

Abstract
Significance: Insulin resistance and its related diseases, obesity and type 2 diabetes mellitus (T2DM), have been linked to changes in aerobic metabolism, pointing to a possible role of mitochondria in the development of insulin resistance. Recent Advances: Refined methodology of ex vivo high-resolution respirometry and in vivo magnetic resonance spectroscopy now allows describing several features of mitochondria in humans. In addition to measuring mitochondrial function at baseline and after exercise-induced submaximal energy depletion, the response of mitochondria to endocrine and metabolic challenges, termed mitochondrial plasticity, can be assessed using hyperinsulinemic clamp tests. While insulin resistant states do not uniformly relate to baseline and post-exercise mitochondrial function, mitochondrial plasticity is typically impaired in insulin resistant relatives of T2DM, in overt T2DM and even in type 1 diabetes mellitus (T1DM). Critical Issues: The variability of baseline mitochondrial function in the main target tissue of insulin action, skeletal muscle and liver, may be attributed to inherited and acquired changes in either mitochondrial quantity or quality. In addition to certain gene polymorphisms and aging, circulating glucose and lipid concentrations correlate with both mitochondrial function and plasticity. Future Directions: Despite the associations between features of mitochondrial function and insulin sensitivity, the question of a causal relationship between compromised mitochondrial plasticity and insulin resistance in the development of obesity and T2DM remains to be resolved. Antioxid. Redox Signal. 19, 258–268.
Introduction
Obesity is dramatically increasing in various regions of the world and has evolved as one of the leading risk factors for premature mortality, mainly due to accelerated atherosclerosis and macrovascular disease. Impaired insulin action (i.e., insulin resistance) and inadequate insulin secretion are frequently observed in obesity. Particularly, insulin resistance also precedes the onset of type 2 diabetes mellitus (T2DM) by many years (78), indicating its predominant role in linking obesity, T2DM, and cardiovascular disease. The pathogenesis of obesity-related insulin resistance is a complex and multifactorial process involving the sequential interplay of several tissues. The current key hypotheses explaining the underlying mechanisms are: (i) the fatty acids hypothesis (lipotoxicity); (ii) the endocrine/inflammatory hypothesis; (iii) the overflow hypothesis, and (iv) mitochondrial hypothesis.
Increased availability of lipids and inflammatory processes contribute to the pathogenesis of obesity-related insulin resistance and T2DM (40). Moreover, proper mitochondrial functioning seems to contribute to the regulation of insulin sensitivity and secretion. Consequently, processes impairing mitochondrial function would lead to disturbed energy homeostasis with insulin resistance and deficiency (42). Indeed, impaired mitochondrial function has been associated with alterations of (i) glucose and fatty acid metabolism, (ii) production of electron transport system (ETS)-related reactive oxygen species (ROS), (iii) ATP-mediated insulin secretion from β-cells, (iv) synthesis of ATP for energy-consuming functions (i.e., insulin-stimulated glucose uptake), (v) exercise-induced production of ATP and aerobic capacity and, finally, (vi) calcium homeostasis which impacts on exercise-mediated glucose uptake (51).
Despite these associations supporting a critical role of mitochondria in glucose metabolism, the literature also provides controversial data, which could result from using different methods and terminologies (75). The present review summarizes the recent data on the associations between alterations in muscular and hepatic mitochondrial plasticity and insulin resistance in humans. In particular, some relevant endocrine and metabolic effects on mitochondrial plasticity will be reviewed, while the molecular regulation during aging and exercise training is beyond the scope of this review and will not be addressed.
Mitochondrial Plasticity
In animals, mitochondria are responsible for oxidative phosphorylation (OXPHOS), the main process of energy and fuel homeostasis regulation. Normally, mitochondria are “plastic” (i.e., they respond rapidly and adequately to metabolic alterations to meet the actual needs of the respective tissues). Recently, the term mitochondrial plasticity was introduced to define changes of mitochondrial activity or oxidative capacity in response to various metabolic conditions created by diet, exercise, insulin, and drugs (75). Of note, these changes could result from alterations of individual mitochondrial function and/or overall mitochondrial content.
Mitochondrial plasticity can be assessed by comparing the following three parameters under basal conditions and during stimulation (e.g., by exogenous lipids or exercise training) (75):
(i) In vivo mitochondrial activity, assessed by 31P-magnetic resonance spectroscopy (MRS) (69), represents unidirectional flux through ATP synthase and thereby resting oxidative phosphorylation at low ADP concentrations, depending on substrate/oxygen availability and demand.
(ii) In vivo submaximal ADP-stimulated oxidative phosphorylation, also assessed by 31P-MRS, represents the rate of phosphocreatine (PCr) re-synthesis (35) upon submaximal exercising and is also influenced by substrate/oxygen flux controlling processes.
(iii) Ex vivo oxidative capacity, assessed from oxygen consumption rates in isolated mitochondria or permeabilized fibers, represents maximal ADP-stimulated oxidative phosphorylation reflecting maximal energy demand at unlimited substrate/oxygen supply (23).
Mitochondrial Plasticity in Insulin-Resistant States
As insulin resistance is predicting and preceding the onset of T2DM (78), it is of interest to compare mitochondrial function with insulin sensitivity in cohorts of variable insulin sensitivity and at increased risk of T2DM.
Mitochondrial plasticity during aging
The aging-related decline in insulin sensitivity is held responsible for the greater risk of T2DM in the elderly (52, 70). Impaired insulin sensitivity was found in some elderly cohorts (52, 76), who were matched for physical activity to younger controls, but physical activity was not assessed using the state-of-the-art method, maximal oxygen uptake (Vo2max). However, studies successfully matching the groups for Vo2max revealed no difference in insulin sensitivity between young and elderly (14), supported by observation that insulin sensitivity as well as mitochondrial oxidative capacity improve after exercise in elderly insulin-resistant patients with T2DM and age-matched normoglycemic controls (55). In contrast to insulin sensitivity, mitochondrial function is consistently associated with aging, even after matching groups for physical activity. In vivo mitochondrial activity (52), insulin-stimulated mitochondrial plasticity (76), as well as ex vivo mitochondrial oxidative capacity (70), are impaired in elderly, which is in some cases accompanied with increased intra-myo/hepatocellular (52) or body fat content (70). Other reports on the impact of age on mitochondrial plasticity at different levels have been recently reviewed (25) and suggest that decreased mitochondrial function directly results from biological aging. Indeed, mitochondrial hypotheses of aging postulate that oxidative damage of mitochondrial DNA and enzymes accumulates during life, with subsequent negative effects on mitochondrial processes such as ETS and ATP synthesis (24). Thus, unlike mitochondrial plasticity, impaired insulin sensitivity in elderly seems to result from decreased physical activity and increased body fat content, suggesting that compromised mitochondrial function and insulin resistance are not necessarily causally interrelated in aging.
