

Abstract
Presently, there are no FDA-approved medications to treat cocaine addiction. Active vaccination has emerged as one approach to intervene through the rapid sequestering of the circulating drug, thus terminating both psychoactive effects and drug toxicity. Herein, we report our efforts examining two complimentary, but mechanistically distinct active vaccines, i.e., noncatalytic and catalytic, for cocaine treatment. A cocaine-like hapten GNE and a cocaine transition-state analogue GNT were used to generate the active vaccines, respectively. GNE-KLH was found to elicit persistent high-titer, cocaine-specific antibodies, and blunt cocaine induced locomotor behaviors. Catalytic antibodies induced by GNT-KLH were also shown to produce potent titers and suppress locomotor response in mice; however, upon repeated cocaine challenges the vaccine’s protecting effects waned. In depth kinetic analysis suggested that loss of catalytic activity was due to antibody modification by cocaine. The work provides new insights for the development of active vaccines for the treatment of cocaine abuse.
Keywords: Active immunization, Anti-cocaine vaccine, Hapten synthesis, Catalytic antibodies, Psychomotor stimulant effects
Introduction
Cocaine is a powerful psychoactive substance whose abuse remains a prevalent health and societal crisis.1 Furthermore, numerous medical complications including cardiovascular toxicity, brain damage and death often accompany cocaine abuse. In addition, the association of the drug with the spread of AIDS is also of great concern.2 Presently, there is no proven medications to treat cocaine addiction, and while a number of direct and indirect agonists or antagonists have been examined for the treatment of cocaine dependence, overall, these have met limited success.3 Over the past two decades, immunopharmacotherapy has been explored sporadically as an alternative strategy for the treatment of cocaine abuse. Cocaine vaccines either active or passive have been created to induce the production of anti-cocaine antibodies (Abs).4 Immune stimulation and antibody production is engaged to bind and/or modify peripherally circulating cocaine, thus blocking the passage of cocaine across the blood-brain barrier (BBB) into the central nervous system (CNS), where the drug exerts its addictive effects.5 Two vaccine strategies have been targeted: the use of simple binding antibodies or catalytic antibodies, both of which invoke the tenet of preventing cocaine from crossing the blood brain barrier, thus terminating the drug-induced efforts.
A number of anti-cocaine vaccines based upon simply sequestering the drug have been disclosed. Their origins include the linking of various cocaine-like haptens to carrier proteins, such as bovine serum albumin (BSA), keyhole limpet hemocyannin (KLH) and cholera toxin, indeed, one termed TA-CD has reached phase II clinical trials.6 Our group reported a first-generation hapten termed GNC appended to KLH, which elicited potent titers of anti-cocaine antibodies with high affinity and specificity for cocaine (Figure 1). The induced antibodies significantly sequestered cocaine in the periphery of rodents and suppressed cocaine-induced psychomotor stimulation.7 A second-generation hapten termed GND was developed to increase hapten stability by replacing the relatively labile C2 and C3 ester bonds with amides (Figure 1). Accordingly, GND-KLH conjugates provided greater and longer-lasting protection against cocaine.8 However, from a synthetic perspective, GND presents a unique challenge, therefore GND is impractical for a viable clinical vaccine. In order to prioritize access to a stable antigen, we synthesized a third-generation cocaine hapten termed GNE, via a C2 ester-amide interchange. We hypothesized that an ester-amide interchange at this position would prevent loss of the cocaine scaffold from the carrier protein in the same way as seen for GND. By this approach we have easier synthetic access to a structure that would still maximize anti-cocaine immune response. Indeed, an anti-cocaine vaccine based on coupling GNE, 3, to the highly immunogenic adenovirus capsid protein has been reported to evoke persistent, high titer anti-cocaine antibodies.9
Figure 1.

The structure of (−)-cocaine and cocaine-like haptens GNC, GND, GNE, SNC and GNT.
In addition to active vaccine sequestering antibodies, catalytic antibodies have also emerged as a powerful tool with the potential to treat cocaine addiction. The unique feature of a catalytic antibody is that after it hydrolyzes its substrate and releases its metabolites, the antibody becomes free for further binding, hydrolysis and thus turnover. Seminal research by Landry and coworkers demonstrated how a transition-state-analogue strategy for hapten design elicited several cocaine-hydrolyzing mAbs.10 Here, the transition-state analogue used was a phenylphosphonate, which granted antibodies that hydrolyzed the benzoyl ester of cocaine, resulting in the inactive psychometabolite ecgonine methyl ester. A second approach reported by Basmadjian and coworkers used again transition-state analogue haptens, but now polyclonal sera was examined for antibody catalysis.11 Although cocaine hydrolysis was observed, no kinetic data was reported and no correlation was seen between titers and catalysis. Preparations of efficient catalytic antibodies that readily catalyze cocaine’s degradation, to a great extent, rely on the design of the transition state moiety embedded within the hapten used for imumnization.12 Yet, previous studies have also shown that the linker, including length, site of attachment, and heteroatom choice can all be very critical for producing efficient catalytic antibodies.10(b),13 Based on the chemical scaffold of the only hapten that has reached clinical trials, succinyl norcocaine (SNC),6(d)(e) we sought a new transition-state hapten, termed GNT, 10, Figure 1, in which a succinyl linker is attached at the tropane nitrogen. In contemplating this hapten design, we were aware of the lability of the C-2 methyl ester mediated by the adjacent tropane nitrogen, which is predominately protonated at physiologic pH.14 We anticipated this lability would be nullified in GNT through the bridge-head amide of the installed linker. Furthermore, the distal linker site in relationship to the anticipated benzoyl ester dominant epitope might better expose the unique transition-state epitope for added immune recognition.
Cocaine vaccines have been examined from many different perspectives for treating addiction and overdose, but to our knowledge, there has been no investigation examining active vaccination with a transition-state analogue hapten in a behavioral model of cocaine’s psychostimulant actions. In this study, we prepared two distinct haptens, GNE and GNT, and examined their active vaccines in an animal behavioral model. We compared their corresponding immunological properties and efficacy as judged from reduction of the psychomotor activation produced by acute cocaine administration. We demonstrated that both GNE-KLH and GNT-KLH evoked potent immune response with rapid generation of robust polyclonal antibody titers. The sequestering antibodies from GNE vaccination were shown to possess high cocaine binding specificity and confer significant protection against cocaine-induced spontaneous locomotion. Intriguingly, although the GNT active vaccine suppressed locomotor behavioral responses to cocaine initially, it slowly lost its protection with repeated cocaine injections. To investigate the loss of efficacy in the locomotor behavioral model, GNT polyclonal antibodies were examined pre- and post-cocaine challenge through kinetic analysis, which indicated that the catalytic antibody component was modified in vivo by cocaine.
