

Abstract
New methodologies are discussed that allow for several commonly used transition-metal-catalyzed coupling reactions to be conducted within aqueous micellar nanoparticles at ambient temperatures.
Keywords: green chemistry, micellar catalysis, designer surfactants, cross-couplings, PTS, TPGS-750-M
1. Introduction
Green chemistry recently took a front row seat on the world stage. Unfortunately, it was not associated with any special technological advance; rather, the 200-million-plus gallons (ca. 5 million barrels) of oil that had leaked into the Gulf of Mexico focused attention on methods for dispersing such a huge quantity of localized hydrocarbons.1 British Petroleum (BP) addressed this catastrophe by injecting dispersants at the site, the key ingredient being a mix of surfactants. While this tactic raised more than a few eyebrows with respect to additional pollutants having been introduced into the ecosphere, the presumption was that sodium dioctylsulfosuccinate (1), Span™ 80 (3), and a mixture of TWEEN® 80 (4) and 85 (5)—present in dispersants COREXIT® EC9500 and COREXIT® EC95272—would help to “solubilize” the oil by forming micelles, thereby relocating over time the oil deposited within their lipophilic cores (Figure 1). While a disaster of this magnitude needs the tincture of time to assess its full impact on the environment, from a purely chemical perspective, tremendous hope has been placed on these surfactants in anticipation that they will prevent further damage to wildlife and shores.
Figure 1.

Ingredients of COREXIT® EC9500 (a Mixture of 1–5) and COREXIT® EC9527 (Includes 1–6). (Ref. 2b)
This micellar chemistry undertaken on a grand scale by the petroleum industry highlights the potential for simple amphiphilic molecules to “solubilize” organic materials in a purely aqueous medium. Many other industries use surfactants routinely; examples include paint, cosmetics, cleaning, leather, carpet, asphalt, and pulp & paper companies.3 Relatively small amounts have been used for decades in highly controlled environments, e.g., as “excipients” in the pharmaceutical arena, to help increase dissolution of otherwise water-insoluble drugs in aqueous media. But where are the studies on their usage in synthesis? Why not apply the same concepts of solubilization within micelles to reactants and catalysts that become, albeit transiently, the occupants? Of course, some synthetic chemistry can be, and has been, done in micelles.4 Interestingly, this approach, oftentimes referred to as “micellar catalysis”, is technically a misnomer since the micelle is not participating as a catalyst in the reaction itself. Nonetheless, why should these, or thousands of other surfactants created by industry and designed for a narrow range of specialized applications, be the most appropriate for use in state-of-the-art transition-metal-mediated cross-couplings? Since organic reactions are oftentimes very sensitive to solvent effects,5 and since the lipophilic portions of micelles are functioning as the reaction medium, which surfactant should be used in this capacity to best assist metal-catalyzed reactions in water at room temperature? No one knows. Perhaps it is time for synthetic chemists to start designing surfactants for synthetic chemistry.
The situation just a few years ago was not encouraging in that there were virtually no general studies of the effect of varying micellar conditions on the most commonly used transition-metal-catalyzed couplings. As green chemistry continues to expand, critical reviews have appeared highlighting micellar media in which the “hydrophobic effect”—the tendency of nonpolar groups to cluster so as to shield themselves from contact with an aqueous environment6—assists with organometallic processes in water, such as in oxidation and reduction reactions,7 as well as in selected C–C-bond forming reactions (e.g., hydroformylations). And so it was in recognition of the potential offered by micellar catalysis, performed in water as the gross reaction medium (and not the solvent), that we set out to develop “designer” surfactants for use in transition-metal-catalyzed cross-coupling reactions.
2. Background
In our previous review in this journal,8 we introduced PTS (7; Figure 2)9 as a useful (nonionic) surfactant; a nanomicelle-forming species in which a variety of Pd- and Ru-catalyzed couplings took place in water at room temperature. The idea behind the choice of PTS was simple: in the Paul Anastas sense, it follows the “12 Principles of Green Chemistry”,10a as explained in Benign by Design.10b That is—in being composed of racemic vitamin E, sebacic acid, and PEG-600—neither PTS nor any of its three components is environmentally of concern. Indeed, PTS is FDA GRAS affirmed for use in dietary supplements.11 By itself, PTS is a provitamin; a modified version of “ester-E.”
Figure 2.

Polyoxyethanyl-α-tocopheryl Sebacate (PTS, 7) and TPGS-1000. (Ref. 8,13)
By comparison, readily available alternative amphiphiles, such as Triton® X-100 and those in the Brij® series,12 only on occasion lead to comparable results in cross-coupling reactions. Even the closely structurally related TPGS (TPGS-1000, Figure 2)13 affords quite different outcomes in cross-couplings under otherwise identical conditions. This seems particularly odd, since both PTS and TPGS-1000 share the same micellar lipophilic interior in the form of α-tocopherol. So what's responsible for the commonly observed greater rates of conversion in PTS? The answer seems to be that both the size and shape of their nanoparticles matter (Figure 3).8,14 For PTS, both 8–10-nm spheres and larger worm- or rod-like particles are present (together averaging ca. 25 nm) according to cryo-TEM analysis (see Section 5). By contrast, TPGS in water forms very sharp 12–13-nm spherical micelles as indicated by microscopy and Dynamic Light Scattering (DLS) measurements.14
Figure 3.

Cryo-TEM Data Comparing Nanoparticles of PTS (A) and TPGS-1000 (B). (Ref. 8,14)
3. Chemistry in PTS-H2O, an Update: a 1st-Generation Amphiphile for Transition-Metal-Catalyzed Cross-Couplings
It is not uncommon for synthetic chemistry to advance at a far greater pace than does our understanding of why the chemistry goes as observed. This is certainly true here as well, involving non-ionic surfactants that self-aggregate to form nanoreactors,15 or what Fujita and co-workers refer to as “functional molecular flasks.”16 Early work focused mainly on a series of Pd-catalyzed “name” reactions (e.g., Heck,17 Suzuki–Miyaura,18 and copper-free Sonogashira19 couplings), as well as olefin cross-20 and ring-closing metathesis21 reactions, all of which could be carried out in water at room temperature (Figure 4).
Figure 4.

PTS-Enabled Reactions in Water at Room Temperature.
Since these initial reports, considerable progress has been made on many related Pd- or Ru-catalyzed couplings. In most cases, the species involved are tolerant of the presence of water as the gross reaction medium.22 Although these couplings are likely occurring within the lipophilic portions of the micelles, some of a micelle's occupants may be exposed at any time to the surrounding water, given its dynamic character.23 That is, there is constant exchange of monomeric units of surfactant between micellar arrays. This motion creates a mechanism by which educts, catalysts, and reaction product(s) can enter and exit a micelle. But the exchange phenomenon also leads to exposure to an aqueous environment through which they must traverse. This can have major (beneficial) consequences for the desired transformation; that is, the content of the water can be altered to great synthetic advantage. Foreshadowing associated with these effects was noted in the previous Aldrichimica Acta review,8 where couplings run in seawater led, in some cases, to faster reactions than had been observed in pure (HPLC grade) water. Changes in the ionic strength24 and pH of the aqueous medium have now been studied and, indeed, there are benefits to be had from such perturbations resulting from the simple addition of selected salts to water (vide infra).
