

Abstract
Peroxisome-proliferator-activated receptors (PPARs) comprise three subtypes (PPARα, δ and γ) to form a nuclear receptor superfamily. PPARs act as key transcriptional regulators of lipid metabolism, mitochondrial biogenesis, and anti-oxidant defense. While their roles in regulating lipid metabolism have been well established, the role of PPARs in regulating redox activity remains incompletely understood. Since redox activity is an integral part of oxidative metabolism, it is not surprising that changes in PPAR signaling in a specific cell or tissue will lead to alteration of redox state. The effects of PPAR signaling are directly related to PPAR expression, protein activities and PPAR interactions with their coregulators. The three subtypes of PPARs regulate cellular lipid and energy metabolism in most tissues in the body with overlapping and preferential effects on different metabolic steps depending on a specific tissue. Adding to the complexity, specific ligands of each PPAR subtype may also display different potencies and specificities of their role on regulating the redox pathways. Moreover, the intensity and extension of redox regulation by each PPAR subtype are varied depending on different tissues and cell types. Both beneficial and adverse effects of PPAR ligands against cardiovascular disorders have been extensively studied by many groups. The purpose of the review is to summarize the effects of each PPAR on regulating redox and the underlying mechanisms, as well as to discuss the implications in the cardiovascular system.
Keywords: Peroxisome-proliferator-activated receptor, Redox, Cardiovascular disorders, Oxidative stress, Antioxidant
Core tip: Numerous studies have shown that peroxisome-proliferator-activated receptors (PPARs) ligands can modulate antioxidants via various mechanisms. Importantly, direct transcriptional regulation of antioxidant genes, such as thioredoxin-1, glutathione peroxidase 3, sestrin-1, catalase, superoxide dismutase (SOD)1, SOD2, and heme oxygenase, is established by identifying functional PPAR responsive element in promoter regions of the above genes. This review summarizes how these important antioxidant genes are regulated by each subtype of PPARs in response to oxidative stress in the cardiovascular system and how oxidative stress affects PPAR function, as well as the biological implications in the cardiovascular system.
INTRODUCTION
Mitochondria are the powerhouse for cells and are vulnerable targets of oxidative damage. The maintenance of redox homeostasis is critical for normal cellular function. The utilization of oxygen for ATP generation in the mitochondria accompanies the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) from electron transport chain complex I, II and III[1]. Mitochondrial ROS further triggers ROS production from other sources, such as Ang II, hyperglycemia, hypoxia, oxidized low-density lipoprotein, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs)[2,3]. NOXs are membrane-bound enzyme complexes that generate superoxide by transferring electrons from intracellular NADPH across the membrane and coupling these to molecular oxygen[4]. In general, the balance between ROS formation and endogenous antioxidant defenses enable redox homeostasis in cells. Under normal conditions, ROS and RNS also serve as signaling molecules[5]. Oxidative stress occurs when the balance between ROS/RNS production and the endogenous antioxidant defense. Oxidative stress is associated with major pathological development of cardiovascular disease[5]. Macrophage-derived ROS contribute to the initiation and development of atherosclerosis. Vascular dysfunction in response to reactive ROS plays an important role in the pathological development and progression of atherosclerotic lesions and heart failure. Oxidative damages are also the main features during the pathological development of cardiac hypertrophy, ischemia/reperfusion and heart failure[6,7].
Several key endogenous antioxidants play crucial roles in maintaining cellular homeostasis, especially in those cells with actively oxidative metabolism. Superoxide dismutase (SOD) is a major superoxide-scavenging enzyme converting superoxide (O2-.) to O2 and hydrogen peroxide (H2O2), which is further converted into H2O by catalase[8], thioredoxin (Trx)[9], or glutathione peroxidases[10-12]. In mammals, three isoforms of SOD have been reported: the cytosolic Cu/Zn SOD (SOD1)[13], the mitochondrial manganese SOD (SOD2 or MnSOD)[8,12,14], and the extracellular form of Cu/Zn-SOD (SOD3 or ecSOD)[15]. Trx reduces the oxidized form of Trx peroxidase, and this reduced form of Trx peroxidase scavenges ROS in both cytosol and nucleus, where it modifies the activity of transcription factors[9]. In addition, heme oxygenase (HO) is an antioxidant enzyme family, consisting three isoforms: the oxidative stress-inducible HO-1 (HSP32), constitutive HO-2, and less active HO-3. HO protects cells against oxidative stress by degrading the prooxidant heme to carbon monoxide (CO), biliverdin, and ferrous iron[16]. These multiple endogenous antioxidants are crucial in maintaining cellular redox balance. If this balance is interrupted, oxidative stress increases, resulting in damage of essential cellular components.
Peroxisome-proliferator-activated receptors (PPARs) including, α, δ and γ, comprise a subfamily of the nuclear receptor superfamily. Similar to other nuclear receptor superfamilies, PPARs share the typical domain structure, including a central DNA-binding domain, an N-terminal ligand-independent activation domain and a C-terminal ligand binding domain[17]. Each PPAR subtype mostly forms heterodimer with the retinoid X receptor (RXR) before binding to PPAR responsive element (PPRE) of their target genes and the subsequent synergistic activation of these genes[18]. Fatty acids and lipid metabolites serve as endogenous PPAR ligands, which exert adaptive metabolic responses to changes in metabolic status in various tissues[18]. In addition to those identified synthetic compounds via high throughput screening, many compounds of natural products and modern medicine have been identified as exogenous PPAR ligands with a wide range of specificities and potencies. Despite the past success of a few of those PPAR subtype specific ligands in the clinical treatment of type II diabetes and dyslipidemia in patients, the potential side effects of these compounds have become a major concern. In-depth understanding of the molecular mechanisms underlying PPAR’s action has become even more important in our effort to rescue and improve this class of drugs with clinically proven effectiveness.
In the past decades, numerous studies elucidated the main functional role of PPAR as key transcriptional regulators of lipid metabolism, mitochondrial biogenesis, and anti-oxidant defense. While PPAR’s role in regulating lipid metabolism is well-established, their role in regulating redox activity remains incompletely understood. Since redox activity is an integral part of oxidative metabolism, changes in PPAR signaling in a specific cell or tissue will lead to alterations of redox state. PPAR’s regulation of cellular redox states appears to be a highly diversified function that is mostly dependent on the specific subtype at a particular tissue under different metabolic and stress conditions. The molecular mechanisms underlying PPAR’s role in redox regulation are not fully understood. While numerous studies demonstrate the indirect influence of PPAR activation on the redox state, the role of PPAR in direct transcriptional regulation of redox has emerged. Given the importance of redox regulation in the pathophysiology of the cardiovascular system, this review will give an overview of PPAR’s function in regulating redox activity, particularly in the cardiovascular system.
