

Abstract
The discovery and application of a new branching pathway synthesis strategy that rapidly produces skeletally diverse scaffolds is described. Two different scaffold types, one a bicyclic iodo-vinylidene tertiary amine/tertiary alcohol and the other, a spirocyclic 3-furanone, are each obtained using a two-step sequence featuring a common first step. Both scaffold types lead to intermediates that can be orthogonally diversified using the same final components. One of the scaffold types was obtained in sufficiently high yield that it was immediately used to produce a 97-compound library.
Introduction
In recent years high throughput organic synthesis has gained an increasingly prominent role for lead generation in the service of drug discovery. The accelerated creation of innovative compound collections with high degrees of structural diversity is now seen as a necessary component of the larger biomedical research landscape. Coupled with increasingly efficient high throughput screening and assay models that seek to be more relevant to biological reality the impetus for developing versatile methods to create chemical diversity continues to increase. Skeletal diversification represents one of the most versatile concepts in high throughput organic synthesis.1 One manifestation of this concept is the derivation of profoundly different molecular scaffolds from a common precursor by employing different reagents, sometimes referred to as the “branching pathway” strategy.2
Our laboratory’s mission involves the development of innovative chemistry with the goal of using it to produce unique compound libraries by high-throughput synthesis. Deriving more than one scaffold from the same substrate is clearly advantageous in terms of both compound diversity and productivity. This report deals with the preparation of two distinct families of scaffolds so derived.
Results and Discussion
Our interest in exploring chemical space by creating structural diversity led us to survey the literature to find under-represented compound types. One such substructure search centered on diversifiable spirocycles. Spirocyclic scaffolds offer an opportunity to create chemical diversity by combining structural complexity with rigidity. Substituents can be moved around the scaffold in discrete, predictable increments thus enabling the controlled exploration of the surrounding space.3 These and other characteristics of spirocycles have been exploited in recent reports.4 Surveying the number of highly saturated spirocycles derived from diversifiable ketones like 4-piperidone with the corresponding ones derived from 3-piperidone revealed that the latter structural class contained comparatively few members. We therefore initiated studies to discover a methodology that affords rapid access to spirocyclic scaffolds from 3-piperidone.
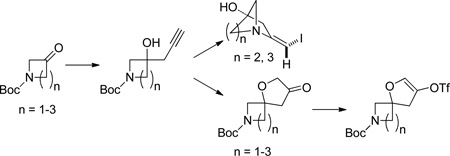
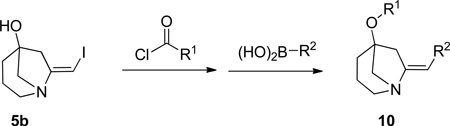
As part of a larger spirocycle synthesis effort we started by noting the precedents of Larock and others who used electrophilic iodoetherification for the efficient preparation of benzofurans from ortho-alkyne substituted anisoles.5 We sought to use iodoetherification to obtain spirocycles bearing a dihydrofuran that is not fused to an aromatic ring. We imagined that treatment of homopropargyl tertiary alcohol 3 with iodine monochloride (ICl) would affect electrophilic cyclization to afford the previously unreported spirocyclic β-iodo-enol ether 4 with possible concomitant loss of the Boc group (Scheme 1). Loss of the protecting group would facilitate isolation of 4 as it could be extracted into 0.1 N HCl and lyophilized to afford its salt. This substrate would be ready for orthogonal diversification via the unprotected amine and the vinyl iodide.
Scheme 1.

Synthesis of bicyclo [3.2.1] β-iodo enamines from homopropargyl tertiary alcohols derived from N-Boc-3-piperidone
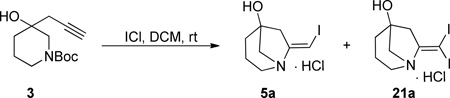
Synthesis of compound 3 was envisioned as a two-step sequence starting from N-Boc-3-piperidone 1. Addition of lithiated 1-TMS-1-propyne to 1 using TMEDA as additive afforded 2 in poor to modest yields that diminished on larger scale.6 Modification of the organolithium with CeCl3 gave 2 in a range of yields that was higher.7 Basic hydrolysis of 2 afforded the cyclization precursor 3 in generally excellent yields and purity. A more direct method using 1, propargyl bromide and Zn/Cu couple with sonication afforded 3 accompanied by a minor amount of its inseparable allene isomer (not shown).8 The allene byproduct did not introduce any apparent complications in the subsequent steps. Treatment of 3 with ICl afforded, not the anticipated 4, but the bicyclic [3.2.1] β-iodo-enamine HCl salt 5a after extraction and lyophilization. β-iodo-enamine HCl salt 5a was accompanied by a minor amount (~10 A% by HPLC) of a di-iodo species which could be separated by pHPLC. Compound 5a was elaborated to the more complex derivative 10{10,1} as described below (Scheme 4). X-ray crystallography of 10{10,1} confirmed the depicted bicyclic core structure of its precursor 5a. The presence of the di-iodo species did not affect subsequent steps. Enamine 5a was stable and could be handled using standard techniques presumably because the bridgehead position of the nitrogen precludes interaction between it and the olefin.
Scheme 4.

Skeletal diversification including introduction of peripheral substituents
The conversion of 3 to 5a is envisioned as proceeding via an unsaturated iodonium intermediate Int1 (Scheme 2). Instead of the iodonium being attacked by the free tertiary alcohol via a 5-endo cyclization (path a) and leading to 4 it is captured by the nitrogen via a 5-exo mechanism (path b, Int2).9 This latter mode of cyclization is precedented for unsubstituted terminal alkynes although not in the presence of a wellpositioned free hydroxyl.10 Also β-iodoenamines contained within a bicyclic structure have been previously reported.11 We were curious about whether removing the possibility of participation by the piperidine nitrogen would lead to the desired cyclization. To test this hypothesis we prepared tertiary alcohols 6 and 8 from 1-Boc-4-piperidone and 1-PhSO2-3-piperidone respectively (Scheme 3). In the case of 6 we predicted that nitrogen would be unable to participate due to distance and/or unfavorable conformation. In the case of 8 we expected the protecting group to survive the reaction conditions and prevent nitrogen unmasking. This would demonstrate that the loss of the Boc group is required for iodoamination to occur. In the event treatment of 6 and 8 with ICl gave only complex mixtures with no detectable formation of 7 or 9.
Scheme 2.

Alternative mechanisms for cyclization of the iodonium derived from 3.
Scheme 3.

Attempted iodoetherification of homopropargyl tertiary alcohols
We then undertook to perform the originally desired spirocyclization using the precedent of Zhang (Scheme 4).12 Thus exposure of 3 to catalytic Au(PPh3)(NTf2) in the presence of a substituted pyridine-N-oxide as co-oxidant under acidic conditions gave the spirocyclic 3-furanone 11.13 Moving further we prepared a pair of potential final library compounds featuring the same peripheral substituents. Salt 5a was neutralized and the free amine 5b was treated with p-Cl-benzoyl chloride/TEA in the presence of DMAP. This method afforded the intermediate ester (not shown) which was subjected to Suzuki coupling to give the orthogonally diversified derivative 10{10,1}, confirmed by X-ray crystallography (see SI, Figure S1). Moving in the other direction spirocycle 11 was converted to its enol triflate 12.14 Enol triflate 12 was subjected to Suzuki coupling then deprotection and amide coupling with p-Cl-benzoyl chloride/TEA/DMAP. This alternate sequence gave the skeletally divergent derivative 13.
We deferred further optimizing the spirocycle route and instead elected to first use the [3.2.1] scaffold 5 for library production. Compounds like 10, having both the bridgehead ester and an exo-arylenamine, are previously unreported and so we used this serendipitous discovery to prepare a novel compound library. A 14×10 diversity component library of 140 members was designed (Figure 1). As a matter of routine, component subsets were used to validate the methodology and the synthesis platform. Components A6 and B9 were poor performers and thus removed. The remaining 117 compounds were prepared using miniblocks.
Figure 1.

Components for proposed 140-member library from scaffold 5.
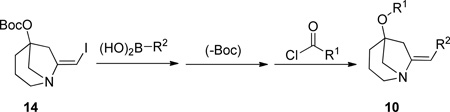
Some of the compounds were obtained by an alternative sequence as shown in Scheme 5. Treatment of free amine 5b with Boc2/TEA in the presence of DMAP gave the O-protected vinyl iodide 14. At this stage any di-iodo species present could be readily removed by silica gel chromatography. Suzuki coupling, deprotection and acylation gave the library compounds.
Scheme 5.

Alternate sequence for library compounds
Table 1 shows the isolated yields of the compounds. Of the 117 compounds proposed 97 were obtained in our target purities of ≥90%.
Table 1.
Yields of library compounds 10 after reverse phase preparative Mass Directed Fractionation (MDF)
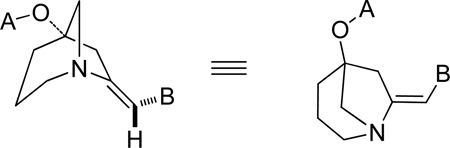 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Library analogs of 10 | ||||||||||
| Boronic Acids | ||||||||||
| B1 | B2 | B3 | B4 | B5 | B6* | B7 | B8 | B10 | ||
| Acid Chlorides | A1 | 30 | 36 | 34 | 19 | 13 | 14 | 31 | 28 | 24 |
| A2 | 37 | 35 | 33 | 25 | 32 | 18 | 39 | 38 | 26 | |
| A3 | 11 | 13 | 13 | - | 6 | 10 | 14 | - | 14 | |
| A4 | 41 | 32 | 40 | 32 | 41 | 23 | 41 | 46 | 43 | |
| A5 | 34 | 27 | 35 | 29 | 35 | 15 | 34 | 41 | 42 | |
| A7 | 19 | 16 | - | 13 | 17 | 9 | 23 | 14 | 19 | |
| A8 | - | 10 | - | 7 | - | - | 14 | - | - | |
| A9 | 4 | 1 | 1 | 4 | - | - | 4 | - | ||
| A10 | 35 | 34 | 26 | 31 | 28 | 23 | 30 | 20 | 32 | |
| A11 | 28 | 21 | 23 | 19 | 32 | 15 | 23 | 21 | 26 | |
| A12 | 31 | 28 | 22 | 34 | 27 | 10 | 37 | 31 | 20 | |
| A13 | 32 | 43 | 42 | 31 | 36 | 26 | 47 | 42 | 50 | |
| A14 | 30 | 14 | - | - | - | - | - | - | - | |
Compounds incorporating B6 were reprocessed by normal phase preparative TLC after MDF to attain 90% purity by HPLC.
Finally, we explored the limits of the both the iodoamination and spirocyclization. For this purpose we employed homopropargyl alcohols 16a and 16b (Scheme 6). Both of these substrates were obtained from the precursor ketones 15a and 15b by the action of propargyl bromide and Zn/Cu couple. Treatment of 16a with ICl afforded no detectable 17a. By contrast 16b gave the corresponding bicyclic [2.2.1] amine 17b in reduced yield. We surmise that the diminished yields obtained in these systems largely arise from increased ring strain.
Scheme 6.