3.1. Amination
The use of PTS is an effective enabling technology for two types of amination, both taking place in water at room temperature. Unsymmetrical di- and triarylamines can be constructed using aryl bromides and aniline derivatives. In the presence of [PdCl(allyl)]2, Takasago's ligand, cBRIDP® (14, Figure 5), was the most effective among several catalysts (8–16) screened. Although these transformations appear to be general in that a variety of reaction partners can be used and yields tend to be good, perhaps the most intriguing aspect of this methodology is the influence of the base (eq 1).25 Thus, while KOH (1.5 equiv) is oftentimes sufficient, reaction times can vary and may take up to a full day to reach completion. That KOH functions well in this capacity is rather interesting, since the coupling is occurring within the lipophilic core of the PTS micelle, where presumably polar species are not to be found. It is certainly possible that KOH remains in the aqueous phase, and that as species exchange between nanoreactors they are exposed to the surrounding water, and it is here when the proton may be abstracted from the participating nitrogen.26 Likely to be more effective, rate-wise, at finding a protonated amine intermediate would be a more lipophilic base, capable of penetrating the micelle into the hydrophobic pocket (eq 2).25 A switch, therefore, to KOt-Bu, made a favorable difference in this regard, notwithstanding the fact that this base in water is mainly KOH. Hence, the same outcome could be achieved by simply adding tert-butyl alcohol to the original mixture containing KOH. Even better, was addition of the commercially available potassium trimethylsilanolate (KOSiMe3),27 which reduced reaction times by almost an order of magnitude. Still more dramatic was inclusion of the far more lipophilic potassium triisopropylsilanolate [(KOSi(i-Pr)3, or KO-TIPS], readily formed in situ from KOH and TIPS-OH.
Figure 5.
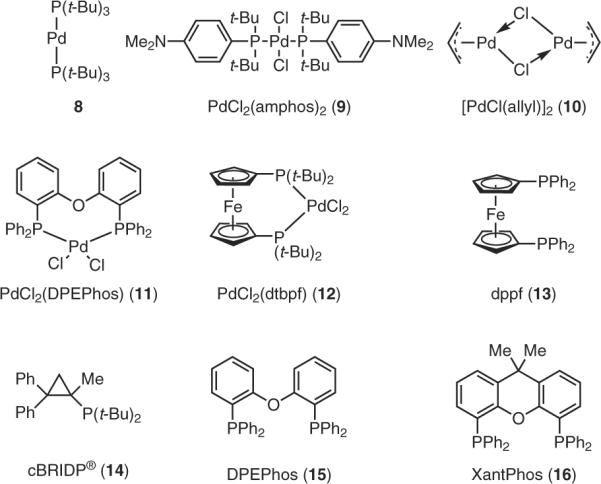
Ligands and/or Catalysts Used in Coupling Reactions. (Ref. 25)
Allylic aminations in water at room temperature can also be accomplished using allylic phenyl ethers as substrates.28 These are unconventional partners in Pd-catalyzed couplings, where activation of the hydroxyl group in an allylic alcohol typically takes place in the form of an acetate, carbonate, sulfonate, or phosphate.29 Nonetheless, under the influence of the hydrophobic effect, and in the presence of excess K2CO3 and methyl formate, couplings result in amination predominantly at the least hindered, terminal site and give high E:Z ratios (eq 3).28 DPEPhos (15)30 was found to be the ligand of choice, while the more rigid analogue XantPhos (16)31 led to only traces of allylic amines.
Perhaps more intriguing is the same net amination, …but with allylic alcohols (eq 4).32 While in situ activation has been achieved previously using a variety of protic or Lewis acids (e.g., SnCl2,33a Et3B,33b RCO2H,33a and CO233b), efforts toward the exclusion of water are common. Those reactions run in pure water typically require harsh conditions.33c–e Under similar micellar conditions applied to allylic ethers (above), couplings give highly favored linear rather than
 |
eq 1 |
(Ref. 25)
 |
eq 2 |
(Ref. 25)
 |
eq 3 |
(Ref. 28)
 |
eq 4 |
(Ref. 32) branched products, also strongly favoring E isomers. Here again, K2CO3 is the preferred base, while HCO2Me in excess is required. Since none of the intermediate resulting from transesterification between the alcohol and methyl formate is observed at any point during the reaction, the proposed mechanism involves Pd(0), the educt, and the formate–reasonable given the presumed high concentration of species within the micelles.
3.2. Suzuki–Miyaura Coupling
As previously described,18 biaryls can be constructed from precursor aryl halides or pseudo-halides Ar–X (X = I, Br, Cl, OSO2R) and boronic acids in 1% PTS in water mainly at ambient temperatures. Subsequent to this work, various heteroaromatic halides were also shown to be amenable.34 Bromide was found to be the preferred leaving group (eq 5)34 and, like chlorides, their reactions occasionally required mild heating to 40 °C (eq 6).34 The choice of catalyst also varied by leaving group: bromides were best accommodated by PdCl2(dtbpf), 12, while chlorides seemed more responsive to PdCl2(amphos)2, 9. A direct comparison with a literature case35 showed the potential for this green chemistry to be highly competitive with traditional organic media (eq 7).34 A review on the Suzuki–Miyaura cross-coupling as an entry to biaryls under green conditions has recently appeared, focusing on water as the reaction medium.36
Carbon–carbon bond formation between usually unreactive, acid- and base-stable allylic phenyl ethers and arylboronic acids is
 |
eq 5 |
(Ref. 34)
 |
eq 6 |
(Ref. 34) also possible under micellar conditions.37 As with allylic aminations (vide supra), there is a strong tendency to generate linear products where conjugation is maintained. Aliphatic ethers, on the other hand, favor branched products presumably due to the known faster rate of reductive elimination from a more hindered Pd(II) intermediate.38 In addition to examples 17–19 (eq 8),37 linchpin 20 bearing both acetate and phenyl ether moieties can be sequentially coupled, where, e.g., amination is followed by Suzuki–Miyaura coupling, all in one pot, all in water at room temperature (eq 9).34
3.3. Silylation
Palladium catalysis in PTS–water can also be extended to the formation of allylic silanes, likewise employing allylic phenyl ethers as substrates, in the presence of Et3N (eq 10).39 Such reactions do not occur in organic solvents (e.g., MeOH) at room temperature. Moreover, whereas silylations of the corresponding acetates require heating in organic solvents (e.g., DMF) for activation of a disilane (R3Si–SiR3),40 cross-couplings in PTS nanoparticles take place at ambient temperatures in ≤20 hours at a global concentration of 0.16 M. The most effective catalyst source is PdCl2(DPEPhos), 11, as monodentate ligands on the metal (e.g., Ph3P), or bidentate chelation of palladium by dtppf or dtbpf, afford only moderate levels of conversion (40–60%). Of the various products possible, linear over branched and E over Z isomers are both favored.39
3.4. C–H Activation
With more than a handful of reviews on the topic of aromatic C–H activation reactions in just the past few years,41 there is now a plethora of methods for introducing C–C bonds mainly ortho- to heteroatom-directing groups… but not in water at room temperature. This can be done, however, employing micellar catalysis.42 Thus, aryl ureas, especially those bearing electron-donating groups, can be cross-coupled with aryl iodides in an aqueous medium containing any one of several surfactants; including PTS, TPGS, Triton® X-100, Solutol,43 Brij® 30, and Brij® 35; where yields in optimization studies varied between 65 and 76%. The corresponding “on water” experiment led to only a 35% yield in the same time frame. Since the best results were obtained in nanomicelles containing Brij® 35, several additional examples were studied in this medium, along with ingredients Pd(OAc)2, AgOAc, and HBF4. Yields were in the 70–97% range (eq 11),42 and products derived from double arylation were rarely observed, presumably reflecting the mild conditions involved.
Arylations with Pd(OAc)2 are typically run in highly acidic media; e.g., HBF4, TFA, TsOH, or HOAc; which facilitate loss of HOAc from the catalyst and lead to electrophilic attack on an aromatic ring.44 Alternatively, it has been shown that Fujiwara–Moritani coupling reactions with acrylates can be carried out in PTS–water (eq 12)45 by using a cationic source of palladium, as in commercially available [Pd(MeCN)4](BF4)2,46 Both benzoquinone (1 equiv) and AgNO3 (2 equiv) are required. Double functionalization ortho to the amide directing group is not observed here as well.