Results
Synthesis of cocaine haptens and hapten-protein immunoconjugates
The synthesis of cocaine hapten GNE is illustrated in Scheme 1. The synthesis commenced with (−)-ecgonine, which was coupled with amine 4 in the presence of EDC and 4-methyl morpholine in DCM to afford amide 1 in 45% yield. Compound 4 was prepared by the protection of the commercially available Boc-6-aminohexanoic acid with benzyl alcohol followed by the removal of the Boc-protecting group. Benzoylation of the hydroxyl group of compound 1 was achieved in 40% yield by the use of benzoyl chloride, Et3N and DMAP in DCM. The benzylated compound 2 was subjected to hydrogenolysis using 1 atm of H2 and 10% Pd-C in MeOH to generate the desired compound 3 (GNE). The new cocaine transition-state analogue GNT was designed and synthesized as shown in Scheme 2. The synthesis commenced with ecgonine methyl ester 5, which was prepared from (−)-cocaine hydrochloride in two steps.12,15 Ecgonine methyl ester 5 was treated with lithium dipropylamide in THF, followed by the addition of compound 612 at 0 °C to provide the required phosphonate diester 7 in 60% yield. Demethylation of 7 was achieved by forming a carbamate intermediate before treatment with zinc dust, providing norcocaine derivative 8 in 41% yield over two steps. Amide 9 was prepared by N-acylation of amine 8 with succinic anhydride in the presence of Et3N in DMF. Finally, the desired phenylphosphonate 10, (GNT), was obtained in 85% yield by catalytic hydrogenolysis.
Scheme 1.

Synthesis of cocaine hapten GNE
Scheme 2.

Synthesis of cocaine hapten GNT
Following by the synthesis of cocaine haptens GNE and GNT, the immunoconjugates GNE-KLH and GNT-KLH were prepared for vaccination. Cocaine haptens were first activated by the use of EDC/sulfo-NHS in DMF, followed by the addition of carrier protein KLH in PBS solution. The resulting mixtures were allowed to stand at 4 °C overnight, and then subjected to dialysis to remove excess reagents. Prior to administration, each protein conjugate as well as the control vehicle, KLH, were formulated with SAS (Sigma Adjuvant System®), a stable oil-in-water emulsion derived from bacterial and mycobacterial cell walls. In addition, both GNE and GNT were also coupled to bovine serum albumin (BSA) for ELISA microtiter plate coating as well as to monitor the coupling efficiency using MALDI-TOF mass spectrometry. The molecular haptenation ratios (number of moles of hapten per mole of carrier protein BSA) were found to be about 20 for both GNE and GNT.
Titers and affinity of cocaine-specific antibodies generated by active immunization in mice
The efficacy of GNE-KLH and GNT-KLH immunoconjugates was assessed by vaccination into groups of 8 Swiss Webster mice. We have chosen Swiss Webster mice, since they have been reported to show locomotor habituation in activity monitors and significant cocaine-induced hyperactivity once habituated.16 Test groups included KLH negative control, GNE-KLH and GNT-KLH. Vaccine was administered on days 0, 21, and 42 with bleeds taken one week post-vaccination. This vaccination schedule has previously been reported by our laboratory.7,8 Titer levels were assessed by ELISA. Titer is defined as the inverse of the serum dilution needed to generate a half-maximal response when binding to the antigen. All bleeds from the KLH control group showed no significant titer either to GNE or GNT. For GNE-KLH group and GNT-KLH group, a significant titer response to the corresponding hapten was observed after the third injection (t = 42 days) compared to that after the second injection (t = 21 days). As shown in Table 1, immunization of mice with either GNE-KLH or GNT-KLH evoked a high-titer response to cocaine with both titer levels in the range of ~1:30000.
Table 1.
Average ELISA titer and average relative affinity of antisera from immunized mice against cocaine as determined by equilibrium dialysisa
| immunoconjugate | titer |
Kd (nM) |
[Ab] (μg/mL) |
|---|---|---|---|
| GNE-KLH | 31,200 | 6.36 ± 1.56 | 85.12 ± 17.34 |
| GNT-KLH | 29,600 | NDb | NDb |
All assays were performed in triplicate.
ND, not detectable.
Another important parameter in determining the antibody efficacy is the ability of the polyclonal response to bind cocaine. Accordingly, cocaine affinity of the antibodies was determined by a soluble radioimmunoassay (RIA) using 3H-cocaine (Table 1).17 Antibodies induced by GNE-KLH in the second bleed provided a Kd of 6.36 ± 1.56 nM (Figure 2). IgG was assumed to have a molecular weight of 150 kDa and two cocaine-binding sites per molecule. This cocaine specific IgG concentration in serum corresponds to a cocaine binding capacity in serum of 85.12 ± 17.34 μg/mL. Strikingly, the antibodies elicited by GNT-KLH showed no binding affinity for cocaine. We suspected that this could be due to the catalytic antibodies elicited from the GNT vaccine and the time course required for the RIA assay. Indeed, LC/MS analysis of the solution presented only cocaine metabolite ecgonine methyl ester (see LC/MS data, Supporting Information).
Figure 2.
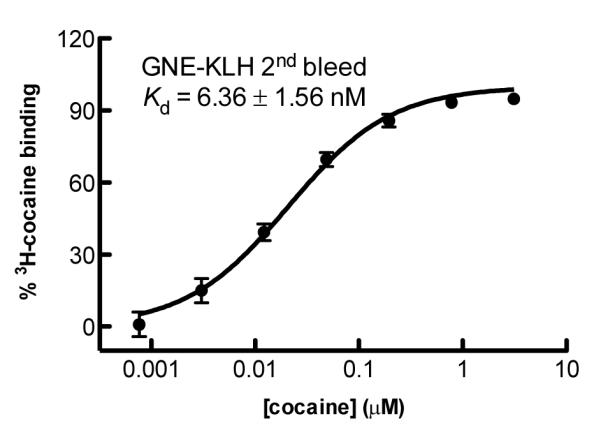
Affinity of GNE polyclonal antibody sera.
Psychomotor activation effects of cocaine in mice immunized with GNT and GNE vaccines
Three groups of vaccinated mice (KLH negative control, GNE-KLH and GNT-KLH) were assessed for the psychomotor stimulant effects of cocaine seven days after the second immunization boost. Following three days of habituation and intraperitoneal injections of saline, the mice were challenged with injections of 15 mg/kg cocaine and evaluated for drug-induced locomotor hyperactivity each day for an additional three days (Supplemental Figures 1–4). In response to cocaine challenge, mice vaccinated with KLH showed significant increases in the number of beam breaks (Figure 3A, session effect F (3,6)=5.344, p<0.01) and crossovers (Figure 3B, session effect F (3,6)=4.522, p<0.05) compared to saline injection. Beam breaks indicate when either one of the two photocell beams across the long access to the cage are broken; crossovers reflect beam 1 being broken after beam 2 and vice versa showing actual movement across the length of the cage. The anti-cocaine antibodies elicited by GNE-KLH vaccination completely prevented the cocaine-induced locomotor hyperactivity in response for the duration of the testing period. In contrast, the antibodies generated by the GNT-KLH vaccinated mice had significant protection against cocaine during the first and second days. However, the antibodies lost some of their protecting effects against cocaine administration in the third day of cocaine challenge, where the number of beam breaks (Figure 3A, session effect F (3,7)=4.174, PBS<0.01), but not crossovers, was significantly increased compared to saline injection.
Figure 3.
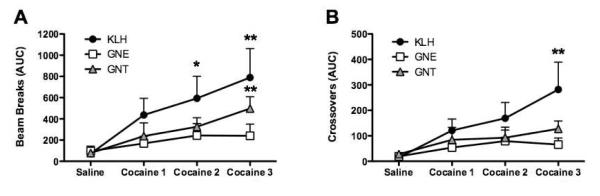
Catalytic vaccine and noncatalytic vaccines prevent the psychomotor stimulating effects of repeated cocaine treatment in mice. * p < 0.05 and ** p < 0.01.
