

Abstract
Multiple Sclerosis (MS) is an immune-mediated demyelinating disease of the central nervous system (CNS) and CD8 T-cells are the predominant T-cell population in MS lesions. Given that transfer of CNS-specific CD8 T-cells results in an attenuated clinical demyelinating disease in C57BL/6 mice with immunization-induced experimental autoimmune encephalomyelitis (EAE), we investigated the cellular target(s) and mechanism(s) of autoreactive regulatory CD8 T-cells. We now report that myelin oligodendrocyte glycoprotein peptide (MOG35–55)-induced CD8 T-cells could also attenuate adoptively transferred, CD4 T-cell-mediated EAE. Whereas CD8−/− mice exhibited more severe EAE associated with increased autoreactivity and inflammatory cytokine production by myelin-specific CD4 T-cells, this was reversed by adoptive transfer of MOG-specific CD8 T-cells. These autoregulatory CD8 T-cells required in vivo MHC Class Ia (KbDb) presentation. Interestingly, MOG-specific CD8 T-cells could also suppress adoptively-induced disease using wildtype MOG35–55-specific CD4 T-cells transferred into KbDb−/− recipient mice, suggesting direct targeting of encephalitogenic CD4 T-cells. In vivo trafficking analysis revealed that autoregulatory CD8 T-cells are dependent on neuro-inflammation for CNS infiltration and their suppression/cytotoxicity of MOG-specific CD4 T-cells is observed both in the periphery and in the CNS. These studies provide important insights into the mechanism of disease suppression mediated by autoreactive CD8 T-cells in EAE.
Keywords: CD8, T-cells, MS, EAE, autoregulatory, autoreactive
INTRODUCTION
MS is an immune-mediated inflammatory demyelinating disease of the central nervous system (CNS) with unclear etiology (1–4). While it is generally believed that the disease is predominantly mediated by pathogenic, autoreactive CD4 T-cells, previous investigations have shown an important role for CD8 T-cells in MS, as highlighted by their enrichment in cerebral spinal fluid (CSF) of MS patients (5), their predominance and oligoclonal expansion in CNS lesions of MS (6, 7) and the prevalence of CNS-specific CD8 T-cell responses in MS patients (8). Studies conducted in murine experimental autoimmune encephalomyelitis (EAE) models, using CD8− or MHC Class I-deficient mice, have suggested both a pathogenic (9–13), as well as regulatory role for CD8 T-cells (9, 14–16). The antigenic specificity of immune regulatory CD8 T-cells remains somewhat unclear, except for specific circumstances, such as TCR-peptide-targeted CD8 T-cells (17, 18). The best characterized immune regulatory CD8 T-cells appear to be Qa-1 restricted, with the capacity to directly recognize CD4 T-cells, as well as activated antigen presenting cells (APC) (19, 20).
Studies focusing on CNS-specific CD8 T-cells have shown their ability to mediate disease pathogenesis, as demonstrated in the C3H mouse strain using MBP-specific CD8 T-cell clones (11), MOG-specific CD8 T-cells in C57BL/6 mice (10, 21) and MOG35–55-specific transgenic TCR-bearing (1C6) CD8 T-cells from NOD mice (22). We have recently demonstrated the unexpected disease regulating ability of CNS-specific CD8 T-cells in wildtype EAE (23). MOG-specific CD8 T-cells could significantly ameliorate EAE in B6 mice (23). Moreover, our recent studies in human MS also suggest an important, clinically relevant immune regulatory function for CNS-targeted autoreactive CD8 T-cells (24). Both MS patients and healthy subjects harbor neuroantigen-specific CD8 T-cell responses (25) with TCR homology to published sequences from CNS-infiltrating T-cells in MS lesions (26). However, the suppressive ability of these cells is dramatically reduced during an acute disease relapse (24). Thus, it is critical to understand the biology of these cells and dissect their mechanism of action.
In the current study, we demonstrate that CNS-specific autoreactive regulatory CD8 T-cells are restricted by classical MHC Class Ia molecules and are capable of directly targeting and suppressing previously activated pathogenic CD4 T-cells. Thus, these studies demonstrate a novel population of disease modulating CD8 T-cells that could be harnessed for adoptive immunotherapy in the future.
MATERIALS AND METHODS
Mice
All experiments used female, six-eight weeks old mice which were housed in climate-controlled pathogen-free facilities under the supervision of certified veterinarians, maintained on a twelve-hour lights on/off cycle, and allowed food and water ad libitum at the UT Southwestern Medical Center Animal Resource Center and used according to approved IACUC protocols. B6.129 CD8−/−, B6.129 β2m−/−, B6.129 IL4−/−, B6.129 IFN-gR−/−, B6.129 IL10−/−, C57BL/6-Tg(Tcra2D2,Tcrb2D2)1Kuch/J and C57BL/6 Prf−/− were purchased from Jackson Laboratory (Bar Harbor, ME). B6.129 IFN-γ−/− were purchased from Jackson Laboratory and kindly provided by Dr. Jerry Niederkorn (UT Southwestern Medical Center, Dallas, TX). B6.129 Tap−/− mice were kindly provided by Dr. James Forman (UT Southwestern Medical Center, Dallas TX). C57BL/KbDb−/− mice were purchased from Taconic (Hudson, NY). Wildtype (WT) C57BL/6 (B6) mice were purchased from Taconic and the UT Southwestern Mouse Breeding Core Facility (Dallas, TX). B6 Ly5.2/Cr mice were purchased from National Cancer Institute (Bethesda, MD).
Active EAE and evaluation
Neuropeptidemyelinoligodendrocyteglycoprotein35–55-peptide(MOG35–55, MEVGWYRSPFSRVVHLYRNGK), and control peptide ovalbumin 323–339-peptide (OVA323–339, ISQAVHAAHAEINEAGR) were synthesized by UT Southwestern Protein Chemistry Technology Center. On day 0, B6 mice were subcutaneously immunized with 100 μg of MOG35–55 in complete freud’s adjuvant (CFA) supplemented with 4 mg/mL mycobacterium tuberculosis (MTB, H37Ra, Difco, Detroit, MI). Additionally, at day 0 and 2, mice were administered 250 ng of pertussis toxin (PTX, List Biological Laboratories, Campbell, CA) via intraperitoneal (i.p.) injection. Clinical EAE disease was assessed using the following criteria; 0, no paralysis; 1, loss of tone in the tail; 2, mild hind limb weakness; 3, significant hind limb paralysis; 4, complete hind limb paralysis; 5, hind limb paralysis and forelimb weakness or moribund/death. Mice that showed grade 5 disease were sacrificed as part of the protocol and were counted as grade 5 through the remainder of the disease course. When appropriate, each experimental condition was represented across multiple cages and the evaluator was blinded to experimental condition, i.e. 2-way blinded EAE scoring.
Adoptive EAE
Lymph node cells from day 10 post-MOG35–55 immunized B6 mice were harvested and incubated for 72 hours at 37°C in EAE culture media (RPMI medium supplemented with 10% fetal calf serum (FSC), L-glutamine, penicillin, streptomycin, HEPES buffer, non-essential amino acids, sodium pyruvate and β-mercaptoethanol) containing 20 μg MOG35–55 and murine rIL-12 (10 ng/mL). CD4 T-cells were obtained using anti-CD4 (L3T4) microbeads (Miltenyi Biotech, Germany) and a total of 5×106 live CD4 T-cells were injected i.p. into naive, wildtype B6 mice at day 0. Pertussis toxin was administered on day 0 and 2 and EAE disease monitored daily.
Autoregulatory CD8 T-cell adoptive transfer experiment
Lymph nodes and splenocytes were harvested from 20–25 days-post immunized mice and viable lymphocytes isolated using Lympholyte-M (CedarlaneLabs, North Carolina) treatment as per manufacturers instructions. Next, cells were stimulated with cognate antigen and murine rIL-2 (10 ρg/mL) for 72 hours at 37°C in a culture flask at 7.5 × 106/mL concentration. Highly purified (TRCβ+CD4−CD8+) CD8 T-cells were obtained using anti-CD8 (Ly-2) microbeads (Miltenyi Biotech, Germany) and a total of 5×106 live CD8 T-cells were injected via intravenous injection tail vein injection (purity ≥ 95%, data not shown). After 24 hours, primary or adoptive EAE was induced and clinical disease evaluated.