3.5. Negishi-like Couplings on the Fly…in Water
According to reference works on the topic, organozinc halides are not moisture-tolerant.47 Indeed, Knochel and co-workers have spent, literally, decades elucidating the extent to which RZnX reagents can be used in the presence of protons of varying acidity.48 Free alcohols, in general, require protection; the presence of even tert-butyl alcohol takes its toll on couplings run in the presence of this additive.48a Water is nowhere to be found among the various listings; hence, the message
 |
eq 7 |
(Ref. 34)
 |
eq 8 |
(Ref. 37)
 |
eq 9 |
(Ref. 37)
 |
eq 10 |
(Ref. 39)
 |
eq 11 |
(Ref. 42)
 |
eq 12 |
(Ref. 45)
 |
eq 13 |
(Ref. 50a) is clear: whether it's “on water”, “in water”, or “with water”,49 the only conditions applicable to zinc reagents involve “no water”.47
Could the textbooks be “wrong”? Not likely. But in this obvious acid–base chemistry between RZnX and H2O two assumptions are implied: (i) that RZnX and H2O find each other, and (ii) that a stoichiometric amount (or more) of RZnX is used; otherwise, with lesser amounts, the anticipated yield must suffer. If both of these conditions are removed; that is, if RZnX could be “insulated” from any water present, and if the amount of RZnX at any given time is minimized and yet, over time, a stoichiometric level of RZnX is formed in the pot, then the desired Pd-catalyzed cross-couplings of organzinc reagents, in water, might be possible. Well, they are, thanks to micellar catalysis.
For both aryl bromides50 and alkenyl halides,51 couplings with primary and secondary alkyl halides can now be accomplished in a remarkably straightforward fashion. The recipe calls for the two precursor halides, a specific (commercially available) palladium catalyst, and importantly, TMEDA. These are combined in water (ca. 0.3 M) containing 2 wt % PTS to which is added inexpensive zinc metal in the form of powder or dust; and the whole mixture is then stirred. After a reasonable period of time, which is substrate-dependent, the cross-coupled product is obtained in good yield. Representative examples involving aromatic and heteroaromatic bromides are illustrated in eq 13.50a
This remarkable process can be envisioned as depending entirely on the precise timing of the various events that need to take place, just like the gears of a fine-tuned pocket watch (Figure 6). Within the nanoparticles of PTS are housed the various reaction components, densely packed due to the hydrophobic effect; no organometallic (RZnX) is present. As nanoparticles collide with zinc metal on its surface, preferential insertion of Zn into the sp3 halide (R–X) takes place in a likely successive one-electron-transfer sequence. The resulting water-sensitive RZnX is insulated from the surrounding water. RZnX is also stabilized by the TMEDA present, which may assist in shuttling it into the micelle where both the Pd catalyst and sp2 halide await, in relatively high concentrations. If the rate of formation of RZnX is too fast for subsequent passage into the hydrophobic micellar core, the reagent escapes and is rapidly quenched by water, as expected. Thus, while electron transfer en route to RZnX can be controlled for both 1° and 2° alkyl halides, reactions involving 3° precursors are far too rapid; thus, only quenched material (R–H) results.50
Figure 6.

Pictorial Account of Couplings of RZnX in Water: Timing Is Key.
The corresponding couplings with alkenyl iodides and bromides, rather than aryl bromides, also give the anticipated products with retention of stereochemical integrity (eq 14).51 With both types of educts (aryl and alkenyl), the choice of catalyst is absolutely crucial: PdCl2(amphos)2(9)52 is the only species screened to date that affords high levels of conversion and, thus, good isolated yields. Even the parent species; i.e., the bis(des-dimethylamino) analogue, was not nearly as effective. (Note that the nomenclature for “amphos” as used for this ligand52is seemingly inconsistent with prior literature.53)
4. New Insights into Micellar Catalysis for Organic Synthesis
The observation that several of these Pd-catalyzed couplings not only can be run in seawater,54 but actually take place in this medium at a greater rate than in HPLC grade water, has led to some very exciting developments of potential practical value. The presence of salts in the water can influence the chemistry in two significant ways:55 (1) the size and shape of the nanoparticles in which these couplings take place are altered, and (2) the pH of the aqueous medium which can impact the nature of the catalysts involved. With NaCl, a “salting out” effect exists,56 which removes water from the PEGylated portions of the particles, tending to increase micellar size and/or modify particle shape as the PEG expands into the water. This effect is due to the anion, and several salts examined lead to similar particle changes, although the corresponding coupling chemistry in the presence of each salt has yet to be examined. Particle shape can also be dramatically altered. These effects can be shown by cryo-TEM analysis, e.g., on PTS with and without NaCl (Figure 7).54 The “salting in” effect, which tends to decrease micellar size, is observed, e.g., with iodides NaI and KI.57
Figure 7.

Cryo-TEM Image of (A) Aqueous PTS (50-nm Scale), (B) Aqueous PTS in the Presence of 3 M NaCl (100-nm Scale), and (C) TPGS-750-M. (Ref. 54)
Changes in pH resulting from addition of small amounts of a salt such as KHSO4 can impact couplings due to the dynamic nature of micelles; i.e., they are constantly in flux.23 Their amphiphiles traverse a sea of surrounding water for the exchange phenomenon to occur. For catalysts that contain phosphines, e.g., ruthenium carbenes, as used routinely in olefin metathesis,58 their phosphine ligands can be protonated upon exposure to aqueous acid,59 and hence, may arrive at the micelle as a coordinatively unsaturated species (and thus, quite “hot”). These phenomena are illustrated below by Heck couplings and olefin metathesis reactions.
4.1. Heck Coupling in PTS-3 M NaCl (aq)
Heck reactions in micellar PTS–H2O are especially responsive to changes in the ionic strength of the medium.54 This effect is not to be expected in related reactions that, albeit conducted in water, rely on alternative phenomena. For example, supported catalysts such as
 |
eq 14 |
(Ref. 51)
 |
eq 15 |
(Ref. 54) Pd(OAc)2 mounted on a reverse-phase N,N-diethylaminopropylated (NDEAP) silica gel impregnated with ionic liquid [bmim]PF6 have been used (Scheme 1).60 Other advances include applications of new Pd-complexed cationic bipyridyl ligands (L-1),61 and new P–N ligands in the form of diazadiphosphetidines (L-2).62 In each case involving an acrylate partner, while water is the reaction solvent, heating is needed to improve conversion.
Scheme 1.

Two examples of reactions subject to the influence of salts under micellar conditions are worth noting. In the first, cinnamate 26 is formed in PTS–water to a limited extent after 14 hours (Scheme 2, top reaction).54 However, in the presence of NaCl (3 M) and at an identical global concentration (0.50 M), the reaction reaches full conversion in the same time period, with an associated high yield. In the second example, the formation of 27 requires mild heating to 50 °C to reach completion after 8 hours; in PTS–3 M aqueous NaCl, however, the reaction was complete at room temperature in 3 hours (Scheme 2, bottom).54
Scheme 2.

Two Examples of the Influence of Added Salt on the Outcome of the Heck Coupling in PTS–H2O. (Ref. 54)
4.2. Olefin Metathesis at pH 2–3
In 2006, Hong and Grubbs reported on ring-opening polymerization (ROMP) reactions in aqueous acid, and showed that the 2nd-generation Grubbs catalyst has its phosphine sequestered via protonation.59 The same protonation is presumably responsible for accelerating cross-metathesis under micellar conditions in water, most visibly in reactions involving tough type-2 olefins such as methyl vinyl ketone (MVK) and acrylonitrile (eq 15).54 For related ring-closing reactions, e.g., forming a 7-membered ring, the net effect of added KHSO4 was equivalent to that seen using 3 M NaCl.
5. Designing a Better Micelle: TPGS-750-M, a 2nd-Generation Amphiphile
Notwithstanding the progress made utilizing the first-generation designer surfactant PTS, the many lessons learned insofar as altering the nature of the surfactant to maximize reaction rates and reagent or catalyst stability encouraged the design of a second-generation amphiphile, TPGS-750-M.63 Experience had shown that micelle size and shape matter; that improvements in rate in going from TPGS-1000 to PTS, where particle size increases from 13 to (on average) 25 nm,54 seems to be an important clue. Since differences between the two were best visualized by cryo-TEM (see Figure 3), showing the longer rods or worms present only with PTS, TPGS-750-M (Figure 7C) was engineered to have a higher percentage of larger particles, preferably in the 50–100-nm range. A shorter PEG chain requiring less volume in its coiled state, and hence micelles able to accommodate more molecules per particle, should expand the particle's radius. A structural comparison between TPGS, PTS, and TPGS-750-M is illustrated in Figure 8.