Kinetic analysis of GNT polyclonal antibody sera
A group of 20 Swiss Webster mice was vaccinated on days 0, 21 and 42 with a suspension of GNT-KLH in PBS in formulation with SAS adjuvant. The first set of ten mice were sacrificed on day 50, and the sera were collected and purified by protein G affinity chromatography to obtain the polyclonal antibody before cocaine challenge (pre-treated GNT pAbs). Following two days of habituation, the second set of ten mice were challenged with injections of 15 mg/kg cocaine on days 50, 51 and 52 and sacrificed. The corresponding sera were collected at the time of sacrifice, and further purified by protein G affinity chromatography to obtain polyclonal antibodies, which had been subjected to cocaine challenges (post-treated GNT pAbs). The pre-treated GNT pAbs and post-treated GNT pAbs were tested for their ability to hydrolyze cocaine at a concentration of 1 μM of pAbs and varying concentrations of cocaine. Their catalytic activity was examined through LC/MS detection of benzoic acid. Michaelis-Menten saturation curves were observed (Supplemental Figure 5). Comparison of the Michaelis-Menten kinetics of the pre-treated and post-treated GNT pAbs (Table 2) shows that treatment of cocaine in vivo resulted in a lower catalytic rate and higher apparent Km. The catalytic rate of the post-treated pAbs was 40% below that of pre-treated pAbs, while apparent Km increased two-fold. The catalytic efficiency kcat/Km of pre-treated GNT pAbs was about 4.18 times higher than that of the post-treated GNT pAbs (84.69 M−1s−1versus 16.36 M−1s−1).
Table 2.
Kinetic data for GNT pAbs (pre-treated pAbs) and cocaine-treated GNT pAbs (post-treated pAbs)
| pAbs |
kcat (min−1)a |
Km (μM) |
kcat/Km (M−1 s−1) |
kcat/k’uncatb |
|---|---|---|---|---|
| Pre-treated pAbs | 0.3143 | 61.85 | 84.69 | 7.67×104 |
| Post-treated pAbs | 0.1989 | 121.60 | 16.36 | 4.85×104 |
Reactions were carried out in PBS (pH 7.4) at 23 °C.
k’uncat = 4.1 × 10−6 min−1.18
GNT vaccine’s reduction in catalysis upon cocaine exposure
3H-cocaine was incubated with pools of GNT antibodies that had been previously untreated or treated with cocaine under physiological conditions for 16 hours and 64 hours. After the removal of free 3H-cocaine by dialysis, a portion of each sample was separated by SDS/PAGE. The band at 150 kDa, corresponding to IgG, was excised and the protein was extracted and assayed by liquid scintillation counting. As presented in Figure 4, active counts, and thus cocaine incorporation, increased over time, wherein the radioactivity of the pre-treated GNT pAbs was more intense than that of the post-treated GNT pAbs after either 16 hours or 64 hours.
Figure 4.
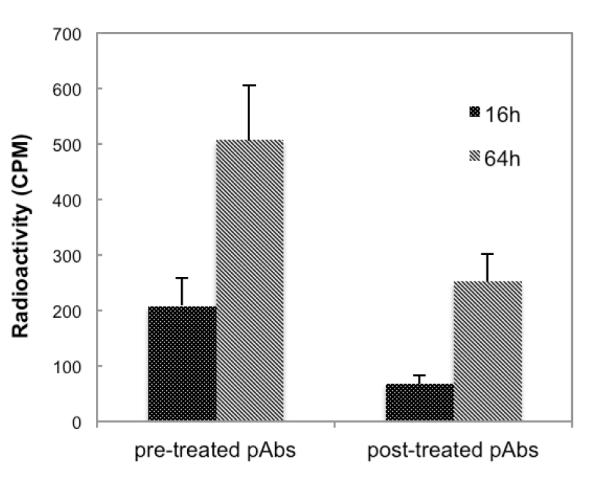
Radioactivity (counts/min) of GNT pAbs (pre-treated pAbs) and cocaine-treated GNT pAbs (post-treated pAbs) after incubation with 3H-cocaine for 16 and 64 hours, respectively.
Discussion
Significant effort has been devoted to the preparation of anti-cocaine vaccines in the hopes of increasing antibody titer, affinity and specificity. Most elaborations within cocaine vaccine preparations can be seen in hapten design. Alternation to scaffolding of the cocaine hapten and the positioning of the linker, including linker length and its chemical composition, have made significant impact on vaccine efficacy. To expand upon the current arsenal of haptens for cocaine vaccine development we were mindful of the spontaneous hydrolysis of the methyl and benzoyl esters embedded within this tropane alkaloid framework.19 Thus, our second generation GND hapten obviates hydrolytic lability issues, however, its synthetic sequence diminishes the robust efficacy seen within animal behavioral studies. To simplify the synthesis of the former reported hapten GND (Figure 1),8 we designed the new hapten GNE, in which we replaced the C2-ester with a C2-amide while maintaining the C3-benzoyl ester (Scheme 1). In collaboration with the Crystal laboratory, we have demonstrated that GNE-based cocaine vaccine dAd5GNE, in which GNE was attached to the disrupted serotype 5 adenovirus (Ad) gene transfer vector, evoked persistent high-titer and high affinity IgG anti-cocaine antibodies.9 To further probe the value of our GNE hapten design, we simply linked GNE to the immunogenic carrier protein KLH using SAS as the adjuvant. Our immunological assays presented evidence that the GNE-KLH vaccine was able to evoke potent-titer and highly specific cocaine antibodies (Figure 2, Table 1). In addition, GNE’s performance as an immunotherapeutic vaccine was examined in a mouse locomotor test and the antibodies elicited were found capable of completely blunting cocaine-induced locomotor hyperactivity over the entire testing period (Figure 3). These results demonstrate that replacement of the C2-ester with the corresponding C2-amide affords a stable hapten with sustainable and robust immunogenicity, thus hapten GNE represents a promising hapten candidate for anti-cocaine vaccination development.
To examine the potential of an active catalytic antibody vaccine, we synthesized a new cocaine transition-state analogue, GNT (Scheme 2). Although hapten GNT appeared equally immunogenic as GNE using titer as the metric, the antibodies elicited by GNT showed no appreciable affinity to cocaine as measured by RIA (Table 1). This finding while seemingly unexpected is readily interpreted. Most likely, cocaine was hydrolyzed during the time course required for the equilibrium dialysis assay (22 hrs); and indeed LC/MS analysis of the solution showed only cocaine metabolized products (LC/MS data in Supporting Information). In the animal behavioral test, catalytic antibodies produced by GNT-KLH provided significant protection against cocaine’s induced hyperlocomotor activity in the first two days of testing but slowly lost their protection with repeated cocaine challenges (Figure 3). Landry et al. reported how cocaine was able to covalently modify a protein through an acylation reaction of the lysine ε-amino groups (Figure 5).20 We posit that the loss of vaccine potency upon repeated exposure to cocaine could be due to modification of the antibody catalysts produced in response to GNT. Using LC/MS analysis, we determined the initial rates of hydrolysis in the presence of the pre-treated and post-treated GNT pAbs as a function of substrate concentration (Table 2). Initial rates of antibody-based hydrolysis displayed saturation or Michaelis-Menten kinetics with respect to cocaine concentration. To rationalize the GNT vaccine’s loss of efficacy in our locomotor behavioral model, we performed a kinetic study comparing the catalytic activity of pre-treated and post-treated pAbs. Although, we did not observe a high enzymatic efficiency for either of these two pAbs, it is evident that repeated cocaine administration compromised the catalysis observed for the GNT vaccine (Table 2). Based on radiolabeling data (Figure 4), cocaine-protein labeling appears plausible for the time dependent loss of catalytic activity. Interestingly, post-treated cocaine sera antibodies were less affected versus the pre-treated cohort, which aligns with protein-cocaine modification in that available modification sites have already been partially blocked. It is tempting to speculate that the catalytic sites of the GNT vaccine are being modified since simple sequestering antibodies evoked by GNE appeared not to be duly affected as viewed by RIA analysis (Table 1), or locomotor behavior (Figure 3). Future analysis of GNT sera where catalysts are physically separated from simple binding antibodies could shed light on this hypothesis.