Mitochondrial plasticity in first-degree relatives of patients with T2DM
First-degree relatives (FDR) are frequently insulin resistant and at greater risk of T2DM. Young, lean, but severely insulin-resistant FDR have ∼30% lower in vivo flux through muscular ATP synthase than insulin-sensitive otherwise matched controls in line with impaired basal mitochondrial activity (53). FDR with marginally lower insulin sensitivity only show a nonsignificant tendency of lower in vivo submaximal oxidative phosphorylation and ex vivo mitochondrial oxidative capacity than age-matched controls (56). Likewise, we reported that insulin-sensitive FDR have similar in vivo mitochondrial activity rather than carefully matched controls (29). Of note, both studies found that associations of mitochondrial function with Vo2max were strong, but weaker (29) or absent (56) with insulin sensitivity. Also, higher age and different gender distribution, both known to modulate mitochondrial function (25, 32), could contribute to the heterogeneous results obtained in FDR. Furthermore, lower mitochondrial function related to lower mitochondrial content assessed by electron microscopy (45), but not to changes in mitochondrial DNA content (45, 56) or citrate synthase activity (56). The key regulators of mitochondrial biogenesis, PGC-1α and PGC-1β, were decreased in older and overweight FDR (50), but not altered in young and lean FDR (45). Thus, other factors need to be taken into account to explain lower mitochondrial content in insulin resistant FDR. Noteworthy, lean young FDR have lower mRNA and protein levels of muscle lipoprotein lipase (LPL), which further positively correlates with mitochondrial density (46). Moreover, knocking-down either LPL or peroxisome proliferator–activated receptor (PPAR)-δ in human myocytes reduces mitochondrial density, expression of mitochondrial proteins, and oxidative capacity (46), suggesting that decreased free fatty acid (FFA)-mediated PPAR-δ activation could contribute to impaired mitochondrial function in FDR (Fig. 8A). Impairment of myocellular mitochondrial activity also associates with decreased mitochondrial substrate oxidation, TCA activity and augmented intramyocellular content of lipids such as diacylglycerols (4, 53), which in turn relates to increased serine phosphorylation of insulin receptor substrate (IRS) (45). This led to the hypothesis that an inherited defect in the activity of mitochondrial oxidative phosphorylation causes dysregulation of myocellular fatty acid metabolism with subsequently impaired insulin signalling (4, 65) (Fig. 1).
FIG. 8.
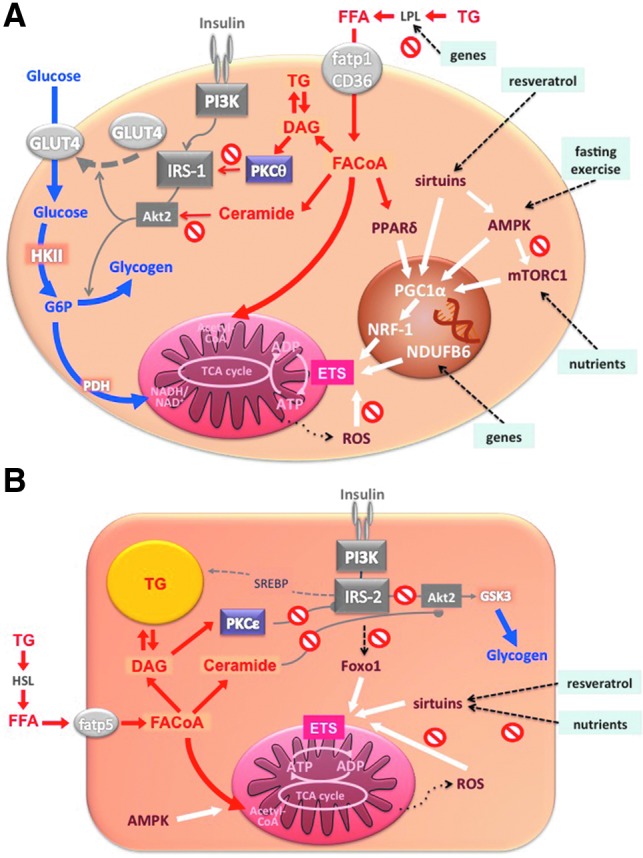
Potential regulators of mitochondrial biogenesis and function. In the myocyte (A), glucose is taken up via glucose transporter 4 (glut4), activated to glucose-6-phosphate (G6P), and then oxidized in the mitochondria or stored as glycogen. Free fatty acids are taken up via LPL and fatty acid transporter proteins (fatp1, CD36) and activated to fatty acyl coenzyme A (FACoA). FACoA can be oxidized in the mitochondria or stored as triglycerides or can favor the formation of diacylglycerols (DAG) and/or ceramides, thus inhibiting insulin signaling by protein kinase C-θ (PKCθ)/IRS-1 pathway and/or protein kinase B-2 (Akt2) phosphorylation, respectively. Both glucose and lipid oxidation fuel the tricarboxylic acid cycle and serve to produce ATP. Black dashed arrows represent genetic predispositions and lifestyle interventions affecting mitochondrial biogenesis/function via different mechanisms (white arrows): inherited factors associate with decreased LPL activity and PPARδ-mediated mitochondrial biogenesis; single nucleotide polymorphism of NDUFB6 gene predisposes to impaired mitochondrial plasticity in response to exercise; and resveratrol, fasting/exercise, and nutrients increase mitochondrial biogenesis/function by increasing PGC1α activity via sirtuins, AMPK, and mTORC1, respectively. ROS have been associated with decreased mitochondrial function. In hepatocytes (B), free fatty acids are taken up via fatty acid transporter protein 5 (fatp5), activated to FACoA, and undergo similar metabolic pathway as in the myocyte. Decreased activity of IRS-2 associates with lower Foxo1 and mitochondrial function. Resveratrol increases while overloading with nutrients decreases sirtuins and mitochondrial biogenesis. Finally, ROS impact negatively on the function of mitochondria. Acetyl-CoA, acetyl coenzyme A; ADP, adenosine diphosphate; GSK3, glycogen synthase kinase 3; HKII, hexokinase II; HSL, hormone sensitive lipase; PDH, pyruvate dehydrogenase; PI3K, phosphatidylinositol 3 kinase; SREBP, sterol regulatory element binding protein; TG, triglyceride. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 1.
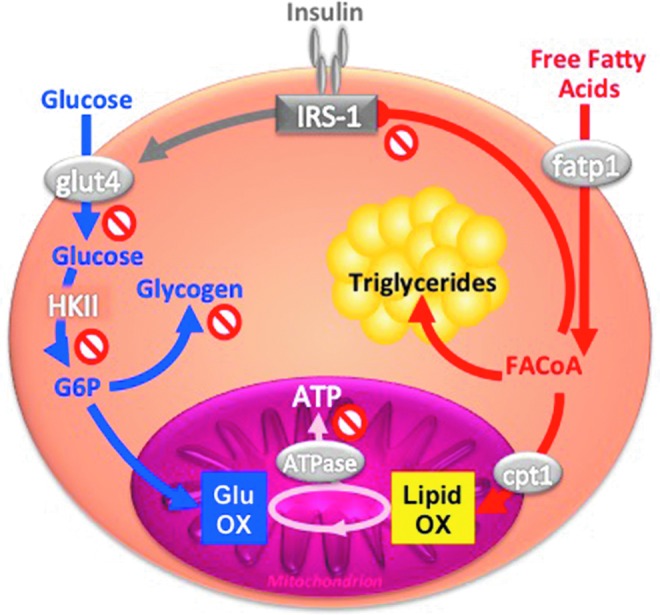
Interaction between glucose and lipids at the level of insulin signaling and mitochondrial function in skeletal muscle. Glucose is taken up via glucose transporter 4 (glut4) into the myocyte, activated to glucose-6-phosphate (G6P), and then oxidized in the mitochondria or stored as glycogen. Free fatty acids are taken up via fatty acid transporter protein 1 (fatp1) into the myocyte, activated to fatty acyl coenzyme A (FACoA), and then transported by the carnitine palmitoyltransferase 1 (cpt1) into mitochondria for oxidation (OX), or stored as triyglycerides, or inhibit insulin signaling by serine phosphorylation of IRS-1. Both glucose and lipid OX fuel the tricarboxylic acid cycle and serve to produce ATP via ATP synthase (ATPase). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Insulin-resistant FDR also showed lower mitochondrial plasticity from diminished insulin-stimulated in vivo flux through ATP synthase, which further associated with lower insulin-stimulated rises of myocellular Pi but not PCr/ATP concentrations (54). This suggests that reduced Pi supply, possibly due to decreased phosphate transport, accounts for the compromised mitochondrial plasticity in insulin-resistant FDR. We further studied mitochondrial plasticity in a cohort of FDR with low degree of insulin resistance employing short- and long-term exercise training (29, 30). In vivo flux through ATP synthase increased with both training interventions, while insulin sensitivity improved only upon short-term exercise training. The beneficial effects on mitochondrial plasticity were present only in some FDR and did not depend on common single nucleotide polymorphisms of PGC1α but on the G/G gene polymorphism of the NADH dehydrogenase (ubiquinone) 1 ß subcomplex (NDUFB6) rs540467 gene, a component of complex I of the ETS. These data show that the role of mitochondrial plasticity for the adaptation of insulin sensitivity to environmental demand may depend on inherited factors. However, it remains unclear whether impaired plasticity results from primary inherited mitochondrial defects or from cellular accumulation of deleterious lipids (Fig. 8A).