CFSE-based proliferation and cytokine assay
Antigen specific responses were evaluated using the CFSE-based dilution assay using bulk splenocyte and lymph node cells from myelin peptide immunized mice. Bulk cells were suspended at a 1 × 106/mL concentration in phosphate buffered saline (PBS) and incubated for 7 minutes with 0.25 μM CFSE. Next, these cells were washed twice with serum-containing media and resuspended at 2×106 cells/mL concentration in EAE culture media. Cells were activated with cognate antigen (MOG35–55, or OVA323–339) at 20 μg/mL and mIL-2 at 10 ρg/mL at 37°C in 5% CO2 for 5 days. Subsequently, cells were washed with staining buffer, incubated for 5 minutes at 4°C with mouse FcR blocking reagent (Miltenyi Biotec) and labeled with eFluor-605NC-anti-CD8 (eBioscience), APC-Cy7-anti-CD4, PE-Cy7-anti-CD25, PE-Cy5-anti-TCRβ (BD Bioscience). After a 45 minute incubation at 4°C, cells were washed with staining buffer and fixed in 1% paraformaldehyde (PFA, Electron Microscopy Sciences, Hatfield, PA.). Flow cytometric data was acquired using a BD LSR II flow cytometer using FACS Diva 5.0 software. FlowJo 9.0 (Tree Star, Ashland OR) software was used to gate on LiveGate+TCRβ+CD4−CD8+ or LiveGate+TCRβ+CD8−CD4+ T-cell subsets and cognate antigen specific responses within the CFSE low population. Responses were considered positive when the two following conditions were met: delta proliferation value (ΔPF) of the cognate antigen stimulated condition was 1% greater and the stimulation index was 2X greater than the no antigen condition.
Intracellular cytokine staining
Following in vitro stimulation with cognate antigen, cells were re-stimulated with 25 ng/mL of phorbol 12-myristae 13-acetate (PMA, Sigma, St, Louis, MO), 1μg/mL of Ionomycin (IO, Sigma) and 10 μg/mL of brefeldin-A (BFA, Sigma) at 37°C in 5% CO2 for 5 hours. Next, cells were permeabilized and fixed using murine Foxp3 staining buffer set (Miltenyi Biotec) as per manufacture’s instructions. Cells were then stained with PE-Cy7-anti-IFN-γ, PE-Cy7-anti-TNF-α, PE-anti-IL17A, APC-anti-IL10, APC-anti-IL4, PE-anti-GM-CSF (BD Biosciences) PE-anti-perforin and APC-anti-Foxp3 (eBioscience) fluorescent antibodies, fixed with 1% paraformaldehyde and flow cytometric data acquired within 24 hours.
CNS Trafficking assay
Congenic Ly5.2+ (CD45.1+) B6 mice were immunized with MOG35–55 emulsion and PTX, as previously described. At day 20, draining lymph nodes and spleens were harvested and single-cell suspension prepared. These cells were then placed into culture for 3 days in EAE culture media with stimulating antigen (MOG35–55 or OVA323–339) at 20 ug/mL and murine rIL-2 (10 pg/ml). Post in vitro stimulation, dead cells were removed using Dead Cell removal kit (Miltenyi Biotec) and CD4+ or CD8+ T-cells isolated using CD4 (L3T4) or CD8 (Ly-2) microbeads (Miltenyi Biotec). The purity of T-cells was consistently higher than 95%. A total of 5 × 106 CD4 or CD8 T-cells were injected intravenously into naive, wild-type B6 mice at day 0. Subsequently, at day 10 and 20, mice were anesthetized with 400 μl of 1.5% Avertin and perfused with 20 mL of cold PBS via left ventricular puncture. Brain and spinal cord tissue was harvested and processed via 30% Percoll (GE Healthcare, New Jersey) gradient. Cervical and inguinal lymph nodes and spleens were harvested and processed using EAE washing media, followed by RBC lysis buffer and subsequent wash. Once all tissues where processed into single-cell suspension, cells were washed with FACS buffer and incubated for 5 minutes at 4°C with mouse FcR blocking reagent (Miltneyi Biotec). Cells were then stained with APC-anti-45.1, Percp5.5-anti-CD45.2, Pacblue-anti-CD4, PE-Cy7-anti-CD8 (BD Biosciences), incubated for 45 mins. in 4°C, washed with staining buffer and fixed with 1% PFA.
In vitro killing assay
As described previously (27) but adapted for murine cells, cytotoxic MOG-specific CD8 T cells were obtained using splenocytes from day 12-immunized mice, which were in vitro activated and expanded for 7 days in MOG35–55 at 20 μg/mL and purified using negative selection CD8 T-cell Isolation Kit (Miltenyi Biotec, Germany). Target splenocytes were harvested from naive WT mice at day 18 and incubated with MOG35–55 (20 μg/mL) and concanavalin A (ConA) at 0.5 μg/mL in 37°C and 5% CO2 and the following day CFSE stained. CFSE stained (targets) cells were resuspended in a 96-well plate at 5000 cell/well. Effector cells were suspended with targets at 0:1; 1:1; 4:1; 16:1; 64:1 ratio in 200 μL of EAE culture media. Following 24 hour incubation at 37°C and 5% CO2, wells were seeded with fluorescent APC beads (BD Biosciences) and data immediately collected on a BD FACS Calibur flow cytometer. For controls purposes, a redirected cell lysis using a mastocytoma cell line (P815) was incubated with murine anti-CD3 (1 μg/mL) and effector CD8 T-cells at indicated ratios. Percent killing was calculated as previously described (27).
Data analysis
Statistical analyses between groups were performed using GraphPad Prism 5.0c. Difference in disease severity, peak and onset were evaluated using two-tailed Student’s t-test. A p value ≤ 0.05 was considered statistically significant.
RESULTS
Autoregulatory CD8 T-cells suppress CD4 T-cell-mediated autoimmune disease
We have recently demonstrated the unexpected finding that autoreactive MOG35–55-induced CD8 T-cells could attenuate EAE in MOG35–55-immunized B6 mice (23). To dissect the mechanisms of this suppression, we first asked whether this attenuation was restricted to actively induced EAE or could be seen in EAE transferred by pre-primed CD4 T-cells. Similar to our prior findings, the transfer of MOG35–55-induced CD8 T-cells (MOG-CD8) one day prior to active EAE induction resulted in significant reduction of disease severity compared to mice receiving OVA323–339-induced CD8 T-cells (OVA-CD8) as controls (Fig. 1A).
Figure 1. Autoreactive myelin-specific CD8 T-cells suppress CD4 T-cell mediated autoimmune demyelinating disease.
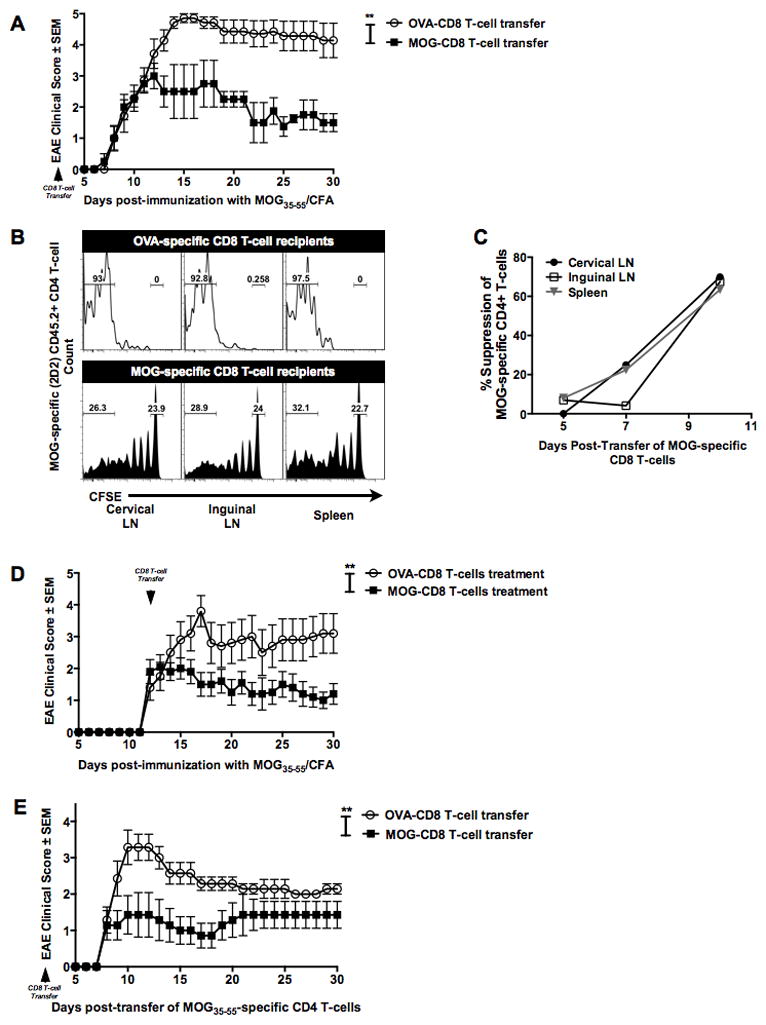
(A) MOG- (or OVA)-specific CD8 T-cells (purity ≧95%) were transferred one day prior to primary EAE induction. EAE clinical severity was evaluated in a blinded manner and the two groups were compared. Mean clinical scores ± SEM are shown on the y-axis versus days post-immunization on the x-axis. Data are representative of at least three independent experiments (n=15 per condition).