Figure 5.
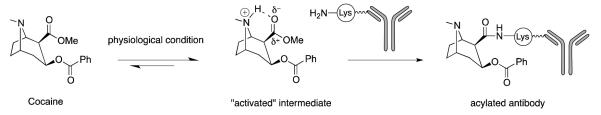
Proposed mechanism of antibody acylation by cocaine.
Conclusions
In summary, we have synthesized a cocaine-like hapten GNE and a cocaine transition-state analogue GNT as well as their KLH immunoconjugates. Both GNE-KLH and GNT-KLH cocaine vaccines were found to elicit high-titer antibodies. While the GNE vaccine granted complete protection from the cocaine stimulatory effects in a locomotor model, GNT-KLH vaccine lost protection after repeated cocaine challenges in vivo. We hypothesize that the reduction of catalytic activity of GNT pAbs response could be due to the acylation of the active site upon repeated cocaine challenges. We note that the catalytic efficiency of catalytic antibodies generated by hapten GNT is modest as judged by enzyme standards, yet, this response was competent enough to provide protection in a mouse locomotor model, albeit it does wane upon repeated cocaine challenges. With these findings it is warranted to examine other catalytic antibody hapten design principles to see if this loss of catalysis can be tempered or if it is a general liability of an active catalytic antibody vaccine. Finally, inspired by a recent report on the combination treatments of cocaine hydrolase and anti-cocaine antibodies,21 we speculate that a combination of sequestering and catalytic antibody active vaccines may also provide a potent dual vaccine approach for examining cocaine induced behavior in animal models.
Experimental Section
All reactions involving air or moisture-sensitive reagents or intermediates were performed under an argon atmosphere. Chemicals and solvents were of reagent grade and were used without further purification. The (−)-cocaine hydrochloride was supplied by the National Institute on Drug Abuse (NIDA). Flash chromatography was performed on silica gel 60 (230-400 mesh) and analytical TLC was carried out on glass plates coated with a 0.25-mm layer of silica gel 60 F-254. HPLC separations were performed on a Vydac 218TP C18 reversed phase preparative (10–15 μm) HPLC column using a gradient of acetonitrile and water. The LC/MS analysis was performed using an Agilent G-1956D single quadrupole mass spectrometer equipped with an 1100 Series LC system from Agilent Technologies. 1H and 13C NMR spectra were obtained using a Bruker 500 MHz or 600 MHz instrument. Chemical shifts were reported in parts per million (ppm, δ) referenced to the residual 1H resonance of the solvent (CDCl3, 7.26 ppm) or (CD3OD, 3.31 ppm). 13C spectra were referenced to the residual 13C resonance of the solvent (CDCl3, 77.0 ppm) or (CD3OD, 49.0 ppm). Splitting patterns were designated as follows: s, singlet; br, broad; d, doublet; dd, doublet of doublets; t, triplet; q, quartet; m, multiplet. High resolution mass spectra were obtained in the Scripps Center for Mass Spectrometry. Hapten protein conjugates were analyzed using MALDI-TOF MS. The purity of all synthetic compounds was higher than 95% as determined by HPLC and/or LC/MS.
Benzyl 6-((1R,2R,3S,5S)-3-hydroxy-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxamido)hexanoate (1)
To a solution of 44.3 mg (0.20 mmol) of (−)-ecgonine in 4 mL of DCM was added 22.0 μL (0.50 mmol) of N-methylmorpholine and 49.7 mg (0.26 mmol) of EDC, followed by 48.6 mg (0.22 mmol) of 4 in 1mL of DCM and 7.3 mg (0.06 mmol) of DMAP at 0 °C. The reaction mixture was maintained at 0 °C and stirred for another 3 h. The solvent was removed under diminished pressure and the crude products were purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 1 as TFA salt: yield 37.4 mg (45%); 1H NMR (CDCl3) δ 1.32-1.35 (m, 2H), 1.52 (quin, 2H, J = 6.0 Hz), 1.62 (quin, 2H, J = 6.0 Hz), 2.06-2.17 (m, 4H), 2.27-2.35 (m, 4H), 2.74 (s, 3H), 3.15-3.28 (m, 3H), 3.81-3.87 (m, 2H), 4.32 (quin, 1H, J = 6.0 Hz), 5.08 (s, 2H), 7.25-7.36 (m, 5H) and 8.40-8.45 (br, 2H); 13C NMR (CDCl3) δ 24.42, 25.15, 26.93, 29.13, 34.86, 36.44, 39.02, 40.43, 48.39, 61.11, 63.93, 67.00, 128.87, 128.89, 129.01, 129.39, 136.87, 173.75, 174.56 and 174.58; mass spectrum (ESI), m/z 389.2446 (M+H)+ (C22H33N2O4 requires m/z 389.2435).
(1R,2R,3S,5S)-2-((6-(Benzyloxy)-6-oxohexyl)carbamoyl)-8-methyl-8-azabicyclo[3.2.1]octan-3-yl benzoate (2)
To a solution of 37.4 mg (0.096 mmol) of 1 in 2 mL of DCM was added 29.5 μL (0.21 mmol) of Et3N followed by 12.3 μL (0.11 mmol) of benzoyl chloride and 1.2 mg (0.06 mmol) of DMAP at 0 °C. The reaction mixture was slowly warmed to room temperature and stirred for another 3 h. The solvent was removed under diminished pressure and the crude products were purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 2 as TFA salt: yield 19.0 mg (40%); 1H NMR (CD3OD) δ 1.15-1.25 (m, 2H), 1.27-1.36 (m, 3H), 1.46-1.50 (m, 2H), 2.02-2.09 (m, 2H), 2.21-2.23 (m, 3H), 2.34-2.42 (m, 3H), 2.67 (s, 3H), 3.06-3.17 (m, 2H), 3.21-3.25 (m, 1H), 3.81-3.91 (m, 2H), 5.09 (s, 2H), 5.46 (quin, 1H, J = 6.0 Hz), 7.33-7.35 (m, 5H), 7.39-7.44 (m, 2H) and 7.93-7.95 (m, 3H); 13C NMR (CD3OD) δ 24.09, 24.98, 26.82, 29.44, 34.21, 38.67, 39.60, 65.38, 66.57, 95.73, 100.43, 128.21, 128.66, 128.99, 129.09, 129.74, 130.09, 130.27, 130.99, 134.15, 137.17, 137.67, 166.15, 172.50 and 174.35; mass spectrum (ESI), m/z 493.2697 (M+H)+ (C29H37N2O5 requires m/z 493.2697).