Mitochondrial Plasticity in Overt T2DM
T2DM is a heterogeneous disease, which is currently defined by a certain degree of hyperglycemia and explained by insulin resistance and relative insulin deficiency. Here, we focus on mitochondrial function in liver and muscle (75).
Skeletal muscle
Already 40 years ago, evidence was provided for decreased activity of mitochondrial TCA enzymes in skeletal muscle of humans with T2DM (3). However, not all subsequent studies found compromised mitochondrial function in muscles of insulin-resistant patients with T2DM. While mitochondrial area and activity of ETS enzymes were decreased in T2DM, and the size of mitochondria positively correlated with insulin sensitivity (33), such correlation disappeared after matching subjects for their physical activity (13), known to stimulate mitochondrial density (79). Human muscle biopsies revealed that mRNA expression of genes encoding key enzymes of oxidative metabolism and mitochondrial function, such as PGC-1α, PGC-1β, and nuclear respiratory factor-1 (NRF-1)-dependent genes (50), as well as activity of NADH oxidase (63), are lower in patients with T2DM (Fig. 8A). Another modulator of mitochondrial activity is mammalian target of rapamycin complex 1 (mTORC1) pathway. Inhibition of mTORC1 resulted in decreased mRNA of PGC-1α and its target genes along with lower oxygen consumption in C2C12 myotubes (15). Patients with T2DM have increased muscle mTORC1 levels, again implying that mitochondrial function can dissociate from insulin sensitivity and/or is under control of other signaling pathways (39). Interestingly, fasting (22) or administration of resveratrol (59) induce PGC-1α, mitochondrial genes, and fatty acid oxidation in myotubes by activating AMP-activated protein kinase (AMPK) and sirtuins, a family of NAD+-dependent acyltransferases (Fig. 8A). Although resveratrol improves insulin sensitivity (7), the relationship between insulin resistance in human T2DM and sirtuins or AMPK is not yet clear.
In addition to studies on enzyme activities and expression levels, ex vivo studies reported lower oxidative capacity in T2DM. Also, ADP-stimulated state 3 respiration was lower in permeabilized muscle fibers from patients with T2DM than in matched controls (56), but not after normalizing the rate of oxygen consumption to mitochondrial DNA copy number (5). But even mitochondria isolated from muscles of patients with T2DM had decreased pyruvate plus malate-driven state 3 mitochondrial oxidative capacity (44). While the majority of these data suggest impaired mitochondrial density and/or function in overt T2DM, ex vivo rates of ATP synthesis and OXPHOS capacity were similar in mitochondria isolated from muscles of healthy humans and Asian Indians with T2DM (48). Although the Asian Indians were more insulin resistant, their OXPHOS capacity was even greater compared with Northern European Americans, again indicating dissociation of mitochondrial function and insulin sensitivity.
Further studies measured features of myocellular mitochondria in vivo, which offer the possibility to examine mitochondrial function in their intact cellular and metabolic environment. Submaximal oxidative phosphorylation was also decreased in T2DM compared to age- and BMI-matched controls (56) and associated with insulin resistance, but not with intramyocellular lipids. PCr recovery kinetics did not differ between controls and patients at early and advanced stages of T2DM (16). Of note, submaximal oxidative phosphorylation positively correlated with insulin sensitivity across a larger cohort, but this relationship was lost within T2DM (2). Discrepancies between these studies may result from differences in exercise tolerance or load used to deplete PCr, variable degree of muscular acidosis, which may affect the rate of PCr re-synthesis. Of note, in vivo flux through ATP synthase at rest was lower in T2DM than in young, but not carefully matched humans without T2DM (76). Nevertheless, the observed heterogeneity could reflect the multifactorial pathogenesis of T2DM.
These studies did not clarify whether mitochondrial function per se or density might be impaired or compensatory altered in T2DM. But even normalization of mitochondrial function for mitochondrial content revealed functional impairment in some (44, 56, 63) but not all studies in T2DM (5, 33). These discrepancies might be due to the use of indirect markers of density such as mitochondrial DNA content or citrate synthase activity, which is affected by acute exercise (18) and hyperinsulinemia (73). Direct quantification of mitochondrial content in situ by transmission electron microscopy showed lower mitochondrial content in insulin-resistant humans with as well as without T2DM. This was explained by reduced intermyofibrillar mitochondrial subpopulations (13), in contrast to a study previous showing reduced subsarcolemmal mitochondrial content in T2DM (64). Again, the observed discrepancy could be the result of methodological differences (13).
Only a few studies specifically address mitochondrial plasticity (e.g., by measuring the effects of insulin on myocellular mitochondrial function) in T2DM. Mitochondria isolated from skeletal muscle of young, lean, healthy subjects respond to high physiologic insulin concentrations with increases in ATP synthesis and expression of several ETS enzymes, both of which were absent in mitochondria obtained from T2DM patients (73). Similarly, we observed that insulin does not stimulate unidirectional flux through ATP synthase in vivo in insulin-resistant patients with T2DM compared to both body mass- and physical activity-matched controls at comparable or younger age (76). Myocellular ATP synthetic rate did not rise during hyperinsulinemic-hyperglycemic clamp conditions, which doubled rates of whole-body glucose uptake and myocellular glucose-6-phosphate (G6P) levels (Fig. 2A and 2B). Thus, impaired stimulation of ATP turnover did not simply result from lower substrate availability but rather from diminished mitochondrial plasticity in T2DM (Fig. 2C) (76).
FIG. 2.

Glucose uptake and ATP synthesis in skeletal muscle of humans with type 2 diabetes mellitus. Insulin-stimulated whole body glucose disposal is about 50% lower in type 2 diabetes than in humans with normal glucose tolerance, which might lead to lower myocellular concentrations of substrates for oxidation such as glucose-6-phosphate (G6P) and thereby lower rates of ATP synthesis (white columns). However, when plasma glucose levels were increased from 5.5 to 9.5 mmol/l during hyperinsulinemic-hyperglycemic clamp tests, the doubling to myocellular G6P, insulin-stimulated ATP synthesis (mitochondrial plasticity) did not improve (black columns) (76).