Skeletal diversification of lower homologs of 3
Using catalytic Au(PPh3)(NTf2), both 16a and 16b were successfully employed in spirocyclization. Thus both substrates afforded spiro-3-furanones 18a and 18b.13 These products were advanced to the enol triflates 19a and 19b by LDA promoted enolization and trapping on oxygen.
Conclusion
We have developed a divergent synthetic strategy that, from N-Boc-3-piperidone, rapidly affords two profoundly different scaffolds, exemplified by compounds 5 and 12, each of which was shown to be orthogonally diversifiable using the same diversity components. The previously unreported scaffold 5 was formed via an unanticipated iodoamination reaction. Compound 5 was used to produce an innovative 97 member library. Work continues to further optimize the preparation and use of spirocyclic scaffold 12 with the goal of using it for production of other libraries of compounds.
Experimental Section
General Methods
All air and moisture sensitive reactions were carried out in flame- or oven-dried glassware under argon atmosphere using standard gas tight syringes, cannula, and septa. Stirring was achieved with oven-dried, magnetic stir bars. CH2Cl2 was purified by passage through a purification system employing activated Al2O3. Flash column chromatography was performed with SiO2 from Sorbent Technology (30930M-25, Silica Gel 60A, 40–63 um) or by using an automated chromatography instrument with an appropriately sized column. Thin layer chromatography was performed on silica gel 60F254 plates (EM-5717, Merck). Deuterated solvents were purchased from commercial sources. 1H and 13C NMR spectra were recorded on instruments operating at 400 or 500 MHz and 100 or 125 MHz respectively. High-resolution mass spectrometry (HRMS) spectra were obtained on a spectrometer operating on ESI. Library synthesis was carried out on a mini-block platform in 17×100 mm tubes with parallel evaporation. Automated preparative reverse-phase HPLC purification was performed using a mass-directed fractionation system with UV-DAD detection and a quadrapole spectrometer using a C18 column (19 × 150mm, 5um, w/ 19 × 10mm guard column). Samples were diluted in DMSO and purified utilizing an elution of water (modified to pH 9.8 through addition of NH4OH) and CH3CN, with a gradient increasing by 20% in CH3CN over 4 minutes at a flow rate of 20ml/min. The starting and ending points of the corresponding preparative CH3CN/water gradient, triggering thresholds, and UV wavelength were selected based on the HPLC analysis of each crude sample. Analytical analysis of each sample after purification employed an HPLC system with UV and mass detection using an ESI-TOF mass spectrometer. The analytical method utilized a Waters Aquity BEH C18 column (2.1 × 50mm, 1.7um) eluting with a linear gradient of 95% water (modified to pH 9.8 through addition of NH4OH) to 100% CH3CN at 0.6 mL/min flow rate where purity was determined using UV peak area at 214 nm. Melting points were determined using an automated apparatus with digital imaging capability. Alkyllithiums were titrated using N-benzylbenzamide.15
t-Butyl 3-hydroxy-3-(3-(trimethylsilyl)prop-2-yn-1-yl)piperidine-1-carboxylate 2
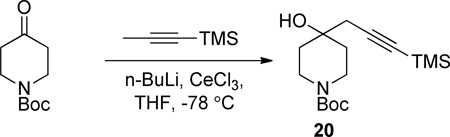
Using a procedure similar to that of Imamoto,7 a 3-necked, 5 L flask equipped with an overhead stirrer and nitrogen inlet was purged with nitrogen for 40 min. Anhydrous cerium(III) chloride (100.0g, 405.6 mmol) pellets were added to the flask and suspended in anhydrous THF (810 mL). The mixture was stirred for 20 h at 120 rpm under nitrogen resulting in a thickly turbid white suspension. A second, 3 L, 3-necked flask equipped with an overhead stirrer, nitrogen inlet, and addition funnel was purged with nitrogen for 40 min. 1-(Trimethylsilyl)-1-propyne (45.52 g, 405.6 mmol) was added to this flask and dissolved in THF (810 mL). This solution was cooled to −78 °C and n-BuLi (181.0 mL of a 2.24 M solution in hexanes, 405.6 mmol) was added via the addition funnel over 15 m. This solution was stirred for 1 h. The initial CeCl3 mixture was cooled to −78 °C and the solution from the second 3-necked flask was added to it via cannula over 1.5 h. The new combined mixture was stirred for 1 h and a solution of N-Boc-3-piperidone 2 (40.40 g, 202.8 mmol) in THF (290 mL) was added via addition funnel over 25 m. The final reaction mixture was stirred for 2 h then sat’d aqueous NH4Cl (1 L) was added. The cold bath was removed and the reaction mixture warmed to rt overnight. The mixture was diluted with 1 M HCl (500 mL) and MTBE (800 mL). The mixture was drawn in portions from the flask by suction through a polypropylene tube and filtered through celite to remove flocculent salts. The two-phase filtrate was returned to the reaction flask and partitioned by siphoning the aqueous via a suction tube. The combined organics were dried over MgSO4, filtered, and concentrated under vacuum using a continuous-feed rotovapor. The crude material (59.00 g) was chromatographed on silica gel (15 to 30% EtOAc in hexanes) to afford the product as a white waxy solid (24.30 g, 38%). A yield of 58% was obtained using 1.350 g (6.760 mmol) of N-Boc-3-piperidone 1.
1H NMR (DMSO-d6, 400 MHz 85 °C) δ 4.15 (s, 1H), 3.46 (m, 1H), 3.38 (d, J = 13.1 Hz, 1H), 3.16 (d, J = 13.1 Hz, 1H), 3.05 (m, 1H), 2.37 (d, J = 17.1 Hz, 1H), 2.32 (d, J = 17.1 Hz, 1H), 1.69 (m, 2H), 1.55 (m, 1H), 1.42 (s, 9H), 1.37 (m, 1H), 0.14 (s, 9H); 13C NMR (DMSO-d6, 100 MHz, 85 °C) δ 153.9, 103.9, 86.1, 77.9, 67.7, 52.2, 42.8, 34.3, 30.3, 27.7, 20.6, −0.39; IR: 3426, 2954, 2176, 1669 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H-Boc)+ (C11H22NOSi)+ 212.1471, found 212.1492.
t-Butyl 4-hydroxy-4-(3-(trimethylsilyl)prop-2-yn-1-yl)piperidine-1-carboxylate 20
Following the procedure for the synthesis of compound 2 N-Boc-4-piperidone (1.000 g, 5.020 mmol) was reacted with cerium trichloride (3.710 g, 15.06 mmol), n-butyllithium (6.350 mL of a 2.37 M solution in hexanes, 15.06 mmol), and 1-(trimethylsilyl)-1-propyne (1.700 g, 15.15 mmol) to afford the product 20 (0.966 g, 62%) as a white granular solid. MP = 98.8–102.4 °C. 1H NMR (DMSO-d6, 400 MHZ, 85 °C) δ 4.28 (s, 1H), 3.78 - 3.58 (m, 2H), 3.09 (td, J = 13.2, 2.9 Hz, 2H), 2.42 - 2.32 (m, 2H), 1.62 (td, J = 12.7, 4.7 Hz, 2H), 1.52 - 1.37 (m, 11H), 0.14 (s, 9H); 13C NMR (DMSO-d6, 100 MHZ, 85 °C) δ 153.6, 104.3, 86.0, 77.9, 67.6, 34.9, 33.7, 27.7, 27.6, −0.4; IR: 3428, 2960, 2175, 1664 cm−1; HRMS (ESI-TOF) m/z calculated for (M+M+H)+ (C32H59N2O6Si2)+ 623.3912, found 623.3908.
t-Butyl 3-hydroxy-3-(prop-2-yn-1-yl)piperidine-1-carboxylate 3
To a 3-liter, 3 necked flask equipped with an overhead stirrer was added t-butyl 3-hydroxy-3-(3-(trimethylsilyl)prop-2-yn-1-yl)piperidine-1-carboxylate 2 (24.30 g, 78.01 mmol), methanol (780 mL) and K2CO3 (12.90 g). The mixture was stirred at rt for 2.5 h, drawn from the flask via suction, concentrated under vacuum, re-dissolved in MTBE (300 mL) and adsorbed onto silica gel (80 g) by evaporation. The dried silica gel was put over celite in a fritted funnel and eluted with MTBE (1500 mL). The filtrate was concentrated under vacuum to give the product (16.70 g, 89%) as a granular white solid which was used without further purification. MP = 63.6–67.0 °C. 1H NMR (DMSO-d6, 400 MHZ, 85 °C) δ 4.22 (s, 1H), 3.39 (m, 1H), 3.32 (d, J = 13.1 Hz, 1H), 3.22 (d, J = 13.1 Hz, 1H), 3.13 (m, 1H), 2.62 (t, J = 2.5 Hz, 1H), 2.30 (m, 2H), 1.78 - 1.62 (m, 2H), 1.55 (m, 1H), 1.41 (s, 9H), 1.36 (m, 1H); 13C NMR (DMSO-d6, 100 MHz, 85 °C) δ 153.9, 80.6, 78.0, 72.0, 67.6, 52.2, 42.8, 34.2, 28.8, 27.7, 20.7; IR: 3427, 2934, 2119, 1670 cm−1; HRMS (ESI-TOF) m/z calculated for (M+NH4)+ (C13H25N2O3)+257.1865, found 257.1877.
t-Butyl 4-hydroxy-4-(prop-2-yn-1-yl)piperidine-1-carboxylate 6
Using the above K2CO3 hydrolysis procedure for compound 3, t-butyl 4-hydroxy-4-(3-(trimethylsilyl)prop-2-yn-1-yl)piperidine-1-carboxylate 20 (0.913 g, 2.93 mmol) was reacted with K2CO3 (0.486g, 3.520 mmol). The crude material was chromatographed on silica gel (5 to 30% EtOAc in hexanes) to afford the product 6 (0.530 g, 76%) as a powdered white solid. MP = 41.9–45.1 °C. 1H NMR (DMSO-d6, 400 MHZ, 85 °C) δ 4.29 (s, 1H), 3.67 (dt, J = 13.0, 3.6 Hz, 2H), 3.11 (m, 2H), 2.62 (t, J = 2.5 Hz, 1H), 2.32 (d, J = 2.5 Hz, 2H), 1.59 (m, 2H), 1.50 (m, 2H), 1.42 (s, 9H); 13C NMR (DMSO-d6, 100 MHZ, 85 °C) δ 153.7, 80.9, 78.0, 72.0, 67.4, 35.0, 32.2, 27.7; IR: 3418, 2934, 2119, 1662 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C13H22NO3)+ 240.1600, found 240.1587.
t-Butyl 3-hydroxy-3-(prop-2-yn-1-yl)piperidine-1-carboxylate 3
directly from N-Boc-3-piperidone 1. (CAUTION: This reaction can be accompanied by a vigorous exotherm and should be properly cooled and vented). Using a procedure modified from that of Wang,8 to a solution of N-Boc-3-piperidone (10.13 g, 50.80 mmol) and propargyl bromide (8.49 mL of an 80 wt% solution in toluene, 76.00 mmol) in THF (10.2 mL) at 0 °C was added Zn/Cu couple (4.550 g, 55.90 mmol). The mixture was sonicated for 10 min then diluted with sat’d aqueous NH4Cl and ethyl acetate. (On a scale >1 g of ketone filtration of the mixture through paper at this point facilitates isolation). The layers were separated and the aqueous layer extracted with ethyl acetate (x3). The combined organic layers were dried over MgSO4, filtered, and concentrated under vacuum. The crude material was chromatographed on silica gel (5 to 30% EtOAc in hexanes) to afford the product (7.100 g, 58%) as a white solid. The product contains an inseparable, but small amount of the allene isomer (~10:1, alkyne:allene as observed by 1H NMR). This causes no perceivable issues in subsequent reactions. Spectral data is reported above.
