure of PTCH1 resulting in constitutive pathway activation [69]. Intriguingly, this biallelic SNP has recently been associated with a higher risk for development of breast and nonmelanoma skin cancers [69]–[71]. The significance of this SNP with respect to etiopathogenesis of MMe, however, requires further study.
We also detected a SMO mutation in one MMe cell line which was also present in a tumor sample. SMO acts as a signal transducer of the HH pathway, mediating communication between transmembrane PTCH1 receptor and transcriptional activators GLI1 and GLI2. Xie et al. found activating somatic mutations in SMO in 3 out of 47 (6.4%) patients with sporadic BCC [37]. Xie et al. went on further to show that overexpression of these mutant SMO proteins in mouse skin produced BCC-like tumors [37]. In this study, the identified mutation, 23L_24GinsL, lies in the signal peptide region of SMO and has been detected in two cases of human gastric cancer {Wang, 2013 #657}. This mutation might affect the processing of the SMO precursor and in turn potentially interfere with protein targeting to the cell membrane.
One of the most intriguing results of this analysis is the identification of a mutation in SUFU. SUFU was originally identified as a negative regulator of the HH pathway in early embryonic development [72]. Taylor et al. first implicated SUFU in the tumorigenesis of childhood MB in 2002 [73]. They identified germline and somatic SUFU nonsense mutations in 8.7% (4 of 46) of desmoplastic MBs. Subsequently, SUFU mutations were found in 4.8% (2 of 42) and 2.5% (2 of 83) of sporadic BCCs and MBs, respectively [33], [74]. We found a missense mutation affecting Thr411 in SUFU in one of the 11 cell lines. Importantly, the same mutation was recently detected in a patient with colorectal cancer as part of the Cancer Genome Atlas [75], supporting a role of this mutation in tumorigenesis. This mutation was predicted by two web-based programs, SIFT and PolyPhen2, that predict the potential functional impact of altered amino acid sequences on encoded protein function, to result in a major change in amino acid class (from small, polar to large, hydrophobic) in the N-terminal region located close to the GLI1 binding domain, thereby implying a negative impact on protein function. SUFU acts as a classic tumor suppressor gene, with mutations leading to the inability of SUFU to transport GLI1 out of the nucleus to the cytoplasm, thereby resulting in aberrant activation of HH signaling in MB [73]. However, functional characterization showed that the p.T411M mutation does not alter the negative regulatory function of SUFU in a Gli1 luciferase reporter assay.
In addition to PTCH1, SUFU and SMO, we screened our MMe cell line panel for mutations in 9 other HH pathway genes PTCH2, DHH, IHH, SHH, GLI1, GLI2, GLI3, KIF7, STK36 and HHIP. We found no evidence for mutations in any of these genes in our cell lines. However, we did find a number of previously reported SNPs, of which 16 were shown to result in non-synonymous codon substitution. To explore the functional significance of these SNPs, all those that are non-synonymous were analyzed by SIFT and PolyPhen2. There was considerable difference between the predictions from different algorithms: a KIF7 SNP rs8179065 was predicted by SIFT to have altered protein function whereas the PolyPhen2 program predicted the PTCH1 SNP rs357564, STK36 SNPs rs1344642 and rs1863704, KIF7 SNP rs138354681 and GLI1 SNP rs2228224 to be deleterious amino acid substitution. Algorithmic differences aside, the real functional significance of these non-synonymous SNPs on HH signaling needs to be validated by biochemical studies.
Recently, SNPs in SHH, GLI2 and GLI3 genes have been reported to be associated with clinical outcome of trans-urethral resection and Bacillus Calmette-Guerin intravesical therapy for non-muscle-invasive bladder cancer patients (NMIBC) [76]. The GLI2 SNP rs11685068 and SHH SNP rs1233560 demonstrated significant associations with recurrence in NMIBC patients who received trans-urethral resection. On the other hand, the NMIBC patients receiving Bacillus Calmette-Guerin treatment who carried the GLI3 SNPs rs3801192 and rs6463089 were found to have a higher cancer recurrence rate and shorter recurrence-free survival time compared to those carrying the wildtype genotype. In a separate genome-wide association study, common variants in the SHH, BTRC and HHIP genes have been associated with the risk of pancreatic cancer [77]. These studies demonstrated a link between common genetic variations in the Hedgehog pathway and cancer risk, thus highlighting the importance of examining the association between SNPs in HH pathway genes and the risk of MMe.