Liver
Proteomic analysis revealed distinct tissue-dependent variability in mitochondrial protein composition, suggesting that mitochondria may also differ functionally (21). Indeed, both ex vivo studies in isolated mitochondria and in vivo 31P-MRS showed that the OXPHOS pathway is thermodynamically more efficient (10) with 3-fold greater ATP turnover in liver than in skeletal muscle (68). Deterioration of mitochondrial OXPHOS capacity could lower lipid oxidation and raise lipid accumulation, thereby causing or contributing to hepatic insulin resistance and steatosis. Using a noninvasive 31P/1H-MRS technique, we found that hepatic energy metabolism is impaired in longstanding T2DM (74). Impaired whole-body insulin sensitivity in these subjects was associated with decreased liver ATP content compared to young, lean as well as age- and BMI-matched controls (Fig. 3A and 3B). Furthermore, basal ATP synthesis was also lower in T2DM (69) and negatively correlated with hepatic insulin resistance (Fig. 3C). Obese patients with T2DM also have lower hepatic expression of OXPHOS genes and exhibit a negative correlation of γ-subunit of ATP synthase with hepatic fat content (57). In other obese subjects, insulin resistance not only correlated with hepatic diacylglycerol content but also with markers of endoplasmic reticulum stress (38), which can impair mitochondrial function (41). However, less obese patients with T2DM can have greater hepatic OXPHOS expression than glucose-tolerant humans (43). Although this does not necessarily reflect greater hepatic mitochondrial function, more studies are needed to study the dynamic role of hepatic mitochondrial function during the development of obesity and T2DM. Of note, animal studies provide evidence for various modulators of hepatic mitochondrial function; for example, impaired insulin signaling in the liver-specific IRS-1/IRS-2 knock-down mice dysregulates mitochondrial biogenesis, morphology, and function by inhibition of forkhead box O1 (Foxo1) (12) and high-fat diet (HFD)-induced steatosis associates with decreased activity of sirtuins and components of ETS (36) (Fig. 8B).
FIG. 3.

Glucose uptake and hepatocellular ATP content and synthesis in humans with type 2 diabetes mellitus (T2DM), in age, sex, and body mass-matched (mCON) and in young (yCON) healthy humans. Whole body glucose uptake is about 50% lower in T2DM (black column) than in mCON (gray column), and substantially lower than in yCON (white column). Myocellular ATP content is reduced in T2DM and ATP synthesis correlates inversely with hepatic insulin resistance as given by endogenous glucose production during suppression by insulin (iEGP) (69, 74).
Independent of differences resulting from measurement, genes, or aging, the metabolic environment could contribute to alterations of mitochondrial function in insulin-resistant states. In detail, in vivo insulin-stimulated myocellular ATP synthesis correlates inversely with both degree of hyperglycemia as assessed from hemoglobin A1c levels (Fig. 4A) and of hyperlipidemia, as assessed from postabsorptive plasma FFA concentrations (Fig. 4B).
FIG. 4.

Correlation of mitochondrial plasticity with glycemia and lipidemia. Insulin-stimulated myocellular unidirectional ATP synthase flux (mitochondrial plasticity) negatively correlates with (A) glycemic control as assessed from hemoglobin A1c (28), and with (B) plasma concentrations of free fatty acids (FFA) in the fasted state (76).
Glucose-Induced Changes in Mitochondrial Plasticity
Chronic hyperglycemia is currently held responsible for the vast majority of microvascular and, to a minor degree, for macrovascular consequences of diabetes, but also for promoting insulin resistance and ß-cell dysfunction (67). Uncontrolled (excessively hyperglycemic) patients with type 1 diabetes mellitus (T1DM) display lower ex vivo rates of ATP production and altered mitochondrial gene expression profile in skeletal muscle (31). Of note, ex vivo mitochondrial respiration negatively correlates with fasting plasma glucose levels and near-normalization of glycemia by insulin improved respiration in T2DM patients with severe hyperglycemia (>15 mmol/l) (61). Recently, we showed that even near-normoglycemic patients with longstanding T1DM show substantial insulin resistance with impaired insulin-stimulated myocellular G6P concentrations, as well as compromised mitochondrial plasticity (28) (Fig. 5A and 5B). Moreover, in vivo myocellular ATP synthesis correlated positively with insulin sensitivity and negatively with glycemic control, as assessed from hemoglobin A1c levels (Fig. 4A). Thus, chronic hyperglycemia per se may deteriorate mitochondrial plasticity and thereby contribute to insulin resistance.
FIG. 5.

Effect of chronic hyperglycemia per se on glucose uptake and ATP synthesis in skeletal muscle of T1DM. Insulin-stimulated whole body glucose disposal is about 50% lower in T1DM (dark bars) than in humans with normal glucose tolerance (Non-DM, light bars). In Non-DM, stimulation by insulin markedly increases myocellular concentrations of substrates for oxidation such as glucose-6-phosphate (G6P) and thereby rates of ATP synthesis (mitochondrial plasticity). In T1DM, both G6P and ATP synthesis do not adequately rise during insulin stimulation (28).
Lipid- and Obesity-Induced Changes in Mitochondrial Plasticity
Hypercaloric nutrition and obesity are frequently associated with hyperlipidemia (i.e., elevation of plasma triglycerides and FFA) and insulin resistance, and may lead to T2DM (65). Specifically, circulating FFA and/or their intracellular metabolites inhibit insulin signaling (Fig. 1) by several mechanisms including lipotoxic and inflammatory pathways (65, 66). Because of their critical role for fat oxidation, mitochondria may also be involved in lipid-induced insulin resistance, which occurs rapidly in the presence of high FFA (acute hyperlipidemia) or may evolve slowly during the development of obesity (chronic hyperlipidemia).
Effect of acute hyperlipidemia
In vitro, primary human myocytes respond to physiologically increased palmitate concentrations with decreased ATP synthesis, oxygen consumption, mitochondrial complex activities, and membrane potential (1). Likewise, palmitate-overloading impaired β-oxidation and promoted intramyocellular lipid accumulation in myotubes from patients with T2DM compared with controls (37). In order to examine the time course of FFA-dependent changes in insulin sensitivity and mitochondrial plasticity, we monitored whole body glucose uptake (Fig. 6A), in vivo G6P levels and rates of ATP synthesis (Fig. 6B) in skeletal muscle during 6-hour lipid infusions in healthy humans. Within 3 hours, plasma FFA elevation impaired insulin sensitivity by inhibiting insulin-stimulated glucose uptake and myocellular G6P but did not affect ATP synthesis (9). Only at 6 hours, insulin-stimulated ATP synthesis was ∼24% lower, indicating that impaired mitochondrial plasticity is a consequence of insulin resistance during acute hyperlipidemia (8). Furthermore, 48 hours of lipid infusion decreased the expression of PGC-1 and nuclear encoded mitochondrial genes, along with decreased glucose uptake (62) and 3-day HFD downregulated muscular OXPHOS genes, ETS components, mitochondrial carrier proteins, and stimulators of mitochondrial biogenesis, even in lean humans (72). Intermediate duration of HFD revealed inconsistent data, showing greater muscular expression of genes regulating lipid oxidation after 5 days (6) and no effect on in vivo submaximal ADP-stimulated oxidative phosphorylation after 7 days (17). However, these dietary interventions also failed to show effects on insulin sensitivity. Thus, the available evidence indicates that acute hyperlipidemia first causes insulin resistance followed by impaired mitochondrial plasticity in skeletal muscle of lean humans.
FIG. 6.

Effect of hyperlipidemia on glucose uptake and myocellular ATP synthesis in healthy humans. Whole body glucose uptake increases depending on the concentration of the plasma insulin (insulin sensitivity) under control conditions (white columns), but is about 50% lower in the presence of elevated serum triglycerides and free fatty acids (insulin resistance, black columns). Myocellular ATP synthesis (mitochondrial plasticity) increases only at a high insulin concentrations (8, 9).