6-((1R,2R,3S,5S)-3-(Benzoyloxy)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxamido)hexanoic acid (3)
A mixture of 23.0 mg (0.05 mmol) of 2 and 15 mg of 10% Pd/C in 1 mL of ethanol was stirred overnight under a H2 atm at room temperature. The catalyst was removed by filtration and the crude products were purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 3 as TFA salt: yield 11.2 mg (60%); (c 0.65, MeOH); 1H NMR (CD3OD) δ 1.12-1.21 (m, 2H), 1.27-1.36 (m, 3H), 1.37-1.46 (m, 2H), 2.12 (t, 2H, J = 6.0 Hz), 2.15-2.24 (m, 2H), 2.33-2.37 (m, 1H), 2.33-2.37 (m, 1H), 2.47-2.52 (m, 2H), 2.58 (td, 1H, J = 12.0, 6.0 Hz), 2.84 (s, 3H), 3.04-3.10 (m, 1H), 3.21-3.26 (m, 2H), 4.00-4.01 (m, 1H), 4.15 (d, 1H, J = 6.0 Hz), 5.52-5.56 (m, 1H), 7.49 (t, 2H, J = 6.0 Hz), 7.63 (t, 1H, J = 6.0 Hz), 7.98 (d, 2H, J = 6.0 Hz) and 8.43 (br, 1H); 13C NMR (CD3OD) δ 23.71, 24.26, 24.94, 26.80, 29.37, 33.68, 34.02, 38.14, 39.91, 46.64, 63.70, 65.00, 65.87, 129.20, 130.02, 130.12, 134.36, 166.02, 172.39 and 176.73; mass spectrum (ESI), m/z 403.2222 (M+H)+ (C22H31N2O5 requires m/z 403.2227).
Benzyl 6-aminohexanoate (4)
To a solution of 0.5 g (2.16 mmol) of Boc-6-aminohexanoic acid in 15 mL of DCM was added 497 mg (2.29 mmol) of EDC followed by 269 μL (2.59 mmol) of benzyl alcohol and 26.4 mg (0.22 mmol) of DMAP at 0 °C. The reaction mixture was slowly warmed to room temperature and stirred for another 16 h. The reaction mixture was quenched by the addition of 10 mL of sat aq NH4Cl. The mixture was extracted with EtOAc. The combined organic layer was washed with brine, dried (MgSO4) and concentrated under diminished pressure. The residue was purified by flash chromatography on a silica gel column (25 × 3.2 cm). Elution with 10:1 hexanes/ethyl acetate gave the product as a yellow oil: yield 0.64 g (92%); silica gel TLC Rf 0.25 (8:2 hexanes/ethyl acetate); 1H NMR (CDCl3) δ 1.45-1.49 (m, 2H), 1.58 (s, 9H), 1.60-1.64 (m, 1H), 1.76-1.84 (m, 3H), 2.50 (t, 2H, J = 8.0 Hz), 3.21-3.25 (m, 2H), 4.63 (br, 1H), 5.25 (s, 2H) and 7.44-7.53 (m, 5H). To 0.64 g (1.99 mmol) of the obtained benzylated product in 10 mL of DCM at 0 °C was added 5 mL of TFA. The reaction was stirred at 0 °C for 2 h before the solvent was removed under diminished pressure to give 4 as light yellow oil: yield 408 mg (85% over two steps); 1H NMR (CDCl3) δ 1.49-1.52 (m, 2H), 1.75-1.82 (m, 4H), 2.49 (t, 2H, J = 7.2 Hz), 3.06-3.11 (m, 2H), 5.24 (s, 2H), 7.41-7.90 (m, 5H) and 7.92 (br, 2H); 13C NMR (CDCl3) δ 24.34, 25.88, 27.28, 34.08, 40.15, 66.79, 128.46, 128.66, 128.94, 136.12 and 174.15; mass spectrum (ESI), m/z 222.1492 (M+H)+ (C13H19NO2 requires m/z 222.1488).
(1R,2R,3S,5S)-Methyl 3-(((benzyloxy)(phenyl)phosphoryl)oxy)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate (7)
To a solution of 39.0 mg (0.20 mmol) of 5 in 1 mL of THF was added 120 μL of LDA (0.22 mmol, 1.8 M in cyclohexane) of LDA followed by 52.2 (0.23 mmol) of 6 at 0 °C. The reaction mixture was slowly warmed to room temperature and stirred for another 16 h. The reaction mixture was concentrated under diminished pressure and purified by flash chromatography on a silica gel column (25 × 3.2 cm). Elution with 40:1 chloroform/MeOH gave the product 7 as a yellow oil: yield 51.5 mg (60%); silica gel TLC Rf 0.5 (9:1 chloroform/MeOH); 1H NMR (CD3OD) δ 1.48-1.54 (m, 2H), 1.65-1.69 (m, 0.4H), 1.82-1.86 (m, 0.6H), 1.98-2.05 (m, 0.6H), 2.15 (s, 3H), 2.36 (td, J = 12, 6 Hz, 0.4H), 2.49 (td, J = 12, 6 Hz, 0.6H), 2.77-2.79 (t, J = 6 Hz, 0.6H), 2.95-2.97 (t, J = 6Hz, 0.4H), 3.14-3.16 (m, 0.4H), 2.96-3.06 (m, 1H), 3.19-3.21 (m, 0.6H), 3.38-3.40 (m, 0.6H), 3.44-3.64 (m, 0.4H), 3.58 (s, 1.8H), 3.67 (s, 1.2H), 4.61-4.69 (m, 1H), 4.92-5.10 (m, 2H), 7.28-7.35 (m, 5H), 7.39-7.45 (m, 2H), 7.51-7.54 (m, 1H) and 7.74-7.84 (m, 2H); The product was shown to exist as a mixture of benzyl ester diastereomers. 13C NMR (CD3OD) δ 25.45, 25.74, 30.12, 37.84, 41.41, 51.71, 51.79, 50.02, 52.05, 52.28, 61.87, 61.97, 65.17, 65.17, 65.21, 67.78, 67.82, 70.00, 70.04, 128.21, 128.65, 128.69, 128.73, 128.81, 128.85, 128.89, 132.04, 132.12, 132.21, 132.29, 132.77, 132.84, 136.65, 136.70, 170.71 and 170.91; mass spectrum (MALDI-TOP), m/z 430.5 (M + H)+ (theoretical 430.2).
(1R,2R,3S,5S)-Methyl 3-(((benzyloxy)(phenyl)phosphoryl)oxy)-8-azabicyclo[3.2.1]octane-2-carboxylate (8)
To a solution of 46.6 mg (0.11 mmol) of amide 7 in 1 mL of benzene was added 134.4 μL (0.98 mmol) of Troc-Cl and 4.5 mg (0.033 mmol) of K2CO3. After being refluxed at 80 °C overnight, the reaction mixture was quenched with saturated NH4Cl aqueous solution and then extracted with EtOAc. The combined organic layer was washed with brine, dried (MgSO4) and concentrated under diminished pressure. The filtrate was concentrated under diminished pressure. The remaining residue was placed under high vacuum for 2 h, followed by the addition of 2 mL of DMF, 11.1 mg (0.17 mmol) of Zn dust, 2.1 μL (0.056 mmol) of formic acid. The resulted mixture was stirred at room temperature overnight. The reaction mixture was quenched with saturated NH4Cl aqueous solution and then extracted with EtOAc. The combined organic layer was washed with brine, dried (MgSO4) and concentrated under diminished pressure to give the product 8, which was used for the next step promptly without any purification or characterization.