Figure 8.

Structures of TPGS, PTS, and the Newly Engineered TPGS-750-M. (Ref. 63)
From the standpoint of synthesis, a switch to four-carbon succinic acid as found in TPGS-1000,13 rather than the ten-carbon sebacic acid as in PTS, makes a huge difference in overall efficiency of the two-step sequence (Scheme 3). That is, opening of succinic anhydride by vitamin E allows for a virtually quantitative esterification involving the most expensive component in the process. By contrast, PTS relies on sebacoyl chloride, a diacid chloride that reacts at both termini with α-tocopherol, thereby affording mixtures of the mono- and the diester.8 Conversion of monoester 28 to TPGS-750-M via traditional esterification can be smoothly done in toluene in the presence of catalytic acid (TsOH), another close to quantitative event.63 Here, use of a mono-methylated polyethylene glycol, or MPEG, rather than PEG (a diol) is the key to avoiding reactions at both ends. This is yet another problem in the route to PTS, which uses PEG-600 (and not an MPEG analogue). The `net-net' of these changes is that the targeted amphiphile can be prepared in an overall yield that exceeds 95%, while the efficiency of making PTS is ca. 45%.
Scheme 3.
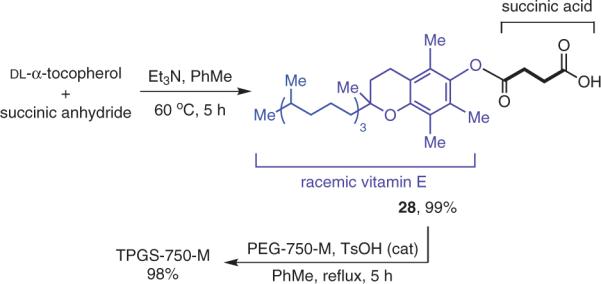
Straightforward, High-Yield Synthesis of TPGS-750-M. (Ref. 63)
The greatly improved cost implies that couplings enabled by TPGS-750-M–H2O need only be as good as those done in PTS. Of course, faster reactions might also be anticipated due to larger particle sizes. By virtue of increases in binding constants of the substrates and catalysts within the micellar environment (i.e., longer time spent within a micelle), reactions should reach higher states of conversion more rapidly.64 What has been found in this regard is that most types of cross-couplings in TPGS-750-M do, in fact, lead to isolated yields that are equal to, or better than, those seen in aqueous PTS. Some direct comparisons are illustrated in Scheme 4, with each pair of reactions being run at the identical concentration and time frame.
Scheme 4.

Cross-Couplings in PTS–H2O and in TPGS-750-M–H2O. (Ref. 63)
Likewise gold-catalyzed cycloisomerizations of allenols take place readily in both aqueous PTS and TPGS-750-M, where the presence of NaCl was found to shorten reaction times (eq 16).65 In a number of cases, yields were better with the second-generation surfactant, although some substrate dependence was noted.
5.1. CuH-Catalyzed Asymmetric Hydrosilylation
The in situ generation and use of copper hydride, derived from precursors such as CuCl–NaOt-Bu, CuF2, or Cu(OAc)2, routinely takes place in organic media: toluene, THF, etc.66 Although the reagent is stable to water and, in fact, was used long ago by Stryker to enhance the rate of quenching of copper(I) intermediates (e.g., enolates resulting from 1,4 addition of hydride from (Ph3P)CuH,67 its use in a strictly aqueous medium is not among the available protocols. It might even be argued that water as “solvent” presents a likely major
 |
eq 16 |
(Ref.65)
 |
eq 17 |
(Ref. 70)
 |
eq 18 |
(Ref.70a) limitation, since the literature on these conjugate reductions clearly suggests that lower reaction temperatures tend to maximize ee's.68 Thus, while eliminating organic solvents from these reactions would impart an element of “greenness”, the tradeoff in substrate insolubility and potentially lower ee's might not favor a “green” approach. On the other hand, such reactions being run at high concentrations within micelles at room temperature may not follow the same rules, in which case the use of organic solvent and consumption of energy for cooling would both be averted.
It came with some surprise, therefore, that treatment of isophorone with CuH ligated by Takasago's (R)-DTBM-SEGPHOS® (29)69(Figure 9) in 2 wt % PTS–H2O at room temperature afforded the product of 1,4 addition in high yield and in 99% ee (eq 17).70 This result is comparable to the best that has been seen to date, albeit done in toluene at low temperatures.71 Unfortunately, this conversion required far too much PhSiH3 (12 equiv, or 36 equiv of hydride), as this reagent is competitively decomposed in aqueous solution. Eventually, the best conditions identified focused on the use of polymethylhydrosiloxane (PMHS)72 as hydride donor (⅓ equiv, 9 equiv of hydride) in 2 wt % TPGS-750-M, giving the same product in 94% ee. By way of comparison, the identical reaction run in water only gave 18% conversion after the same 18-h reaction time.
Figure 9.
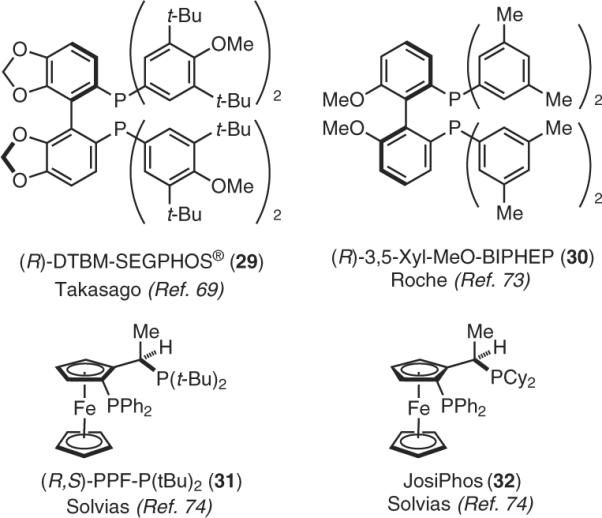
Nonracemic Ligands Used in CuH-Catalyzed Asymmetric Hydrosilylation.
Other ligands, including Roche's 3,5-Xyl-MeO-BIPHEP (30)73 and Solvias's JosiPhos ligands (31 and 32)74 are also effective, depending upon the nature of the substrate.70 Particularly interesting is enone 34, where in combination with ligand 31 and at %#x2212;78 °C in toluene, an 87% ee was reported.71 The identical reaction run in water at room temperature gave a comparable yield; however, the ee was higher: 93% (eq 18).70a Likewise, butyrolactone 35 reacted at room temperature and led to a similar yield and ee relative to that seen earlier, where the reaction was run at 0 °C. That ee's can be obtained that are on par with, or even exceed, those normally realized at low temperatures may be a consequence of restricted reagent and/ or substrate movement within tightly packed micellar arrays, where energetic differences between diastereomeric transition states are accentuated, and less favorable orientations are minimized. The nature of the micelle interior also plays a crucial role, as documented by these asymmetric hydrosilylations run under otherwise identical conditions using alternative amphiphiles (Brij® 30, Cremophor®, SDS, and Solutol®)–water solutions (eq 19).70 Clearly, the newly designed TPGS-750-M affords the most effective medium for this type of transformation. That high ee's can be realized at room temperature within a micellar array without normal recourse to low temperatures is particularly striking.