(B) MOG- (or OVA)-specific CD8 T-cells were transferred into naive CD45.1+ mice on day −2. On day −1, all mice received CFSE-labeled MOG35–55-specific transgenic (2D2) CD45.2+ CD4 T-cells and were immunized with MOG35–55/CFA on day 0. At days 5, 7 and 10 post-immunization, cervical and inguinal lymph nodes and spleens were harvested and CFSE dilution of CD45.2+ TCR β + CD8− CD4+ T-cells measured. Representative histograms of 2D2 CD4 T-cells CFSE dilution at day 10 in control CD8 T-cell recipient mice (top panel) and MOG-specific CD8 T-cells (bottom panel) are shown. Numbers on the histograms indicate the percentage of gated cells that were either in the CFSE-lowest or CFSE-high fractions in the OVA-CD8 and MOG-CD8 recipients.
(C) The graph indicates % suppression, calculated as % suppression = 100% − ((test condition/control condition)*100). Data are representative of three independent experiments. Cumulative graphs from days 5, 7, and 10 are shown.
(D) Mice were immunized with MOG35–55/CFA, followed by transfer of MOG-specific (or control) CD8 T-cells at day 12 post-immunization. Data are representative of two independent (n=20 per condition).
(E) MOG- (or OVA-) CD8 T-cells were transferred into naïve mice. The next day, adoptive EAE was induced using purified activated CD4 from MOG35–55-immunized mice. Data are representative of 5 experimental replicates (n=25 per condition).
In this setting, we wanted to test whether transfer of MOG-specific CD8 T-cells had an effect on MOG35–55-specific CD4 T-cell responses. We addressed this by adoptively transferring transgenic MOG-specific (2D2) CFSE-stained naive CD4 T-cells (CD45.2+) into CD45.1+ congenic mice treated with either MOG- or OVA-specific CD8 T-cells (CD45.1+) and subsequent immunization with MOG35–55 peptide. CFSE dilution of gated 2D2 CD45.2+ CD4+ T-cells was evaluated at various time points post-immunization. Unimpeded 2D2 CD4 T-cell responses to MOG35–55 were observed in control-CD8 T-cell recipients, whereas MOG35–55-induced CD8 T-cell-treated mice showed a significant portion of undiluted CFSE-high 2D2 CD4 T-cells (0% vs 23.9% in CLN, 0.2% vs 24% in ILN and 0% vs 22.7% in SPL, Fig. 1B). In fact, in vivo encephalitogenic CD4 T-cell suppression by autoregulatory CD8 T-cells could be weakly detected as early as day 5-post transfer, although this suppression was most robustly observed on days 7 and 10 post-immunization (Fig. 1C).
We next observed that transfer of MOG-CD8 T-cells following the onset of disease also attenuated the clinical course of EAE (Fig. 1D), suggesting the ability of these cells to interfere with CD4 T-cell responses even after initial CD4 T-cell priming. To confirm this finding, we studied the effect of CD8 T-cell transfer on disease induced by the adoptive transfer of purified MOG35–55-specific CD4 T-cells. Again, to control for cell numbers, OVA-specific CD8 T cells were used as controls, as in prior experiments these cells did not affect the course of active or adoptive disease ((23) and data not shown). Relative to controls (and similar to our findings in active disease), autoreactive regulatory (autoregulatory) CD8 T-cells could also suppress adoptively transferred EAE (AT-EAE) (Fig. 1E), suggesting that they could target/suppress pathogenic CD4 T-cells.
CD8−/− mice exhibit augmented CD4 T-cell autoreactivity, which can be reversed by autoregulatory CD8 T-cells
To study the effect of transferred CD8 T-cells without contribution from endogenous CD8 T-cells, we used CD8−/− mice, which are known to have an increase in disease severity in this model (15). We hypothesized that in CD8-deficient mice, EAE severity increases due to the unfettered activation of encephalitogenic CD4 T-cells. First, we induced EAE in CD8−/− B6 mice using a range of immunizing dose of MOG35–55 (200, 100 and 50 μg per mouse). As expected, disease was significantly more severe with greater incidence in CD8−/− mice compared to littermate controls, with the differences more obvious when sub-optimal doses were used (Table I). Disease curves at the 100 μg dose demonstrated a significant effect at the recovery/chronic stages of disease (Fig. 2A). These observations were also confirmed in experiments where CD8 T-cells were depleted using anti-CD8 antibody vs. IgG control injections (Fig. S1A and S1B). CD8-depleted mice showed an increase in mean maximum score and decrease in mean day onset (Fig. S1C).
Table I.
Increased EAE disease susceptibility in CD8−/− B6 mice
| % of incidence | Mean Day of Onset | Mean Max Score | ||||
|---|---|---|---|---|---|---|
| WT | CD8−/− | WT | CD8−/− | WT | CD8−/− | |
| 200ug | 10/10 (100%) | 10/10 (100%) | 9.5 +/− 0.2 | 9.4 +/− 0.3* | 4.3 +/− 0.2 | 3.6 +/− 0.1 |
| 100ug | 8/10 (80%) | 10/10 (100%) | 15 +/− 2.3 | 10 +/− 0.3* | 3.05 +/− 0.5 | 3.95 +/− 0.2* |
| 50ug | 8/10 (80%) | 10/10 (100%) | 15.3 +/− 2.8 | 9.5 +/− 0.3* | 3.05 +/− 0.6 | 4.35 +/− 0.2* |
Clinical disease data statistically compared between WT and CD8−/− mice.
p<0.05
Figure 2. Autoregulatory CD8 T-cells are sufficient in reversing augmented disease and CD4 autoreactivity in CD8−/− mice.
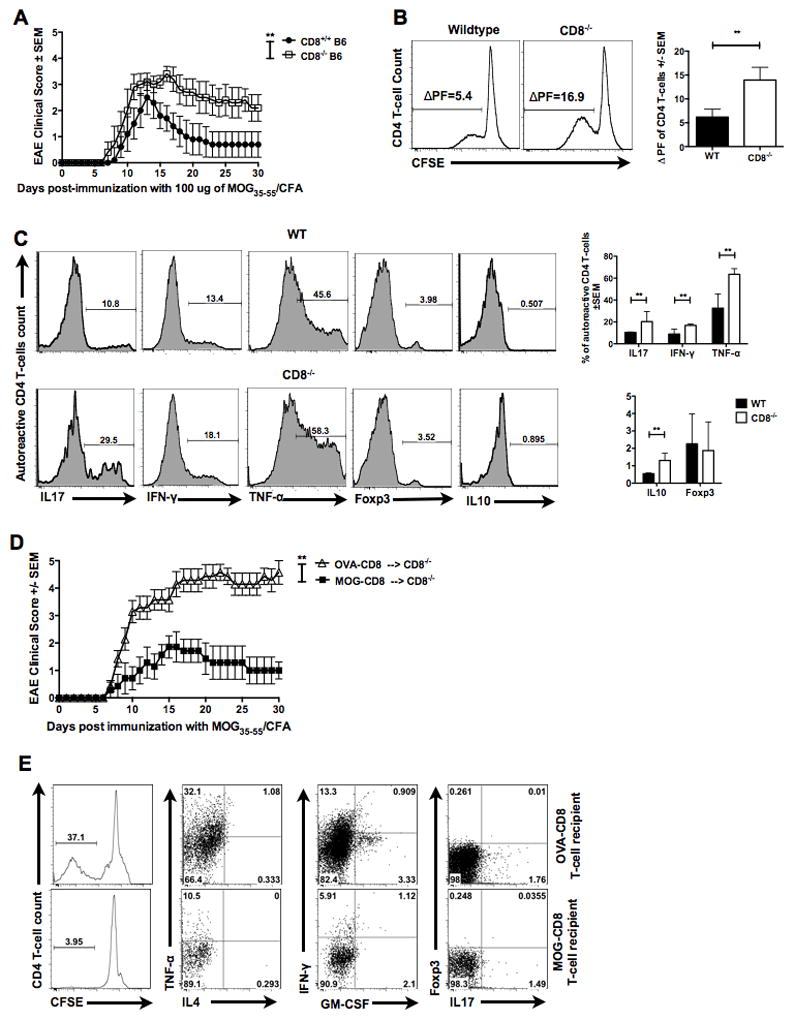
(A) CD8+/+ and CD8−/− mice were immunized with 100 μg of MOG35–55 and disease course evaluated for 30 days. Representative of three independent experiments (n=15 per condition).