EFFECTS OF OXIDATIVE STRESS ON PPAR SIGNALING
PPAR expression, PPAR activities and PPAR interactions with their coregulators are the factors that directly determine the effects of PPAR signaling. Oxidative stress is a common cellular stress condition that can trigger a series of responses leading to altered PPAR expression and activity by different mechanisms. Increased oxidative stress regulates a variety of signaling pathways that subsequently affect gene expression by modulating a large number of transcription factors, including PPARs. Additionally, redox states may also regulate PPAR signaling via transcriptional regulation and post-translational modification. PPAR expression and functional activity have recently been observed in the vasculature such as endothelial cells[19] and vascular smooth muscle cells (VSMCs)[20], suggesting that PPARs could be redox sensitive transcription factors in the vasculature and could be selectively activated by oxidized-fatty acids. PPARγ is abundantly expressed in macrophage/foam cells of atherosclerotic lesions[21,22]. In vitro experiments confirmed that ROS-related increase of oxidized low-density lipoprotein in macrophage upregulates PPARγ expression[21]. In contrast, H2O2 induced oxidative stress in vascular endothelial cells attenuates PPARγ expression and activity through suppression of PPARγ transcription, potentially via activating inhibitory redox-regulated transcription factors[19]. Similar ROS-related alteration of PPARγ expression may occur in other tissues too. Increased lipid oxidation not only causes oxidative stress, but also increases PPARγ expression in the skeleton (osteoblasts)[23]. Moreover, peroxidized polyunsaturated fatty acids promote PPARγ-mediated transcription and binding of PPARγ to specific target genes, including PPARγ itself[23]. PPARγ is inhibited by histone deacetylase 4 in cortical neurons under oxidative stress in neurons[24]. Treating the cultured cells with a glutathione-depleting agent diethylmaleate reduces DNA-binding activity of PPARα[25]. Supplement of an antioxidant, vitamin E, can effectively restore PPARα expression in aged mice to levels seen in younger mice[26]. This may also occur in cells of the cardiovascular system. This observation implicates that balancing the cellular redox state may serve as an essential transcriptional regulation for PPARα. ERK1/2 activation is one of the common consequences of oxidative stress[27]. Activation of ERK1/2 signaling can induce the expression of PPARγ during the differentiation of 3T3-L1 preadipocytes[28]. Another oxidative stress induced factor, Platelet-derived growth factor (PDGF), has been shown to upregulate PPARδ gene expression in VSMCs by the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway[29]. Therefore, it appears that oxidative stress may influence individual PPAR activity in a tissue specific manner.
The transcriptional activity of PPARs can be regulated by post-translational modifications such as phosphorylation, SUMOylation, and ubiquitination[30,31]. Increase in ROS levels is accompanied by p38 mitogen-activated protein kinase and 5’AMP-activated protein kinase (AMPK) activation in the heart[32-34]. The PPARα phosphorylation by the p38 MAPK decreases the transcriptional activity of PPARα[35].
As summarized in Figure 1, PPAR expression and activity may be altered by the status of cellular energy metabolism (redox), and oxidative stress is attributed to altered PPAR expression and activity as an adaptive feedback or a maladaptive feedback that leads to a vicious cycle.
Figure 1.
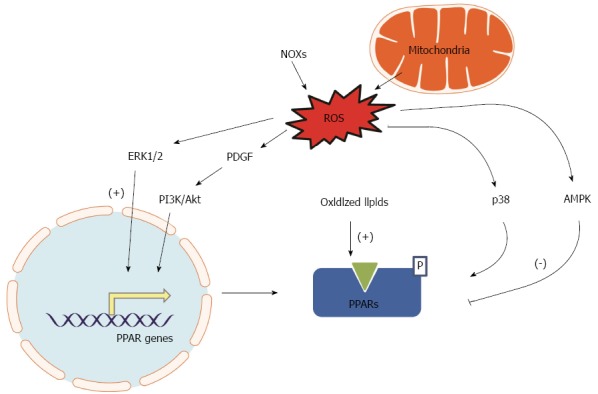
Oxidative stress-induced signaling pathways affecting peroxisome-proliferator-activated receptor transcript and protein activity. Oxidative stress triggers activation of ERK1/2, platelet-derived growth factor (PDGF), and phosphatidylinositol 3-kinase (PI3K)/Akt resulting in increased transcription of peroxisome-proliferator-activated receptors (PPARs) as a defense mechanism. Oxidized lipids activate transcription and activation of PPARs. Oxidative stress increases p38 mitogen-activated protein kinase and 5’AMP-activated protein kinase (AMPK) resulting in the phosphorylation of PPAR proteins resulting in suppressed transcription of PPARs. NOX: Nicotinamide adenine dinucleotide phosphate oxidase; ROS: Reactive oxygen species.
SUBTYPE SPECIFIC ROLE OF PPARS ON THE REGULATION OF REDOX PATHWAY
The three subtypes of PPARs regulate cellular lipid and energy metabolism in most tissues in the body with overlapping and preferential effects on different metabolic steps depending on a specific tissue. Adding to the complexity, specific ligands of each PPAR subtype may also display different potencies and specificities of their role on regulating the redox pathways. Each PPAR subtype regulates redox status with various intensity and extension in a tissue and cell type specific manner. Many studies revealed the beneficial effects of PPAR ligands against cardiovascular disorders, although recent studies demonstrate potential adverse effects of synthetic PPAR ligands on cardiovascular disease models. This section summarizes how each PPAR regulates redox mainly in the cardiovascular system.
PPARα and oxidative stress
PPARα is expressed in various cell types related to the cardiovascular system, including cardiomyocytes[36], endothelial[37,38], smooth muscle cells[20] and monocytes/macrophages[39]. The role of PPARα in lipid and lipoprotein metabolism is well established[36,40]. Fibrates are drugs that selectively target PPARα and have been clinically used to lower hypertriglyceridemia, a risk factor of cardiovascular disease. Moreover, PPARα is a critical regulator of intra- and extracellular lipid metabolism. In addition, the therapeutic efficacy of fibrates in inhibiting atherogenesis may also in part attribute to its capacity to regulate cholesterol efflux[41] and redox signaling. Activation of PPARα protects the heart from ischemia/reperfusion injury[40,42,43].
Evidence supporting the role of PPARα in regulating redox pathways remains relatively superficial and paradoxical. Most of the early studies are based on in vitro and in vivo experiments using PPARα-selective ligands, such as clofibrate, fenofibrate and Wy14643. It has been shown that clofibrate protects rat hearts from coronary artery occlusion-induced myocardial ischemia by reducing ROS production and lipid peroxidation. These protective effects are mainly attributed to significantly increased expression and activity of SOD1, SOD2, and catalase in the heart tissue[43]. Furthermore, clofibrate is able to suppress the upregulation of Ang II, Ang II AT1-receptor and subsequently the related oxidative stress in the heart, at least partially contributing to the improved cardiac function[43]. Another synthetic ligand of PPARα, Wy14643, also protects rabbit hearts from ischemia/reperfusion injury by increasing HO-1 expression and decreasing caspase-3 activation[44]. In human macrophage, PPARα activation by another subtype selective ligand, GW647, can upregulate the transcript and protein expression of Trx-1[45]. Moreover, PPARα activation could also enhance the Trx-1 activity by indirect down-regulation of the natural Trx-1 inhibitor, vitamin D3 up-regulated protein 1[45]. Therefore, stimulation of PPARα could exert a beneficial effect against the development of atherosclerosis. The remaining questions are whether PPARα ligand treatments could alleviate specifically oxidative stress and contribute to their beneficial effects via a PPARα dependent or independent mechanism. Studies on mouse models of transgenic overexpression and/or knockout of PPARα specifically in the cardiovascular system solved some of the puzzles. However, discrepancies among different studies exist. PPARα knockout mice showing minimal phenotypic changes in the heart are probably in different genetic backgrounds from those showing major phenotypic changes. In a study reporting cardiac contractile dysfunction in PPARα null mice, a mechanism of oxidative damage in sarcomere proteins and lipid peroxidation was proposed based on the observation that a significant decrease of SOD2 protein and the corresponding activity with no change of other antioxidant enzymes such as Cu/ZnSOD (SOD1), catalase, and glutathione peroxidase (GPx) was reported[46]. It is not clear if the repression of SOD2 protein level and activity also occurred at the transcript level. Moreover, another study could not confirm the downregulation of SOD2 transcript and protein in the PPARα null heart[47]. Therefore, strong evidence of a direct interplay between cardiac SOD2 and PPARα is still lacking and whether the observation recorded is due to a lack of genuine regulation by PPARα or a long-term developmental adaptation to the absence of PPARα remains yet to be established.