Effect of chronic hyperlipidemia associated with obesity
Unlike lean, obese people are metabolic inflexible in that they do not respond appropriately to metabolic stimuli such as dietary fat intake with greater fat oxidation (34). Thus, HFD neither increases whole-body lipid oxidation nor expression of genes of lipid oxidation (6) in obese humans. Similar to some T2DM cohorts, obese individuals display decreased size (33) and content of muscular mitochondria (13, 64) without evidence for changes in one specific subpopulation of mitochondria. Transcription levels of genes encoding components of OXPHOS were also lower in adipose tissue of obese than in their non-obese monozygotic twins (47), pointing to the predominant role of acquired factors driving abnormalities in energy metabolism. Reduced mitochondrial function was confirmed in obese humans ex vivo in some studies on oxidative capacity (11) but not by bioluminescence assays (32). Interestingly, already obese children may have reduced in vivo submaximal ADP-stimulated oxidative phosphorylation, which associates with insulin resistance but not with obesity per se (20).
Aside from longstanding obesity, the large variety of diets due to unlimited combinations of dietary FFA species will have various effects on lipid bioavailability and distribution with complex consequences for cellular energy metabolism. There is growing evidence from mouse studies that lipid class, saturation index, and/or chain length of FFA differently affects insulin sensitivity and mitochondrial plasticity. For example, treatment of rat muscle cells with fatty acids of varying degree of saturation revealed that only saturated fatty acids, such as palmitate, impair both insulin response and ATP synthesis (26). The mono-unsaturated fatty acid oleate dose-dependently protected from palmitate-induced inhibition of insulin signaling and induction of ROS production in rat hepatocytes (49). Combining HFD enriched in n-3 polyunsaturated fatty acids (n-3 PUFA) and caloric restriction resulted in synergistic increase in mitochondrial oxidative capacity and lipid catabolism in adipocytes of mice (19). Eicosapentaenoic fatty acid rescued impaired mitochondrial oxidative capacity in LPL-deficient myotubes, probably via PPAR-δ-mediated activation of mitochondrial biogenesis (46). We showed that raising the ratio of dietary n-3 PUFA prevents HFD-induced reduction of palmitate oxidation in mouse hepatocytes (Fig. 7), which was dependent on (AMPK) and associated with improvements of insulin sensitivity and liver lipid content (27).
FIG. 7.

Diverse effects of high-fat diets differing in the degree of saturation on palmitate oxidation in hepatocytes and involvement of AMPK. Stimulation of palmitate oxidation by AMPK activator 5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR) is decreased in primary hepatocytes isolated from wild-type mice fed high-fat diet (cHF, black columns) for 9 weeks compared to low-fat diet (Chow, white columns) fed mice. Addition of polyunsaturated fatty acids into diet (cHF+F, dashed columns) prevents this decrease. Beneficial effects of feeding by cHF+F diet are lost in mice lacking α2 subunit of AMPK (AMPKα2-/- mice) (27).
In summary, short-and long-term hyperlipidemia can impair muscular mitochondrial function and plasticity. But these changes do not seem to cause lipid-mediated insulin resistance but rather arise as a consequence of the altered cellular metabolism.
Conclusions and Future Directions
Some nondiabetic but insulin-resistant cohorts such as severely insulin-resistant FDR show reduced myocellular mitochondrial density, function, and plasticity (4, 45, 53, 54, 56). In less insulin-resistant FDR (29, 56) or women with a history of gestational diabetes, pGDM (60) do not show abnormal basal mitochondrial function. Thus, abnormalities of mitochondrial function do not necessarily precede insulin resistance or T2DM, which is supported by findings that muscle- and liver-specific deficiency of OXPHOS does not cause insulin resistance, at least in mice (58, 80). However, it is uncertain whether insulin-sensitive FDR and pGDM also show altered mitochondrial plasticity or will develop T2DM later in their lives. At least, in some certain variants of mitochondrial diabetes, the inherited mtDNA mutation seems to cause both impaired myocellular mitochondrial plasticity and insulin resistance (77). Of note, patients with congenital mutations of insulin receptor and severe insulin resistance can secondarily develop impaired in vivo mitochondrial function (71). Similarly, mice with liver-specific downregulation of IRS-1/IRS-2 are insulin resistant and show dysregulated mitochondrial biogenesis and function (12), supporting the hypothesis that insulin resistance leads to impaired mitochondrial function (Fig. 8B).
Acutely, excessive lipid availability induces insulin resistance, which can be followed by lower mitochondrial plasticity (Fig. 6) (8, 9). Chronic elevation of lipid availability during the development of obesity seems to induce an adaptation of mitochondrial oxidative capacity in that it increases proportionally with the degree of obesity in insulin-sensitive humans but falls in the most obese at the onset of insulin resistance (11). The concept of such adaptation has been supported by findings in animal models on HFD and in human athletes in whom myocellular lipid accumulation is associated with high oxidative capacity and insulin sensitivity. HFD- and obesity-induced activity of mTORC1, a positive regulator of mitochondrial biogenesis in muscle (15), could be a link between nutrient overload and increased mitochondrial function (39) (Fig. 8A). However, all these models do not mirror human hyperlipidemia and obesity, which may exhibit differences in amount and composition of cellular lipids inhibiting insulin signaling (e.g., diacylglycerols or ceramides), and in production of ROS and lipid peroxides. For example, ex vivo treatment of skeletal muscle cells with physiological concentrations of palmitate suppressed, while lower palmitate levels improved ATP production (1).
Finally, the contributions of various insulin-sensitive tissues to the pathogenesis of T2DM may vary. In glucose-tolerant women with pGDM, liver lipid storage rather than muscular mitochondrial activity correlated with insulin sensitivity (60), indicating that abnormal hepatic rather than muscular energy metabolism might be present in certain groups at increased risk of T2DM, as suggested for overt T2DM (69, 74).
In conclusion, insulin resistance can impair mitochondrial function or vice versa, but both abnormalities are not always causally related. The different findings cannot be exclusively attributed to differences in the employed methods or protocols, but also result from the multifactorial alterations underlying the heterogeneous condition, currently defined as T2DM. Alterations of the intracellular lipid profile, maybe resulting from inadequate fatty acid oxidation, can impair insulin signaling and thereby serve as a causal link between abnormal mitochondrial function and insulin resistance.