(B) Draining lymph node cells from WT or CD8−/− MOG35–55-immunized mice were CFSE stained and cultured for 5 days with cognate antigen. CFSE dilution in TCRβ+CD8−CD4+ T-cells was evaluated using flow cytometry. Representative histogram (left panels) and cumulative data (right panel) of three independent experiments are shown (n=9 per condition). Δ PF represents difference between proliferation in the presence of antigen and background.
(C) Five-day CFSE cultures were stimulated with PMA/ionomycin/brefeldin-A and stained for surface and intracellular markers. Representative histograms (left panels) and cumulative graphs (right panels) of gated CFSE-low (proliferating) TCRβ+CD8−CD4+ T-cells are shown for the indicated functional molecule (n=10 per condition).
(D) OVA- or MOG-specific CD8 T-cells were transferred into CD8−/− B6 mice i.v. One day later, all mice where immunized with MOG35–55/CFA and EAE clinical disease evaluated for 30 days. Representative of two independent experiments (n=11 per condition).
(E) Flow cytometry data of MOG35–55-responding CD4 T-cells from CD8−/− recipients of MOG-specific or OVA-specific CD8 T-cells. Representative of two independent experiments (n=11 per condition)
Next, we determined if the increase in disease susceptibility in CD8−/− mice correlated with an altered functional profile of the neuroantigen-specific CD4 T-cells. First, we compared MOG35–55-specific CD4 T-cells recall responses between CD8−/− and WT mice using the CFSE dilution assay. Briefly, WT and CD8−/− mice were immunized with MOG35–55, followed by harvesting of draining LN cells, spleen cells and CNS-infiltrating cells at day 20. Cells were stained with CFSE and cultured in vitro for five days in the presence or absence of cognate antigen. Subsequently, cells were stained with fluorophore conjugated anti-TCRβ, CD4 and CD8 antibodies, and CFSE dilution measured within the TCRβ+CD8−CD4+ population. Replicate cultures were stimulated on day 5 with PMA/ionomycin/brefeldin-A mixture for 4 hours for characterization of cytokine profiles. Both peripheral (Fig. 2B) and CNS (Fig. S2) CD4 T-cells from CD8−/− mice were found to have a significantly higher recall response to MOG35–55 peptide, as compared to WT. Moreover, evaluation of intracellular cytokine production within the MOG35–55-responding (CFSE-low) CD4 T-cells revealed an increase in IL17A, IFN-γ and TNF-α producing MOG35–55-specific CD4 T-cells in CD8−/− mice as compared to WT (Fig. 2C). Interestingly, IL10+ MOG35–55-specific CD4 T-cells were found to be increased in CD8−/− mice. Foxp3+ MOG35–55-specific CD4 T-cells were found to be comparable between CD8−/− and WT cohorts.
We next determined if autoregulatory CD8 T-cells were sufficient in reversing the increased EAE severity in CD8−/− mice. We performed a rescue experiment by reconstituting CD8−/− mice with MOG-specific autoregulatory CD8 T-cells (versus OVA-specific controls), followed by induction of primary EAE. In CD8−/− mice, transfer of MOG-specific CD8 T-cells was sufficient to significantly suppress EAE (Fig. 2D). At the same time, recall CD4 T-cell responses from “protected” mice revealed a decrease in TNF-α, IFN-γ, IL17A and GM-CSF producing encephalitogenic MOG35–55-specific CD4 T-cells (Fig. 2E).
Myelin antigen-specific CD8 T-cell activation, as well as disease suppression is MHC Class Ia-dependent
Next, we delineated the requirement of MHC Class I molecules in CD8 T-cell-mediated suppression of autoimmune demyelinating disease. First, we performed an in vitro blocking assay where CFSE-stained bulk splenocytes from MOG35–55-immunized mice were cultured with IgG isotype control, anti-KbDb antibody or anti-Qa-1b antibody and stimulated with either MOG35–55 peptide or concanavalin A (ConA) for 5 days. Relative to the isotype control, KbDb blockade (but not Qa-1b blockade) showed a reduction in MOG-specific CD8 T-cell response (Fig. 3A). ConA stimulation revealed no appreciable loss of responses with either KbDb or Qa-1b blockade (data not shown). To confirm our antibody blockade findings, we ascertained if MHC I-deficient APC could stimulate autoregulatory CD8 T-cells. In vitro stimulation with cognate antigen revealed a significant response from MOG-specific CD8 T-cells in the presence of WT APC, which was absent in KbDb−/− APC culture condition (Fig. S3A).
Figure 3. Autoregulatory CD8 T-cells require MHC Class Ia in vivo for disease amelioration.
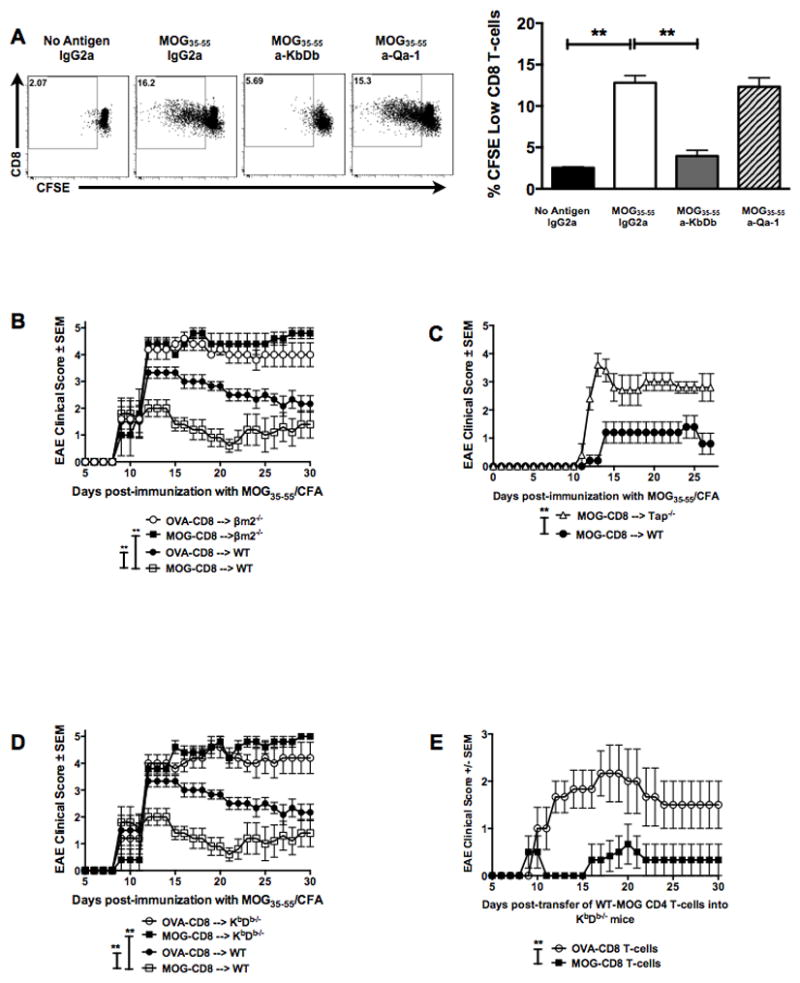
(A) In vitro blocking experiments using antibodies to classical and non-classical MHC Class I molecules were performed in order to evaluate the role MHC Class I in CD8 T-cell mediate disease suppression. Representative CFSE dilution histograms of gated TCRβ+CD4−CD8+ T-cells are shown (left panels) as well as the cumulative data (right panel). Numbers indicate CFSE-low fraction of CD8 T-cells in the presence of IgG control, anti-KbDb or anti-Qa-1b antibody.