5.2. Borylation of Aryl Halides
Aryl boronates, in particular those derived from pinacol using bis(pinacolato)diboron (B2pin2), figure prominently as coupling partners in Suzuki–Miyaura reactions.75 Aryl bromides are common educts, although borylations in organic media (e.g., dioxane, DMSO, or THF) at room temperature are very rare.76 Couplings “on water” do lead to product, but to a limited and unpredictable extent. However, in water containing 2–3 wt % TPGS-750-M, aryl pinacolatoboranes (36) could be smoothly prepared usually within three hours at a global concentration of 0.25 M (eq 20).77 A Pd(0) catalyst, Pd(Pt-Bu3)2 (8, 3 mol %), afforded the highest yields over several alternatives examined (e.g., Pd(OAc)2–XPhos, PdCl2(dppf), Pd2(dba)3–2PCy3, etc.). Given that palladium is already present in the form of Pd(Pt-Bu3)2, as well as its known use in Suzuki–Miyaura couplings,78 introduction of a second aryl bromide into the reaction mixture ultimately leads to biaryl 37 (eq 21). Compound 37 is formally the net cross-coupling product of two aryl bromides, achieved in one pot in water at room temperature.79
6. Summary and Outlook
Surfactants have an especially rich history of service to many areas of organic chemistry.3 But most amphiphiles listed in catalogs perused by organic chemists today are from decades ago; they are rarely “green”, nor are they matched in any way to the nanoscale environments that maximize the quality of transition-metal-catalyzed reactions. However, starting to appear are newly engineered “designer” surfactants that better accommodate reaction partners, additives, and catalysts characteristic of modern organic synthesis. In the interest of sustainability, the goal in designing new surfactants for synthesis is to develop reproducible and scalable processes that minimize involvement of organic solvents, at least from the standpoint of the reaction medium. Historically, however, “surfactant science & technology as pursued in academia has not overlapped well with the mindset of industrial chemists in this area”.80 That is, while academicians tend to strive for purified, homogeneous materials that are readily subject to analytical techniques (e.g., surfactants such as Triton® X-100 and SDS), industrial researchers oftentimes are faced with a “Make it work and don't worry about why!” approach. As author Drew Meyers unabashedly continues in his monograph,80 “The sad fact of life is that real surfactant systems are almost always composed of mixed chemical isomers, contaminants, and added materials that can alter the effects of a given surfactant on a system…” This is precisely the current state of affairs surrounding PTS, since, in fact, it is a mix of many components: PEG-600 contains a broad range of polyethylene units, and the many byproducts from its preparation, as discussed earlier, are likely to be present in varying amounts. Its chromatographic purification using peak-shaving techniques led to the unexpected observation that, remarkably, PTS of >95% purity is actually insoluble in water!81
The switch to TPGS-750-M, therefore, provides an amphiphile that avoids many of these contaminants by virtue of its design. While remaining benign, it offers the community a better economic profile, along with enhanced reaction rates for the cross-couplings taking place within its nanomicelles. As with PTS, it also usually leads to a very attractive impurity profile, given the room temperature conditions for the vast majority of reactions in this aqueous medium. For the intended industrial uses, time in the kettle (i.e., throughput) is another virtue, since there is no time (or energy) investment due to heating and/or cooling of these coupling reactions. While this second-generation surfactant has yet to be fully evaluated, it would be naïve to assume that future generations of designed nanomicelles that offer even better matches between reaction ingredients and micellar interiors will not be forthcoming.
Part of the insight yet to be gained will come from just hard work; making and testing surfactants that address variables that may provide additional clues as to how to improve metal-catalyzed couplings. For example, the surfactant “Nok” (Figure 10) is currently being studied as an analogue of TPGS-750-M; it relies on a “healthy” phytosterol, β-sitosterol, in keeping with the theme that any newly created surfactant should pose no potential environmental insult regardless of scale of usage. The point to be tested, however, is whether in providing a hydrophobic interior as solvent composed of a cyclic hydrocarbon, as opposed to the linear hydrocarbon found in the vitamin E portion of TPGS-750-M, along with the anticipated control of size in the 50–100-nm range, there are further improvements in any or all of the cross-coupling chemistry of interest. Moreover, let's
 |
eq 19 |
(Ref. 70)
 |
eq 20 |
(Ref. 77)
 |
eq 21 |
(Ref. 77) appreciate that micellar catalysis, by virtue of synthetic design and manipulation, de facto offers a virtually unlimited array of potential reaction media tailored to best match a given transformation, each to be used catalytically as a nanoreactor.82 By contrast, consider just how few the choices of traditional organic solvents there really are.
Figure 10.
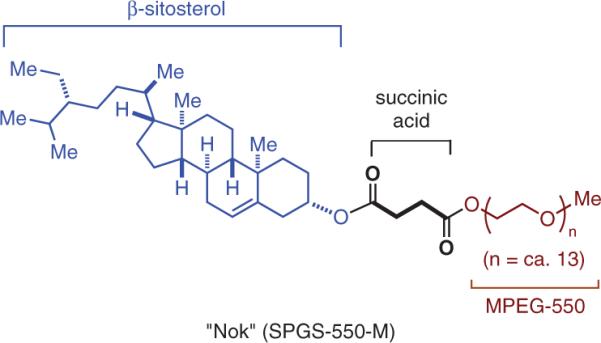
Structure of “Nok” (SPGS-550-M) Currently under Study.
New discoveries of potential major consequence in synthesis that take advantage of the hydrophobic effect await us; two of these have been discussed herein: organozinc-mediated cross-couplings in water (see Section 3.5),50,51 and unexpectedly high ee's from asymmetric hydrosilylations with CuH at room temperature (see Section 5.1).70 More are on the way; e.g., homogeneous catalytic organocopper chemistry that forms C–Cbonds in water at room temperature.83 This latter discovery is seemingly difficult to accept, given the many reviews on copper chemistry written by one of the authors. This is especially true in the Schlosser Manuals,84 advising readers to be extremely cautious about the sensitivity of carbon-based copper reagents to moisture! One interpretation of these data could be that the “rules” for doing chemistry at high concentrations in nanoparticles may well be different from those accumulated over the past 40–50 years of modern organic synthesis. This notion was foreshadowed by Lindström and Andersson in their Angewandte Chemie article “Hydrophobicity Directed Organic Synthesis”,85 in which these authors suggest that, “The design of stereoselective reactions based on hydrophobic interactions is an area of great potential that is still largely unexplored.” After all, Nature does not do chemistry by matching substrates and catalysts to organic solvents as we know them. Nature's macroscopic medium is water, which is used either as a legitimate solvent, or to force molecular organization to create hydrophobic pockets. Isn't that precisely what we call “micellar catalysis”?
Acknowledgements
The chemistry discussed herein has been the culmination of tremendous efforts by the PI's graduate students (Ben Taft, Alex Abela, David Chung, Ralph Moser, Karl Voigtritter, Shenlin Huang, and Zarko Boskovic), postdoctoral students (Subir Ghorai, Takashi Nishikata, Christophe Duplais, and Arkady Krasovskiy), and group associates (Wendy Leong, Isabelle Thome, Julian Graff, Paul Konopelski, and Valeria Krasovskaya). These co-workers deserve the credit for the progress we have made in the past few years since our 2008 review in a previous Aldrichimica Acta issue.8 We are also most appreciative of the support extended by the NIH (GM 86485), and the especially generous gifts of both ligands and catalysts used in our studies by Dr. Tom Colacot (Johnson Matthey); Dr. Richard Pedersen (Materia); Ms. Astrid Metzger (Umicore AG & Co.); Drs. Takao Saito and Hideo Shimizu, and Mr. Izuru Nagasaki (Takasago); Dr. Benoit Pugin (Solvias); and Drs. Rudolf Schmid and Michelangelo Scalone (Roche). The author also warmly acknowledges the intellectual contributions of Dr. Volker Berl (Mycell Technologies) toward the creation of TPGS-750-M, and Prof. Norbert Krause, along with several past and present group members for their comments on this manuscript prior to publication.
Biographies

Bruce Lipshutz has been on the faculty at UC Santa Barbara for the past 33 years. His training initially as an undergraduate with Howard Alper (SUNY at Binghamton), then Harry Wasserman (Yale), and finally as a postdoctoral student with E. J. Corey (Harvard) set the stage for his interest in organic synthesis and, in particular, organometallics. From his early contributions in the form of reagents such as SEM-Cl and higher order cyanocuprates to heterogeneous catalysts in the form of nickel- and copper-in-charcoal, the focus has been on providing technologies that are broadly applicable to synthetic problems. More recently, he and his co-workers have turned their attention in large measure to “green” chemistry, in appreciation of the major problems now facing society from the standpoint of sustainability, and, more specifically, issues associated with the reduction of organic waste, much of which is solvent-related. Hence, the Lipshutz group has introduced the concept of “designer” surfactants, utilizing micellar catalysis as an environmentally innocuous means of carrying out important transition-metal-catalyzed cross-coupling reactions, as well as several other reaction types (e.g., organocatalysis), in water at room temperature.