1-(Phenylsulfonyl)-3-(prop-2-yn-1-yl)piperidin-3-ol 8
Using the above Zn/Cu/propargyl bromide procedure for compound 3, N-benzensulfonyl-3-piperidone (1.024 g, 4.280 mmol) was reacted with propargyl bromide (0.715 mL, 6.420 mmol) and Zn/Cu couple (0.383 g, 4.710 mmol) under sonication for 50 m to afford the product 8 (0.610 g, 51%) as a viscous, orange oil. The product contains an inseparable, but small amount of the allene isomer (~8.3:1, alkyne:allene as observed by 1H NMR). 1H NMR (DMSO-d6, 400 MHZ, 85 °C) δ 7.75 (m, 2H), 7.66 (m, 3H), 3.04 (m, 1H), 2.95 (d, J = 11.6 Hz, 1H), 2.87 (d, J = 11.5 Hz, 1H), 2.80 (m, 1H), 2.63 (t, J = 2.6 Hz, 1H), 2.41 (dd, J = 16.9, 2.6 Hz, 1H), 2.34 (dd, J = 16.9, 2.6 Hz, 1H), 1.79 (m, 1H), 1.61 (m, 1H), 1.56 - 1.43 (m, 2H); 13C NMR (DMSO-d6, 100 MHZ, 85 °C) δ 136.4, 132.3, 128.7, 126.7, 80.2, 72.3, 67.4, 54.0, 45.3, 33.4, 28.8, 20.5; IR: 3497, 2940, 2115, 1446, 1334 cm−1; HRMS (ESI-TOF) m/z calculated for (C14H18NO3S)+ (M+H)+, 280.1007 found 280.1021.
t-Butyl 3-hydroxy-3-(prop-2-yn-1-yl)azetidine-1-carboxylate 16a
Using the above Zn/Cu/propargyl bromide procedure for compound 3, N-Boc-3-azetidinone (1.673 g, 9.77 mmol) was reacted with propargyl bromide (1.633 mL, 14.66 mmol) and Zn/Cu couple (0.875 g, 10.75 mmol) under sonication for 30 m to afford the product 16a (1.674 g, 81%) as a white solid. The product contains an inseparable but small amount of the allene isomer (~8.3:1, alkyne:allene as observed by 1H NMR. This causes no perceivable issues in subsequent reactions). MP = 83.8–90.0 °C 1H NMR (CDCl3, 400 MHZ) δ 3.92 (d, J = 9.6 Hz, 2H), 3.87 (d, J = 9.6 Hz, 2H), 3.53 (s, 1H), 2.62 (d, J = 2.6 Hz, 2H), 2.06 (t, J = 2.6 Hz, 1H), 1.42 (s, 9H); 13C NMR (CDCl3, 100 MHZ) δ 156.4, 79.9, 79.0, 71.1, 68.9, 61.0, 29.4, 28.3; IR: 3384, 2977, 1669 cm−1; HRMS (ESI-TOF) m/z calculated for (C11H18NO3)+ (M+H)+ 212.1287, found 212.1287.
t-Butyl 3-hydroxy-3-(prop-2-yn-1-yl)pyrrolidine-1-carboxylate 16b
Using the above Zn/Cu/propargyl bromide procedure for compound 3, N-Boc-3-pyrrolidinone 15b (4.062 g, 21.93 mmol) was reacted with propargyl bromide (3.660 mL, 32.90 mmol) and Zn/Cu couple (1.964 g, 24.12 mmol) under sonication for 45 m to afford the product 16b (2.535 g, 51%) as a white solid. The product contains an inseparable, but small amount of the allene isomer (~8:1, alkyne:allene as observed by 1H NMR. This causes no perceivable issues in subsequent reactions). MP = 75.3–79.8 °C 1H NMR (DMSO-d6, 500 MHZ, 90 °C) δ 4.54 (s, 1H), 3.45 - 3.36 (m, 2H), 3.32 (m, 1H), 3.27 (d, J = 11.2 Hz, 1H), 2.54 (s, 1H), 2.49 (m, 2H), 1.96 (m, 1H), 1.84 (m, 1H), 1.44 (s, 9H); 13C NMR (DMSO-d6, 125 MHZ, 90 °C) δ 153.2, 80.6, 77.5, 76.2, 70.9, 56.1, 43.9, 35.9, 28.0, 27.6; IR: 3379, 2978, 2109, 1658 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C12H20NO3)+ 226.1443, found 226.1452.
(E)-7-(iodomethylene)-1-azabicyclo[3.2.1]octan-5-ol (HCl salt) 5a
To a 5 L, 3-necked flask equipped with an overhead stirrer was added t-butyl 3-hydroxy-3-(prop-2-yn-1-yl)piperidine-1-carboxylate 3 (16.70 g, 69.78 mmol) and dichloromethane (DCM, 700 mL). To this was added a solution of iodine monochloride (34.00 g, 209.3 mmol) in DCM (700 mL) over 30 m via addition funnel. The reaction mixture was stirred for 22 h, then quenched with sat’d aqueous Na2SO3 (1000 mL) and made basic (pH paper checking) with sat’d aqueous NaHCO3 (1000 mL). The aqueous layer was removed via siphon and the organic layer washed in this fashion with deionized water (300 mL × 2). Approximately 250 mL of deionized water was added to the organic layer and the mixture acidified to pH 2 with 1 M HCl as determined by pH meter. The layers were separated and the aqueous layer lyophilized to afford the amine salt 5a (15.80 g, ~75%) as a yellow powder which was used without further purification. In all cases, and as determined by LC-MS, the product contains the diiodo-species 21a as a 10 area% impurity. MP (HCl salt 5a as mixture with 21a) = 175.7–177.4 °C (dec). The two products could be separated via reverse-phase HPLC as their free bases. The stationary phase was a Waters Sunfire™ C18-OBD 5 µM 30 × 150 mm column. The mobile phase was an CH3CN/water (0.02% TFA) gradient starting at 0% CH3CN and ending at 100% CH3CN over 13 min. Both products were obtained as viscous brown oils. Remaining data provided for free base 5b: 1H NMR (CDCl3, 500 MHZ) δ 6.96 (t, J = 2.5 Hz, 1H), 3.55 (d, J = 10.2 Hz, 1H), 3.35 (dd, J = 12.4, 4.8 Hz, 1H), 3.25 - 3.11 (m, 2H), 2.66 (d, J = 18.0 Hz, 1H), 2.61 (dd, J = 18.0, 2.7 Hz, 1H), 2.08 - 1.85 (m, 4H); 13C NMR (CDCl3, 125 MHz) δ 145.1, 75.0, 73.2, 62.8, 55.6, 43.3, 35.2, 18.1; HRMS (ESI-TOF) m/z calculated for (C8H13INO)+ (M+H)+ 266.0042, found 266.0054. Data provided for free base 21b: 1H NMR (DMSO-d6, 500 MHZ) δ 3.20 (dd, J = 12.8, 5.7 Hz, 1H), 2.99 (d, J = 10.0 Hz, 1H), 2.85 (d, J = 9.5 Hz, 1H), 2.70 (m, 1H), 2.44 - 2.35 (m, 2H), 2.19 (d, J = 16.8 Hz, 1H), 1.69 - 1.54 (m, 3H), 1.47 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 155.7, 75.9, 65.2, 52.0, 48.8, 35.9, 20.5, 10.1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C8H12I2NO)+ 391.9008, found 391.9029.
(E)-2-(iodomethylene)-1-azabicyclo[2.2.1]heptan-4-ol (HCl salt) 17b
Following the above procedure for compound 5a, t-butyl 3-hydroxy-3-(prop-2-yn-1-yl)pyrrolidine-1-carboxylate 16b (0.530 g, 2.353 mmol) was reacted with iodine monochloride (1.146 g, 7.060 mmol) to afford 17b (0.217 g, 32%) as a yellow powder. MP = 155.9–159.1 °C (dec). 1H NMR (DMSO-d6, 400 MHZ) δ 12.63 (s, 1H), 6.97 (s, 1H), 6.41 (s, 1H), 3.72 (td, J = 11.1, 6.2 Hz, 1H), 3.34 - 3.22 (m, 2H), 3.19 (d, J = 8.1 Hz, 1H), 2.48 (m, 2H), 1.99 (m, 2H); 13C NMR (DMSO-d6, 100 MHZ) δ 146.7, 78.4, 74.6, 60.5, 54.4, 42.5, 31.3; IR: 3247, 2824, 2503, 1356 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C7H11INO)+ 251.9885, found 251.9906.
(E)-7-(iodomethylene)-1-azabicyclo[3.2.1]octan-5-ol
5b by neutralization of 5a. A solution of salt 5a (5.008 g, 16.61 mmol) in water (150 mL) was made basic to pH > 7 (pH paper) using sat’d aqueous sodium bicarbonate. The solution was extracted into ethyl acetate (x6) and the combined organic layers were dried over MgSO4, filtered, and concentrated under vacuum to give the free base as a viscous, brown oil (3.900 g, 89%).
(E)-7-benzylidene-1-azabicyclo[3.2.1]octan-5-yl 4-chlorobenzoate 10{10,1}
To a solution of (E)-7-(iodomethylene)-1-azabicyclo[3.2.1]octan-5-ol 5b (0.064 mg, 0.242 mmol), triethyl amine (0.200 mL, 1.45 mmol), and dimethylaminopyridine (0.015 g, 0.121 mmol) in CH3CN (1.00 mL) was added 4-chlorobenzoyl chloride (0.16 mL, 1.21 mmol). The reaction mixture was heated to 50 °C for 22 h. The reaction mixture was diluted with diethyl ether, filtered through celite, and concentrated under vacuum. The crude residue was chromatographed on silica gel (15 to 30% EtOAc in hexanes) to give the purified intermediate (E)-7-(iodomethylene)-1-azabicyclo[3.2.1]octan-5-yl 4-chlorobenzoate (0.078 g, 79%). This was taken directly on to the next step.