(B) In vivo MHC requirements were evaluated by transferring WT MOG- (or OVA-reactive) CD8 T-cells into either WT or β2m−/− mice. MOG35–55 EAE was induced the following day and clinical disease evaluated in all four groups. Representative of two independent experiments (n=20 per condition).
(C) Disease curves for MHC Class I deficient (Tap−/−) versus control group (WT). Both groups received WT MOG-specific CD8 T-cells at day -1 and were immunized with MOG35–55/CFA at day 0. Representative of two independent experiments (10 mice per condition).
(D) MOG- (or OVA-) CD8 T-cells were transferred into either WT or KbDb−/− mice, followed by disease induction. Data are representative of two independent experiments (n=20 per condition).
(E) WT MOG- or OVA-CD8 T-cells were transferred into KbDb−/− mice, followed by induction of adoptive EAE using WT MOG35–55-specific CD4 T-cells. Representative EAE data of two independent experiments (n=14 per condition).
Next, we evaluated the requirement of in vivo MHC Class I presentation during CD8 T-cell-mediated disease regulation. WT B6 MOG-specific CD8 T-cells were transferred into β2m−/− (MHC Class I deficient) or WT mice, followed by MOG35–55 EAE induction. Compared to the protection seen when WT MOG-specific CD8 T-cells were transferred into WT recipient mice, these same cells had no effect on EAE disease in β2m−/− recipients (Fig. 3B). Similarly, WT MOG-specific CD8 T-cells were incapable of suppressing disease when transferred into Tap−/− (MHC Class I deficient) mice (Fig. 3C). However, in β2m−/− and Tap−/− mice, there is loss of CD8 T cells over time, which may explain these findings. Therefore, we further elucidated the requirement of classical (Class Ia) vs non-classical (Class Ib) MHC Class I, by using the KbDb−/− (MHC Class Ia deficient and Class Ib competent) mice, where we observed >50% survival of transferred CD8 T-cells, compared to WT hosts (data not shown). Even in this setting, the transferred CD8 T-cells were ineffective in disease amelioration (Fig. 3D), indicating an in vivo requirement of classical MHC Class Ia-restricted presentation. Similar to prior reports (16), we also observed an augmentation of disease in MHC Class I-deficient mice when comparing the control CD8 T-cell-treated groups (Fig. 3B and 3D).
Thus, having observed that autoregulatory CD8 T-cells could suppress CD4 T-cell-mediated adoptive disease (Fig. 1E) and that disease suppression by these cells was MHC Class Ia-dependent (Fig. 3D), we next investigated whether targeting of MHC Class I-replete CD4 T-cells by autoregulatory CD8 T-cells may be a sufficient mechanism for EAE amelioration. For this, we used the AT-EAE model, such that, only the inducing CD4 T-cells were the source of MHC Class Ia molecules. This was done by modifying our adoptive EAE protocol and transferring purified WT MOG35–55-specific CD4 T-cells into KbDb−/− host mice. Interestingly, autoregulatory CD8 T-cells were capable of suppressing WT adoptively-transferred EAE when the recipient host was devoid of MHC Class Ia molecules (Fig. 3E). Hence, we concluded that focused targeting of encephalitogenic CD4 T-cells was sufficient in modulating disease severity.
Suppression of EAE by autoregulatory CD8 T-cells is dependent on IFN-γ and perforin but not IL4 or IL10
The phenotypic characteristics of autoregulatory CD8 T-cells were examined by combining the CFSE dilution assay and evaluating the expression of various functional molecules on MOG-specific (proliferating) CD8 T-cells. A significant proportion of autoregulatory CD8 T-cells were found to produce IFN-γ, TNF-α and perforin, whereas a negligible fraction showed IL17A, IL10, Foxp3 or IL4 positivity (Fig.S4A). To test potential cytotoxic function, we directly determined if autoregulatory CD8 T-cells could be cytotoxic by performing an in vitro killing assay. ConA-stimulated MOG-loaded splenocytes were used as target cells and cultured with an increasing number of MOG-specific CD8 T-cells in media alone or with cognate antigen for 48 hours. Killing was measured by evaluating the number of target cells normalized to control beads. For control purposes, MOG-specific CD8 T-cells were cultured with P815 cells decorated with murine anti-CD3 (re-directed lysis). Autoregulatory CD8 T-cell killing increased as effectors to target cell ratio increased (Fig. S4B).
In view of these functional observations, we next evaluated the relevance of these cytokines to in vivo disease suppression. It has been previously shown that IFN-γ producing CD8 T-cells may act in a suppressive manner (17, 28, 29). Thus, we evaluated the role of IFN-γ in autoregulatory CD8 T-cell disease inhibition. We obtained IFN-γ-deficient MOG-specific CD8 T-cells from MOG35–55-immunized IFN-γ−/− B6 mice. We transferred these CD8 T-cells into WT naive recipient mice at day -1. As controls, we also transferred MOG- and OVA-specific CD8 T-cells from WT mice. At day 0, EAE was induced in all three groups and clinical disease evaluated. Recipients of IFN-γ-deficient MOG-specific CD8 T-cells showed no protection from disease, exhibiting significantly more severe disease at the acute and chronic phases, compared to the WT MOG-specific CD8 T-cell recipients (Fig. 4A). Tajima and others have shown that IL12 can augment the activation of CD8 T-cells (30, 31). Thus we supplemented our cultures with IL12 and observed both an increase in IFN-γ+ autoreactive CD8 T-cells, as well as an increase in IFN-γ production per cell (Fig. S4C). These increases in IFN-γ production and IFN-γ+ cells correlated with an enhanced disease-suppressing role (Fig. S4D). We also evaluated the role of the cytotoxic molecule perforin, cytokines IL-4, IL-10 and the requirement of IFNγ-R on MOG-specific CD8 T-cells. We observed that in vivo protection by these cells was also dependent on perforin (Fig. 4B), but not the other molecules (Fig. 4C–E).
Figure 4. Autoregulatory CD8 T-cell ameliorate EAE via IFN-γ and perforin.
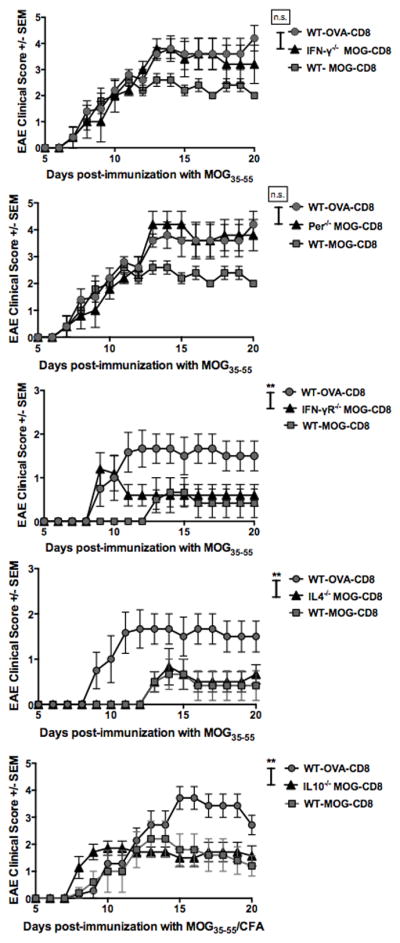
In all experiments, WT MOG- and OVA-specific CD8 T-cells were transferred i.v. into WT B6 mice. Additional groups received MOG-specific CD8 T-cells derived from IFN-γ−/− (A), perforin−/− (B), IFN-γR−/− (C), IL10−/− (D) or IL4−/− (E) mice. The next day, EAE was induced in all groups by MOG35–55/CFA immunization. Clinical scores are shown. Each graph is representative of two independent experimental replicates (n=14 per condition).