4-((1R,2R,3S,5S)-3-(((Benzyloxy)(phenyl)phosphoryl)oxy)-2-(methoxycarbonyl)-8-azabicyclo[3.2.1]octan-8-yl)-4-oxobutanoic acid (9)
To a solution of 50.9 mg (0.12 mmol) of amine 8 in 2 mL of DMF was added 34.2 μL (0.25 mmol) of Et3N, and 24.5 mg (0.25 mmol) of succinic anhydride. The reaction mixture was then heated to 45 °C, and stirred for 16 h. The reaction mixture diluted with 5 mL of H2O and extracted with three 10-mL portions of EtOAc. The combined organic layer was washed with brine, dried (MgSO4) and concentrated under diminished pressure. The crude products were purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 9 as TFA salt: yield 39.8 mg (63%); 1H NMR (CD3OD) δ 1.55-1.74 (m, 2H), 1.77-2.05 (m, 2H), 2.11-2.38 (m, 1H), 2.45-2.75 (m, 4H), 2.95-3.12 (m, 1H), 3.49 (s, 1H), 3.53 (s, 0.5H), 3.57 (2, 1H), 3.64 (s, 0.5H), 4.41-4.45 (m, 0.6H), 4.55-4.60 (m, 0.4H), 4.65-4.70 (m, 0.4H), 4.86-4.86 (m, 2H), 4.97-5.13 (m, 2.6H), 7.32-7.37 (m, 5H), 7.49-7.52 (m, 2H), 7.60-7.63 (m, 1H), and 7.68-7.75 (m, 2H); The product was shown to exist as a mixture of amide rotamers of benzyl ester diastereomers, and the ratio of two rotamers was estimated to be approximately 2:3. 13C NMR (CD3OD) δ 26.52, 26.94, 26.95, 28.38, 28.53, 28.60, 28.62, 28.81, 51.47, 51.51, 51.75, 51.84, 51.88, 51.91, 53.86, 53.88, 54.27, 54.32, 56.32, 68.74, 68.76, 68.87, 70.26, 70.30, 70.33, 70.37, 70.30, 128.66, 128.70, 129.20, 129.29, 129.39, 131.96, 132.03, 132.04, 132.07, 132.12, 133.76, 136.86, 169.71, 170.79, 170.89, 170.94, 171.01, 175.65 and 175.70; mass spectrum (ESI), m/z 516.1773 (M + H)+ (C26H31NO8P requires 516.1782).
4-((1R,2R,3S,5S)-3-((hydroxy(phenyl)phosphoryl)oxy)-2-(methoxycarbonyl)-8-azabicyclo[3.2.1]octan-8-yl)-4-oxobutanoic acid (10)
A mixture of 11.2 mg (0.02 mmol) of 9 and 5 mg of 10% Pd/C in 1 mL of ethanol was stirred overnight under a H2 atm at room temperature. The catalyst was removed by filtration and the crude products were purified on a VYDAC® C18 reversed phase semi-preparative (250 × 22 mm, 10–15 μm) HPLC column using water and 0.1% TFA in CH3CN mobile phases. A linear gradient was employed (90:10 H2O/0.1%TFA in CH3CN → 10:90 H2O/0.1%TFA in CH3CN) over a period of 40 min at a flow rate of 10 mL/min. Fractions containing the desired product were collected, frozen and lyophilized to give 10 as TFA salt: yield 7.8 mg (85%); TFA salt: yield 7.8 mg (85%) (c 1.15, MeOH); 1H NMR (CD3OD) δ 1.66-2.34 (m, 6H), 2.45-2.76 (m, 4H), 2.96-3.06 (m, 1H), 3.34 (s, 1.2H), 3.56 (s, 1.8H), 3.63-3.65 (m, 1H), 4.44-4.46 (m, 0.6H), 4.57-4.59 (m, 0.4H), 4.67-4.69 (m, 0.4H), 4.80-4.81 (m, 1.6H), 7.47-7.50 (m, 2H), 7.56-7.59 (m, 1H) and 7.72-7.76 (m, 2H); The product was shown to exist as a mixture of amide rotamers of benzyl ester diastereomers, and the ratio of two rotamers was estimated to be approximately 2:3. 13C NMR (CD3OD) δ 26.59, 27.03, 28.46, 28.54, 28.61, 28.86, 29.32, 29.62, 35.61, 35.64, 36.59, 36.61, 51.20, 51.22, 51.40, 51.58, 51.68, 51.75, 53.87, 53.89, 54.40, 56.32, 68.93, 68.97, 128.97, 129.07, 131.71, 131.78, 132.80, 132.82, 169.69, 169.75, 171.06, 171.16, 175.66 and 175.72; mass spectrum (ESI), m/z 426.1310 (M + H)+ (C19H25NO8P requires 426.1312).
Hapten-Protein Immunoconjugates
Haptens GNT and GNE were activated, respectively, at room temperature for 6 h using standard EDC/sulfo-NHS (2 equivalent each) coupling procedure in DMF. After DMF removal under reduced pressure, to the residue was slowly added the corresponding amount of KLH protein (1 mg of hapten versus 1 mg of protein) in 1 mL of 100 mM PBS, pH 7.2. The resultant solution was allowed to stand for 12 h at 4 °C. Coupling efficiencies were monitored using MALDI-TOF MS, save for KLH, which cannot be directly analyzed.
Enzyme-Linked Immunosorbent Assay (ELISA)
Production of cocaine-specific IgG was monitored by ELISA using a GNE-BSA or GNT-BSA conjugate as the coating antigen. Titers were calculated from the plot of absorbance versus log dilution, as the dilution corresponding to an absorbance reading 50% of the maximal value. BSA conjugate was added to COSTAR 3690 microtiter plates and allowed to dry at 37 °C overnight. Following methanol fixation, nonspecific binding was blocked with a solution of 5% nonfat powdered milk in PBS for 0.5 h at 37 °C. Next, mouse serum was serially diluted in a 1% BSA solution across the plate and allowed to incubate for 1–2 h at 37 °C in a moist chamber. Plates were then washed with DI H2O and treated with goat anti-mouse-HRP antibody for 0.5 h at 37 °C. Following another wash cycle, plates were developed with the TMB 2-step kit (Pierce; Rockford, IL).
Radioimmunoassay (Equilibrium Dialysis)
Refined values of antibody affinity and cocaine binding capacity were determined via a soluble radioimmunoassay (RIA). A modified version of Müller’s method19 was followed as it allows for determination of both affinity constant and concentration of specific antibody in serum. The RIA was carried out in a 96-well equilibrium dialyzer MWCO 5000 Da (Harvard Apparatus, Holliston, MA) to allow easy separation of bound and free l-[benzoyl1-3,4-3H(N)]-cocaine tracer; specific activity = 26 Ci/mmol (PerkinElmer, Boston, MA). Mice serum was diluted in RIA buffer (sterile filtered 2% BSA in 1× PBS, pH 7.4) to a concentration that would bind 50% of ~30,000 decays/min of 3H-cocaine tracer. A 75 μL aliquot of serum was combined with 75 μL of radiolabeled tracer (~ 30,000 decays/min); 150 μL of unlabeled cocaine at varying concentrations in RIA buffer (sterile filtered 1% BSA in 1× PBS, pH 7.4) was added to the solvent chamber and the samples were allowed to reach equilibrium on a plate rotator (Harvard Apparatus, Holliston, MA) at room temperature for at least 22 h. A 75 μL aliquot from each sample/solvent chamber was slowly aspirated and suspended in 5 mL of scintillation fluid (Ecolite, ICN, Irvine, CA), and the radioactivity of each sample was determined by liquid scintillation assay.