A solution of (E)-7-(iodomethylene)-1-azabicyclo[3.2.1]octan-5-yl 4-chlorobenzoate (0.046 g, 0.110 mmol), cesium carbonate (0.22 mL of a 2 M aqueous solution, 0.440 mmol), phenyl boronic acid (0.015 g, 0.120 mmol), and tetrakis(triphenylphosphine)palladium(0) (0.008 g, 0.007 mmol) in DMF (1.10 mL) was heated to 90 °C for 90 min. The reaction mixture was diluted with EtOAc and filtered through a 1g silica gel solid phase extraction column (SPE). The filtrate was concentrated under vacuum and the residue chromatographed on reversed phase C18 (10 to 100% CH3CN in water) to give the product 10{10,1} (0.026 mg, 68%) as a brown solid. 1H NMR (CDCl3, 500 MHZ) δ 7.97 (m, 2H), 7.43 (m, 2H), 7.38 - 7.31 (m, 4H), 7.21 (m, 1H), 6.34 (t, J = 2.3 Hz, 1H), 3.49 (dt, J = 16.8, 2.1 Hz, 1H), 3.29 (dd, J = 10.7, 2.1 Hz, 1H), 3.21 (dd, J = 10.7, 3.2 Hz, 1H), 3.16 (dd, J = 13.4, 6.0 Hz, 1H), 3.12 - 2.99 (m, 2H), 2.39 (m, 1H), 2.22 (tdd, J = 12.4, 5.8, 1.3 Hz, 1H), 2.02 (m, 1H), 1.68 (m, 1H); 13C NMR (CDCl3, 125 MHZ) δ 164.8, 149.5, 139.5, 136.6, 131.0, 128.9, 128.7, 128.5, 127.8, 126.2, 117.1, 84.1, 62.7, 58.1, 42.2, 33.2, 21.5; IR: 2942, 1718 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C21H21ClNO2)+ 354.1261, found 354.1254.
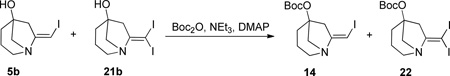
(E)-t-butyl (7-(iodomethylene)-1-azabicyclo[3.2.1]octan-5-yl) carbonate 14 and t-butyl (7-(diiodomethylene)-1-azabicyclo[3.2.1]octan-5-yl) carbonate 22
To a solution of the alcohol mixture 5b (3.538 g, 13.35 mmol) and 21b (579 mg, 1.48 mmol) in DCM (62 mL) was added N,N-dimethylaminopyridine (DMAP, 0.379 g, 3.110 mmol), triethylamine (TEA, 4.330 mL, 31.10 mmol), and di-t-butyldicarbonate (Boc2O, 4.070 g, 18.64 mmol). The reaction mixture was stirred for 3 h then diluted with water and DCM and the layers separated. The aqueous layer was extracted with DCM (x2), and the combined organic layers were washed with 10% aqueous citric acid (x1) and brine (x1), dried over MgSO4, filtered and concentrated under vacuum. The crude material was chromatographed on silica gel (5 to 30% EtOAc in hexanes) to give the separated final products: (E)-t-butyl (7-(iodomethylene)-1-azabicyclo[3.2.1]octan-5-yl) carbonate 14 as a light brown powder (4.131 g, 11.31 mmol) and t-butyl (7-(diiodomethylene)-1-azabicyclo[3.2.1]octan-5-yl) carbonate 22 as a granular brown solid (0.393 g, 0.80 mmol). Overall amount: 12.11 mmol, 82%. Mono-iodide 14: MP = 61.1–64.8 °C. 1H NMR (CDCl3, 400 MHZ) δ 5.89 (m, 1H), 3.14 (dd, J = 11.0, 2.3 Hz, 1H), 3.10 (dd, J = 11.0, 3.0 Hz, 1H), 3.07 - 2.94 (m, 2H), 2.84 (td, J = 13.1, 4.5 Hz, 1H), 2.56 (ddd, J = 17.2, 2.5, 1.9 Hz, 1H), 2.31 - 2.21 (m, 1H), 2.05 (tdd, J = 12.3, 5.9, 1.6 Hz, 1H), 1.86 (m, 1H), 1.64 (m, 1H), 1.47 (s, 9H); 13C NMR (CDCl3, 100 MHZ) δ 158.1, 151.9, 83.4, 82.3, 64.9, 64.3, 57.7, 45.0, 32.9, 27.7, 21.5; IR: 2940, 1738 cm−1; HRMS (ESI-TOF) m/z calculated for (C13H21INO3)+ (M+H)+ 366.0566, found 366.0596. Di-iodide 22: MP = 98.2–137.7 °C (slowly decomposes over reported range). 1H NMR (CDCl3, 400 MHZ) δ 3.45 - 3.32 (m, 1H), 3.27 - 2.96 (m, 3H), 2.69 (m, 1H), 2.60 (dd, J = 17.2, 1.6 Hz, 1H), 2.28 (dd, J = 11.2, 1.6 Hz, 1H), 2.05 (m, 1H), 1.73 (m, 2H), 1.47 (s, 9H); 13C NMR (CDCl3, 100 MHZ) δ 159.5, 151.7, 85.2, 82.4, 64.4, 52.5, 47.4, 32.6, 27.7, 22.1, 0.6; IR: 2977, 1737 cm−1; HRMS (ESI-TOF) m/z calculated for (C13H20I2NO3)+ (M+H)+ 491.9533, found 491.9532.
t-Butyl 3-oxo-1-oxa-7-azaspiro[4.5]decane-7-carboxylate 11
Using the method of Zhang,12 to a solution of t-butyl 3-hydroxy-3-(prop-2-yn-1-yl)piperidine-1-carboxylate 3 (0.927 g, 3.870 mmol) in DCE (31.0 mL) was added 3,5-dichloropyridine-N-oxide (1.270 g, 7.750 mmol) and methane sulfonic acid (0.302 mL, 4.650 mmol). In a separate flask, triphenylphosphinegold(I) bis(trifluoromethanesulfonyl)imidate (0.143 g, 0.194 mmol, 5 mol%) was weighed in a glove box. The gold catalyst was placed under argon, removed from the glove box and dissolved in DCE (46.50 mL). The contents of the first reaction flask were added to the gold catalyst-containing flask via cannula and the reaction mixture stirred for 4 h. The reaction mixture was concentrated under vacuum and chromatographed on silica gel (5 to 30% EtOAc in hexanes) to give the product 11 (0.511 g, 52%) as a colorless oil. 1H NMR (DMSO-d6, 500 MHZ, 90 °C) δ 4.02 (d, J = 17.0 Hz, 1H), 3.96 (d, J = 17.0 Hz, 1H), 3.51 - 3.39 (m, 2H), 3.38 - 3.23 (m, 2H), 2.38 (d, J = 18.2 Hz, 1H), 2.34 (d, J = 18.1 Hz, 1H), 1.89 - 1.69 (m, 3H), 1.49 – 1.36 (m, 1H), 1.44 (s, 9H); 13C NMR (CDCl3, 125 MHZ, 90 °C) δ 212.7, 153.5, 78.5, 78.2, 68.7, 50.0, 44.6, 42.6, 34.1, 27.4, 21.0; IR: 2936, 1760, 1687 cm−1; HRMS (ESI-TOF) m/z calculated for (M-Boc+H)+ (C8H14NO2)+ 156.1025, found 156.1053.
t-Butyl 7-oxo-5-oxa-2-azaspiro[3.4]octane-2-carboxylate 18a
Following the above procedure for compound 11, t-butyl 3-hydroxy-3-(prop-2-yn-1-yl)azetidine-1-carboxylate 16a (1.138 g, 5.39 mmol) was reacted with 3,5-dichloropyridine-N-oxide (1.767 g, 10.77 mmol), methane sulfonic acid (0.419 mL, 6.460 mmol), and triphenylphosphinegold(I) bis(trifluoromethanesulfonyl)imidate (0.199 g, 0.269 mmol) to afford the product 18a (0.606 g, 50%) as a granular white solid. MP = 70.6–80.5 °C. 1H NMR (CDCl3, 400 MHZ) δ 4.10 (d, J = 9.4 Hz, 2H), 4.00 – 3.97 (t, J = 5.9 Hz, 4H), 2.70 (s, 2H), 1.43 (s, 9H); 13C NMR (CDCl3, 100 MHZ) δ 212.2, 156.1, 80.0, 77.3, 70.8, 61.0, 45.5, 28.2; IR: 2978, 1765, 1694 cm−1; HRMS (ESI-TOF) m/z calculated for (C11H16NO4)− (M−H)−, 226.1085 found 226.1062.
t-Butyl 3-oxo-1-oxa-7-azaspiro[4.4]nonane-7-carboxylate 18b
Following the above procedure for compound 11, t-butyl 3-hydroxy-3-(prop-2-yn-1-yl)pyrrolidine-1-carboxylate 16b (2.470 g, 10.96 mmol) was reacted with 3,5-dichloropyridine-N-oxide (3.600 g, 21.93 mmol), methane sulfonic acid (0.854 mL, 13.16 mmol), and triphenylphosphinegold(I) bis(trifluoromethanesulfonyl)imidate (0.405 g, 0.548 mmol) to afford the product 18b (1.653 g, 63%) as a granular pale yellow solid. MP = 70.8−76.2 °C. 1H NMR (DMSO-d6, 500 MHZ, 90 °C) δ 4.01 (m, 2H), 3.54 (d, J = 11.7 Hz, 1H), 3.48 - 3.40 (m, 2H), 3.36 (d, J = 11.7 Hz, 1H), 2.66 (d, J = 18.1 Hz, 1H), 2.57 (d, J = 18.1 Hz, 1H), 2.19 - 2.08 (m, 1H), 1.99 (m, 1H), 1.45 (s, 9H); 13C NMR (DMSO-d6, 125 MHZ, 90 °C) δ 212.4, 153.1, 85.8, 77.9, 69.2, 54.6, 43.8, 42.9, 34.8, 27.6; IR: 2975, 1762, 1688 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C12H20NO4)+ 242.1392, found 242.1374.
t-Butyl 3-(((trifluoromethyl)sulfonyl)oxy)-1-oxa-7-azaspiro[4.5]dec-2-ene-7-carboxylate 12
To a solution of diisopropylamine (0.373 mL, 2.660 mmol) in THF (8.87 mL) at −78 °C was added n-butyllithium (1.142 mL of a 2.330 M solution in hexanes). The reaction mixture was warmed to 0 °C and stirred for 30 m then cooled to −78 °C. A solution of t-butyl 3-oxo-1-oxa-7-azaspiro[4.5]decane-7-carboxylate 11 (0.227 g, 0.887 mmol) in THF (4.44 mL) was then added via cannula and the mixture stirred for 30 min. A solution of 1,1,1-trifluoro-N-phenyl-N-(trifluoromethylsulfonyl)methanesulfonamide (0.634 g, 1.775 mmol) in THF (7.10 mL) was then added via cannula and the solution allowed to warm to rt and stirred overnight for 18 h. The reaction mixture was quenched with sat’d, aqueous NH4Cl, diluted with water and MTBE and the layers separated. The aqueous layer was extracted with MTBE (x3) and the combined organic layers were dried over MgSO4, filtered through a pad of silica gel and concentrated under vacuum. The crude residue was chromatographed on reverse-phase C18 (0 to 100% CH3CN in pH 9.4 ammonia/water) to give the product 12 (0.196 g, 57%) as an orange/brown oil. 1H NMR (DMSO-d6, 500 MHZ, 85 °C) δ 6.09 (t, J = 1.9 Hz, 1H), 4.64 (m, 2H), 3.43 - 3.36 (m, , 2H), 3.37 (d, J = 13.3 Hz, 1H), 3.23 (m, 1H), 1.82 (m, 1H), 1.73 (m, 2H), 1.52 (m, 1H), 1.43 (s, 9H); 13C NMR (CDCl3, 125 MHZ, 85 °C) δ 153.5, 144.2, 117.8, 117.6 (q, JC-F = 322.12 Hz), 85.1, 78.2, 67.7, 50.7, 42.3, 33.9, 27.4, 20.7; IR: 2938, 1692 cm−1; HRMS (ESI-TOF) m/z calculated for (M-Boc+H)+ (C9H13F3NO4S)+ 288.0517, found 288.0532.