It is well established that PPARα specific ligands activate fatty acid oxidation. However, it remains unclear, at least in cardiovascular tissues, whether this increased lipid oxidation would subsequently lead to oxidative stress due to the augmented respiration. There is evidence that activation of PPARα in the heart either by transgenic overexpression or PPARα selective ligand treatment causes increased fatty acid oxidation[48,49]. Activation of PPARα may be associated with the repression of estrogen related receptors (ERRs). ERRs are members of another nuclear receptor subfamily that governs mitochondrial biogenesis[50], thus exacerbating pressure overload-induced cardiac hypertrophy and heart failure due to mitochondrial dysfunction[51]. However, the subsequent changes of ROS in the heart have not been well characterized. In human and murine macrophages, different PPARα, but not PPARγ, agonists increase the production of ROS (H2O2 and superoxide)[52]. Most importantly, this study excluded the potential off-target effects of the tested PPARα ligands by showing the mediating role of PPARα agonists. PPARα agonists induce ROS production by increasing NOXs expression and stimulating its activity, which will generate more endogenous PPARα ligands. This vicious cycle will lead to augmented oxidative stress in macrophages. It has become obvious that the activation of PPARα in macrophage could have opposite effects on regulating redox state in other tissues showing increase in the expression and activity of either Trx-1 or NOXs. The consequences of PPARα activation on ROS production in macrophage are not clear. Overwhelming evidence supports the fact that PPARα activation suppresses atherogenic inflammation in macrophage[39,53]. It is likely that ROS will be reduced in PPARα ligand treated macrophages.
Overall, it is likely that PPARα plays certain roles in regulating redox state in the cardiovascular system. However, supporting evidence will be needed to further address the mechanistic aspects of PPARα related redox changes.
PPARγ and oxidative stress
PPARγ is a primary regulator of lipid storage and adipogenesis mainly in adipose tissue. However, it also plays an important role in other tissues and cells of the cardiovascular system, since PPARγ is expressed in the heart and vasculature[42,54]. Activation of PPARγ may exert anti-atherogenic[41,55] and anti-hypertrophic effects[56]. Despite the relatively low expression in the myocardium, cardiomyocyte-restricted PPARγ knockout in mice leads to cardiac hypertrophy and even heart failure[57,58]. On the other hand, PPARγ activation substantially reduces myocardial infarct size, significantly improve aortic flow during reperfusion in both normal and diabetic hearts and substantially ameliorate post-ischemic functional recovery in rats[42,59,60]. These observations suggest an important role of PPARγ in the heart.
While the transcriptional transrepression of nuclear factor kappa-B (NF-κB) signaling by PPARγ has been suggested as a mechanism in most of above studies, the role of PPARγ as a transcriptional regulator of endogenous antioxidants is another mechanism. Our previous study on the cardiomyocyte-restricted PPARγ knockout mice unveiled that oxidative stress plays an essential role in the development of progressive cardiac hypertrophy and dilated cardiomyopathy in these mice[58]. However, further study from our group on adult mice with short term cardiac-specific PPARγ knockout showed only modest cardiac hypertrophy without oxidative stress, though fatty acid utilization was impaired and cardiac performance was compromised. Neither the mitochondrial ultrastructure nor mitochondrial copy number was altered compared with control mice[61]. These two contrast outcomes of PPARγ knockout suggest that cardiac oxidative stress may cause chronic damage instead of acute lethality. The deteriorating phenotype of PPARγ knockout was prevented by administration of MnTBAP, which mimics SOD by scavenging superoxide. Therefore, both PPARα and PPARγ are involved in the regulation of mitochondrial SOD2 under specific conditions, playing a crucial role in cardiac redox balance.
Treatment of PPARγ-specific ligands, rosiglitazone and pioglitazone, can ameliorate H2O2-induced oxidative damages in the newborn rabbit heart. These oxidative damages feature repressed left ventricular developed pressure, sarcomere shortening, decreased catalase expression level, and increases lactate dehydrogenase. The protective effect of PPARγ ligands against oxidative damage seems to be mediated by catalase, since the effect is abolished by PPARγ blocker or catalase inhibitor, indicating that the PPARγ-regulated catalase is crucial for cardioprotective effect of PPARγ ligands[62]. Studies also have shown that PPARγ ligands have protective effects against hypertrophy, induced by ischemia/reperfusion or angiotensin II, in rodents via various mechanisms[63]. Ang II inhibits PPARγ transcriptional activity, which in turn suppresses expression of antioxidant enzymes. PPARγ agonists, pioglitazone and 15d-PGJ2, reverse the Ang II-induced suppression of catalase in adventitial fibroblasts of rat aorta[64]. Pioglitazone also attenuates atrial fibrillation, in which oxidative stress plays an important role in the pathophysiology and often complicated by ischemic heart disease, valvular disease, and left ventricular hypertrophy[65]. PPARγ plays an important role in macrophage inflammatory homeostasis, partly by regulating cholesterol efflux[41]. On the other hand, lipopolysaccharide and IFN-γ in macrophages upregulates PPARγ activity and attenuates the oxidative burst[66]. PPAR-γ ligands can directly alter vascular endothelial function by enhancing endothelial NO bioavailability, in part by altering endothelial superoxide metabolism through suppression of NOXs and induction of SOD1[67]. This effect is also found in the vasculature of diabetic mice independent of correction of diabetic metabolic derangements[68]. Moreover, another PPARγ ligand rosiglitazone can attenuate high glucose induced oxidative stress and subsequent monocyte-endothelial interactions by attenuating NF-κB/p65 activation and NOX4 expression, thus favorably modulating endothelial responses in the diabetic vasculature[69]. Therefore, it is clear that PPARγ is an essential regulator of redox signaling in the cardiovascular system and can protect against many cardiovascular disorders via transcriptional activation of antioxidant genes.
PPARδ and oxidative stress
PPARδ is ubiquitously expressed with differential expression abundances in various tissues depending on pathophysiological condition. PPARδ is abundantly expressed in the heart and plays an essential role in regulating fatty acid oxidation in cardiomyocytes[70]. The essential role of PPARδ in the heart is further demonstrated by the striking cardiac pathological development in mice with cardiomyocyte-restricted knockout of PPARδ[71]. Several other studies confirmed the myocardial protective effects either with the treatment of PPARδ ligands in rats[72,73] or cardiomyocyte-restricted overexpression in transgenic mice[74,75]. PPARδ is also expressed in VSMCs and up-regulated after vascular injury[76]. PPARδ activation facilitates VSMC proliferation causing matrix modulation and vascular remodeling. This is an opposite outcome to the activation of PPARα and PPARγ by which inflammation is decreased[76,77]. A PPARδ-specific ligand compound promotes lipid accumulation in human macrophages by increasing the expression of genes involved in lipid uptake and storage, whereas this treatment represses lipid metabolism and efflux[78]. However, another PPARδ-specific ligand GW501516 increases expression of the reverse cholesterol transporter, ATP-binding cassette A1, and induced apolipoprotein A1-specific cholesterol efflux in macrophages[79]. This observation of PPARδ activation is similar to the effects of PPARα and PPARγ activation in macrophages by which cholesterol is removed from foam cells[41]. In addition, it has been reported that PPARδ ligand L-165041 inhibits VCAM-1 expression and cytokine-induced MCP-1 secretion in endothelial cells and increases high-density lipoprotein (HDL) levels in db/db mice[80]. Since the increased HDL levels are well associated with decreased risk of atherosclerosis, it appears PPARδ activation may inhibit atherogenesis.