Clearly, the frequency of mutations in key HH pathway genes in MMe is less than in BCC and MB where the mutations lead to hyperactivation of the pathway. However, the importance of these and other unidentified mutations in HH pathway genes in the pathogenesis of a subset of MMe tumors cannot be discounted and needs further investigation in a larger sample set.
In summary, we report mutations in SMO and SUFU and a novel multi-exonic deletion in PTCH1 in MMe cell lines and tumors. Our data suggest that unlike BCC, MB and rhabdomyosarcoma in Gorlin syndrome, aberrant activation of HH signaling in MMe is unlikely to be driven by mutations in the majority of tumors but instead activated through autocrine signaling as suggested by Shi et al. [54] This pathway may represent a novel therapeutic target in MMe for the recently developed HH pathway inhibitors.
Supporting Information
List of primers used for the PCR amplification and sequencing of Hedgehog pathway genes.
(XLSX)
PCR conditions used in this study.
(XLSX)
List of primers used for the detection of PTCH1 exon deletions.
(XLSX)
Acknowledgments
We thank Winthrop Professor Bruce Robinson and Professor Steven Albelda for MMe cells; Dr Brian McCaughan for patient samples; Professor Rune Toftgård for the Gli luciferase construct; Dr David Chandler, Dr Shane Herbert and Mr Matthew Davis (AGRF) for support with sequencing and Faang Cheah for helpful discussion.
Funding Statement
This study was supported by grants from the National Health and Medical Research Council, Australia (Project ID number 572676) and the Mesothelioma Applied Research Foundation, USA. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Robinson BWS, Chahinian AP (2002) Mesothelioma. London: M. Dunitz. xiii, 366 p.
- 2. Moolgavkar SH, Meza R, Turim J (2009) Pleural and peritoneal mesotheliomas in SEER: age effects and temporal trends, 1973–2005. Cancer Causes Control 20: 935–944. [DOI] [PubMed] [Google Scholar]
- 3. Price B, Ware A (2004) Mesothelioma trends in the United States: an update based on Surveillance, Epidemiology, and End Results Program data for 1973 through 2003. Am J Epidemiol 159: 107–112. [DOI] [PubMed] [Google Scholar]
- 4. Musk AW, de Klerk NH (2004) Epidemiology of malignant mesothelioma in Australia. Lung Cancer 45 Suppl 1S21–23. [DOI] [PubMed] [Google Scholar]
- 5. Peto J, Decarli A, La Vecchia C, Levi F, Negri E (1999) The European mesothelioma epidemic. Br J Cancer 79: 666–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Musk AW, Olsen N, Alfonso H, Reid A, Mina R, et al. (2011) Predicting survival in malignant mesothelioma. Eur Respir J 38: 1420–1424. [DOI] [PubMed] [Google Scholar]
- 7. Lee AY, Raz DJ, He B, Jablons DM (2007) Update on the molecular biology of malignant mesothelioma. Cancer 109: 1454–1461. [DOI] [PubMed] [Google Scholar]
- 8. Ingham P, McMahon A (2001) Hedgehog signaling in animal development: paradigms and principles. Genes Dev 15: 3059–3087. [DOI] [PubMed] [Google Scholar]
- 9. Jiang J, Hui C (2008) Hedgehog signaling in development and cancer. Dev Cell 15: 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Varjosalo M, Taipale J (2008) Hedgehog: functions and mechanisms. Genes Dev 22: 2454–2472. [DOI] [PubMed] [Google Scholar]
- 11. Yang L, Xie G, Fan Q, Xie J (2010) Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 29: 469–481. [DOI] [PubMed] [Google Scholar]
- 12. Carpenter D, Stone DM, Brush J, Ryan A, Armanini M, et al. (1998) Characterization of two patched receptors for the vertebrate hedgehog protein family. Proc Natl Acad Sci U S A 95: 13630–13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taipale J, Cooper MK, Maiti T, Beachy PA (2002) Patched acts catalytically to suppress the activity of Smoothened. Nature 418: 892–897. [DOI] [PubMed] [Google Scholar]
- 14. Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R (2010) The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev 24: 670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kogerman P, Grimm T, Kogerman L, Krause D, Unden AB, et al. (1999) Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nature cell biology 1: 312–319. [DOI] [PubMed] [Google Scholar]