Abbreviations Used
- AICAR
5-aminoimidazole-4-carboxyamide ribonucleoside
- AMP
adenosine monophosphate
- AMPK
AMP-activated protein kinase
- ATP
adenosine triphosphate
- ATPase
ATP synthase
- BMI
body mass index
- cpt1
carnitine palmitoyltransferase 1
- ETS
electron transport system
- FACoA
fatty acyl coenzyme A
- fatp1
fatty acid transporter protein 1
- FDR
first-degree relatives
- FFA
free fatty acids
- Foxo1
forkhead box O1
- G6P
glucose-6-phosphate
- glut4
glucose transporter 4
- HFD
high-fat diet
- iEGP
insulin-supressed endogenous glucose production
- IGT
impaired glucose tolerance
- IRS-1
insulin receptor substrate-1
- LPL
lipoprotein lipase
- mCON
body mass-matched controls
- MRS
magnetic resonance spectroscopy
- mTOR C1
mammalian target of rapamycin complex 1
- n3-PUFA
n-3 polyunsaturated fatty acids
- NADH
nicotinamide adenine dinucleotide
- NAFLD
non-alcoholic fatty liver disease
- NGT
normal glucose tolerance
- NRF-1
nuclear respiratory factor-1
- OX
oxidation
- OXPHOS
oxidative phosphorylation
- Pi
phosphate
- PCr
phosphocreatine
- PGC
peroxisome proliferator-activated receptor-gamma coactivator
- pGDM
previous gestational diabetes mellitus
- PPAR
peroxisome proliferator–activated receptor
- ROS
reactive oxygen species
- SFA
saturated fatty acids
- T1DM
type 1 diabetes mellitus
- T2DM
type 2 diabetes mellitus
- TCA
tricarboxylic acid cycle
- UFA
unsaturated fatty acids
- Vo2max
maximal oxygen uptake
- yCON
young controls
Acknowledgments
The studies by the authors were supported in part by a DAAD-Leibniz scholarship and by a grant from the Czech Science Foundation (P301/10/1420; Grant recipient: Martin Rossmeisl) to TJ, and by grants from the European Foundation for the Study of Diabetes (EFSD), Juvenile Diabetes Foundation (JDRF), Schmutzler-Stiftung, Skröder-Stiftung, German Research Foundation (DFG; SFB 512) to MR, and from the German Federal Ministry of Education and Research (BMBF) to the German Center for Diabetes Research (DZD e.V.).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Abdul-Ghani MA. Muller FL. Liu Y. Chavez AO. Balas B. Zuo P. Chang Z. Tripathy D. Jani R. Molina-Carrion M. Monroy A. Folli F. Van Remmen H. DeFronzo RA. Deleterious action of FA metabolites on ATP synthesis: Possible link between lipotoxicity, mitochondrial dysfunction, and insulin resistance. Am J Physiol Endocrinol Metab. 2008;295:E678–685. doi: 10.1152/ajpendo.90287.2008. [DOI] [PubMed] [Google Scholar]
- 2.Bajpeyi S. Pasarica M. Moro C. Conley K. Jubrias S. Sereda O. Burk DH. Zhang Z. Gupta A. Kjems L. Smith SR. Skeletal muscle mitochondrial capacity and insulin resistance in type 2 diabetes. J Clin Endocrinol Metab. 2011;96:1160–1168. doi: 10.1210/jc.2010-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bass A. Vondra K. Rath R. Vitek V. Havranek T. Metabolic changes in the quadriceps femoris muscle of obese people. Enzyme activity patterns of energy-supplying metabolism. Pflugers Arch. 1975;359:325–334. doi: 10.1007/BF00581443. [DOI] [PubMed] [Google Scholar]
- 4.Befroy DE. Petersen KF. Dufour S. Mason GF. de Graaf RA. Rothman DL. Shulman GI. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56:1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boushel R. Gnaiger E. Schjerling P. Skovbro M. Kraunsoe R. Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 2007;50:790–796. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyle KE. Canham JP. Consitt LA. Zheng D. Koves TR. Gavin TP. Holbert D. Neufer PD. Ilkayeva O. Muoio DM. Houmard JA. A high-fat diet elicits differential responses in genes coordinating oxidative metabolism in skeletal muscle of lean and obese individuals. J Clin Endocrinol Metab. 2011;96:775–781. doi: 10.1210/jc.2010-2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brasnyo P. Molnar GA. Mohas M. Marko L. Laczy B. Cseh J. Mikolas E. Szijarto IA. Merei A. Halmai R. Meszaros LG. Sumegi B. Wittmann I. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br J Nutr. 2011;106:383–389. doi: 10.1017/S0007114511000316. [DOI] [PubMed] [Google Scholar]
- 8.Brehm A. Krssak M. Schmid AI. Nowotny P. Waldhausl W. Roden M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes. 2006;55:136–140. [PubMed] [Google Scholar]
- 9.Brehm A. Krssak M. Schmid AI. Nowotny P. Waldhausl W. Roden M. Acute elevation of plasma lipids does not affect ATP synthesis in human skeletal muscle. Am J Physiol Endocrinol Metab. 2010;299:E33–38. doi: 10.1152/ajpendo.00756.2009. [DOI] [PubMed] [Google Scholar]
- 10.Cairns CB. Walther J. Harken AH. Banerjee A. Mitochondrial oxidative phosphorylation thermodynamic efficiencies reflect physiological organ roles. Am J Physiol. 1998;274:R1376–1383. doi: 10.1152/ajpregu.1998.274.5.R1376. [DOI] [PubMed] [Google Scholar]
- 11.Chanseaume E. Barquissau V. Salles J. Aucouturier J. Patrac V. Giraudet C. Gryson C. Duche P. Boirie Y. Chardigny JM. Morio B. Muscle mitochondrial oxidative phosphorylation activity, but not content, is altered with abdominal obesity in sedentary men: Synergism with changes in insulin sensitivity. J Clin Endocrinol Metab. 2010;95:2948–2956. doi: 10.1210/jc.2009-1938. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Z. Guo S. Copps K. Dong X. Kollipara R. Rodgers JT. Depinho RA. Puigserver P. White MF. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med. 2009;15:1307–1311. doi: 10.1038/nm.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chomentowski P. Coen PM. Radikova Z. Goodpaster BH. Toledo FG. Skeletal muscle mitochondria in insulin resistance: Differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J Clin Endocrinol Metab. 2011;96:494–503. doi: 10.1210/jc.2010-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crane JD. Devries MC. Safdar A. Hamadeh MJ. Tarnopolsky MA. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J Gerontol A Biol Sci Med Sci. 2010;65:119–128. doi: 10.1093/gerona/glp179. [DOI] [PubMed] [Google Scholar]
- 15.Cunningham JT. Rodgers JT. Arlow DH. Vazquez F. Mootha VK. Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–40. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 16.De Feyter HM. van den Broek NM. Praet SF. Nicolay K. van Loon LJ. Prompers JJ. Early or advanced stage type 2 diabetes is not accompanied by in vivo skeletal muscle mitochondrial dysfunction. Eur J Endocrinol. 2008;158:643–653. doi: 10.1530/EJE-07-0756. [DOI] [PubMed] [Google Scholar]
- 17.Edwards LM. Murray AJ. Holloway CJ. Carter EE. Kemp GJ. Codreanu I. Brooker H. Tyler DJ. Robbins PA. Clarke K. Short-term consumption of a high-fat diet impairs whole-body efficiency and cognitive function in sedentary men. FASEB J. 2011;25:1088–1096. doi: 10.1096/fj.10-171983. [DOI] [PubMed] [Google Scholar]
- 18.Fernstrom M. Tonkonogi M. Sahlin K. Effects of acute and chronic endurance exercise on mitochondrial uncoupling in human skeletal muscle. J Physiol. 2004;554:755–763. doi: 10.1113/jphysiol.2003.055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flachs P. Ruhl R. Hensler M. Janovska P. Zouhar P. Kus V. Macek Jilkova Z. Papp E. Kuda O. Svobodova M. Rossmeisl M. Tsenov G. Mohamed-Ali V. Kopecky J. Synergistic induction of lipid catabolism and anti-inflammatory lipids in white fat of dietary obese mice in response to calorie restriction and n-3 fatty acids. Diabetologia. 2011;54:2626–2638. doi: 10.1007/s00125-011-2233-2. [DOI] [PubMed] [Google Scholar]