t-Butyl 7-(((trifluoromethyl)sulfonyl)oxy)-5-oxa-2-azaspiro[3.4]oct-6-ene-2-carboxylate 19a
Following the above procedure for compound 12, t-Butyl 7-oxo-5-oxa-2-azaspiro[3.4]octane-2-carboxylate 18a (0.125 g, 0.550 mmol) was reacted with LDA (0.231 mL, 1.650 mmol of diisopropyl amine with 0.743 mL, 1.650 mmol of a 2.220 M solution of butyllithium in hexanes) and N-(5-chloropyridin-2-yl)-1,1,1-trifluoro-N-((trifluoromethyl)sulfonyl)methanesulfonamide (Comins’ reagent, 0.432 g, 1.100 mmol) in place of 1,1,1-trifluoro-N-phenyl-N-(trifluoromethylsulfonyl)methanesulfonamide. The reaction was quenched with 5 M aqueous NH4OH sat’d with NH4Cl and washed again (x1) with this solution after dilution with MTBE. The work up then proceeds as for compound 12. Chromatography on silica gel (0 to 25% EtOAc in hexanes) afforded the product 19a (0.075 g, 38%) as an granular off-white solid. MP = 46.2–52.5 °C. 1H NMR (CDCl3, 400 MHZ) δ 5.98 (s, 1H), 4.66 (d, J = 2.1 Hz, 2H), 4.20 - 4.09 (m, 2H), 4.07 - 3.96 (m, 2H), 1.43 (s, 9H); 13C NMR (CDCl3, 100 MHZ) δ 156.0, 145.8, 118.4 (q, JC-F = 321.1 Hz), 114.4, 83.8, 80.0, 70.2, 62.0, 28.3; IR: 2978, 1702, 1667 cm−1; HRMS (ESI-TOF) m/z calculated for (C24H33F6N2O12S2)+ (M+M+H)+, 719.1379 found 719.1380.
t-Butyl 3-(((trifluoromethyl)sulfonyl)oxy)-1-oxa-7-azaspiro[4.4]non-2-ene-7-carboxylate 19b
Following the above procedure for compound 12, t-butyl 3-oxo-1-oxa-7-azaspiro[4.4]nonane-7-carboxylate 18b (0.186 g, 0.771 mmol) was reacted with LDA (0.324 mL, 2.313 mmol of diisopropyl amine with 1.042 mL, 2.313 mmol of a 2.220 M solution of butyllithium in hexanes) and 1,1,1-trifluoro-N-phenyl-N-(trifluoromethylsulfonyl)methanesulfonamide (0.551 g, 1.542 mmol). In this case chromatography on silica gel (0 to 25% EtOAc in hexanes) afforded the product 19b (0.112 g, 39%) as an orange/brown oil. 1H NMR (DMSO-d6, 500 MHZ, 90 °C) δ 6.15 (s, 1H), 4.65 (m, 2H), 3.48 (m, 1H), 3.45 - 3.34 (m, 3H), 2.11 (dt, J = 13.0, 9.1 Hz, 1H), 2.02 (ddd, J = 13.0, 7.1, 3.1 Hz, 1H), 1.45 (s, 9H); 13C NMR (DMSO-d6, 125 MHZ, 90 °C) δ 152.9, 144.1, 122.3, 117.6 (q, JC-F = 321.7 Hz), 92.3, 77.9, 67.8, 54.5, 43.7, 35.4, 27.5; IR: 2979, 1693, 1671 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C13H19F3NO6S)+ 374.0885, found 374.0901.
(4-chlorophenyl)(3-phenyl-1-oxa-7-azaspiro[4.5]dec-2-en-7-yl)methanone 13
A solution of t-butyl 3-(((trifluoromethyl)sulfonyl)oxy)-1-oxa-7-azaspiro[4.5]dec-2-ene-7-carboxylate 12 (0.044 g, 0.113 mmol), phenylboronic acid (0.015 g, 0.125 mmol), and Cs2CO3 (0.227 mL of a 2 M aqueous solution, 0.453 mmol) in DMF (1.11 mL) was purged with argon for 5 min. Tetrakis triphenylphosphine palladium(0) (0.008 g, 0.007 mmol) was added and the reaction mixture heated to 90 °C for 110 min. The reaction mixture was cooled and diluted with ethyl acetate and water and the layers separated. The aqueous layer was extracted with ethyl acetate (x3) and the combined organic layers were washed with brine (x1), dried over MgSO4, filtered, and concentrated under vacuum. The crude material was chromatographed on silica gel (0 to 10% EtOAc in hexanes) to give the intermediate arylated product, t-butyl 3-phenyl-1-oxa-7-azaspiro[4.5]dec-2-ene-7-carboxylate (0.018 mg, 51%) as a yellow oil. This was taken directly to the next step.
To a solution of t-butyl 3-phenyl-1-oxa-7-azaspiro[4.5]dec-2-ene-7-carboxylate (0.018 g, 0.057 mmol) in DCM (0.300 mL) was added activated 4 Å molecular sieves and trifluoroacetic acid (TFA, 0.300 mL). After 5 min the reaction mixture was filtered and the filtrate was diluted with toluene and concentrated under vacuum (flushed) three times to remove TFA. The residue was then dissolved in DCM (1.000 mL). To this solution was added TEA (0.020 mL, 0.143 mmol), DMAP (~1 mg, ~0.006 mmol), and 4-chlorobenzoyl chloride (0.011 mL, 0.086 mmol). The reaction mixture was stirred for 2 h at rt, concentrated under vacuum and directly chromatographed on silica gel (20 to 40% EtOAc in hexanes) to give the product 13 (0.016 g, 78%) as a white, amorphous solid. 1H NMR (DMSO-d6, 500 MHZ, 90 °C) δ 7.53 - 7.24 (m, 9H), 6.30 (s, 1H), 4.95 (dd, J = 12.6, 1.8 Hz, 1H), 4.81 (d, J = 12.6 Hz, 1H), 3.82 (m, 1H), 3.54 (d, J = 13.2 Hz, 1H), 3.44 (d, J = 13.2 Hz, 1H), 3.23 (m, 1H), 1.88 - 1.84 (m, 3H), 1.67 (m, 1H); 13C NMR (CDCl3, 125 MHZ, 90 °C) δ 168.0, 138.6, 135.0, 133.3, 131.5, 128.1, 127.8, 127.6, 127.5, 125.2, 124.8, 87.6, 72.7, 52.4, 43.0, 34.0, 21.0; IR: 2941, 1628 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C21H21ClNO2)+ 354.1261, found 354.1254.
General Procedure for library synthesis, Route 1
The following describes the synthesis of a subset of the library consisting of 48 (8×6) members. Stock solutions were prepared to accommodate 52 members in order to provide for potential handling losses.
Step 1: Stock solutions were prepared as follows:
(E)-7-(iodomethylene)-1-azabicyclo[3.2.1]octan-5-ol 5b: 4.461 g (16.83 mmol) in acetonitrile (CH3CN, 58.40 mL, 0.288 M)
TEA (8.320 mL, 59.69 mmol) and DMAP (572.0 mg, 4.322 mmol) in CH3CN (7.800 mL)
To each of 48 17×100 mm tubes equipped with stirring bars on two 24-place mini-block platforms on stirring plates was added: 1.000 mL of stock solution 1 (0.288 mmol amine), 0.320 mL of stock solution 2 (1.150 mmol TEA and 0.090 mmol DMAP), and the appropriate volume of each neat acid chloride (0.864 mmol, 8 acid chlorides were used 6 times each in this first step). The reactor blocks were heated to 50 °C, as judged by thermocouple inserted into each block, for 22 h. Each reaction mixture was then diluted with MTBE (10 mL) and filtered into new 17×100 mm reaction tubes through a 1 g silica gel SPE using a mini-block filtration platform. The filtrates were concentrated in parallel using a parallelpersonal evaporator. The crude material was taken on to the final step.
Step 2: Using the same mini-block set up described above, to each reaction tube was added dimethylformamide (2.900 mL), cesium carbonate (0.580 mL of a 2 M solution in water, 1.15 mmol), the appropriate amount of each bornic acid (0.320 mmol), and tetrakis(triphenylphosphine)palladium(0) (20.00 mg, 0.017 mmol, added using the mini-block powder dispenser). The reactor blocks were heated to 90 °C, as judged by thermocouple inserted into each block, for 90 min. Each reaction mixture was then diluted with ethyl acetate (7 mL) and filtered through 1 g silica gel SPE tubes into bar-coded 16×100 mm screw cap tubes. The filtrates were concentrated under vacuum using a parallel evaporator and the crude products purified using high-throughput MDF as described in the general procedures section above.
General Procedure for library synthesis, Route 2
The following describes the synthesis of a subset of the library consisting of 18 (9×2) members. Stock solutions were prepared in a 10% excess to provide for potential handling losses.
Step 1: Stock solutions were prepared as follows:
(E)-t-butyl (7-(iodomethylene)-1-azabicyclo[3.2.1]octan-5-yl) carbonate 21 (2.530 g, 6.930 mmol) in DMF (69.30 mL, 0.100 M).
Using the same mini-block set up described above to each reaction tube was added 3.5 mL of stock solution 1 (0.350 mmol of 21), cesium carbonate (0.700 mL of a 2 M solution in water, 1.400 mmol), the appropriate boronic acid (0.385 mmol, 9 boronic acids were used twice each in this first step), and tetrakis(triphenylphosphine)palladium(0) (24.00 mg, 0.021 mmol, added using the mini-block powder dispenser). The reactor blocks were heated to 90 °C, as judged by thermocouple inserted into each block, for 3 h. Each reaction mixture was then diluted with ethyl acetate (8 mL) and filtered into new 17×100 mm reaction tubes through a 1 g silica gel SPE using a mini-block filtration platform. The filtrates were concentrated in parallel using a parallel evaporator.
Step 2: A stock solution of 1:1 DCM:TFA (1.80 mL) was added to each of the 18 reaction tubes. The mixtures were stirred for 30 min and then toluene (4.000 mL) was added to each reaction. After removal of the solvent on the parallel personal evaporator, another 4.00 mL of toluene was added to each and once again removed under vacuum in the same manner.
Step 3: A stock solution of TEA and DMAP was prepared:
TEA (6.80 mL, 48.79 mmol) and DMAP (260.0 mg, 2.128 mmol) in CH3CN (35.00 mL)
To each reaction tube was added 1.750 mL of CH3CN, 2.100 mL of stock solution 1, and the appropriate acid chloride (1.050 mmol). The reactor blocks were either heated to 50 °C, as judged by thermocouple, (A8, isoxazole derived acid chloride) for 3 h, or kept at room temperature (A14, mesyl chloride) for 1 h. In the case of A8, each reaction mixture was diluted with MTBE (5 mL), while the A14 mixtures were diluted with 1:1, DCM:CH3CN (5 mL). All reaction mixtures were filtered through 1 g silica gel SPE tubes into bar-coded 16×100 mm screw cap tubes. The filtrates were concentrated under vacuum using a parallel evaporator and the crude products purified using high-throughput MDF as described in the general procedures section above.