- 16. Murone M, Luoh SM, Stone D, Li W, Gurney A, et al. (2000) Gli regulation by the opposing activities of fused and suppressor of fused. Nature cell biology 2: 310–312. [DOI] [PubMed] [Google Scholar]
- 17. Chuang PT, McMahon AP (1999) Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature 397: 617–621. [DOI] [PubMed] [Google Scholar]
- 18. Varjosalo M, Li S, Taipale J (2006) Divergence of hedgehog signal transduction mechanism between Drosophila and mammals. Dev Cell 10: 177–186. [DOI] [PubMed] [Google Scholar]
- 19. Cheung HO, Zhang X, Ribeiro A, Mo R, Makino S, et al. (2009) The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci Signal 2: ra29. [DOI] [PubMed] [Google Scholar]
- 20. Katoh Y, Katoh M (2005) Hedgehog signaling pathway and gastric cancer. Cancer Biol Ther 4: 1050–1054. [DOI] [PubMed] [Google Scholar]
- 21. Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, et al. (1996) Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85: 841–851. [DOI] [PubMed] [Google Scholar]
- 22. Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, et al. (1996) Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272: 1668–1671. [DOI] [PubMed] [Google Scholar]
- 23. Gailani MR, Stahle-Backdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, et al. (1996) The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nature genetics 14: 78–81. [DOI] [PubMed] [Google Scholar]
- 24. Xie J, Johnson RL, Zhang X, Bare JW, Waldman FM, et al. (1997) Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer research 57: 2369–2372. [PubMed] [Google Scholar]
- 25. Vorechovsky I, Unden AB, Sandstedt B, Toftgard R, Stahle-Backdahl M (1997) Trichoepitheliomas contain somatic mutations in the overexpressed PTCH gene: support for a gatekeeper mechanism in skin tumorigenesis. Cancer research 57: 4677–4681. [PubMed] [Google Scholar]
- 26. Maesawa C, Tamura G, Iwaya T, Ogasawara S, Ishida K, et al. (1998) Mutations in the human homologue of the Drosophila patched gene in esophageal squamous cell carcinoma. Genes, chromosomes & cancer 21: 276–279. [PubMed] [Google Scholar]
- 27. Ping XL, Ratner D, Zhang H, Wu XL, Zhang MJ, et al. (2001) PTCH mutations in squamous cell carcinoma of the skin. The Journal of investigative dermatology 116: 614–616. [DOI] [PubMed] [Google Scholar]
- 28. McGarvey TW, Maruta Y, Tomaszewski JE, Linnenbach AJ, Malkowicz SB (1998) PTCH gene mutations in invasive transitional cell carcinoma of the bladder. Oncogene 17: 1167–1172. [DOI] [PubMed] [Google Scholar]
- 29. Barreto DC, Gomez RS, Bale AE, Boson WL, De Marco L (2000) PTCH gene mutations in odontogenic keratocysts. Journal of dental research 79: 1418–1422. [DOI] [PubMed] [Google Scholar]
- 30. Sun LS, Li XF, Li TJ (2008) PTCH1 and SMO gene alterations in keratocystic odontogenic tumors. Journal of dental research 87: 575–579. [DOI] [PubMed] [Google Scholar]
- 31. Song YL, Zhang WF, Peng B, Wang CN, Wang Q, et al. (2006) Germline mutations of the PTCH gene in families with odontogenic keratocysts and nevoid basal cell carcinoma syndrome. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine 27: 175–180. [DOI] [PubMed] [Google Scholar]
- 32. Michimukai E, Kitamura N, Zhang Y, Wang H, Hiraishi Y, et al. (2001) Mutations in the human homologue of the Drosophila segment polarity gene patched in oral squamous cell carcinoma cell lines. In vitro cellular & developmental biology Animal 37: 459–464. [DOI] [PubMed] [Google Scholar]
- 33. Reifenberger J, Wolter M, Knobbe CB, Kohler B, Schonicke A, et al. (2005) Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. The British journal of dermatology 152: 43–51. [DOI] [PubMed] [Google Scholar]
- 34. Reifenberger J, Wolter M, Weber RG, Megahed M, Ruzicka T, et al. (1998) Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer research 58: 1798–1803. [PubMed] [Google Scholar]
- 35. Lam CW, Xie J, To KF, Ng HK, Lee KC, et al. (1999) A frequent activated smoothened mutation in sporadic basal cell carcinomas. Oncogene 18: 833–836. [DOI] [PubMed] [Google Scholar]