While the role of PPARδ activation in protecting against pathogenesis in the cardiovascular system is established, how much of the protective mechanisms are involved in the anti-oxidation effects remains obscure. However, the role of PPARδ activation in anti-oxidant defense of the cardiovascular system is unraveling. A recent study demonstrated that the PPARδ counteracts Ang II-induced ROS production in VSMCs. A PPARδ-specific ligand GW501516 significantly reduced Ang II-induced ROS generation in VSMCs via inhibiting PTEN-mediated modulation of PI3K/Akt/Rac1 signaling[81,82]. Activation of PPARδ suppresses the translocation of Rac1 to the plasma membrane, a key step in NOXs-induced ROS production, in VSMCs[82]. Another recent study focusing on human endothelial cells demonstrates similar findings. PPARδ activation by GW501516 inhibits angiotensin II-induced premature senescence featured with elevated ROS production in human coronary artery endothelial cells[83]. These results illustrate that ligand-activated PPARδ plays an important role in the cellular response to oxidative stress by decreasing Ang II-induced ROS in vascular cells. In addition, we have recently demonstrated that PPARδ is essential for not only the constitutive function of fatty acid metabolism and mitochondrial biogenesis, but also in maintaining antioxidant defense of the heart[84]. Cardiomyocytes-restricted PPARδ knockout from adult heart leads to oxidative damages with repressed expression of SOD1 and SOD2[84]. Interestingly, the PPARα null mice with additional PPARδ knockout from the heart showed similar results. Both the transcript and protein expression of SOD1 and SOD2 was repressed in PPARδ, but not PPARα deficient hearts[47]. In this study, none of the endogenous antioxidants appears to be affected at basal condition in the PPARδ null heart. Therefore, it appears repressed antioxidant expression is the main reason for the major oxidative damages in the hearts of PPARδ knockout and PPARδ/PPARα double knockout mice. The effects of PPARδ activation in regulating cardiac antioxidant defense have also been proven in mouse models with cardiomyocyte-restricted overexpression of a constitutively active PPARδ. The enhanced antioxidant defense in these mice enables them to have improved cardiac performance under left ventricular pressure overload condition[75]. However, the transcriptional regulation of antioxidants by PPARδ in the heart may depend on various metabolic conditions with different pathological development. The myocardial protective effects of PPARδ ligand treatment and transgenic PPARδ overexpression have been attributed to their roles in ameliorating lipid profile by increasing fatty acid β-oxidation[72] and in enhancing myocardial glucose utilization[74]. Nevertheless, it is likely that the PPARδ mediated upregulation of the antioxidants defense may also contribute to the beneficial effects.
The effects of each of the three PPARs on redox signaling in the cardiovascular system are generally beneficial in the cardiovascular system. However, contradicting results from various studies exist. It is far from clear as how each of the three PPARs differentially regulates redox signaling in various tissues and cells of the cardiovascular system.
MECHANISMS OF ACTION OF PPARS IN REGULATING REDOX
The molecular mechanisms underlying the PPAR-mediated regulation of redox signaling have been extensively studied. There is emerging evidence supporting that PPAR activation exerts direct transcriptional regulation on the expression of several key endogenous antioxidants, including SOD1[13], SOD2[14], catalase[58,85,86], GPx, OH-1 and Trx-1[45].
Rat SOD1, which is analogous to human SOD1, has PPRE consensus sequence in its promoter region[87]. SOD1 is induced in HepG2 human hepatoma cells by arachidonic acid (polyunsaturated fatty acid), one of the peroxisome proliferators, as a defense system. A promoter analysis of SOD1 gene revealed a conserved PPRE sequence located in -797 and -792 nt, which is able to induce CAT reporter gene activity. The HepG2 nuclear extract showed PPRE-binding in gel mobility shift assay and nuclear extract from retinoic acid-treated HepG2 cells increased the intensity of the DNA-protein complex indicating SOD1 gene is induced by arachidonic acid through the binding of PPAR to the PPRE of the SOD1 gene[85]. PPARγ ligands have been shown to reduce superoxide by stimulating both activity and expression of SOD1 in human umbilical vein endothelial cell and suppressing NOXs[67].
Our previous study showed that PPARγ is essential for the full expression of SOD2 transcript in the heart[58]. SOD2 expression level in cardiac-specific PPARγ knockout mice heart is significantly decreased, and mitochondrial superoxide production was significantly increased compared to that in control mice. We have further confirmed that the PPRE sequence found between -985 and -935 nt in the SOD2 promoter region is functioning. The truncated promoter fragments did not transactivate luciferase reporter gene by rosiglitazone[58]. Therefore, both SOD1 and SOD2 are regulated by direct interaction of PPARs on the PPRE of their promoters under specific conditions.
Cat (catalase; EC 1.11.1.6) plays an important role in cellular protection against oxidative stress by scavenging H2O2 generated from peroxisomal fatty acid β-oxidation. Numerous studies have shown that ligands of PPARα, PPARδ, and PPARγ increase catalase expression level and activity. Catalase has been characterized as PPARγ target gene with a PPRE consensus sequence on its promoter region. One study reported that in vitro translated protein PPARγ/RXRα heterodimer binds 5’-proximal promoter region (5 kb) of catalase. The putative PPRE fragment increased reporter gene activity in the presence of PPARγ ligands and deletion of the region containing PPRE abolished the response to the ligand. Further promoter deletion assay revealed that the PPRE was located between -1027 and -1015 nt. Tandem repeated 3 × PPRE significantly increased PPARγ-stimulated promoter activity, suggesting that PPRE alone is enough to induce transactivation of target gene[86].
The selective PPARα ligand GW647 significantly increases Trx-1 expression and activity in human macrophage. A luciferase reporter assay on human macrophage and detailed computer analysis revealed that PPRE is located between -2185 and -2198 nt of Trx-1 promoter. Mutated PPRE abolished transactivation activity on luciferase reporter assay. In an electrophoretic mobility shift assay, in vitro translated RXRα and PPARα proteins bind this PPRE by heterodimerization.
The GPx3 expression level in the skeletal muscles is significantly decreased in db/db relative to control mice[88]. Additionally, the plasma GPx3 levels are significantly decreased in type 2 diabetic patients compared to normal subjects. PPARγ ligands troglitazone, rosiglitazone, and pioglitazone decrease extracellular H2O2 levels and prevent H2O2-induced insulin resistance by increasing the expression of GPx3 in human skeletal muscle cells[88]. This increase of GPx3 is PPARγ-specific and exclusive to GPx3, but not other GPx family. Whereas the PPARγ siRNA represses TZD-induced GPx3 expression, GPx3 siRNA inhibits the H2O2 scavenging antioxidant effect of TZD. These data indicate that GPx3 is regulated by PPARγ playing cellular protective role against oxidative stress. In the luciferase reporter assay GPx3 promoter -2294 nt region shows strong trans-activation of reporter gene and PPRE is found between -2186 and -2174 nt[88].
In addition to the direct transcriptional effects of PPARs, the interaction of PPAR signaling with many other cell signals can also mediate PPAR’s effects on regulating redox state in cells of the cardiovascular system. PPARδ can act through inhibiting the PI3K/Akt signaling pathway to suppress the marked increase in ROS levels induced by Ang II. Ligand-activated PPARδ also blocked Ang II-induced translocation of Rac1 to the cell membrane, inhibiting the activation of NOXs and consequently ROS generation[82]. The activation of NF-κB is the main signaling event that triggering the inflammatory responses and the subsequent oxidative stress. It has been well recognized that PPAR can exert transrepression effects on the NF-κB signaling and suppress inflammatory responses; hence oxidative stress is ameliorated[89-92]. Therefore, it appears that PPARs may exert their antioxidant effects by direct transcriptional regulation of endogenous antioxidants and by directly or indirectly interfering/coordinating the related signaling transduction pathways to reduced ROS production (Figure 2). However, the mechanisms of how PPARs as a transcription factor would perturb these signaling pathways remain incompletely understood. This is especially the case in understanding how each of the three PPARs exerts antioxidant effects. While PPARδ activation appears to exert most of the direct and indirect inhibitory effects on ROS production in the cardiovascular system, the effects of PPARα and PPARγ appear to be more complicated depending on cell types and specific conditions. Investigations on the potential beneficial effects of duel or triple PPAR ligands are emerging. This novel class of PPAR ligands may be able to avoid potential side effects of each single ligand[93]. Phase II clinical trials for a dual PPARα/γ agonist, aleglitazar, validate their hypoglycemic and hypolipidemic effects[94]. An ongoing phase III clinical trial[95] will reveal the efficacy against cardiovascular event. However, a few dual agonists, such as ragaglitazar, MK-0767, and naveglitazar, cause bladder cancer in rodents suggesting tissue-specific response should be meticulously tested. However, whether these upcoming new agonists modulate oxidative stress in cardiovascular system remains unclear. Further studies should be conducted.