MOG-specific CD8 T-cells decrease both peripheral and CNS MOG-specific CD4 T-cell numbers
We next wanted to ascertain the in vivo location of CD8 T-cell-mediated immune regulation (i.e., peripheral immune compartment vs. CNS). First, we evaluated whether MOG-CD8 T-cells could traffic to the CNS. After transferring congenic CD45.1+ MOG-CD8 donor T-cells i.v., we attempted to detect them in the CNS at days 5, 10 and 20. While we could consistently detect cells in the cervical and inguinal lymph nodes, as well as spleen, we could not detect MOG-specific CD8 T-cells in the CNS (Fig. 5A). In contrast (and as expected), CD45.1+ MOG-CD4 T-cells readily trafficked into naïve CNS, as part of establishing adoptive disease (Fig. 5A). We next evaluated the tissue infiltration of autoregulatory CD8 T-cells during active CNS inflammation. We transferred CD45.1+ MOG-specific CD8 T-cells, followed by induction of primary EAE. During active disease, congenic CD8 T-cells were detectable in the CNS at days 10 (data not shown) and 20 (Fig. 5B). These data demonstrate CNS infiltration by autoregulatory CD8 T-cells only during CNS pathology, further reducing the possibility that they could be independently pathogenic and strongly suggesting that they may potentially have their regulatory effect at the site of pathology.
Figure 5. Autoregulatory CD8 T-cells suppress MOG-CD4 T-cells numbers in the peripheral and CNS compartments.

(A) In vivo trafficking studies were performed using MOG-specific CD45.1+ CD8+ (top panels) or CD4+ (bottom panels) T-cells, which were transferred into naive (CD45.2+) mice and indicated tissues harvested at (day 10 data not shown) and day 20. Numbers indicate percentage of CNS-specific CD8 or CD4 T-cells that were CD45.1+. Representative of two independent experiments (n=10 per condition).
(B) After adoptive cell transfer of MOG-specific CD45.1 CD8 T-cells, EAE was induced in recipient CD45.2 mice. Indicated tissues were harvested at day 10 (similar results at day 20, data not shown). Numbers indicate percentage of CNS-specific CD8 T-cells that were CD45.1+. Representative of two independent experiments (n=10 per condition).
(C) In vivo tracking of MOG-specific TCR-transgenic (2D2) CD45.2+ CD4 T-cells was performed by transferring these cells into CD45.1+ recipients of OVA- or MOG-specific CD8 T-cells. Indicated tissues were analyzed 10 days later. Percentages and absolute counts of CD45.2+ CD4 T-cells in tissues are indicated in representative dot plots (top) and cumulative graphs (bottom) of three independent experiments (n=24 per condition).
To address the effect of autoregulatory CD8 T-cells on CNS-infiltrating CD4 T cells, we transferred MOG- or OVA-CD8 T-cells into CD45.1+ B6 mice, followed by transfer of MOG35–55-specific transgenic (2D2) CD45.2+ CD4 T-cells, with subsequent induction of primary EAE. 2D2 CD4 T-cell numbers were evaluated in the CNS and the periphery. We observed reduction in the numbers of MOG35–55-specific CD4 T-cells both in the periphery (inguinal LN and spleen) as well as in the CNS (Fig. 5C), suggesting that the disease suppression could be explained by either a peripheral mechanism alone or a combination.
DISCUSSION
As thymic negative selection is not perfect, the presence of peripheral autoreactive CD4 and CD8 T-cells is the norm rather than the exception. By corollary, peripheral control of potentially pathogenic autoreactive T-cells is important in controlling immune-mediated disease. It is becoming clearer that several immune-mediated diseases, such as MS, are characterized by perturbation of immune regulation (27, 32–38). In the murine model of MS (EAE), there is ample evidence that neuroantigen-specific CD4 T-cells can initiate and sustain neuroinflammation and pathology. In contrast, the role of CD8 T-cells, particularly CNS-specific CD8 T-cells, is still poorly understood and somewhat controversial. Some studies have demonstrated a potential pathogenic role for CNS-targeted CD8 T-cells (10, 11, 21, 22, 39), and this makes intuitive sense in that CNS antigens would be presented to these cells by resident CNS cells in the context of MHC Class I. In that regard, recent mouse models based on transgenically expressed CNS-sequestered antigens combined with TCR-transgenic CD8 T-cells or HLA-transgenic mice also suggest that these cells may have pro-inflammatory potential (40–42). While these studies do demonstrate the pathogenic potential of certain CD8 T-cells, many of them involved either induced homeostatic expansion of CD8 T-cells in T-cell-deficient mice or the use of transgenic manipulation, potentially selecting for either a rare self-reactive clone(s) or a clone(s) of T-cells that does not evolve through usual thymic selection.
Using wildtype B6 mice, we have recently demonstrated the unexpected disease regulatory role of CNS-targeted CD8 T-cells (23). As a population, MOG-specific CD8 T-cells are able to suppress MOG35–55-induced primary EAE. We have observed similar suppressive function for CD8 T-cells of other CNS specificities as well (unpublished data, manuscript in preparation). In the current study, we further confirm that these autoregulatory CD8 T-cells can suppress ongoing primary EAE, as well as EAE induced with adoptively transferred, pre-activated CD4 T-cells. Using different variants of MHC Class I-deficient mice, we demonstrate that these cells are restricted by classical MHC Class I molecules (KbDb) and their function is mediated through a combination of IFN-γ-mediated regulation and perforin-mediated cytotoxicity. We further show that in the absence of CD8 T-cells, CD4 T-cells show enhanced pro-inflammatory phenotype, which is reversed by the transfer of CNS-specific CD8 T-cells. Importantly, using the adoptive transfer model where only the transferred CD4 T-cells had MHC Class Ia, we provide evidence that these autoregulatory CD8 T-cells are capable of directly targeting and suppressing encephalitogenic CD4 T-cells.
CNS-specific autoregulatory CD8 T-cells appear to be a unique class of T-cells that have some features of cytotoxic CD4-targeting Qa-1-restricted regulatory CD8 T-cells (19, 20), at the same time being classical MHC I-restricted, much like effector CD8 T-cells. Thus, they are distinct from therapeutically induced regulatory CD8 T-cells (20, 27, 33, 43, 44). Moreover, they are specific for target tissue antigen (CNS), requiring the in vivo priming with their cognate antigenic peptide (unpublished data). In that regard, they appear to be similar to other autoantigen-specific CD8 T-cells described in diabetes models (45). It is interesting to note that tissue-specific CD8 T-cells can target CD4 T-cells in an MHC class I-restricted manner. This requires that CNS antigen get presented on the CD4 T-cells. Several mechanisms may result in this ability, including passive loading of digested myelin components at site of pathology or trogocytosis (i.e., membrane exchange) between APC and CD4 T-cells, as proposed in prior studies (46–50). Future studies are needed to elucidate these mechanisms in the context of EAE. However, it is notable that effects on CD4 T-cell numbers and function were noted both in the periphery as well as in the CNS, suggesting that the mechanism of suppression could be operative at both sites.
Two important mechanisms of these autoregulatory CD8 T-cells are noteworthy. First, these cells produce IFN-γ and this production is necessary for in vivo disease amelioration. It is known that IFN-γ has pleiotropic properties, including alteration of indoleamine 2,3-dioxygenase (IDO) production, augmentation of the regulatory capacity of DC (51), modulation of MHC Class I expression on APC (52) and conversion of effector CD4 T-cells to CD4 Tregs (28), all potential avenues which might be used for inhibiting the presentation of myelin antigen and decreasing encephalitogenic CD4 T-cell activity. Similarly, perforin was found to be necessary for in vivo disease amelioration and we hypothesize that its primary role is to target pathogenic CD4 T-cells. Again, unlike the regulatory Qa-1-restricted CD8 T-cells, which secrete IL10 in order to exert their inhibitory activity, EAE-generated autoregulatory CD8 T-cells did not need to produce IL-10 to mediate their effects. While we have shown than CD4 T-cell targeting by autoregulatory CD8 T-cells is sufficient for some of their function in a reductionist approach, this does not rule out their effects on APC, either through IFN-γ-mediated modulation or cytotoxic killing of activated, pro-inflammatory APC. This question is being actively addressed in ongoing studies.