- 20.Fleischman A. Kron M. Systrom DM. Hrovat M. Grinspoon SK. Mitochondrial function and insulin resistance in overweight and normal-weight children. J Clin Endocrinol Metab. 2009;94:4923–4930. doi: 10.1210/jc.2009-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forner F. Foster LJ. Campanaro S. Valle G. Mann M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol Cell Proteomics. 2006;5:608–619. doi: 10.1074/mcp.M500298-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Gerhart-Hines Z. Rodgers JT. Bare O. Lerin C. Kim SH. Mostoslavsky R. Alt FW. Wu Z. Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: New perspectives of mitochondrial physiology. Int J Biochem Cell Biol. 2009;41:1837–1845. doi: 10.1016/j.biocel.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 24.Harman D. Free radical theory of aging: Dietary implications. Am J Clin Nutr. 1972;25:839–843. doi: 10.1093/ajcn/25.8.839. [DOI] [PubMed] [Google Scholar]
- 25.Hebert SL. Lanza IR. Nair KS. Mitochondrial DNA alterations and reduced mitochondrial function in aging. Mech Ageing Dev. 2010;131:451–462. doi: 10.1016/j.mad.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirabara SM. Curi R. Maechler P. Saturated fatty acid-induced insulin resistance is associated with mitochondrial dysfunction in skeletal muscle cells. J Cell Physiol. 2010;222:187–194. doi: 10.1002/jcp.21936. [DOI] [PubMed] [Google Scholar]
- 27.Jelenik T. Rossmeisl M. Kuda O. Jilkova ZM. Medrikova D. Kus V. Hensler M. Janovska P. Miksik I. Baranowski M. Gorski J. Hebrard S. Jensen TE. Flachs P. Hawley S. Viollet B. Kopecky J. AMP-activated protein kinase alpha2 subunit is required for the preservation of hepatic insulin sensitivity by n-3 polyunsaturated fatty acids. Diabetes. 2010;59:2737–2746. doi: 10.2337/db09-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kacerovsky M. Brehm A. Chmelik M. Schmid AI. Szendroedi J. Kacerovsky-Bielesz G. Nowotny P. Lettner A. Wolzt M. Jones JG. Roden M. Impaired insulin stimulation of muscular ATP production in patients with type 1 diabetes. J Intern Med. 2011;269:189–199. doi: 10.1111/j.1365-2796.2010.02298.x. [DOI] [PubMed] [Google Scholar]
- 29.Kacerovsky-Bielesz G. Chmelik M. Ling C. Pokan R. Szendroedi J. Farukuoye M. Kacerovsky M. Schmid AI. Gruber S. Wolzt M. Moser E. Pacini G. Smekal G. Groop L. Roden M. Short-term exercise training does not stimulate skeletal muscle ATP synthesis in relatives of humans with type 2 diabetes. Diabetes. 2009;58:1333–1341. doi: 10.2337/db08-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kacerovsky-Bielesz G. Kacerovsky M. Chmelik M. Farukuoye M. Ling C. Pokan R. Tschan H. Szendroedi J. Schmid AI. Gruber S. Herder C. Wolzt M. Moser E. Pacini G. Smekal G. Groop L. Roden M. A single nucleotide polymorphism associates with the response of muscle ATP synthesis to long-term exercise training in relatives of type 2 diabetic humans. Diabetes Care. 2012;35:350–357. doi: 10.2337/dc11-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karakelides H. Asmann YW. Bigelow ML. Short KR. Dhatariya K. Coenen-Schimke J. Kahl J. Mukhopadhyay D. Nair KS. Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes. 2007;56:2683–2689. doi: 10.2337/db07-0378. [DOI] [PubMed] [Google Scholar]
- 32.Karakelides H. Irving BA. Short KR. O'Brien P. Nair KS. Age, obesity, and sex effects on insulin sensitivity and skeletal muscle mitochondrial function. Diabetes. 2010;59:89–97. doi: 10.2337/db09-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelley DE. He J. Menshikova EV. Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 34.Kelley DE. Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: A reexamination. Diabetes. 2000;49:677–683. doi: 10.2337/diabetes.49.5.677. [DOI] [PubMed] [Google Scholar]
- 35.Kemp GJ. Radda GK. Quantitative interpretation of bioenergetic data from 31P and 1H magnetic resonance spectroscopic studies of skeletal muscle: An analytical review. Magn Reson Q. 1994;10:43–63. [PubMed] [Google Scholar]
- 36.Kendrick AA. Choudhury M. Rahman SM. McCurdy CE. Friederich M. Van Hove JL. Watson PA. Birdsey N. Bao J. Gius D. Sack MN. Jing E. Kahn CR. Friedman JE. Jonscher KR. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011;433:505–514. doi: 10.1042/BJ20100791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitzmann M. Lantier L. Hebrard S. Mercier J. Foretz M. Aguer C. Abnormal metabolism flexibility in response to high palmitate concentrations in myotubes derived from obese type 2 diabetic patients. Biochim Biophys Acta. 2011;1812:423–430. doi: 10.1016/j.bbadis.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 38.Kumashiro N. Erion DM. Zhang D. Kahn M. Beddow SA. Chu X. Still CD. Gerhard GS. Han X. Dziura J. Petersen KF. Samuel VT. Shulman GI. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2011;108:16381–16385. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laplante M. Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee DE. Kehlenbrink S. Lee H. Hawkins M. Yudkin JS. Getting the message across: Mechanisms of physiological cross talk by adipose tissue. Am J Physiol Endocrinol Metab. 2009;296:E1210–1229. doi: 10.1152/ajpendo.00015.2009. [DOI] [PubMed] [Google Scholar]
- 41.Leem J. Koh EH. Interaction between mitochondria and the endoplasmic reticulum: Implications for the pathogenesis of type 2 diabetes mellitus. Exp Diabetes Res. 20122012:242984. doi: 10.1155/2012/242984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lowell BB. Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 43.Misu H. Takamura T. Matsuzawa N. Shimizu A. Ota T. Sakurai M. Ando H. Arai K. Yamashita T. Honda M. Kaneko S. Genes involved in oxidative phosphorylation are coordinately upregulated with fasting hyperglycaemia in livers of patients with type 2 diabetes. Diabetologia. 2007;50:268–277. doi: 10.1007/s00125-006-0489-8. [DOI] [PubMed] [Google Scholar]
- 44.Mogensen M. Sahlin K. Fernstrom M. Glintborg D. Vind BF. Beck-Nielsen H. Hojlund K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56:1592–1599. doi: 10.2337/db06-0981. [DOI] [PubMed] [Google Scholar]
- 45.Morino K. Petersen KF. Dufour S. Befroy D. Frattini J. Shatzkes N. Neschen S. White MF. Bilz S. Sono S. Pypaert M. Shulman GI. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005;115:3587–3593. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morino K. Petersen KF. Sono S. Choi CS. Samuel VT. Lin A. Gallo A. Zhao H. Kashiwagi A. Goldberg IJ. Wang H. Eckel RH. Maegawa H. Shulman GI. Regulation of mitochondrial biogenesis by lipoprotein lipase in muscle of insulin-resistant offspring of parents with type 2 diabetes. Diabetes. 2012;61:877–887. doi: 10.2337/db11-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mustelin L. Pietilainen KH. Rissanen A. Sovijarvi AR. Piirila P. Naukkarinen J. Peltonen L. Kaprio J. Yki-Jarvinen H. Acquired obesity and poor physical fitness impair expression of genes of mitochondrial oxidative phosphorylation in monozygotic twins discordant for obesity. Am J Physiol Endocrinol Metab. 2008;295:E148–154. doi: 10.1152/ajpendo.00580.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nair KS. Bigelow ML. Asmann YW. Chow LS. Coenen-Schimke JM. Klaus KA. Guo ZK. Sreekumar R. Irving BA. Asian Indians have enhanced skeletal muscle mitochondrial capacity to produce ATP in association with severe insulin resistance. Diabetes. 2008;57:1166–1175. doi: 10.2337/db07-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura S. Takamura T. Matsuzawa-Nagata N. Takayama H. Misu H. Noda H. Nabemoto S. Kurita S. Ota T. Ando H. Miyamoto K. Kaneko S. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J Biol Chem. 2009;284:14809–14818. doi: 10.1074/jbc.M901488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patti ME. Butte AJ. Crunkhorn S. Cusi K. Berria R. Kashyap S. Miyazaki Y. Kohane I. Costello M. Saccone R. Landaker EJ. Goldfine AB. Mun E. DeFronzo R. Finlayson J. Kahn CR. Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patti ME. Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev. 2010;31:364–395. doi: 10.1210/er.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petersen KF. Befroy D. Dufour S. Dziura J. Ariyan C. Rothman DL. DiPietro L. Cline GW. Shulman GI. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petersen KF. Dufour S. Befroy D. Garcia R. Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petersen KF. Dufour S. Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Phielix E. Meex R. Moonen-Kornips E. Hesselink MK. Schrauwen P. Exercise training increases mitochondrial content and ex vivo mitochondrial function similarly in patients with type 2 diabetes and in control individuals. Diabetologia. 2010;53:1714–1721. doi: 10.1007/s00125-010-1764-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Phielix E. Schrauwen-Hinderling VB. Mensink M. Lenaers E. Meex R. Hoeks J. Kooi ME. Moonen-Kornips E. Sels JP. Hesselink MK. Schrauwen P. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes. 2008;57:2943–2949. doi: 10.2337/db08-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pihlajamaki J. Boes T. Kim EY. Dearie F. Kim BW. Schroeder J. Mun E. Nasser I. Park PJ. Bianco AC. Goldfine AB. Patti ME. Thyroid hormone-related regulation of gene expression in human fatty liver. J Clin Endocrinol Metab. 2009;94:3521–3529. doi: 10.1210/jc.2009-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pospisilik JA. Knauf C. Joza N. Benit P. Orthofer M. Cani PD. Ebersberger I. Nakashima T. Sarao R. Neely G. Esterbauer H. Kozlov A. Kahn CR. Kroemer G. Rustin P. Burcelin R. Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 59.Price NL. Gomes AP. Ling AJ. Duarte FV. Martin-Montalvo A. North BJ. Agarwal B. Ye L. Ramadori G. Teodoro JS. Hubbard BP. Varela AT. Davis JG. Varamini B. Hafner A. Moaddel R. Rolo AP. Coppari R. Palmeira CM. de Cabo R. Baur JA. Sinclair DA. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prikoszovich T. Winzer C. Schmid AI. Szendroedi J. Chmelik M. Pacini G. Krssak M. Moser E. Funahashi T. Waldhausl W. Kautzky-Willer A. Roden M. Body and liver fat mass rather than muscle mitochondrial function determine glucose metabolism in women with a history of gestational diabetes mellitus. Diabetes Care. 2011;34:430–436. doi: 10.2337/dc10-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rabol R. Hojberg PM. Almdal T. Boushel R. Haugaard SB. Madsbad S. Dela F. Effect of hyperglycemia on mitochondrial respiration in type 2 diabetes. J Clin Endocrinol Metab. 2009;94:1372–1378. doi: 10.1210/jc.2008-1475. [DOI] [PubMed] [Google Scholar]
- 62.Richardson DK. Kashyap S. Bajaj M. Cusi K. Mandarino SJ. Finlayson J. DeFronzo RA. Jenkinson CP. Mandarino LJ. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem. 2005;280:10290–10297. doi: 10.1074/jbc.M408985200. [DOI] [PubMed] [Google Scholar]
- 63.Ritov VB. Menshikova EV. Azuma K. Wood R. Toledo FG. Goodpaster BH. Ruderman NB. Kelley DE. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am J Physiol Endocrinol Metab. 2010;298:E49–58. doi: 10.1152/ajpendo.00317.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ritov VB. Menshikova EV. He J. Ferrell RE. Goodpaster BH. Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 65.Roden M. Muscle triglycerides and mitochondrial function: Possible mechanisms for the development of type 2 diabetes. Int J Obes (Lond) 2005;29(Suppl 2):S111–115. doi: 10.1038/sj.ijo.0803102. [DOI] [PubMed] [Google Scholar]
- 66.Roden M. Price TB. Perseghin G. Petersen KF. Rothman DL. Cline GW. Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rossetti L. Glucose toxicity: The implications of hyperglycemia in the pathophysiology of diabetes mellitus. Clin Invest Med. 1995;18:255–260. [PubMed] [Google Scholar]
- 68.Schmid AI. Chmelik M. Szendroedi J. Krssak M. Brehm A. Moser E. Roden M. Quantitative ATP synthesis in human liver measured by localized 31P spectroscopy using the magnetization transfer experiment. NMR Biomed. 2008;21:437–443. doi: 10.1002/nbm.1207. [DOI] [PubMed] [Google Scholar]
- 69.Schmid AI. Szendroedi J. Chmelik M. Krssak M. Moser E. Roden M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care. 2011;34:448–453. doi: 10.2337/dc10-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Short KR. Bigelow ML. Kahl J. Singh R. Coenen-Schimke J. Raghavakaimal S. Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci USA. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sleigh A. Raymond-Barker P. Thackray K. Porter D. Hatunic M. Vottero A. Burren C. Mitchell C. McIntyre M. Brage S. Carpenter TA. Murgatroyd PR. Brindle KM. Kemp GJ. O'Rahilly S. Semple RK. Savage DB. Mitochondrial dysfunction in patients with primary congenital insulin resistance. J Clin Invest. 2011;121:2457–2461. doi: 10.1172/JCI46405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sparks LM. Xie H. Koza RA. Mynatt R. Hulver MW. Bray GA. Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54:1926–1933. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- 73.Stump CS. Short KR. Bigelow ML. Schimke JM. Nair KS. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci USA. 2003;100:7996–8001. doi: 10.1073/pnas.1332551100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Szendroedi J. Chmelik M. Schmid AI. Nowotny P. Brehm A. Krssak M. Moser E. Roden M. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology. 2009;50:1079–1086. doi: 10.1002/hep.23093. [DOI] [PubMed] [Google Scholar]
- 75.Szendroedi J. Phielix E. Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2011;8:92–103. doi: 10.1038/nrendo.2011.138. [DOI] [PubMed] [Google Scholar]
- 76.Szendroedi J. Schmid AI. Chmelik M. Toth C. Brehm A. Krssak M. Nowotny P. Wolzt M. Waldhausl W. Roden M. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med. 2007;4:e154. doi: 10.1371/journal.pmed.0040154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Szendroedi J. Schmid AI. Meyerspeer M. Cervin C. Kacerovsky M. Smekal G. Graser-Lang S. Groop L. Roden M. Impaired mitochondrial function and insulin resistance of skeletal muscle in mitochondrial diabetes. Diabetes Care. 2009;32:677–679. doi: 10.2337/dc08-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tabak AG. Jokela M. Akbaraly TN. Brunner EJ. Kivimaki M. Witte DR. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: An analysis from the Whitehall II study. Lancet. 2009;373:2215–2221. doi: 10.1016/S0140-6736(09)60619-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Toledo FG. Watkins S. Kelley DE. Changes induced by physical activity and weight loss in the morphology of intermyofibrillar mitochondria in obese men and women. J Clin Endocrinol Metab. 2006;91:3224–3227. doi: 10.1210/jc.2006-0002. [DOI] [PubMed] [Google Scholar]
- 80.Wredenberg A. Freyer C. Sandstrom ME. Katz A. Wibom R. Westerblad H. Larsson NG. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem Biophys Res Commun. 2006;350:202–207. doi: 10.1016/j.bbrc.2006.09.029. [DOI] [PubMed] [Google Scholar]