Figure 2.
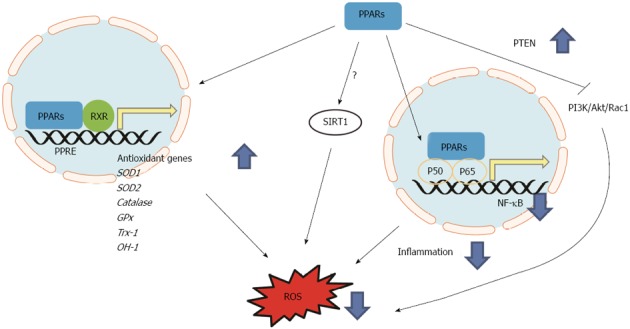
Antioxidant mechanisms of peroxisome-proliferator-activated receptors. Peroxisome-proliferator-activated receptors (PPARs) activate antioxidant genes via transcriptional regulation by binding on PPAR response element (PPRE) of promoter region of target genes. PPARs suppress nuclear factor kappa-B (NF-κB)-light-chain-enhancer of activated B cells via interaction with p50 and p65 resulting in decreased inflammatory response and oxidative stress. PPARs suppress phosphatidylinositol 3-kinase (PI3K)/Akt/Rac1 signaling axis via activation of PTEN resulting in decreased reactive oxygen species (ROS). RXR: Retinoid X receptor; sod: Superoxide dismutase; trx: Thioredoxin; gpx: Glutathione peroxidase; ho: Heme oxygenase.
Another difficulty involved in dissecting the antioxidant role of PPARs is the potent effects of PPAR activation in many other metabolic pathways, which all pose major influences on the redox balance. Therefore, we will have to interpret many of the current findings in contexts of specific cell types, specific animal strain/species and specific disease states.
FUTURE PERSPECTIVES AND CONCLUSIONS
The PPARs are one of the most extensively studied members among the nuclear hormone receptor family. Compounds (fibrate and TZD drugs) targeting PPARα and PPARγ have been used broadly in treating diabetes and dyslipidemia. While the effects of PPAR signaling on redox regulation in the cardiovascular system are major indications of the therapeutic potential, use of clinically available PPAR agonists for heart failure and atherosclerosis remains controversial for major safety concerns[96,97]. The main issues concern whether the risk outweighs the benefit. Current literatures strongly support a key role of PPARs as regulators of redox signaling in response to oxidative stress in the cardiovascular system by exerting antioxidative effects through transcriptional or post-translational regulations. It remains crucial to confirm many of the in vitro findings on the role of PPARs as redox regulators in intact animals under normal physiological and pathological conditions. Preclinical studies on animal models with temporal and spatial genetic manipulations have emerged as powerful tools for preclinical understanding of PPAR’s roles in regulating redox signaling in the cardiovascular system. Most importantly, these studies will provide insights into the potential development of partial PPAR modulators that regulate specific cellular redox state without major unwanted effects.
Footnotes
Supported by Grants from National Institutes of Health, 1R01 HL085499 to Yang Q, NO. 1R01 HL084456, and NO. T32 HL007457 to Kim T and the ADA Basic Science Award, #7-12-BS-208, to Yang Q
P- Reviewers Aggarwal A, Chawla M, Cheng XS S- Editor Wen LL L- Editor A E- Editor Yan JL
References
- 1.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 3.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol. 2009;21:894–899. doi: 10.1016/j.ceb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol. 2002;34:379–388. doi: 10.1006/jmcc.2002.1526. [DOI] [PubMed] [Google Scholar]
- 6.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49:241–248. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Tocchetti CG, Krieg T, Moens AL. Oxidative and nitrosative stress in the maintenance of myocardial function. Free Radic Biol Med. 2012;53:1531–1540. doi: 10.1016/j.freeradbiomed.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 8.Kunau WH, Dommes V, Schulz H. beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: a century of continued progress. Prog Lipid Res. 1995;34:267–342. doi: 10.1016/0163-7827(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka T, Nakamura H, Nishiyama A, Hosoi F, Masutani H, Wada H, Yodoi J. Redox regulation by thioredoxin superfamily; protection against oxidative stress and aging. Free Radic Res. 2000;33:851–855. doi: 10.1080/10715760000301361. [DOI] [PubMed] [Google Scholar]
- 10.Drevet JR. The antioxidant glutathione peroxidase family and spermatozoa: a complex story. Mol Cell Endocrinol. 2006;250:70–79. doi: 10.1016/j.mce.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 11.Takebe G, Yarimizu J, Saito Y, Hayashi T, Nakamura H, Yodoi J, Nagasawa S, Takahashi K. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J Biol Chem. 2002;277:41254–41258. doi: 10.1074/jbc.M202773200. [DOI] [PubMed] [Google Scholar]
- 12.Wallace DC. Animal models for mitochondrial disease. Methods Mol Biol. 2002;197:3–54. doi: 10.1385/1-59259-284-8:003. [DOI] [PubMed] [Google Scholar]
- 13.Francke U, Taggart RT. Assignment of the gene for cytoplasmic superoxide dismutase (Sod-1) to a region of chromosome 16 and of Hprt to a region of the X chromosome in the mouse. Proc Natl Acad Sci USA. 1979;76:5230–5233. doi: 10.1073/pnas.76.10.5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu ML, Ho YC, Yet SF. A central role of heme oxygenase-1 in cardiovascular protection. Antioxid Redox Signal. 2011;15:1835–1846. doi: 10.1089/ars.2010.3726. [DOI] [PubMed] [Google Scholar]
- 17.Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Curr Opin Cell Biol. 1998;10:384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 18.Kliewer SA, Xu HE, Lambert MH, Willson TM. Peroxisome proliferator-activated receptors: from genes to physiology. Recent Prog Horm Res. 2001;56:239–263. doi: 10.1210/rp.56.1.239. [DOI] [PubMed] [Google Scholar]
- 19.Blanquicett C, Kang BY, Ritzenthaler JD, Jones DP, Hart CM. Oxidative stress modulates PPAR gamma in vascular endothelial cells. Free Radic Biol Med. 2010;48:1618–1625. doi: 10.1016/j.freeradbiomed.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature. 1998;393:790–793. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- 21.Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, Witztum JL, Auwerx J, Palinski W, Glass CK. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1998;95:7614–7619. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 23.Almeida M, Ambrogini E, Han L, Manolagas SC, Jilka RL. Increased lipid oxidation causes oxidative stress, increased peroxisome proliferator-activated receptor-gamma expression, and diminished pro-osteogenic Wnt signaling in the skeleton. J Biol Chem. 2009;284:27438–27448. doi: 10.1074/jbc.M109.023572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Y, Qin X, Liu S, Li J, Zhu X, Gao T, Wang X. Peroxisome proliferator-activated receptor γ is inhibited by histone deacetylase 4 in cortical neurons under oxidative stress. J Neurochem. 2011;118:429–439. doi: 10.1111/j.1471-4159.2011.07316.x. [DOI] [PubMed] [Google Scholar]