Subir Ghorai was born in 1977 in Panskura, West Bengal, India. After receiving his B.S. and M.S. degrees in chemistry from Jadavpur University, India, he joined the Indian Institute of Chemical Biology (IICB), Jadavpur, in 2000 as a CSIR research fellow. He received his Ph.D. degree in 2005 from IICB, working under the supervision of Dr. Anup Bhattacharjya on the synthesis of chiral dendrimers and heterocycles from carbohydrate precursors. From 2005 to 2006, he worked on isonitrile chemistry as a postdoctoral fellow with Professor Michael C. Pirrung at the University of California, Riverside. He then moved to UC Santa Barbara as a postdoctoral fellow working with Professor Bruce H. Lipshutz, where he helped initiate “green” chemistry involving transition-metal-catalyzed reactions in aqueous media. Recently, he has taken a position at Sigma-Aldrich in Sheboygan, Wisconsin, as an R&D scientist in the Catalysis and Organometallics group.
Footnotes
Trademarks. BRIDP® and SEGPHOS® (Takasago International Corp.); Brij® and TWEEN® (Uniqema Americas LLC); CAS Registry Number® (The American Chemical Society); COREXIT® (NALCO Co.); BASF®, Cremophor®, and Solutol® (BASF SE); Span™ (Croda International PLC); Triton® (Union Carbide Corp.).
8. References and Notes
- (1). [accessed Jan 2012]; http://en.wikipedia.org/wiki/Deepwater_Horizon_oil_spill.
- (2).(a) [accessed Jan 2012]; http://bpoilspillcrisisinthegulf.webs.com/corexit.htm.; (b) [accessed Jan 2012]; http://www.nalco.com/news-and-events/4297.htm.
- (3).(a) Zana R, editor. Dynamics of Surfactant Self-Assemblies: Micelles, Microemulsions, Vesicles, and Lyotropic Phases. Taylor & Francis; Boca Raton, FL: 2005. (Surfactant Science Series 125). [Google Scholar]; (b) Tadros TF, editor. Applied Surfactants: Principles and Applications. Wiley-VCH; Weinheim, Germany: 2005. [Google Scholar]
- (4).Engberts JBFN. Organic Chemistry in Water: Green and Fast. In: Tundo P, Perosa A, Zecchini F, editors. Methods and Reagents for Green Chemistry: An Introduction. Wiley; Hoboken, NJ: 2007. pp. 159–170. Chapter 7. [Google Scholar]
- (5).Reichardt C, Welton T, editors. Solvents and Solvent Effects in Organic Chemistry. Wiley-VCH; Weinheim, Germany: 2011. [Google Scholar]
- (6). [accessed Jan 2012]; http://www.everythingbio.com/glos/definition.php?word=hydrophobic+effect.
- (7).(a) Scarso A, Strukul G. Adv. Synth. Catal. 2005;347:1227. [Google Scholar]; (b) Selke R, Holz J, Riepe A, Bömer A. Chem.—Eur. J. 1998;4:769. [Google Scholar]; (c) Scarso A. Chimica e l'Industria. 2009;91:142. [Google Scholar]
- (8).Lipshutz BH, Ghorai S. Aldrichimica Acta. 2008;41:59. [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Borowy-Borowski H, Sikorska-Walker M, Walker PR. Water-Soluble Compositions of Bioactive Lipophilic Compounds. U.S. Patent 6,045,826. 2000 Apr 4;; (b) Borowy-Borowski H, Sikorska-Walker M, Walker PR. Water-Soluble Compositions of Bioactive Lipophilic Compounds. U.S. Patent 6,191,172. 2001 Feb 20;; (c) Borowy-Borowski H, Sikorska-Walker M, Walker PR. Water-Soluble Compositions of Bioactive Lipophilic Compounds. U.S. Patent 6,632,443. 2003 Oct 14;
- (10).(a) Anastas PT, Heine LG, Williamson TC, editors. Green Chemical Syntheses and Processes. American Chemical Society; Washington, DC: 2000. (ACS Symposium Series 767). [Google Scholar]; (b) Anastas PT, Farris CA, editors. Benign by Design: Alternative Synthetic Design for Pollution Prevention. American Chemical Society; Washington, DC: 1994. (ACS Symposium Series 557). [Google Scholar]
- (11).U.S. Food and Drug Administration [accessed Jan 2012]; Notice No. GRN 000202. http://www.accessdata.fda.gov/scripts/fcn/fcnNavigation.cfm?rpt=grasListing&displayAll=false&page=5.
- (12).For more information on Brij® 30 (CAS Registry Number® 9002-92-0), see Satkowski WB, Huang SK, Liss RL. Polyoxyethylene Alcohols. In: Schick MJ, editor. Nonionic Surfactants. Vol. 23. Dekker; New York: 1967. pp. 86–141. (Surfactant Science Series)..
- (13).Cawley JD, Stern MH. Water-Soluble Tocopherol Derivatives. U.S. Patent 2,680,749. 1954 Jun 8;
- (14).Borkovec M. Measuring Particle Size by Light Scattering. In: Holmberg K, editor. Handbook of Applied Surface and Colloid Chemistry. Vol. 2. Wiley; Chichester, U.K.: 2002. pp. 357–370. [Google Scholar]
- (15).Vriezema DM, Aragonès MC, Elemans JAAW, Cornelissen JJLM, Rowan AE, Nolte RJM. Chem. Rev. 2005;105:1445. doi: 10.1021/cr0300688. [DOI] [PubMed] [Google Scholar]
- (16).Yoshizawa M, Klosterman JK, Fujita M. Angew. Chem., Int. Ed. 2009;48:3418. doi: 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]
- (17).Lipshutz BH, Taft BR. Org. Lett. 2008;10:1329. doi: 10.1021/ol702755g. [DOI] [PubMed] [Google Scholar]
- (18).Lipshutz BH, Petersen TB, Abela AR. Org. Lett. 2008;10:1333. doi: 10.1021/ol702714y. [DOI] [PubMed] [Google Scholar]
- (19).Lipshutz BH, Chung DW, Rich B. Org. Lett. 2008;10:3793. doi: 10.1021/ol801471f. [DOI] [PubMed] [Google Scholar]
- (20).Lipshutz BH, Aguinaldo GT, Ghorai S, Voigtritter K. Org. Lett. 2008;10:1325. doi: 10.1021/ol800028x. [DOI] [PubMed] [Google Scholar]
- (21).Lipshutz BH, Ghorai S, Aguinaldo GT. Adv. Synth. Catal. 2008;350:953. [Google Scholar]
- (22).Jessop PG. Green Chem. 2011;13:1391. [Google Scholar]
- (23).Malmsten M, editor. Surfactants and Polymers in Drug Delivery. Vol. 122. Marcel Dekker; New York: 2002. p. 27. (Drugs and the Pharmaceutical Sciences Series). [Google Scholar]
- (24).Bharatiya B, Ghosh G, Bahadur P, Mata J. J. Disp. Sci. Tech. 2008;29:696. [Google Scholar]
- (25).Lipshutz BH, Chung DW, Rich B. Adv. Synth. Catal. 2009;351:1717. doi: 10.1002/adsc.200900323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).(a) Hartwig JF. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. Vol. 1. Wiley; New York: 2002. pp. 1051–1096. [Google Scholar]; (b) Louie J, Hartwig JF. Tetrahedron Lett. 1995;36:3609. [Google Scholar]
- (27).Potassium trimethylsilanolate: CAS Registry Number® 10519-96-7
- (28).Nishikata T, Lipshutz BH. Chem. Commun. 2009:6472. doi: 10.1039/b914982a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).(a) Lu Z, Ma S. Angew. Chem., Int. Ed. 2008;47:258. doi: 10.1002/anie.200605113. and references therein. [DOI] [PubMed] [Google Scholar]; (b) Weissman SA, Zewge D. Tetrahedron. 2005;61:7833. [Google Scholar]; (c) Wuts PGM, Greene TW. Greene's Protective Groups in Organic Synthesis. 4th ed. Wiley-Interscience; New York: 2007. p. 16. [Google Scholar]; (d) Takahashi K, Miyake A, Hata G. Bull. Chem. Soc. Jpn. 1972;45:230. [Google Scholar]
- (30).(a) DPEPhos: CAS Registry Number® 166330-10-5. Kranenburg M, van der Burgt YEM, Kamer PCJ, van Leeuwen PWNM, Goubitz K, Fraanje J. Organometallics. 1995;14:3081..