(E)-7-benzylidene-1-azabicyclo[3.2.1]octan-5-yl 4-chlorobenzoate 10{10,1}
1H NMR (CDCl3, 500 MHZ) δ 7.97 (m, 2H), 7.43 (m, 2H), 7.38 - 7.31 (m, 4H), 7.21 (m, 1H), 6.34 (t, J = 2.3 Hz, 1H), 3.49 (dt, J = 16.8, 2.1 Hz, 1H), 3.29 (dd, J = 10.7, 2.1 Hz, 1H), 3.21 (dd, J = 10.7, 3.2 Hz, 1H), 3.16 (dd, J = 13.4, 6.0 Hz, 1H), 3.12 - 2.99 (m, 2H), 2.39 (m, 1H), 2.22 (tdd, J = 12.4, 5.8, 1.3 Hz, 1H), 2.02 (m, 1H), 1.68 (m, 1H); 13C NMR (CDCl3, 125 MHZ) δ 164.8, 149.5, 139.5, 136.6, 131.0, 128.9, 128.7, 128.5, 127.8, 126.2, 117.1, 84.1, 62.7, 58.1, 42.2, 33.2, 21.5; IR: 2942, 1718 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C21H21ClNO2)+ 354.1261, found 354.1254. Crystal Structure CIF file available.
(E)-7-(4-cyanobenzylidene)-1-azabicyclo[3.2.1]octan-5-yl furan-2-carboxylate 10{2,10}
1H NMR (DMSO-d6, 500 MHZ) δ 7.98 (dd, J = 1.7, 0.8 Hz, 1H), 7.78 (d, J = 8.5 Hz, 2H), 7.49 (d, J = 8.5 Hz, 2H), 7.31 (dd, J = 3.5, 0.8 Hz, 1H), 6.70 (dd, J = 3.5, 1.7 Hz, 1H), 6.31 (s, 1H), 3.32 (d, J = 17.0 Hz, 1H), 3.21 (dd, J = 10.6, 2.3 Hz, 1H), 3.11 (d, 17.0 Hz, 1H), 3.05 – 3.00 (m, 3H), 2.21 (m, 1H), 2.14 (m, 1H), 1.83 (m, 1H), 1.59 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 157.1, 155.2, 147.7, 143.9, 141.7, 132.3, 128.1, 119.2, 118.8, 114.7, 112.4, 107.9, 83.9, 61.7, 57.7, 42.5, 32.6, 21.3; IR: 2946, 2223, 1721, 1603, 1301 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C20H19N2O3)+ 335.1390, found 335.1369.
(E)-7-(3-chlorobenzylidene)-1-azabicyclo[3.2.1]octan-5-yl cyclopentanecarboxylate 10{5,7}
1H NMR (DMSO-d6, 500 MHZ) δ 7.36 (t, J = 7.9 Hz, 1H), 7.32 (t, J = 1.7 Hz, 1H), 7.26 (d, J = 7.9 Hz, 1H), 7.22 (ddd, J = 7.9, 2.1, 0.9 Hz, 1H), 6.18 (t, J = 2.1 Hz, 1H), 3.20 (dt, J = 16.9, 1.9 Hz, 1H), 3.06 (dd, J = 10.6, 2.3 Hz, 1H), 2.96 – 2.84 (m, 4H), 2.72 (m, 1H), 2.09 (m, 1H), 2.00 (dt, J = 11.9, 6.0 Hz, 1H), 1.84 – 1.50 (m, 10H); 13C NMR (DMSO-d6, 125 MHz) δ 175.1, 152.9, 139.0, 133.2, 130.3, 127.0, 125.9, 125.6, 114.4, 82.6, 61.8, 57.6, 43.3, 42.1, 32.5, 29.5, 29.4, 25.4, 21.1; IR: 2947, 1730, 1662 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C20H25ClNO2)+ 346.1568, found 346.1550.
(E)-7-(4-methoxybenzylidene)-1-azabicyclo[3.2.1]octan-5-yl 4-fluorobenzoate 10{11,3}
1H NMR (DMSO-d6, 500 MHZ) δ 8.03 (m, 2H), 7.36 (t, J = 9.8 Hz, 2H), 7.25 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 6.16 (t, J = 2.0 Hz, 1H), 3.74 (s, 3H), 3.28 (d, J = 16.7 Hz, 1H), 3.19 (dd, J = 10.6, 2.1 Hz, 1H), 3.03 – 2.95 (m, 4H), 2.24 (m, 1H), 2.16 (dt, J = 12.0, 5.8 Hz, 1H), 1.85 (m, 1H), 1.58 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 165.2 (d, JC-F = 249.9 Hz), 164.1, 157.5, 148.3, 132.2 (d, JC-F = 9.5 Hz), 129.3, 128.7, 126.8 (d, JC-F = 2.8 Hz), 115.9 (d, JC-F = 21.9 Hz), 114.0, 84.0, 61.9, 57.5, 55.1, 41.9, 32.8, 21.3; IR: 2941, 1718, 1280 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C22H23FNO3)+ 368.1656, found 368.1629.
(E)-7-(3-(dimethylamino)benzylidene)-1-azabicyclo[3.2.1]octan-5-yl furan-2-carboxylate 10{2,6}
1H NMR (DMSO-d6, 500 MHZ) δ 7.97 (dd, J = 1.8, 0.9 Hz, 1H), 7.31 (dd, J = 3.6, 0.9 Hz, 1H), 7.14 (t, J = 7.9 Hz, 1H), 6.69 (dd, J = 3.6, 1.8 Hz, 1H), 6.64 (d, J = 7.9 Hz, 1H), 6.61 (m, 1H), 6.57 (dd, J = 8.1, 2.2, 1H), 6.15 (t, J = 2.0, 1H), 3.29 (d, J = 16.7 Hz, 1H), 3.16 (dd, J = 10.6, 2.2 Hz, 1H), 3.01 – 2.94 (m, 4H), 2.88 (s, 6H), 2.22 (m, 1H), 2.14 (dt, J = 12.2, 6.0 Hz, 1H), 1.84 (m, 1H), 1.58 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 157.1, 150.6, 149.9, 147.7, 144.0, 137.2, 128.9, 118.7, 116.7, 115.6, 112.4, 112.0, 110.5, 84.0, 61.8, 57.5, 42.1, 40.2, 32.8, 21.3; IR: 2943, 1714, 1298 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C21H25N2O3)+ 353.1860, found 353.1866.
(E)-7-(3-chlorobenzylidene)-1-azabicyclo[3.2.1]octan-5-yl isoxazole-5-carboxylate 10{8,7}
1H NMR (DMSO-d6, 500 MHZ) δ 8.85 (d, J = 1.9 Hz, 1H), 7.39 – 7.35 (m, 2H), 7.30 – 7.28 (m, 2H), 7.25 (ddd, J = 7.9, 2.0, 0.9 Hz, 1H), 6.24 (t, J = 2.1 Hz, 1H), 3.31 (dt, J = 16.9, 1.9 Hz, 1H), 3.21 (dd, J = 10.6, 2.2 Hz, 1H), 3.14 (d, J = 16.9 Hz, 1H), 3.06 (dd, J = 10.6, 3.0 Hz, 1H), 3.00 – 2.99 (m, 2H), 2.23 (m, 1H), 2.18 (dt, J = 11.8, 6.0 Hz, 1H), 1.86 (m, 1H), 1.59 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 159.2, 155.4, 152.2, 138.9, 133.3, 130.3, 127.1, 126.0, 125.8, 114.7, 109.9, 85.5, 61.4, 57.5, 41.6, 32.5, 21.3; IR: 2946, 1731, 1279 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C18H18ClN2O3)+ 345.1000, found 345.0998.
(E)-7-(4-cyanobenzylidene)-1-azabicyclo[3.2.1]octan-5-yl pivalate 10{7,10}
1H NMR (DMSO-d6, 500 MHZ) δ 7.78 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.4, 2H), 6.29 (t, J = 2.0 Hz, 1H), 3.24 (dt, J = 17.0, 1.9 Hz, 1H), 3.10 (dd, J = 10.6, 2.3 Hz, 1H), 3.02 – 2.88 (m, 4H), 2.08 (m, 1H), 2.00 (dt, J = 12.2, 5.6 Hz, 1H), 1.77 (m, 1H), 1.54 (m, 1H), 1.14 (s, 9H); 13C NMR (DMSO-d6, 125 MHz) δ 176.9, 155.5, 141.7, 132.4, 128.1, 119.2, 114.7, 107.8, 82.5, 61.8, 57.7, 42.5, 38.5, 32.3, 26.9, 21.2; IR: 2961, 2224, 1726 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C20H25N2O2)+ 325.1911, found 325.1887.
(E)-7-(4-cyanobenzylidene)-1-azabicyclo[3.2.1]octan-5-yl 4-chlorobenzoate 10{10,10}
1H NMR (DMSO-d6, 500 MHZ) δ 7.97 (d, J = 8.7 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.7 Hz, 2H), 7.49(d, J = 8.4 Hz, 2H), 6.32 (s, 1H), 3.34 (m, 1H), 3.24 (dd, J = 10.6, 2.2 Hz, 1H), 3.16 (d, J = 17.7 Hz, 1H), 3.08 (dd, J = 10.6, 2.8 Hz, 1H), 3.05 – 3.02 (m, 2H), 2.25 – 2.15 (m, 2H), 1.84 (m, 1H), 1.59 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 164.2, 155.3, 141.7, 138.4, 132.4, 131.2, 129.0, 128.9, 128.1, 119.2, 114.7, 107.9, 84.0, 61.7, 57.7, 42.5, 32.5, 21.3; IR: 2946, 2223, 1717 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C22H20ClN2O2)+ 379.1208, found 379.1209.
(E)-7-(pyridin-4-ylmethylene)-1-azabicyclo[3.2.1]octan-5-yl 4-chlorobenzoate 10{10,4}
1H NMR (DMSO-d6, 500 MHZ) δ 8.49 (d, J = 6.2 Hz, 2H), 7.97 (d, J = 8.7 Hz, 2H), 7.61 (d, J = 8.7 Hz, 2H), 7.28 (d, J = 6.2 Hz, 2H), 6.21 (s, 1H), 3.37 (dd, J = 16.6, 1.9 Hz, 1H), 3.25 (dd, J = 10.6, 2.3 Hz, 1H), 3.17 (d, J = 17.0 Hz, 1H), 3.08 (dd, J = 10.6, 2.8 Hz, 1H), 3.05 – 3.03 (m, 2H), 2.22 (m, 1H), 2.19 (dt, J = 11.7, 5.6 Hz, 1H), 1.85 (m, 1H), 1.59 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 164.2, 156.5, 149.7, 144.0, 138.4, 131.2, 129.0, 128.9, 122.0, 113.6, 83.9, 61.7, 57.8, 42.5, 32.5, 21.3; IR: 2944, 1719 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C20H20ClN2O2)+ 355.1208, found 355.1197.