Locomotor Testing
Sixteen identical hanging wire cages (26 cm wide × 35 cm long × 20 cm high) were used to monitor locomotor activity following daily intraperitoneal injections of saline (1mg/mL) or cocaine (15 mg/kg) in the testing context (n = 7–8 per group). Each cage was equipped with two pairs of infrared emitter–detector photocells that were positioned along the long axis 1 cm from the floor and 8 cm from the front and back of the cage. Photocell interruptions served as a measure of locomotor activity. Beam breaks and cage crossovers were recorded by a PC in 10-minute bins (Supplemental Figures 1–4). Area under the curve (AUC) was calculated using the 30 minutes pre-injection as the threshold, then determining the total peak area in the 60 minutes post-injection. AUCs were compared for each vaccine group using a repeated-measure one-way ANOVA, and a Dunnett’s test was used for post-hoc comparisons.
LC/MS Analysis
The LC/MS analysis was performed using an Agilent G-1956D single quadrupole mass spectrometer equipped with an 1100 Series LC system from Agilent Technologies. Initial rates of antibody-catalyzed degradation of cocaine were determined by monitoring the formation of benzoic acid (BzOH) on a Waters Symmetry C18 column (2.1×150 mm, 3.5 μm particle size) with a guard column. Analytical separation was performed using an isocratic 50:50 mixture of 5 mM ammonium acetate buffer (pH 4.5) and methanol at the flow rate of 0.2 mL/min. Mass calibration curves of BzOH and BzOH-d5 were obtained at six different concentration ratios with 10 μM of BzOH-d5. The kinetic analysis was conducted at ambient temperature in PBS (pH 7.4) with 1.0 μM polyclonal antibody and cocaine at four concentrations from 5 μM to 200 μM. The total volume of all reactions was 100 μL. Experiments were performed by adding the appropriate volume of a 1.0 mM cocaine solution to a solution of polyclonal antibody in PBS. The background assays are conducted at ambient temperature in PBS with the same concentration of cocaine. Ten μL of sample was removed and mixed with 10 μL of 20 μM BzOH-d5 for analysis every 30 min. The concentration of benzoic acid was determined by interpolation of peak height and area relative to standard curves. Initial rates were calculated by linear regression analysis of plots of [benzoic acid] versus time. The kinetic parameters kcat and Km were calculated by fitting plots of initial rate versus cocaine concentration to the Michaelis-Menten equation using Prism Version 5.0b software (GraphPad Software, USA).
Scintillation Method
3H-cocaine (~0.3 mM, 20-50 Ci/mmol, 2 μL) was incubated with GNT antibodies treated or untreated with cocaine in 200 μL of 10 mM PBS, pH 7.4, at 37 °C. After 16 hrs and 64 hrs, 100 μL of the mixture was taken and subjected to dialysis against 1 L of 10 mM PBS (2 exchanges). Ten μL of samples were separated by SDS/PAGE. The band at 150 kDa was excised and placed in a clean microcentrifuge tube. To the tube was added 1 mL of elution buffer (50 mM Tris-HCl, 150 mM NaCl, and 0.1 mM EDTA; pH 7.5). The gel pieces were crushed using a clean pestle and incubated in a rotary shaker at 37 °C overnight, centrifuged at 10,000 × g for 10 minutes. The supernatant was pipetted into a scintillation tube, mixed with 5 mL of scintillation cocktail and assayed by liquid scintillation counting.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge the Skaggs Institute for Chemical Biology and the National Institute on Drug Abuse (DA08590 to K.D.J.) for financial support.
ABBREVIATIONS USED
- Abs
antibodies
- BzOH
benzoic acid
- DMAP
dimethylaminepyridine
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- kcat
turnover number
- Kd
dissociation constant
- KLH
keyhole limpet hemocyannin
- pAbs
polyclonal antibodies
- SAS
Sigma Adjuvant System®
- SNC
succinyl norcocaine
- sulfo-NHS
N-hydroxysulfosuccinimide sodium salt
- Troc-Cl
2,2,2-trichloroethyl chloroformate
Footnotes
Supporting Information. Additional biological assays and compound characterization spectra (1H NMR and 13C NMR). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.National Institute of Drug Abuse . National Household Survey on Drug Abuse, Population Estimates. U.S. Dept. of Health and Human Services; Rockville, MD: 2001. [Google Scholar]
- 2 (a).Des Jarlais DC, Friedman SR. AIDS and i.v. Drug Use. Science. 1989;245:578–579. doi: 10.1126/science.2762809. [DOI] [PubMed] [Google Scholar]; (b) Brody SL, Slovis CM, Wrenn KD. Cocaine-Related Medical Problems: Consecutive Series of 233 Patients. Am. J. Med. 1990;88:325–331. doi: 10.1016/0002-9343(90)90484-u. [DOI] [PubMed] [Google Scholar]
- 3.Carrera MRA, Meijler MM, Janda KD. Cocaine Pharmacology and Current Pharmacotherapies for Its Abuse. Bioorg. Med. Chem. 2004;12:5019–5030. doi: 10.1016/j.bmc.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 4.Moreno A, Janda KD. Immunopharmacotherapy: Vaccination Strategies as a Treatment for Drug Abuse and Dependence. Pharmacol. Biochem. Behav. 2009;92:199–205. doi: 10.1016/j.pbb.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5 (a).Moreno A, Janda KD. Current Challenges for the Creation of Effective Vaccines Against Drugs of Abuse. Expert Rev. Vaccines. 2011;10:1637–1639. doi: 10.1586/erv.11.145. [DOI] [PubMed] [Google Scholar]; (b) Janda KD, Treweek JB. Vaccines Targeting Drugs of Abuse: Is the Glass Half-Empty or Half-Full? Nat. Rev. Immunol. 2012;12:67–72. doi: 10.1038/nri3130. [DOI] [PubMed] [Google Scholar]