- (31).(a) XantPhos: CAS Registry Number® 161265-03-8. Hillebrand S, Bruckmann J, Krüger C, Haenel MW. Tetrahedron Lett. 1995;36:75..
- (32).Nishikata T, Lipshutz BH. Org. Lett. 2009;11:2377. doi: 10.1021/ol900235s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).(a) Muzart J. Eur. J. Org. Chem. 2007:3077. and references therein. [Google Scholar]; (b) Tamaru Y. Eur. J. Org. Chem. 2005:2647. [Google Scholar]; (c) Yang S-C, Hsu Y-C, Gan K-H. Tetrahedron. 2006;62:3949. [Google Scholar]; (d) Yokoyama Y, Hikawa H, Mitsuhashi M, Uyama A, Hiroki Y, Murakami Y. Eur. J. Org. Chem. 2004:1244. [Google Scholar]; (e) Yokoyama Y, Takagi N, Hikawa H, Kaneko S, Tsubaki N, Okuno H. Adv. Synth. Catal. 2007;349:662. [Google Scholar]
- (34).Lipshutz BH, Abela AR. Org. Lett. 2008;10:5329. doi: 10.1021/ol801712e. [DOI] [PubMed] [Google Scholar]
- (35).Billingsley K, Buchwald SL. J. Am. Chem. Soc. 2007;129:3358. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]
- (36).Polshettiwar V, Decottignies A, Len C, Fihri A. ChemSusChem. 2010;3:502. doi: 10.1002/cssc.200900221. [DOI] [PubMed] [Google Scholar]
- (37).Nishikata T, Lipshutz BH. J. Am. Chem. Soc. 2009;131:12103. doi: 10.1021/ja905082c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).(a) Yamamoto Y, Takada S, Miyaura N, Iyama T, Tachikawa H. Organometallics. 2009;28:152. [Google Scholar]; (b) Yamamoto Y, Takada S, Miyaura N. Chem. Lett. 2006;35:1368. [Google Scholar]
- (39).Moser R, Nishikata T, Lipshutz BH. Org. Lett. 2010;12:28. doi: 10.1021/ol9023908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).(a) Tsuji Y, Funato M, Ozawa M, Ogiyama H, Kajita S, Kawamura T. J. Org. Chem. 1996;61:5779. [Google Scholar]; (b) Tsuji Y, Kajita S, Isobe S, Funato M. J. Org. Chem. 1993;58:3607. [Google Scholar]
- (41).(a) Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2010;110:624. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ackermann L, Vicente R, Kapdi AR. Angew. Chem., Int. Ed. 2009;48:9792. doi: 10.1002/anie.200902996. [DOI] [PubMed] [Google Scholar]; (c) Daugulis O, Do H-Q, Shabashov D. Acc. Chem. Res. 2009;42:1074. doi: 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li C-J. Acc. Chem. Res. 2009;42:335. doi: 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]; (f) Kakiuchi F, Kochi T. Synthesis. 2008:3013. [Google Scholar]
- (42).Nishikata T, Abela AR, Lipshutz BH. Angew. Chem., Int. Ed. 2010;49:781. doi: 10.1002/anie.200905967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).For applications and properties of BASF®'s Solutol® HS 15 (CAS Registry Number® 70142-34-6), see [accessed Jan 2012]; http://worldaccount.basf. com/wa/NAFTA/Catalog/Pharma/info/BASF/exact/solutol_hs_15..
- (44).(a) Join B, Yamamoto T, Itami K. Angew. Chem., Int. Ed. 2009;48:3644. doi: 10.1002/anie.200806358. [DOI] [PubMed] [Google Scholar]; (b) Campeau L-C, Stuart DR, Leclerc J-P, Bertrand-Laperle M, Villemure E, Sun H-Y, Lasserre S, Guimond N, Lecavallier M, Fagnou K. J. Am. Chem. Soc. 2009;131:3291. doi: 10.1021/ja808332k. [DOI] [PubMed] [Google Scholar]; (c) Yang F, Wu Y, Li Y, Wang B, Zhang J. Tetrahedron. 2009;65:914. [Google Scholar]; (d) Scarborough CC, McDonald RI, Hartmann C, Sazama GT, Bergant A, Stahl SS. J. Org. Chem. 2009;74:2613. doi: 10.1021/jo802632v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kim J, Jo M, So W, No Z. Tetrahedron Lett. 2009;50:1229. [Google Scholar]; (f) Gorelsky SI, Lapointe D, Fagnou K. J. Am. Chem. Soc. 2008;130:10848. doi: 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]
- (45).Nishikata T, Lipshutz BH. Org. Lett. 2010;12:1972. doi: 10.1021/ol100331h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Tetrakis (acetonitrile) palladium (II) tetraflu or oborate: [Pd(MeCN)4](BF4)2, CAS Registry Number® 21797-13-7.
- (47).(a) Negishi E. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. Vol. 1. Wiley-Interscience; New York: 2002. p. 243. [Google Scholar]; (b) Negishi E, Gagneur S. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. Vol. 1. Wiley-Interscience; New York: 2002. p. 597. [Google Scholar]
- (48).(a) Knoess HP, Furlong MT, Rozema MJ, Knochel P. J. Org. Chem. 1991;56:5974. [Google Scholar]; (b) Manolikakes G, Dong Z, Mayr H, Li J, Knochel P. Chem.—Eur. J. 2009;15:1324. doi: 10.1002/chem.200802349. [DOI] [PubMed] [Google Scholar]; (c) Manolikakes G, Schade MA, Munoz Hernandez C, Mayr H, Knochel P. Org. Lett. 2008;10:2765. doi: 10.1021/ol8009013. [DOI] [PubMed] [Google Scholar]; (d) Manolikakes G, Munoz Hernandez C, Schade MA, Metzger A, Knochel P. J. Org. Chem. 2008;73:8422. doi: 10.1021/jo8015852. [DOI] [PubMed] [Google Scholar]
- (49).(a) Blackmond DG, Armstrong A, Coombe V, Wells A. Angew. Chem., Int. Ed. 2007;46:3798. doi: 10.1002/anie.200604952. [DOI] [PubMed] [Google Scholar]; (b) Narayan S, Muldoon J, Finn MG, Fokin VV, Kolb HC, Sharpless KB. Angew. Chem., Int. Ed. 2005;44:3275. doi: 10.1002/anie.200462883. [DOI] [PubMed] [Google Scholar]; (c) Hayashi Y. Angew. Chem., Int. Ed. 2006;45:8103. doi: 10.1002/anie.200603378. [DOI] [PubMed] [Google Scholar]; (d) Breslow R. Acc. Chem. Res. 1991;24:159. [Google Scholar]; (e) Pihko PM, Laurikainen KM, Usano A, Nyberg AI, Kaavi JA. Tetrahedron. 2006;62:317. [Google Scholar]; (f) Nyberg AI, Usano A, Pihko PM. Synlett. 2004:1891. [Google Scholar]
- (50).(a) Krasovskiy A, Duplais C, Lipshutz BH. J. Am. Chem. Soc. 2009;131:15592. doi: 10.1021/ja906803t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krasovskiy A, Thomé I, Graff J, Krasovskaya V, Konopelski P, Duplais C, Lipshutz BH. Tetrahedron Lett. 2011;52:2203. [Google Scholar]; (c) Duplais C, Krasovskiy A, Lipshutz BH. Organometallics. 2011;30:6090. doi: 10.1021/om200846h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Krasovskiy A, Duplais C, Lipshutz BH. Org. Lett. 2010;12:4742. doi: 10.1021/ol101885t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).PdCl2(amphos)2: CAS Registry Number® 887919-35-9. Amphos = 4-dimethylaminophenyldi(tert-butyl)phosphine. Guram AS, King AO, Allen JG, Wang X, Schenkel LB, Chan J, Bunel EE, Faul MM, Larsen RD, Martinelli MJ, Reider PJ. Org. Lett. 2006;8:1787. doi: 10.1021/ol060268g.. Guram AS, Wang X, Bunel EE, Faul MM, Larsen RD, Martinelli MJ. J. Org. Chem. 2007;72:5104. doi: 10.1021/jo070341w..