(E)-7-(4-chlorobenzylidene)-1-azabicyclo[3.2.1]octan-5-yl 3-fluorobenzoate 10{12,2}
1H NMR (DMSO-d6, 500 MHZ) δ 7.81 (dt, J = 7.6, 1.2 Hz, 1H), 7.70 (ddd, J = 9.5, 2.6, 1.6 Hz, 1H), 7.59 (ddd, J = 13.9, 8.2, 5.8 Hz, 1H), 7.53 (ddt, J = 8.7, 2.6, 1.2 Hz, 1H), 7.39 (d, J = 8.5 Hz, 2H), 7.34 (d, J = 8.5 Hz, 2H) 6.22 (t, J = 2.1 Hz, 1H), 3.30 (m, 1H), 3.22 (dd, J = 10.6, 2.2 Hz, 1H), 3.08 – 3.04 (m, 2H), 3.00 – 2.95 (m, 2H), 2.25 – 2.15 (m, 2H), 1.89 – 1.81 (m, 1H), 1.61 – 1.57 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 163.9 (JCF = 2.5 Hz), 162.0 (d, JCF = 243.8 Hz), 151.7, 135.7, 132.5 (d, JCF = 7.5 Hz), 131.0 (d, JCF = 7.5 Hz); 130.3, 129.2, 128.4, 125.5 (d, JCF = 6.3 Hz), 120.5 (d, JCF = 21.1 Hz), 115.9 (d, JCF = 22.9 Hz), 114.6, 84.2, 61.7, 57.6, 42.0, 32.6, 21.3; IR: 2944, 1720 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C21H20ClFNO2)+ 372.1167, found 372.1146.
(E)-7-(3-methoxybenzylidene)-1-azabicyclo[3.2.1]octan-5-yl acetate 10{13,5}
1H NMR (DMSO-d6, 500 MHZ) δ 7.24 (t, J = 7.8 Hz, 1H), 6.87 (d, J = 7.8 Hz, 1H), 6.82 (m, 1H), 6.76 (m, 1H), 6.15 (t J = 2.2 Hz), 3.75 (s, 3H), 3.20 (dt, J = 16.8, 2.0 Hz, 1H), 3.04 (dd, J = 10.6, 2.3 Hz, 1H), 2.96 – 2.90 (m, 2H), 2.89 – 2.82 (m, 2H), 2.16 – 2.10 (m, 1H), 2.06 – 1.96 (m, 1H), 2.00 (s, 3H), 1.83 – 1.71 (m, 1H), 1.58 – 1.48 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 169.8, 159.3, 151.3, 138.1, 129.4, 119.9, 115.5, 113.1, 111.4, 82.8, 61.9, 57.5, 55.0, 42.1, 32.6, 21.4, 21.2; IR: 2942, 1735 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C17H22NO3)+ 288.1600, found 288.1593.
(E)-7-(pyridin-3-ylmethylene)-1-azabicyclo[3.2.1]octan-5-yl 4-fluorobenzoate 10{11,8}
1H NMR (DMSO-d6, 500 MHZ) δ 8.53 (d, J = 2.1 Hz, 1H), 8.37 (dd, J = 4.7, 1.5 Hz, 1H), 8.08 - 8.00 (m, 2H), 7.74 (dt, J = 8.0, 1.8 Hz, 1H), 7.36 (m, 3H), 6.24 (t, J = 2.0 Hz, 1H), 3.33 (m, 1H), 3.24 (dd, J = 10.6, 2.2 Hz, 1H), 3.15 - 2.99 (m, 4H), 2.28 - 2.13 (m, 2H), 1.95 - 1.76 (m, 1H), 1.67 - 1.52 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 166.2, 165.2 (d, JCF = 251.5 Hz), 164.1, 153.3, 148.8, 146.7, 133.9, 132.2 (d, JCF = 9.6 Hz), 126.7 (d, JCF = 2.8 Hz), 123.5, 115.9 (d, JCF = 22.1 Hz), 112.2, 83.8, 61.8, 57.6, 42.1, 32.6, 21.2; IR: 2947, 1717 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C20H20FN2O2)+ 339.1509, found 339.1483.
(E)-7-benzylidene-1-azabicyclo[3.2.1]octan-5-yl acetate 10{13,1}
1H NMR (DMSO-d6, 500 MHZ) δ 7.36 - 7.31 (m, 2H), 7.29 (m, 2H), 7.21 - 7.14 (m, 1H), 6.18 (t, J = 2.2 Hz, 1H), 3.20 (dt, J = 16.8, 2.1 Hz, 1H), 3.04 (dd, J = 10.6, 2.3 Hz, 1H), 2.97 - 2.81 (m, 4H), 2.18 - 2.10 (m, 1H), 2.06 - 1.96 (m, 1H), 2.00 (s, 3H), 1.84 - 1.70 (m, 1H), 1.53 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 169.8, 150.9, 136.7, 128.5, 127.5, 125.9, 115.6, 82.8, 61.9, 57.5, 42.0, 32.6, 21.4, 21.2; IR: 2942, 1735 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C16H19NO2)+ 258.1494, found 258.1464.
(E)-7-(pyridin-3-ylmethylene)-1-azabicyclo[3.2.1]octan-5-yl 4-chlorobenzoate 10{10,8}
1H NMR (DMSO-d6, 500 MHZ) δ 8.53 (d, J = 2.1 Hz, 1H), 8.37 (dd, J = 4.7, 1.5 Hz, 1H), 7.99 - 7.95 (m, 2H), 7.74 (dt, J = 8.0, 1.8 Hz, 1H), 7.64 - 7.59 (m, 2H), 7.37 (dd, J = 7.9, 4.7 Hz, 1H), 6.24 (s, 1H), 3.38 - 3.29 (m, 1H), 3.24 (dd, J = 10.6, 2.2 Hz, 1H), 3.15 - 2.99 (m, 4H), 2.28 - 2.14 (m, 2H), 1.94 - 1.80 (m, 1H), 1.65 - 1.56 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 164.2, 153.2, 148.8, 146.7, 138.4, 133.9, 132.6, 131.2, 128.9, 123.5, 112.3, 84.0, 61.8, 57.6, 42.1, 32.6, 21.2; IR: 2944, 1718 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C20H20ClN2O2)+355.1213, found 355.1216.
(E)-7-(3-chlorobenzylidene)-1-azabicyclo[3.2.1]octan-5-yl cyclohexanecarboxylate 10{4,7}
1H NMR (DMSO-d6, 500 MHZ) δ 7.36 (t, J = 7.9 Hz, 1H), 7.32 (t, J = 1.7 Hz, 1H), 7.26 (d, J = 7.9 Hz, 1H), 7.23 (ddd, J = 7.9, 2.1, 0.9 Hz, 1H), 6.19 (t, J = 2.1 Hz, 1H), 3.20 (dd, J = 10.3, 8.4 Hz, 1H), 3.07 (dd, J = 10.7, 2.2 Hz, 1H), 2.99 - 2.80 (m, 4H), 2.27 (tt, J = 10.7, 3.6 Hz, 1H), 2.08 (dd, J = 10.3, 5.6 Hz, 1H), 2.00 (td, J = 12.0, 6.1 Hz, 1H), 1.86 - 1.70 (m, 3H), 1.70 - 1.61 (m, 2H), 1.55 (m, 2H), 1.41 - 1.12 (m, 5H); 13C NMR (DMSO-d6, 125 MHz) δ 174.4, 152.9, 139.0, 133.2, 130.3, 127.0, 125.9, 125.6, 114.4, 82.5, 61.8, 57.6, 42.5, 42.1, 32.5, 28.6, 28.5, 25.3, 24.8, 24.7, 21.1; IR: 2934, 1731 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C21H27ClNO2)+ 360.1730, found 360.1711.
(E)-7-benzylidene-1-azabicyclo[3.2.1]octan-5-yl nicotinate 10{9,1}
1H NMR (DMSO-d6, 500 MHZ) δ 9.11 (dd, J = 2.2, 0.7 Hz, 1H), 8.82 (dd, J = 4.8, 1.8 Hz, 1H), 8.34 - 8.28 (m, 1H), 7.60 - 7.56 (m, 1H), 7.38 - 7.32 (m, 4H), 7.19 (m, 1H), 6.23 (t, J = 2.2 Hz, 1H), 3.36 - 3.31 (m, 1H), 3.23 (dd, J = 10.6, 2.2 Hz, 1H), 3.14 - 3.04 (m, 2H), 3.04 - 2.97 (m, 2H), 2.30 - 2.16 (m, 2H), 1.86 (m, 1H), 1.61 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 164.0, 153.7, 150.7, 150.2, 137.0, 136.7, 128.5, 127.5, 126.1, 126.0, 123.9, 115.7, 84.3, 61.7, 57.5, 42.0, 32.69, 21.28; IR: 2943, 1718 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C20H21N2O2)+ 321.1603, found 321.1580.
(E)-7-(4-methoxybenzylidene)-1-azabicyclo[3.2.1]octan-5-yl benzoate 10{1,3}
1H NMR (DMSO-d6, 500 MHZ) δ 8.00 - 7.94 (m, 2H), 7.70 - 7.63 (m, 1H), 7.56 - 7.50 (m, 2H), 7.25 (d, J = 8.8 Hz, 2H), 6.95 - 6.89 (m, 2H), 6.16 (t, J = 2.0 Hz, 1H), 3.75 (s, 3H), 3.29 (d, J = 16.7 Hz, 1H), 3.20 (dd, J = 10.5, 2.0 Hz, 1H), 3.07 - 2.92 (m, 4H), 2.29 - 2.21 (m, 1H), 2.16 (m, 1H), 1.91 - 1.78 (m, 1H), 1.58 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 165.0, 157.5, 148.4, 133.4, 130.2, 129.3, 128.7, 128.7, 115.2, 114.0, 83.8, 61.9, 57.5, 55.1, 41.9, 32.8, 21.3 (one overlap peak in aromatic region); IR: 2939, 1714 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C22H24NO3)+ 350.1756, found 350.1738.
(E)-7-(pyridin-4-ylmethylene)-1-azabicyclo[3.2.1]octan-5-yl furan-2-carboxylate 10{2,4}
1H NMR (DMSO-d6, 500 MHZ) δ 8.48 (dd, J = 4.6, 1.6 Hz, 2H), 7.98 (dd, J = 1.6, 0.8 Hz, 1H), 7.32 (dd, J = 3.5, 0.8 Hz, 1H), 7.27 (dd, J = 4.6, 1.6 Hz, 2H), 6.70 (dd, J = 3.5, 1.6 Hz, 1H), 6.20 (t, J = 2.0 Hz, 1H), 3.39 - 3.31 (m, 1H), 3.21 (dd, J = 10.6, 2.3 Hz, 1H), 3.12 (d, J = 17.6 Hz, 1H), 3.04 (m, 3H), 2.25 - 2.10 (m, 2H), 1.90 - 1.74 (m, 1H), 1.60 - 1.56 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 157.1, 156.4, 149.7, 147.7, 143.9, 122.0, 118.8, 113.6, 112.4, 83.9, 61.7, 57.7, 42.5, 32.6, 21.3 (one overlap in aromatic region); IR: 2945, 1718 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C18H19N2O3)+ 311.1396, found 311.1377.