The clinically relevant role of these autoregulatory CD8 T-cells is revealed by the observations that these T-cells infiltrate their target organ only during ongoing CNS inflammation. These temporal dynamics explain the somewhat delayed effect on EAE, as evidenced in CD8-deficient mice. Moreover, lack of CD8 T-cell infiltration in unimmunized mice also strongly argues against a predominant pathogenic role for these cells. The importance of CNS-specific CD8 T-cells in human MS was highlighted by our recent observations, where we showed that CNS-specific CD8 T-cells showed immune suppressive properties, which were significantly deficient during an active disease relapse, but were recovered during disease quiescence (24). Thus, it is tempting to speculate that the adoptive transfer of a selected population of non-pathogenic, autoregulatory CD8 T-cells may offer a novel immune intervention for these patients. It is encouraging to note that these cells were able to ameliorate ongoing/established EAE. As further pre-clinical evidence, it would be important to delineate methods to enhance the in vitro propagation of the most effective subpopulation of these cells, perhaps using selection based on functional molecules and demonstration of the most effective disease reversal.
To summarize, our studies demonstrate a novel autoregulatory population of CNS-specific CD8 T-cells that ameliorates EAE using suppressor/cytotoxic mechanisms. Although distinct from their MHC Class Ib-restricted siblings, these autoregulatory CD8 T-cells reveal an additional potent immunomodulatory arm of the adaptive immune system, which may in a concerted effort try to ameliorate autoimmune demyelinating disease and may be harnessed for immunotherapeutic intervention.
Supplementary Material
Acknowledgments
These studies were supported, in part, by grants from the National Institutes of Health and the National Multiple Sclerosis Society, including the Harry Weaver Neuroscience Scholar Award of the National MS Society.
The authors would like to thank Thomas Lee, Wallace Baldwin, Jorge Franco, Andrew Benagh and Maycie Garibay for technical assistance. We also wish to thank Fatema Chowdhury and Drs. Chris Ayers, Ethan Baughman, Todd Eagar, Mihail Firan and Sushmita Sinha for helpful discussions and critical reading of this manuscript.
Abbreviations
- β 2m
beta-2-microglobulin
- CD
cluster of differentiation
- CSF
cerebrospinal fluid
- DC
dendritic cell
- EAE
experimental autoimmune encephalomyelitis
- MBP
myelin basic protein
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
Footnotes
DISCLOSURES
The authors have no financial conflict of interest.
References
- 1.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 2.Raine CS, Cross AH. Axonal dystrophy as a consequence of long-term demyelination. Lab Invest. 1989;60:714–725. [PubMed] [Google Scholar]
- 3.Keegan BM, Noseworthy JH. Multiple sclerosis. Annu Rev Med. 2002;53:285–302. doi: 10.1146/annurev.med.53.082901.103909. [DOI] [PubMed] [Google Scholar]
- 4.Stromnes IM, Goverman JM. Passive induction of experimental allergic encephalomyelitis. Nat Protoc. 2006;1:1952–1960. doi: 10.1038/nprot.2006.284. [DOI] [PubMed] [Google Scholar]
- 5.Jilek S, Schluep M, Rossetti AO, Guignard L, Le Goff G, Pantaleo G, Du Pasquier RA. CSF enrichment of highly differentiated CD8+ T cells in early multiple sclerosis. Clin Immunol. 2007;123:105–113. doi: 10.1016/j.clim.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol. 1986;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- 7.Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schröder R, Deckert M, Schmidt S, Ravid R, Rajewsky K. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crawford MP. High prevalence of autoreactive, neuroantigen-specific CD8+ T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood. 2004;103:4222–4231. doi: 10.1182/blood-2003-11-4025. [DOI] [PubMed] [Google Scholar]
- 9.Koh DR, Fung-Leung WP, Ho A, Gray D, Acha-Orbea H, Mak TW. Less mortality but more relapses in experimental allergic encephalomyelitis in CD8−/− mice. Science. 1992;256:1210–1213. doi: 10.1126/science.256.5060.1210. [DOI] [PubMed] [Google Scholar]
- 10.Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, Raine CS. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol. 2001;166:7579–7587. doi: 10.4049/jimmunol.166.12.7579. [DOI] [PubMed] [Google Scholar]
- 11.Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlén C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med. 2001;194:669–676. doi: 10.1084/jem.194.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ford ML, Evavold BD. Specificity, magnitude, and kinetics of MOG-specific CD8+ T cell responses during experimental autoimmune encephalomyelitis. Eur J Immunol. 2005;35:76–85. doi: 10.1002/eji.200425660. [DOI] [PubMed] [Google Scholar]
- 13.Ji Q, Goverman J. Experimental autoimmune encephalomyelitis mediated by CD8+ T cells. Ann N Y Acad Sci. 2007;1103:157–166. doi: 10.1196/annals.1394.017. [DOI] [PubMed] [Google Scholar]
- 14.Jiang H, Zhang SI, Pernis B. Role of CD8+ T cells in murine experimental allergic encephalomyelitis. Science. 1992;256:1213–1215. doi: 10.1126/science.256.5060.1213. [DOI] [PubMed] [Google Scholar]
- 15.Najafian N, Chitnis T, Salama AD, Zhu B, Benou C, Yuan X, Clarkson MR, Sayegh MH, Khoury SJ. Regulatory functions of CD8+CD28− T cells in an autoimmune disease model. J Clin Invest. 2003;112:1037–1048. doi: 10.1172/JCI17935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linker RA, Rott E, Hofstetter HH, Hanke T, Toyka KV, Gold R. EAE in beta-2 microglobulin-deficient mice: axonal damage is not dependent on MHC-I restricted immune responses. Neurobiol Dis. 2005;19:218–228. doi: 10.1016/j.nbd.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 17.Beeston T, Smith TRF, Maricic I, Tang X, Kumar V. Involvement of IFN-γ and perforin, but not Fas/FasL interactions in regulatory T cell-mediated suppression of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010:1–7. doi: 10.1016/j.jneuroim.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou YK, Henderikx P, Jones RE, Kotzin B, Hashim GA, Offner H, Vandenbark AA. Human CD8+ T cell clone regulates autologous CD4+ myelin basic protein specific T cells. Autoimmunity. 1992;14:111–119. doi: 10.3109/08916939209083129. [DOI] [PubMed] [Google Scholar]
- 19.Hu D, Ikizawa K, Lu L, Sanchirico ME, Shinohara ML, Cantor H. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat Immunol. 2004;5:516–523. doi: 10.1038/ni1063. [DOI] [PubMed] [Google Scholar]
- 20.Jiang H, Braunstein NS, Yu B, Winchester R, Chess L. CD8+ T cells control the TH phenotype of MBP-reactive CD4+ T cells in EAE mice. Proc Natl Acad Sci USA. 2001;98:6301–6306. doi: 10.1073/pnas.101123098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bettini M, Rosenthal K, Evavold BD. Pathogenic MOG-reactive CD8+ T cells require MOG-reactive CD4+ T cells for sustained CNS inflammation during chronic EAE. J Neuroimmunol. 2009:1–9. doi: 10.1016/j.jneuroim.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson AC, Chandwaskar R, Lee DH, Sullivan JM, Solomon A, Rodriguez-Manzanet R, Greve B, Sobel RA, Kuchroo VK. A transgenic model of central nervous system autoimmunity mediated by CD4+ and CD8+ T and B cells. The Journal of Immunology. 2012;188:2084–2092. doi: 10.4049/jimmunol.1102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.York NR, Mendoza JP, Ortega SB, Benagh A, Tyler AF, Firan M, Karandikar NJ. Immune regulatory CNS-reactive CD8+T cells in experimental autoimmune encephalomyelitis. J Autoimmun. 2010;35:33–44. doi: 10.1016/j.jaut.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baughman EJ, Mendoza JP, Ortega SB, Ayers CL, Greenberg BM, Frohman EM, Karandikar NJ. Neuroantigen-specific CD8+ regulatory T-cell function is deficient during acute exacerbation of multiple sclerosis. J Autoimmun. 2011;36:115–124. doi: 10.1016/j.jaut.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crawford MP, Yan SX, Ortega SB, Mehta RS, Hewitt RE, Price DA, Stastny P, Douek DC, Koup RA, Racke MK, Karandikar NJ. High prevalence of autoreactive, neuroantigen-specific CD8+ T cells in multiple sclerosis revealed by novel flow cytometric assay. Blood. 2004;103:4222–4231. doi: 10.1182/blood-2003-11-4025. [DOI] [PubMed] [Google Scholar]