- 25.Iannelli P, Zarrilli V, Varricchio E, Tramontano D, Mancini FP. The dietary antioxidant resveratrol affects redox changes of PPARalpha activity. Nutr Metab Cardiovasc Dis. 2007;17:247–256. doi: 10.1016/j.numecd.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Poynter ME, Daynes RA. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998;273:32833–32841. doi: 10.1074/jbc.273.49.32833. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Liu JZ, Hu JX, Wu H, Li YL, Chen HL, Bai H, Hai CX. ROS-activated p38 MAPK/ERK-Akt cascade plays a central role in palmitic acid-stimulated hepatocyte proliferation. Free Radic Biol Med. 2011;51:539–551. doi: 10.1016/j.freeradbiomed.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 28.Prusty D, Park BH, Davis KE, Farmer SR. Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor gamma (PPARgamma ) and C/EBPalpha gene expression during the differentiation of 3T3-L1 preadipocytes. J Biol Chem. 2002;277:46226–46232. doi: 10.1074/jbc.M207776200. [DOI] [PubMed] [Google Scholar]
- 29.Fu M, Zhu X, Wang Q, Zhang J, Song Q, Zheng H, Ogawa W, Du J, Chen YE. Platelet-derived growth factor promotes the expression of peroxisome proliferator-activated receptor gamma in vascular smooth muscle cells by a phosphatidylinositol 3-kinase/Akt signaling pathway. Circ Res. 2001;89:1058–1064. doi: 10.1161/hh2301.099642. [DOI] [PubMed] [Google Scholar]
- 30.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta. 2007;1771:952–960. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wadosky KM, Willis MS. The story so far: post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. Am J Physiol Heart Circ Physiol. 2012;302:H515–H526. doi: 10.1152/ajpheart.00703.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol. 2003;35:1385–1394. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Di Lisa F, Kaludercic N, Paolocci N. β₂-Adrenoceptors, NADPH oxidase, ROS and p38 MAPK: another 'radical' road to heart failure. Br J Pharmacol. 2011;162:1009–1011. doi: 10.1111/j.1476-5381.2010.01130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamberts RR, Onderwater G, Hamdani N, Vreden MJ, Steenhuisen J, Eringa EC, Loer SA, Stienen GJ, Bouwman RA. Reactive oxygen species-induced stimulation of 5’AMP-activated protein kinase mediates sevoflurane-induced cardioprotection. Circulation. 2009;120:S10–S15. doi: 10.1161/CIRCULATIONAHA.108.828426. [DOI] [PubMed] [Google Scholar]
- 35.Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem. 2001;276:44495–44501. doi: 10.1074/jbc.M105945200. [DOI] [PubMed] [Google Scholar]
- 36.Brandt JM, Djouadi F, Kelly DP. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J Biol Chem. 1998;273:23786–23792. doi: 10.1074/jbc.273.37.23786. [DOI] [PubMed] [Google Scholar]
- 37.Inoue I, Goto S, Matsunaga T, Nakajima T, Awata T, Hokari S, Komoda T, Katayama S. The ligands/activators for peroxisome proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu2+,Zn2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism. 2001;50:3–11. doi: 10.1053/meta.2001.19415. [DOI] [PubMed] [Google Scholar]
- 38.Lee H, Shi W, Tontonoz P, Wang S, Subbanagounder G, Hedrick CC, Hama S, Borromeo C, Evans RM, Berliner JA, et al. Role for peroxisome proliferator-activated receptor alpha in oxidized phospholipid-induced synthesis of monocyte chemotactic protein-1 and interleukin-8 by endothelial cells. Circ Res. 2000;87:516–521. doi: 10.1161/01.res.87.6.516. [DOI] [PubMed] [Google Scholar]
- 39.Neve BP, Corseaux D, Chinetti G, Zawadzki C, Fruchart JC, Duriez P, Staels B, Jude B. PPARalpha agonists inhibit tissue factor expression in human monocytes and macrophages. Circulation. 2001;103:207–212. doi: 10.1161/01.cir.103.2.207. [DOI] [PubMed] [Google Scholar]
- 40.Yue TL, Bao W, Jucker BM, Gu JL, Romanic AM, Brown PJ, Cui J, Thudium DT, Boyce R, Burns-Kurtis CL, et al. Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation. 2003;108:2393–2399. doi: 10.1161/01.CIR.0000093187.42015.6C. [DOI] [PubMed] [Google Scholar]
- 41.Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, Teissier E, Minnich A, Jaye M, Duverger N, et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 2001;7:53–58. doi: 10.1038/83348. [DOI] [PubMed] [Google Scholar]
- 42.Wayman NS, Hattori Y, McDonald MC, Mota-Filipe H, Cuzzocrea S, Pisano B, Chatterjee PK, Thiemermann C. Ligands of the peroxisome proliferator-activated receptors (PPAR-gamma and PPAR-alpha) reduce myocardial infarct size. FASEB J. 2002;16:1027–1040. doi: 10.1096/fj.01-0793com. [DOI] [PubMed] [Google Scholar]
- 43.Ibarra-Lara L, Hong E, Soria-Castro E, Torres-Narváez JC, Pérez-Severiano F, Del Valle-Mondragón L, Cervantes-Pérez LG, Ramírez-Ortega M, Pastelín-Hernández GS, Sánchez-Mendoza A. Clofibrate PPARα activation reduces oxidative stress and improves ultrastructure and ventricular hemodynamics in no-flow myocardial ischemia. J Cardiovasc Pharmacol. 2012;60:323–334. doi: 10.1097/FJC.0b013e31826216ed. [DOI] [PubMed] [Google Scholar]
- 44.Yeh CH, Chen TP, Lee CH, Wu YC, Lin YM, Lin PJ. Cardiomyocytic apoptosis following global cardiac ischemia and reperfusion can be attenuated by peroxisome proliferator-activated receptor alpha but not gamma activators. Shock. 2006;26:262–270. doi: 10.1097/01.shk.0000225863.56714.96. [DOI] [PubMed] [Google Scholar]
- 45.Billiet L, Furman C, Cuaz-Pérolin C, Paumelle R, Raymondjean M, Simmet T, Rouis M. Thioredoxin-1 and its natural inhibitor, vitamin D3 up-regulated protein 1, are differentially regulated by PPARalpha in human macrophages. J Mol Biol. 2008;384:564–576. doi: 10.1016/j.jmb.2008.09.061. [DOI] [PubMed] [Google Scholar]
- 46.Guellich A, Damy T, Lecarpentier Y, Conti M, Claes V, Samuel JL, Quillard J, Hébert JL, Pineau T, Coirault C. Role of oxidative stress in cardiac dysfunction of PPARalpha-/- mice. Am J Physiol Heart Circ Physiol. 2007;293:H93–H102. doi: 10.1152/ajpheart.00037.2007. [DOI] [PubMed] [Google Scholar]
- 47.Liu J, Wang P, He L, Li Y, Luo J, Cheng L, Qin Q, Brako LA, Lo WK, Lewis W, et al. Cardiomyocyte-Restricted Deletion of PPARβ/δ in PPARα-Null Mice Causes Impaired Mitochondrial Biogenesis and Defense, but No Further Depression of Myocardial Fatty Acid Oxidation. PPAR Res. 2011;2011:372854. doi: 10.1155/2011/372854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pettersen JC, Pruimboom-Brees I, Francone OL, Amacher DE, Boldt SE, Kerlin RL, Ballinger WE. The PPARα agonists fenofibrate and CP-778875 cause increased β-oxidation, leading to oxidative injury in skeletal and cardiac muscle in the rat. Toxicol Pathol. 2012;40:435–447. doi: 10.1177/0192623311431945. [DOI] [PubMed] [Google Scholar]