- 36. Couve-Privat S, Bouadjar B, Avril MF, Sarasin A, Daya-Grosjean L (2002) Significantly high levels of ultraviolet-specific mutations in the smoothened gene in basal cell carcinomas from DNA repair-deficient xeroderma pigmentosum patients. Cancer research 62: 7186–7189. [PubMed] [Google Scholar]
- 37. Xie J, Murone M, Luoh SM, Ryan A, Gu Q, et al. (1998) Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391: 90–92. [DOI] [PubMed] [Google Scholar]
- 38. Smyth I, Narang MA, Evans T, Heimann C, Nakamura Y, et al. (1999) Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Human molecular genetics 8: 291–297. [DOI] [PubMed] [Google Scholar]
- 39. Zaphiropoulos PG, Unden AB, Rahnama F, Hollingsworth RE, Toftgard R (1999) PTCH2, a novel human patched gene, undergoing alternative splicing and up-regulated in basal cell carcinomas. Cancer research 59: 787–792. [PubMed] [Google Scholar]
- 40. Lee Y, Miller HL, Jensen P, Hernan R, Connelly M, et al. (2003) A molecular fingerprint for medulloblastoma. Cancer research 63: 5428–5437. [PubMed] [Google Scholar]
- 41. Lee Y, Miller HL, Russell HR, Boyd K, Curran T, et al. (2006) Patched2 modulates tumorigenesis in patched1 heterozygous mice. Cancer research 66: 6964–6971. [DOI] [PubMed] [Google Scholar]
- 42. Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, et al. (2002) Mutations in SUFU predispose to medulloblastoma. Nature genetics 31: 306–310. [DOI] [PubMed] [Google Scholar]
- 43. Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH Jr, et al. (1997) Basal cell carcinomas in mice overexpressing sonic hedgehog. Science 276: 817–821. [DOI] [PubMed] [Google Scholar]
- 44. McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ, et al. (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, et al. (2010) Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466: 869–873. [DOI] [PubMed] [Google Scholar]
- 46. Manning LS, Whitaker D, Murch AR, Garlepp MJ, Davis MR, et al. (1991) Establishment and characterization of five human malignant mesothelioma cell lines derived from pleural effusions. Int J Cancer 47: 285–290. [DOI] [PubMed] [Google Scholar]
- 47. Holloway AJ, Diyagama DS, Opeskin K, Creaney J, Robinson BW, et al. (2006) A molecular diagnostic test for distinguishing lung adenocarcinoma from malignant mesothelioma using cells collected from pleural effusions. Clin Cancer Res 12: 5129–5135. [DOI] [PubMed] [Google Scholar]
- 48. Kao SC, Lee K, Armstrong NJ, Clarke S, Vardy J, et al. (2011) Validation of tissue microarray technology in malignant pleural mesothelioma. Pathology 43: 128–132. [DOI] [PubMed] [Google Scholar]
- 49. Jones S, Zhang X, Parsons D, Lin J, Leary R, et al. (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321: 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet 43: 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, et al. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314: 268–274. [DOI] [PubMed] [Google Scholar]
- 52. Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31: 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shi Y, Moura U, Opitz I, Soltermann A, Rehrauer H, et al. (2012) Role of hedgehog signaling in malignant pleural mesothelioma. Clin Cancer Res 18: 4646–4656. [DOI] [PubMed] [Google Scholar]
- 55. Li H, Lui N, Cheng T, Tseng HH, Yue D, et al. (2013) Gli as a novel therapeutic target in malignant pleural mesothelioma. PloS one 8: e57346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang XD, Inzunza H, Chang H, Qi Z, Hu B, et al. (2013) Mutations in the hedgehog pathway genes SMO and PTCH1 in human gastric tumors. PloS one 8: e54415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Scales SJ, de Sauvage FJ (2009) Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends in Pharmacological Sciences 30: 303–312. [DOI] [PubMed] [Google Scholar]
- 58. Jiao X, Wood LD, Lindman M, Jones S, Buckhaults P, et al. (2012) Somatic mutations in the Notch, NF-KB, PIK3CA, and Hedgehog pathways in human breast cancers. Genes, chromosomes & cancer 51: 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lindstrom E, Shimokawa T, Toftgard R, Zaphiropoulos PG (2006) PTCH mutations: distribution and analyses. Human mutation 27: 215–219. [DOI] [PubMed] [Google Scholar]