- 26.Biegler BW, Yan SX, Ortega SB, Tennakoon DK, Racke MK, Karandikar NJ. Clonal composition of neuroantigen-specific CD8+ and CD4+ T-cells in multiple sclerosis. J Neuroimmunol. 2011;234:131–140. doi: 10.1016/j.jneuroim.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tennakoon DK, Mehta RS, Ortega SB, Bhoj V, Racke MK, Karandikar NJ. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J Immunol. 2006;176:7119–7129. doi: 10.4049/jimmunol.176.11.7119. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z, Hong J, Sun W, Xu G, Li N, Chen X, Liu A, Xu L, Sun B, Zhang JZ. Role of IFN-gamma in induction of Foxp3 and conversion of CD4+ CD25− T cells to CD4+ Tregs. J Clin Invest. 2006;116:2434–2441. doi: 10.1172/JCI25826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen M, Yan B, Kozoriz D, Weiner H. Novel CD8(+) regulatory T cells suppress experimental autoimmune encephalomyelitis by TGF-beta- and IFN-gamma-dependent mechanisms. Eur J Immunol. 2009;39:3435. doi: 10.1002/eji.200939441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tajima M, Wakita D, Satoh T, Kitamura H, Nishimura T. IL-17/IFN-γ double producing CD8+ T (Tc17/IFN-γ) cells: a novel cytotoxic T-cell subset converted from Tc17 cells by IL-12. Int Immunol. 2011;23:751–759. doi: 10.1093/intimm/dxr086. [DOI] [PubMed] [Google Scholar]
- 31.Chowdhury FZ, Ramos HJ, Davis LS, Forman J, Farrar JD. IL-12 selectively programs effector pathways that are stably expressed in human CD8+ effector memory T cells in vivo. Blood. 2011;118:3890–3900. doi: 10.1182/blood-2011-05-357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karandikar NJ, Crawford MP, Yan X, Ratts RB, Brenchley JM, Ambrozak DR, Lovett-Racke AE, Frohman EM, Stastny P, Douek DC, Koup RA, Racke MK. Glatiramer acetate (Copaxone) therapy induces CD8(+) T cell responses in patients with multiple sclerosis. J Clin Invest. 2002;109:641–649. doi: 10.1172/JCI14380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dal Ben ERR, Hdo Prado C, Baptista TSA, Bauer ME, Staub HL. Decreased Levels of Circulating CD4+CD25+Foxp3+ Regulatory T Cells in Patients with Primary Antiphospholipid Syndrome. Journal of clinical immunology. 2013 doi: 10.1007/s10875-012-9857-y. [DOI] [PubMed] [Google Scholar]
- 35.Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Bäckström BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sobel ES, Brusko TM, Butfiloski EJ, Hou W, Li S, Cuda CM, Abid AN, Reeves WH, Morel L. Defective response of CD4(+) T cells to retinoic acid and TGFβ in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:R106. doi: 10.1186/ar3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyara M, Gorochov G, Ehrenstein M, Musset L, Sakaguchi S, Amoura Z. Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun Rev. 2011;10:744–755. doi: 10.1016/j.autrev.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Pan X, Yuan X, Zheng Y, Wang W, Shan J, Lin F, Jiang G, Yang YH, Wang D, Xu D, Shen L. Increased CD45RA+ FoxP3(low) regulatory T cells with impaired suppressive function in patients with systemic lupus erythematosus. PLoS ONE. 2012;7:e34662. doi: 10.1371/journal.pone.0034662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol. 2010;11:628–634. doi: 10.1038/ni.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mars LT, Bauer J, Gross DA, Bucciarelli F, Firat H, Hudrisier D, Lemonnier F, Kosmatopoulos K, Liblau RS. CD8 T cell responses to myelin oligodendrocyte glycoprotein-derived peptides in humanized HLA-A*0201-transgenic mice. J Immunol. 2007;179:5090–5098. doi: 10.4049/jimmunol.179.8.5090. [DOI] [PubMed] [Google Scholar]
- 41.Na SY, Cao Y, Toben C, Nitschke L, Stadelmann C, Gold R, Schimpl A, Hünig T. Naive CD8 T-cells initiate spontaneous autoimmunity to a sequestered model antigen of the central nervous system. Brain. 2008;131:2353–2365. doi: 10.1093/brain/awn148. [DOI] [PubMed] [Google Scholar]
- 42.Saxena A, Bauer J, Scheikl T, Zappulla J, Audebert M, Desbois S, Waisman A, Lassmann H, Liblau RS, Mars LT. Cutting edge: Multiple sclerosis-like lesions induced by effector CD8 T cells recognizing a sequestered antigen on oligodendrocytes. The Journal of Immunology. 2008;181:1617–1621. doi: 10.4049/jimmunol.181.3.1617. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Medaer R, Stinissen P, Hafler D, Raus J. MHC-restricted depletion of human myelin basic protein-reactive T cells by T cell vaccination. Science. 1993;261:1451–1454. doi: 10.1126/science.7690157. [DOI] [PubMed] [Google Scholar]
- 44.Volovitz I, Marmor Y, Mor F, Flügel A, Odoardi F, Eisenbach L, Cohen IR. T cell vaccination induces the elimination of EAE effector T cells: analysis using GFP-transduced, encephalitogenic T cells. J Autoimmun. 2010;35:135–144. doi: 10.1016/j.jaut.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 45.Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, Yang Y, Medarova Z, Moore A, Santamaria P. Reversal of Autoimmunity by Boosting Memory-like Autoregulatory T Cells. Immunity. 2010 doi: 10.1016/j.immuni.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 46.Elong Ngono A, Pettré S, Salou M, Bahbouhi B, Soulillou J-P, Brouard S, Laplaud D-A. Frequency of circulating autoreactive T cells committed to myelin determinants in relapsing-remitting multiple sclerosis patients. Clin Immunol. 2012;144:117–126. doi: 10.1016/j.clim.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 47.Hudrisier D, Aucher A, Puaux AL, Bordier C, Joly E. Capture of target cell membrane components via trogocytosis is triggered by a selected set of surface molecules on T or B cells. J Immunol. 2007;178:3637–3647. doi: 10.4049/jimmunol.178.6.3637. [DOI] [PubMed] [Google Scholar]
- 48.Hudrisier D, Riond J, Garidou L, Duthoit C, Joly E. T cell activation correlates with an increased proportion of antigen among the materials acquired from target cells. Eur J Immunol. 2005;35:2284–2294. doi: 10.1002/eji.200526266. [DOI] [PubMed] [Google Scholar]
- 49.Xiang J, Huang H, Liu Y. A new dynamic model of CD8+ T effector cell responses via CD4+ T helper-antigen-presenting cells. J Immunol. 2005;174:7497–7505. doi: 10.4049/jimmunol.174.12.7497. [DOI] [PubMed] [Google Scholar]
- 50.Umeshappa CS, Huang H, Xie Y, Wei Y, Mulligan SJ, Deng Y, Xiang J. CD4+ Th-APC with acquired peptide/MHC class I and II complexes stimulate type 1 helper CD4+ and central memory CD8+ T cell responses. The Journal of Immunology. 2009;182:193–206. doi: 10.4049/jimmunol.182.1.193. [DOI] [PubMed] [Google Scholar]
- 51.Jürgens B, Hainz U, Fuchs D, Felzmann T, Heitger A. Interferon-gamma-triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood. 2009;114:3235–3243. doi: 10.1182/blood-2008-12-195073. [DOI] [PubMed] [Google Scholar]
- 52.Martini M, Testi MG, Pasetto M, Picchio MC, Innamorati G, Mazzocco M, Ugel S, Cingarlini S, Bronte V, Zanovello P, Krampera M, Mosna F, Cestari T, Riviera AP, Brutti N, Barbieri O, Matera L, Tridente G, Colombatti M, Sartoris S. IFN-γ-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine. 2010;28:3548–3557. doi: 10.1016/j.vaccine.2010.03.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.