- 6 (a).Kosten TR, Rosen M, Bond J, Settles M, Roberts JSC, Shields J, Jack L, Fox B. Human Therapeutic Cocaine Vaccine: Safety and Immunogenicity. Vaccine. 2002;20:1196–1204. doi: 10.1016/s0264-410x(01)00425-x. [DOI] [PubMed] [Google Scholar]; (b) Martell BB, Mitchell E, Poling J, Gonsai K, Kosten TR. Vaccine Pharmacotherapy for the Treatment of Cocaine Dependence. Biol. Psychiatry. 2005;58:158–164. doi: 10.1016/j.biopsych.2005.04.032. [DOI] [PubMed] [Google Scholar]; (c) Martell BA, Orson FM, Poling J, Mitchell E, Rossen RD, Gardner T, Kosten TR. Cocaine Vaccine for the Treatment of Cocaine Dependence in Methadone-Maintained Patients: A Randomized, Double-Blind, Placebo-Controlled Efficacy Trial. Arch. Gen. Psychiatry. 2009;66:1116–1123. doi: 10.1001/archgenpsychiatry.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Haney M, Gunderson EW, Jiang H, Collins ED, Foltin RW. Cocaine-Specific Antibodies Blunt the Subjective Effects of Smoked Cocaine in Human. Biol. Psychiatry. 2010;67:59–65. doi: 10.1016/j.biopsych.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Fox BS, Kantak KM, Edwards MA, Black KM, Bollinger BK, Alison JB, French TL, Thompson TL, Schad VC, Greenstein JL, Gefter ML, Exley MA, Swain PA, Briner TJ. Efficacy of a Therapeutic Cocaine Vaccine in Rodent Models. Nat. Med. 1996;2:1129–1132. doi: 10.1038/nm1096-1129. [DOI] [PubMed] [Google Scholar]; (f) Lazer ES, Aggarwal ND, Hite GJ, Nieforth KA, Kelleher RT, Speralman RD, Schuster CR, Wolverton W. Synthesis and Biological Activity of Cocaine Analogs I: N-Alkylated Norcocaine Derivatives. J. Pharm. Sci. 1978;67:1656–1658. doi: 10.1002/jps.2600671204. [DOI] [PubMed] [Google Scholar]
- 7 (a).Carrera MRA, Ashley JA, Parsons LH, Wirsching P, Koob GF, Janda KD. Suppression of Psychoactive Effects of Cocaine by Active Immunization. Nature. 1995;378:727–730. doi: 10.1038/378727a0. [DOI] [PubMed] [Google Scholar]; (b) Carrera MRA, Ashley JA, Zhou B, Wirsching P, Koob GF, Janda KD. Cocaine Vaccines: Antibody Protection Against Relapse in a Rat Model. Proc. Natl. Acad. Sci. U.S.A. 2000;32:6202–6207. doi: 10.1073/pnas.97.11.6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrera MRA, Ashley JA, Wirsching P, Koob GF, Janda KD. A Second-Generation Vaccine Protects Against the Psychoactive Effects of Cocaine. Proc. Natl. Acad. Sci. U.S.A. 2001;98:1988–1992. doi: 10.1073/pnas.041610998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9 (a).Koob G, Hicks MJ, Wee S, Rosenburg JB, De BP, Kaminsky SM, Moreno A, Janda KD, Crystal RG. Anti-Cocaine Analog to a Disrupted Adenovirus. CNS Neurol. Disord. Drug targets. 2011;10:899–1992. doi: 10.2174/187152711799219334. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hicks MJ, Bishnu PD, Rosenberg JB, Davidson JT, Moreno AY, Janda KD, Wee S, Koob GF, Hackett NR, Kaminsky SM, Worgall S, Toth M, Mezey JG, Crystal RG. Cocaine Analog Coupled to Disrupted Adenovirus: A Vaccine Strategy to Evoke High-Titer Immunity Against Addictive Drugs. Mol. Ther. 2011;19:612–619. doi: 10.1038/mt.2010.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10 (a).Landry DW, Zhao K, Yang GX, Glickman M, Georgiadis TM. Antibody-Catalyzed Degradation of Cocaine. Science. 1993;259:1899–1901. doi: 10.1126/science.8456315. [DOI] [PubMed] [Google Scholar]; (b) Yang G, Chun J, Arakawa-Uramoto H, Wang X, Gawinowicz MA, Zhao K, Landry DW. Anti-Cocaine Antibodies: A Synthetic Approach to Improved Antibody Diversity. J. Am. Chem. Soc. 1996;118:5881–5890. [Google Scholar]; (c) Mets B, Winger G, Cabrera C, Seo S, Jamdar S, Yang G, Zhao K, Briscoe RJ, Almonte R, Woods JH, Landry DW. A Catalytic Antibody against Cocaine Prevents Cocaine’s Reinforcing and Toxic Effects in Rats. Proc. Natl. Acad. Sci. U.S.A. 1998;95:10176–10181. doi: 10.1073/pnas.95.17.10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basmadjian GP, Singh S, Sastrodjojo B, Smith BT, Avor KS, Chang F, Mills SL, Searle TW. Generation of Polyclonal Catalytic Antibodies against Cocaine Using Transition State Analogs of Cocaine Conjugated to Diphtheria Toxoid. Chem. Pharm. Bull. 1995;43:1902–1911. doi: 10.1248/cpb.43.1902. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka F. Catalytic Antibodies as Designer Proteases and Esterases. Chem. Rev. 2002;102:4885–4906. doi: 10.1021/cr010180a. [DOI] [PubMed] [Google Scholar]
- 13.Matsushita M, Hoffman TZ, Ashley JA, Zhou B, Wirsching P, Janda KD. Cocaine Catalytic Antibodies: The Primary Importance of Linker Effects. Bioorg. Med. Chem. Lett. 2001;11:87–90. doi: 10.1016/s0960-894x(00)00659-4. [DOI] [PubMed] [Google Scholar]
- 14.Li P, Zhao K, Deng S-X, Landry DW. Noenzymatic Hydrolysis of Cocaine via Intramolecular Acid Catalysis. Helv. Chim. Acta. 1999;82:85–89. [Google Scholar]
- 15.Isomura S, Hoffman TZ, Wirsching P, Janda KD. Synthesis, Properties, and Reactivity of Cocaine Benzoylthio Ester Possessing the Cocaine Absolute Configuration. J. Am. Chem. Soc. 2001;124:3661–3668. doi: 10.1021/ja012376y. [DOI] [PubMed] [Google Scholar]
- 16.Thomsen M, Caine SB. Psychomotor Stimulant Effects of Cocaine in Rats and 15 Mouse Strains. Exp. Clin. Psychopharm. 2011;19:321–341. doi: 10.1037/a0024798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Müller R. Determination of Affinity and Specificity of Anti-Hapten Antibodies by Competitive Radioimmunoassay. Methods Enzymol. 1983;92:589–601. doi: 10.1016/0076-6879(83)92046-3. [DOI] [PubMed] [Google Scholar]
- 18.McKenzie KM, Mee JM, Rogers CJ, Hixon MS, Kaufman GF, Janda KD. Identification and Characterization of Single Chain Anti-Cocaine Catalytic Antibodies. J. Mol. Biol. 2007;365:722–731. doi: 10.1016/j.jmb.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhan CG, Deng SH, Skiba JG, Hayes BA, Tschmpel S, Shields GC, Landry DW. First-Principle Studies of Intermolecular and Intramolecular Catalysis of Protonated Cocaine. J. Comp. Chem. 2005;26:980–986. doi: 10.1002/jcc.20241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng SX, Bharat N, Fischman MC, Landry DW. Covalent Modification of Proteins by Cocaine. Proc. Natl. Acad. Sci. U.S.A. 2002;99:3412–3416. doi: 10.1073/pnas.042700599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carroll ME, Zlebnik NE, Anker JJ, Kosten TR, Orson FM, Shen X, Kinsey B, Parks RJ, Gao Y, Brimijoin S. Combined Cocaine Hydrolase Gene Transfer and Anti-Cocaine Vaccine Synergistically Block Cocaine-Induced Locomotion. PLoS One. 2012;7:e43536. doi: 10.1371/journal.pone.0043536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.