- 60. Lee J, Ekker S, von Kessler D, Porter J, Sun B, et al. (1994) Autoproteolysis in hedgehog protein biogenesis. Science 266: 1528–1537. [DOI] [PubMed] [Google Scholar]
- 61. Lu X, Liu S, Kornberg TB (2006) The C-terminal tail of the Hedgehog receptor Patched regulates both localization and turnover. Genes Dev 20: 2539–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Johnson RL, Milenkovic L, Scott MP (2000) In vivo functions of the patched protein: requirement of the C terminus for target gene inactivation but not Hedgehog sequestration. Mol Cell 6: 467–478. [DOI] [PubMed] [Google Scholar]
- 63. Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ (1996) Biochemical evidence that patched is the Hedgehog receptor. Nature 384: 176–179. [DOI] [PubMed] [Google Scholar]
- 64. Kagawa H, Shino Y, Kobayashi D, Demizu S, Shimada M, et al. (2011) A novel signaling pathway mediated by the nuclear targeting of C-terminal fragments of mammalian Patched 1. PLoS One 6: e18638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ohki K, Kumamoto H, Ichinohasama R, Sato T, Takahashi N, et al. (2004) PTC gene mutations and expression of SHH, PTC, SMO and GLI-1 in odontogenic keratocysts. International Journal of Oral and Maxillofacial Surgery 33: 584–592. [DOI] [PubMed] [Google Scholar]
- 66. Hafner C, Schmiemann V, Ruetten A, Coras B, Landthaler M, et al. (2007) PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum Pathol 38: 1496–1500. [DOI] [PubMed] [Google Scholar]
- 67. Evans T, Boonchai W, Shanley S, Smyth I, Gillies S, et al. (2000) The spectrum of patched mutations in a collection of Australian basal cell carcinomas. Human mutation 16: 43–48. [DOI] [PubMed] [Google Scholar]
- 68. Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G (1997) Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res 57: 2581–2585. [PubMed] [Google Scholar]
- 69. Asplund A, Gustafsson AC, Wikonkal NM, Sela A, Leffell DJ, et al. (2005) PTCH codon 1315 polymorphism and risk for nonmelanoma skin cancer. Br J Dermatol 152: 868–873. [DOI] [PubMed] [Google Scholar]
- 70. Chang-Claude J, Dunning A, Schnitzbauer U, Galmbacher P, Tee L, et al. (2003) The patched polymorphism Pro1315Leu (C3944T) may modulate the association between use of oral contraceptives and breast cancer risk. Int J Cancer 103: 779–783. [DOI] [PubMed] [Google Scholar]
- 71. Liboutet M, Portela M, Delestaing G, Vilmer C, Dupin N, et al. (2006) MC1R and PTCH gene polymorphism in French patients with basal cell carcinomas. J Invest Dermatol 126: 1510–1517. [DOI] [PubMed] [Google Scholar]
- 72. Stone DM, Murone M, Luoh S, Ye W, Armanini MP, et al. (1999) Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. J Cell Sci 112 (Pt 23): 4437–4448. [DOI] [PubMed] [Google Scholar]
- 73. Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, et al. (2002) Mutations in SUFU predispose to medulloblastoma. Nat Genet 31: 306–310. [DOI] [PubMed] [Google Scholar]
- 74. Slade I, Murray A, Hanks S, Kumar A, Walker L, et al. (2011) Heterogeneity of familial medulloblastoma and contribution of germline PTCH1 and SUFU mutations to sporadic medulloblastoma. Fam Cancer 10: 337–342. [DOI] [PubMed] [Google Scholar]
- 75. Cancer Genome Atlas N (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen M, Hildebrandt MA, Clague J, Kamat AM, Picornell A, et al. (2010) Genetic variations in the sonic hedgehog pathway affect clinical outcomes in non-muscle-invasive bladder cancer. Cancer Prev Res (Phila) 3: 1235–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li D, Duell EJ, Yu K, Risch HA, Olson SH, et al. (2012) Pathway analysis of genome-wide association study data highlights pancreatic development genes as susceptibility factors for pancreatic cancer. Carcinogenesis 33: 1384–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of primers used for the PCR amplification and sequencing of Hedgehog pathway genes.
(XLSX)
PCR conditions used in this study.
(XLSX)
List of primers used for the detection of PTCH1 exon deletions.
(XLSX)