- 50.Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem. 2002;277:40265–40274. doi: 10.1074/jbc.M206324200. [DOI] [PubMed] [Google Scholar]
- 51.Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, Sadoshima J. PPARα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab. 2011;14:598–611. doi: 10.1016/j.cmet.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teissier E, Nohara A, Chinetti G, Paumelle R, Cariou B, Fruchart JC, Brandes RP, Shah A, Staels B. Peroxisome proliferator-activated receptor alpha induces NADPH oxidase activity in macrophages, leading to the generation of LDL with PPAR-alpha activation properties. Circ Res. 2004;95:1174–1182. doi: 10.1161/01.RES.0000150594.95988.45. [DOI] [PubMed] [Google Scholar]
- 53.Marx N, Mackman N, Schönbeck U, Yilmaz N, Hombach V, Libby P, Plutzky J. PPARalpha activators inhibit tissue factor expression and activity in human monocytes. Circulation. 2001;103:213–219. doi: 10.1161/01.cir.103.2.213. [DOI] [PubMed] [Google Scholar]
- 54.Law RE, Goetze S, Xi XP, Jackson S, Kawano Y, Demer L, Fishbein MC, Meehan WP, Hsueh WA. Expression and function of PPARgamma in rat and human vascular smooth muscle cells. Circulation. 2000;101:1311–1318. doi: 10.1161/01.cir.101.11.1311. [DOI] [PubMed] [Google Scholar]
- 55.Moore KJ, Rosen ED, Fitzgerald ML, Randow F, Andersson LP, Altshuler D, Milstone DS, Mortensen RM, Spiegelman BM, Freeman MW. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat Med. 2001;7:41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- 56.Asakawa M, Takano H, Nagai T, Uozumi H, Hasegawa H, Kubota N, Saito T, Masuda Y, Kadowaki T, Komuro I. Peroxisome proliferator-activated receptor gamma plays a critical role in inhibition of cardiac hypertrophy in vitro and in vivo. Circulation. 2002;105:1240–1246. doi: 10.1161/hc1002.105225. [DOI] [PubMed] [Google Scholar]
- 57.Duan SZ, Ivashchenko CY, Russell MW, Milstone DS, Mortensen RM. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-gamma both induce cardiac hypertrophy in mice. Circ Res. 2005;97:372–379. doi: 10.1161/01.RES.0000179226.34112.6d. [DOI] [PubMed] [Google Scholar]
- 58.Ding G, Fu M, Qin Q, Lewis W, Kim HW, Fukai T, Bacanamwo M, Chen YE, Schneider MD, Mangelsdorf DJ, et al. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc Res. 2007;76:269–279. doi: 10.1016/j.cardiores.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 59.Yue Tl TL, Chen J, Bao W, Narayanan PK, Bril A, Jiang W, Lysko PG, Gu JL, Boyce R, Zimmerman DM, et al. In vivo myocardial protection from ischemia/reperfusion injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2001;104:2588–2594. doi: 10.1161/hc4601.099403. [DOI] [PubMed] [Google Scholar]
- 60.Khandoudi N, Delerive P, Berrebi-Bertrand I, Buckingham RE, Staels B, Bril A. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma, inhibits the Jun NH(2)-terminal kinase/activating protein 1 pathway and protects the heart from ischemia/reperfusion injury. Diabetes. 2002;51:1507–1514. doi: 10.2337/diabetes.51.5.1507. [DOI] [PubMed] [Google Scholar]
- 61.Luo J, Wu S, Liu J, Li Y, Yang H, Kim T, Zhelyabovska O, Ding G, Zhou Y, Yang Y, et al. Conditional PPARγ knockout from cardiomyocytes of adult mice impairs myocardial fatty acid utilization and cardiac function. Am J Transl Res. 2010;3:61–72. [PMC free article] [PubMed] [Google Scholar]
- 62.Chen T, Jin X, Crawford BH, Cheng H, Saafir TB, Wagner MB, Yuan Z, Ding G. Cardioprotection from oxidative stress in the newborn heart by activation of PPARγ is mediated by catalase. Free Radic Biol Med. 2012;53:208–215. doi: 10.1016/j.freeradbiomed.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 63.Takano H, Komuro I. Peroxisome proliferator-activated receptor gamma and cardiovascular diseases. Circ J. 2009;73:214–220. doi: 10.1253/circj.cj-08-1071. [DOI] [PubMed] [Google Scholar]
- 64.Yang W, Zhang J, Wang H, Shen W, Gao P, Singh M, Fang N. Peroxisome proliferator-activated receptor γ regulates angiotensin II-induced catalase downregulation in adventitial fibroblasts of rats. FEBS Lett. 2011;585:761–766. doi: 10.1016/j.febslet.2011.01.040. [DOI] [PubMed] [Google Scholar]
- 65.Xu D, Murakoshi N, Igarashi M, Hirayama A, Ito Y, Seo Y, Tada H, Aonuma K. PPAR-γ activator pioglitazone prevents age-related atrial fibrillation susceptibility by improving antioxidant capacity and reducing apoptosis in a rat model. J Cardiovasc Electrophysiol. 2012;23:209–217. doi: 10.1111/j.1540-8167.2011.02186.x. [DOI] [PubMed] [Google Scholar]
- 66.Von Knethen A A, Brüne B. Delayed activation of PPARgamma by LPS and IFN-gamma attenuates the oxidative burst in macrophages. FASEB J. 2001;15:535–544. doi: 10.1096/fj.00-0187com. [DOI] [PubMed] [Google Scholar]
- 67.Hwang J, Kleinhenz DJ, Lassègue B, Griendling KK, Dikalov S, Hart CM. Peroxisome proliferator-activated receptor-gamma ligands regulate endothelial membrane superoxide production. Am J Physiol Cell Physiol. 2005;288:C899–C905. doi: 10.1152/ajpcell.00474.2004. [DOI] [PubMed] [Google Scholar]
- 68.Hwang J, Kleinhenz DJ, Rupnow HL, Campbell AG, Thulé PM, Sutliff RL, Hart CM. The PPARgamma ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vascul Pharmacol. 2007;46:456–462. doi: 10.1016/j.vph.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 69.Williams CR, Lu X, Sutliff RL, Hart CM. Rosiglitazone attenuates NF-κB-mediated Nox4 upregulation in hyperglycemia-activated endothelial cells. Am J Physiol Cell Physiol. 2012;303:C213–C223. doi: 10.1152/ajpcell.00227.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng L, Ding G, Qin Q, Xiao Y, Woods D, Chen YE, Yang Q. Peroxisome proliferator-activated receptor delta activates fatty acid oxidation in cultured neonatal and adult cardiomyocytes. Biochem Biophys Res Commun. 2004;313:277–286. doi: 10.1016/j.bbrc.2003.11.127. [DOI] [PubMed] [Google Scholar]
- 71.Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–1250. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 72.Yue TL, Nerurkar SS, Bao W, Jucker BM, Sarov-Blat L, Steplewski K, Ohlstein EH, Willette RN. In vivo activation of peroxisome proliferator-activated receptor-delta protects the heart from ischemia/reperfusion injury in Zucker fatty rats. J Pharmacol Exp Ther. 2008;325:466–474. doi: 10.1124/jpet.107.135327. [DOI] [PubMed] [Google Scholar]
- 73.Jucker BM, Doe CP, Schnackenberg CG, Olzinski AR, Maniscalco K, Williams C, Hu TC, Lenhard SC, Costell M, Bernard R, et al. PPARdelta activation normalizes cardiac substrate metabolism and reduces right ventricular hypertrophy in congestive heart failure. J Cardiovasc Pharmacol. 2007;50:25–34. doi: 10.1097/FJC.0b013e31804b4163. [DOI] [PubMed] [Google Scholar]