- (53).DeVasher RB, Moore LR, Shaughnessy KH. J. Org. Chem. 2004;69:7919. doi: 10.1021/jo048910c. [DOI] [PubMed] [Google Scholar]
- (54).Lipshutz BH, Ghorai S, Leong WWY, Taft BR, Krogstad DV. J. Org. Chem. 2011;76:5061. doi: 10.1021/jo200746y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Schott H. J. Colloid Interface Sci. 1995;173:265. [Google Scholar]
- (56).Anton N, Saulnier P, Béduneau A, Benoit J-P. J. Phys. Chem. B. 2007;111:3651. doi: 10.1021/jp0664768. and references therein. [DOI] [PubMed] [Google Scholar]
- (57).Nishikido N, Matuura R. Bull. Chem. Soc. Jpn. 1977;50:1690. [Google Scholar]
- (58).(a) Grubbs RH, Chang S. Tetrahedron. 1998;54:4413. [Google Scholar]; (b) Fürstner A. Angew. Chem., Int. Ed. 2000;39:3012. [PubMed] [Google Scholar]; (c) Trnka TM, Grubbs RH. Acc. Chem. Res. 2001;34:18. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]; (d) Schrock RR, Hoveyda AH. Angew. Chem., Int. Ed. 2003;42:4592. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; (e) Connon SJ, Blechert S. Angew. Chem., Int. Ed. 2003;42:1900. doi: 10.1002/anie.200200556. [DOI] [PubMed] [Google Scholar]; (f) Grubbs RH, editor. Handbook of Metathesis. Wiley-VCH; Weinheim, Germany: 2003. 3-volume set. [Google Scholar]; (g) Deiters A, Martin SF. Chem. Rev. 2004;104:2199. doi: 10.1021/cr0200872. [DOI] [PubMed] [Google Scholar]; (h) Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem., Int. Ed. 2005;44:4490. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]; (i) Gradillas A, Pérez-Castells J. Angew. Chem., Int. Ed. 2006;45:6086. doi: 10.1002/anie.200600641. [DOI] [PubMed] [Google Scholar]; (j) Schrodi Y, Pederson RL. Aldrichimica Acta. 2007;40:45. [Google Scholar]
- (59).Hong SH, Grubbs RH. J. Am. Chem. Soc. 2006;128:3508. doi: 10.1021/ja058451c. [DOI] [PubMed] [Google Scholar]
- (60).Hagiwara H, Sugawara Y, Hoshi T, Suzuki T. Chem. Commun. 2005:2942. doi: 10.1039/b502528a. [DOI] [PubMed] [Google Scholar]
- (61).Huang S-H, Chen J-R, Tsai F-Y. Molecules. 2010;15:315. doi: 10.3390/molecules15010315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Iranpoor N, Firouzabadi H, Tarassoli A, Fereidoonnezhad M. Tetrahedron. 2010;66:2415. [Google Scholar]
- (63).Lipshutz BH, Ghorai S, Abela AR, Moser R, Nishikata T, Duplais C, Krasovskiy A, Gaston RD, Gadwood RC. J. Org. Chem. 2011;76:4379. doi: 10.1021/jo101974u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Mancin F, Scrimin P, Tecilla P, Tonellato U. Coord. Chem. Rev. 2009;253:2150. [Google Scholar]
- (65).Minkler SRK, Lipshutz BH, Krause N. Angew. Chem., Int. Ed. 2011;50:7820. doi: 10.1002/anie.201101396. [DOI] [PubMed] [Google Scholar]
- (66).(a) Lipshutz BH. Synlett. 2009:509. [Google Scholar]; (b) Deutsch C, Krause N, Lipshutz BH. Chem. Rev. 2008;108:2916. doi: 10.1021/cr0684321. [DOI] [PubMed] [Google Scholar]
- (67).(a) Mahoney WS, Brestensky DM, Stryker JM. J. Am. Chem. Soc. 1988;110:291. [Google Scholar]; (b) Mahoney WS, Stryker JM. J. Am. Chem. Soc. 1989;111:8818. [Google Scholar]
- (68).(a) Czekelius C, Carreira EM. Angew. Chem., Int. Ed. 2003;42:4793. doi: 10.1002/anie.200352175. [DOI] [PubMed] [Google Scholar]; (b) Lee D, Kim D, Yun J. Angew. Chem., Int. Ed. 2006;45:2785. doi: 10.1002/anie.200600184. [DOI] [PubMed] [Google Scholar]
- (69).Saito T, Yokozawa T, Ishizaki T, Moroi T, Sayo N, Miura T, Kumobayashi H. Adv. Synth. Catal. 2001;343:264. [Google Scholar]
- (70).Huang S, Voigtritter KR, Unger JB, Lipshutz BH. Synlett. 2010:2041. [Google Scholar]
- (71).Lipshutz BH, Servesko JM. Angew. Chem., Int. Ed. 2003;42:4789. doi: 10.1002/anie.200352313. [DOI] [PubMed] [Google Scholar]
- (72).Lawrence NJ, Drew MD, Bushell SM. J. Chem. Soc., Perkin Trans. 1999;1:3381. [Google Scholar]
- (73).(a) Schmid R, Broger EA, Cereghetti M, Crameri Y, Foricher J, Lalonde M, Mueller RK, Scalone M, Schoettel G, Zutter U. Pure Appl. Chem. 1996;68:131. [Google Scholar]; (b) Schmid R, Foricher J, Cereghetti M, Schönholzer P. Helv. Chim. Acta. 1991;74:370. [Google Scholar]
- (74).(a) Togni A, Breutel C, Schnyder A, Spindler F, Landert H, Tijani A. J. Am. Chem. Soc. 1994;116:4062. [Google Scholar]; (b) Blaser H-U, Brieden W, Pugin B, Spindler F, Studer M, Togni A. Top. Catal. 2002;19:3. [Google Scholar]
- (75).(a) Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem., Int. Ed. 2005;44:4442. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; (b) Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- (76).For a copper-catalyzed borylation of aryl bromides at ambient temperature, see Kleeberg C, Dang L, Lin Z, Marder TB. Angew. Chem., Int. Ed. 2009;48:5350. doi: 10.1002/anie.200901879..
- (77).Lipshutz BH, Moser R, Voigtritter KR. Isr. J. Chem. 2010;50:691. doi: 10.1002/ijch.201000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Lou S, Fu GC. Adv. Synth. Catal. 2010;352:2081. doi: 10.1002/adsc.201000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Alternatively, direct oxidation to the corresponding phenol should be possible; see Inamoto K, Nozawa K, Yonemoto M, Kondo Y. Chem. Commun. 2011:11775. doi: 10.1039/c1cc14974a..
- (80).Myers D. Surfactant Science and Technology. 3rd ed. Wiley-Interscience; Hoboken, NJ: 2006. [Google Scholar]
- (81).Lipshutz BH. University of California; Santa Barbara, CA: 2007. Unpublished work. [Google Scholar]
- (82).The potential for “designer” surfactants to provide an infinite number of organic solvents was first recognized (2010) by Dr. Zarko Boskovic at UCSB.
- (83).Lipshutz BH, Huang S, Leong WWY, Isley NA. 2012. submitted for publication.
- (84).Lipshutz BH. Organocopper Chemistry. In: Schlosser M, editor. Organometallics in Synthesis: a Manual. 2nd ed. Wiley; Chichester, U.K.: 2002. pp. 665–816. [Google Scholar]
- (85).Lindström UM, Andersson F. Angew. Chem., Int. Ed. 2006;45:548. doi: 10.1002/anie.200502882. [DOI] [PubMed] [Google Scholar]