- 74.Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, Shoghi K, Welch MJ, Kelly DP. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930–3939. doi: 10.1172/JCI32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu J, Wang P, Luo J, Huang Y, He L, Yang H, Li Q, Wu S, Zhelyabovska O, Yang Q. Peroxisome proliferator-activated receptor β/δ activation in adult hearts facilitates mitochondrial function and cardiac performance under pressure-overload condition. Hypertension. 2011;57:223–230. doi: 10.1161/HYPERTENSIONAHA.110.164590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang J, Fu M, Zhu X, Xiao Y, Mou Y, Zheng H, Akinbami MA, Wang Q, Chen YE. Peroxisome proliferator-activated receptor delta is up-regulated during vascular lesion formation and promotes post-confluent cell proliferation in vascular smooth muscle cells. J Biol Chem. 2002;277:11505–11512. doi: 10.1074/jbc.M110580200. [DOI] [PubMed] [Google Scholar]
- 77.Shi Y, Hon M, Evans RM. The peroxisome proliferator-activated receptor delta, an integrator of transcriptional repression and nuclear receptor signaling. Proc Natl Acad Sci USA. 2002;99:2613–2618. doi: 10.1073/pnas.052707099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vosper H, Patel L, Graham TL, Khoudoli GA, Hill A, Macphee CH, Pinto I, Smith SA, Suckling KE, Wolf CR, et al. The peroxisome proliferator-activated receptor delta promotes lipid accumulation in human macrophages. J Biol Chem. 2001;276:44258–44265. doi: 10.1074/jbc.M108482200. [DOI] [PubMed] [Google Scholar]
- 79.Oliver WR, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci USA. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leibowitz MD, Fiévet C, Hennuyer N, Peinado-Onsurbe J, Duez H, Bergera J, Cullinan CA, Sparrow CP, Baffic J, Berger GD, et al. Activation of PPARdelta alters lipid metabolism in db/db mice. FEBS Lett. 2000;473:333–336. doi: 10.1016/s0014-5793(00)01554-4. [DOI] [PubMed] [Google Scholar]
- 81.Kim HJ, Ham SA, Kim MY, Hwang JS, Lee H, Kang ES, Yoo T, Woo IS, Yabe-Nishimura C, Paek KS, et al. PPARδ coordinates angiotensin II-induced senescence in vascular smooth muscle cells through PTEN-mediated inhibition of superoxide generation. J Biol Chem. 2011;286:44585–44593. doi: 10.1074/jbc.M111.222562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee H, Ham SA, Kim MY, Kim JH, Paek KS, Kang ES, Kim HJ, Hwang JS, Yoo T, Park C, et al. Activation of PPARδ counteracts angiotensin II-induced ROS generation by inhibiting rac1 translocation in vascular smooth muscle cells. Free Radic Res. 2012;46:912–919. doi: 10.3109/10715762.2012.687448. [DOI] [PubMed] [Google Scholar]
- 83.Kim MY, Kang ES, Ham SA, Hwang JS, Yoo TS, Lee H, Paek KS, Park C, Lee HT, Kim JH, et al. The PPARδ-mediated inhibition of angiotensin II-induced premature senescence in human endothelial cells is SIRT1-dependent. Biochem Pharmacol. 2012;84:1627–1634. doi: 10.1016/j.bcp.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 84.Wang P, Liu J, Li Y, Wu S, Luo J, Yang H, Subbiah R, Chatham J, Zhelyabovska O, Yang Q. Peroxisome proliferator-activated receptor {delta} is an essential transcriptional regulator for mitochondrial protection and biogenesis in adult heart. Circ Res. 2010;106:911–919. doi: 10.1161/CIRCRESAHA.109.206185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yoo HY, Chang MS, Rho HM. Induction of the rat Cu/Zn superoxide dismutase gene through the peroxisome proliferator-responsive element by arachidonic acid. Gene. 1999;234:87–91. doi: 10.1016/s0378-1119(99)00176-6. [DOI] [PubMed] [Google Scholar]
- 86.Girnun GD, Domann FE, Moore SA, Robbins ME. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol. 2002;16:2793–2801. doi: 10.1210/me.2002-0020. [DOI] [PubMed] [Google Scholar]
- 87.Kim YH, Yoo HY, Chang MS, Jung G, Rho HM. C/EBP alpha is a major activator for the transcription of rat Cu/Zn superoxide dismutase gene in liver cell. FEBS Lett. 1997;401:267–270. doi: 10.1016/s0014-5793(96)01487-1. [DOI] [PubMed] [Google Scholar]
- 88.Chung SS, Kim M, Youn BS, Lee NS, Park JW, Lee IK, Lee YS, Kim JB, Cho YM, Lee HK, et al. Glutathione peroxidase 3 mediates the antioxidant effect of peroxisome proliferator-activated receptor gamma in human skeletal muscle cells. Mol Cell Biol. 2009;29:20–30. doi: 10.1128/MCB.00544-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Westergaard M, Henningsen J, Johansen C, Rasmussen S, Svendsen ML, Jensen UB, Schrøder HD, Staels B, Iversen L, Bolund L, et al. Expression and localization of peroxisome proliferator-activated receptors and nuclear factor kappaB in normal and lesional psoriatic skin. J Invest Dermatol. 2003;121:1104–1117. doi: 10.1046/j.1523-1747.2003.12536.x. [DOI] [PubMed] [Google Scholar]
- 90.Planavila A, Laguna JC, Vázquez-Carrera M. Nuclear factor-kappaB activation leads to down-regulation of fatty acid oxidation during cardiac hypertrophy. J Biol Chem. 2005;280:17464–17471. doi: 10.1074/jbc.M414220200. [DOI] [PubMed] [Google Scholar]
- 91.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ding G, Cheng L, Qin Q, Frontin S, Yang Q. PPARdelta modulates lipopolysaccharide-induced TNFalpha inflammation signaling in cultured cardiomyocytes. J Mol Cell Cardiol. 2006;40:821–828. doi: 10.1016/j.yjmcc.2006.03.422. [DOI] [PubMed] [Google Scholar]
- 93.Bénardeau A, Verry P, Atzpodien EA, Funk JM, Meyer M, Mizrahi J, Winter M, Wright MB, Uhles S, Sebokova E. Effects of the dual PPAR-α/γ agonist aleglitazar on glycaemic control and organ protection in the Zucker diabetic fatty rat. Diabetes Obes Metab. 2013;15:164–174. doi: 10.1111/dom.12006. [DOI] [PubMed] [Google Scholar]
- 94.Henry RR, Lincoff AM, Mudaliar S, Rabbia M, Chognot C, and Herz M. Effect of the dual peroxisome proliferator-activated receptor-alpha/gamma agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (SYNCHRONY): a phase II, randomised, dose-ranging study. Lancet. 2009;374:126–35. doi: 10.1016/S0140-6736(09)60870-9. [DOI] [PubMed] [Google Scholar]
- 95.Wilding JP. PPAR agonists for the treatment of cardiovascular disease in patients with diabetes. Diabetes Obes Metab. 2012;14:973–982. doi: 10.1111/j.1463-1326.2012.01601.x. [DOI] [PubMed] [Google Scholar]
- 96.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 97.Stockl KM, Le L, Zhang S, Harada AS. Risk of acute myocardial infarction in patients treated with thiazolidinediones or other antidiabetic medications. Pharmacoepidemiol Drug Saf. 2009;18:166–174. doi: 10.1002/pds.1700. [DOI] [PubMed] [Google Scholar]