(E)-7-(4-methoxybenzylidene)-1-azabicyclo[3.2.1]octan-5-yl butyrate 10{3,3}
1H NMR (DMSO-d6, 500 MHZ) δ 7.22 (d, J = 8.8 Hz, 2H), 6.96 - 6.84 (m, 2H), 6.12 (t, J = 2.1 Hz, 1H), 3.74 (s, 3H), 3.22 - 3.10 (m, 1H), 3.03 (dd, J = 10.6, 2.1 Hz, 1H), 2.96 - 2.81 (m, 3H), 2.77 (m, 1H), 2.26 (t, J = 7.3 Hz, 2H), 2.17 - 2.08 (m, 1H), 2.00 (td, J = 12.0, 5.7 Hz, 1H), 1.86 - 1.67 (m, 1H), 1.60 - 1.45 (m, 3H), 0.88 (t, J = 7.4 Hz, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 172.2, 157.5, 148.5, 129.3, 128.7, 115.1, 114.0, 82.8, 62.0, 57.4, 55.1, 41.9, 35.9, 32.7, 21.2, 18.0, 13.4; IR: 2958, 1733 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C19H26NO3)+ 316.1913, found 316.1898.
(E)-7-(pyridin-4-ylmethylene)-1-azabicyclo[3.2.1]octan-5-yl cyclohexanecarboxylate 10{4,4}
1H NMR (DMSO-d6, 500 MHZ) δ 8.47 (dd, J = 4.6, 1.6 Hz, 2H), 7.24 (dd, J = 4.7, 1.5 Hz, 2H), 6.17 (t, J = 2.0 Hz, 1H), 3.25 (dt, J = 17.0, 2.0 Hz, 1H), 3.09 (dd, J = 10.7, 2.4 Hz, 1H), 3.03 - 2.84 (m, 4H), 2.27 (tt, J = 10.7, 3.6 Hz, 1H), 2.15 - 2.04 (m, 1H), 2.04 - 1.96 (m, 1H), 1.86 - 1.69 (m, 3H), 1.66 (m, 2H), 1.55 (m, 2H), 1.43 - 1.08 (m, 5H); 13C NMR (DMSO-d6, 125 MHz) δ 174.4, 156.7, 149.7, 144.0, 122.0, 113.5, 82.4, 61.8, 57.7, 42.6, 42.4, 32.4, 28.6, 28.5, 25.3, 24.8, 24.7, 21.2; IR: 2932, 1731 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C20H27N2O2)+ 327.2073, found 327.2058.
(E)-7-(3-methoxybenzylidene)-1-azabicyclo[3.2.1]octan-5-yl cyclopentanecarboxylate 10{5,5}
1H NMR (DMSO-d6, 500 MHZ) δ 7.25 (t, J = 7.9 Hz, 1H), 6.88 (d, J = 7.8 Hz, 1H), 6.82 (m, 1H), 6.76 (m, 1H), 6.15 (t, J = 2.1 Hz, 1H), 3.74 (s, 3H), 3.22 - 3.19 (m, 1H), 3.05 (dd, J = 10.6, 2.2 Hz, 1H), 3.00 - 2.77 (m, 4H), 2.77 - 2.65 (m, 1H), 2.10 (m, 1H), 1.99 (m, 1H), 1.87 - 1.73 (m, 3H), 1.73 - 1.63 (m, 2H), 1.63 - 1.45 (m, 5H); 13C NMR (DMSO-d6, 125 MHz) δ 175.2, 159.3, 151.3, 138.1, 129.5, 119.8, 115.6, 113.3, 111.3, 82.6, 61.9, 57.6, 55.0, 43.3, 42.2, 32.5, 29.5, 29.4, 25.4, 21.1; IR: 2947, 1729 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C21H28NO3)+ 342.2069, found 342.2054.
(E)-7-(4-chlorobenzylidene)-1-azabicyclo[3.2.1]octan-5-yl pivalate 10{7,2}
1H NMR (DMSO-d6, 500 MHZ) δ 7.46 - 7.35 (m, 2H), 7.35 - 7.25 (m, 2H), 6.19 (t, J = 2.1 Hz, 1H), 3.19 (m, 1H), 3.07 (dd, J = 10.6, 2.2 Hz, 1H), 3.02 - 2.89 (m, 2H), 2.86 (dd, J = 10.6, 3.1 Hz, 1H), 2.80 (d, J = 16.9 Hz, 1H), 2.08 (m, 1H), 2.00 (m, 1H), 1.77 (m, 1H), 1.53 (m, 1H), 1.14 (s, 9H); 13C NMR (DMSO-d6, 125 MHz) δ 176.9, 151.9, 135.7, 130.2, 129.1, 128.4, 114.5, 82.5, 61.8, 57.5, 42.0, 38.5, 32.3, 26.9, 21.1; IR: 2957, 1727 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C19H25ClNO2)+ 334.1574, found 334.1571.
(E)-7-(3-methoxybenzylidene)-1-azabicyclo[3.2.1]octan-5-yl pivalate 10{7,5}
1H NMR (DMSO-d6, 500 MHZ) δ 7.26 (t, J = 7.9 Hz, 1H), 6.88 (d, J = 7.9 Hz, 1H), 6.82 (m, 1H), 6.77 (m, 1H), 6.16 (t, J = 2.1 Hz, 1H), 3.75 (s, 3H), 3.21 (m, 1H), 3.06 (dd, J = 10.6, 2.2 Hz, 1H), 2.93 (m, 2H), 2.85 (dd, J = 10.6, 3.1 Hz, 1H), 2.79 (d, J = 16.8 Hz, 1H), 2.09 (m, 1H), 1.99 (m, 1H), 1.77 (m, 1H), 1.53 (m, 1H), 1.14 (s, 9H); 13C NMR (DMSO-d6, 125 MHz) δ 176.9, 159.3, 151.2, 138.1, 129.5, 119.8, 115.6, 113.4, 111.2, 82.5, 61.8, 57.5, 55.0, 42.1, 38.5, 32.4, 26.9, 21.1; IR: 2957, 1726 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C20H28NO3)+ 330.2069, found 330.2070.
(E)-7-(pyridin-3-ylmethylene)-1-azabicyclo[3.2.1]octan-5-yl benzoate 10{1,8}
1H NMR (DMSO-d6, 500 MHZ) δ 8.53 (d, J = 2.1 Hz, 1H), 8.37 (dd, J = 4.7, 1.5 Hz, 1H), 7.97 (m, 2H), 7.74 (dt, J = 8.0, 1.8 Hz, 1H), 7.66 (m, 1H), 7.53 (m, 2H), 7.36 (dd, J = 8.0, 4.7 Hz, 1H), 6.24 (t, J = 2.0 Hz, 1H), 3.39 - 3.30 (m, 1H), 3.24 (dd, J = 10.6, 2.2 Hz, 1H), 3.16 - 2.95 (m, 4H), 2.32 - 2.12 (m, 2H), 1.87 (m, 1H), 1.60 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 165.0, 153.4, 148.8, 146.7, 133.8, 133.4, 132.6, 130.1, 129.3, 128.7, 123.5, 112.2, 83.7, 61.9, 57.6, 42.2, 32.6, 21.3; IR: 2945, 1716 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C20H21N2O2)+ 321.1603, found 321.1608.
(E)-7-(4-chlorobenzylidene)-1-azabicyclo[3.2.1]octan-5-yl butyrate 10{3,2}
1H NMR (DMSO-d6, 500 MHZ) δ 7.38 (m, 2H), 7.30 (m, 2H), 6.18 (t, J = 2.1 Hz, 1H), 3.19 (dt, J = 16.8, 2.2 Hz, 1H), 3.06 (dd, J = 10.6, 2.2 Hz, 1H), 3.01 - 2.76 (m, 4H), 2.26 (t, J = 7.3 Hz, 2H), 2.32 (m, 1H), 2.01 (m, 1H), 1.77 (m, 1H), 1.61 - 1.47 (m, 3H), 0.88 (t, J = 7.4 Hz, 3H); 13C NMR (DMSO-d6, 125 MHz) δ 172.2, 152.0, 135.7, 130.2, 129.1, 128.4, 114.5, 82.7, 61.9, 57.5, 42.1, 35.9, 32.6, 21.2, 18.0, 13.4; IR: 2939, 1732 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C18H23ClNO2)+ 320.1417, found 320.1422.
(E)-7-(3-(dimethylamino)benzylidene)-1-azabicyclo[3.2.1]octan-5-yl 3-fluorobenzoate 10{12,6}
1H NMR (DMSO-d6, 500 MHZ) δ 7.81 (dt, J = 7.6, 1.2 Hz, 1H), 7.71 (ddd, J = 9.5, 2.5, 1.5 Hz, 1H), 7.59 (m, 1H), 7.53 (m, 1H), 7.15 (t, J = 7.9 Hz, 1H), 6.66 (d, J = 7.7 Hz, 1H), 6.62 (m, 1H), 6.58 (dd, J = 8.1, 2.3 Hz, 1H), 6.17 (t, J = 2.0 Hz, 1H), 3.32 (m, 1H), 3.21 (dd, J = 10.6, 2.1 Hz, 1H), 3.10 - 2.93 (m, 4H), 2.89 (s, 6H), 2.31 - 2.11 (m, 2H), 1.86 (m, 1H), 1.59 (m, 1H); 13C NMR (DMSO-d6, 125 MHz) δ 163.9 (d, JCF = 3.0 Hz), 162.0 (d, JCF = 245.0 Hz), 150.6, 149.9, 137.2, 132.5 (d, JCF = 7.3 Hz), 131.0 (d, JCF = 8.0 Hz), 128.9, 125.5 (d, JCF = 2.7 Hz), 120.4 (d, JCF = 21.2 Hz), 116.7, 115.9 (d, JCF = 22.9 Hz), 115.5, 112.1, 110.5, 84.2, 61.7, 57.5, 42.0, 40.2, 32.6, 21.3; IR: 2943, 1718 cm−1; HRMS (ESI-TOF) m/z calculated for (M+H)+ (C23H26FN2O2)+ 381.1978, found 381.1954.
Supplementary Material
Acknowledgements
We gratefully acknowledge the assistance of Prof. Jeff Aubé CMLD Center Director, and Prof. Ryan Altman for kindly reviewing this manuscript. We acknowledge Mr. Patrick Porubsky, KU-CMLD Purification Chemist and Compound Curator as well as Mr. Jared Downard who carried out the pTLC final purification of library compounds indicated in Table 1. Financial support for the KU-CMLD was provided under NIGMS Grant #5P50GM069663. Support for the KU-NMR Center (JTD) was provided under Grant #NIH 1S10RR024664-01. Support for the KU-X-Ray facility (VWD) was provided under NSF-MRI Grant CHE-0923449.
Footnotes
Supporting Information. X-ray structure of compound 10{10,1}, details about library compound yield trend data for individual components, 1H and 13C NMR spectral data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected recent examples of skeletal diversification, see: Nielsen TE, Schreiber SL. Angew. Chem. Int. Ed. 2008;47(1):48–56. doi: 10.1002/anie.200703073. Kelly AR, Wei J, Kesavan S, Marie J-C, Windmon N, Young DW, Marcaurelle LA. Org. Lett. 2009;11(11):2257–2260. doi: 10.1021/ol900562u. Medeiros MR, Narayan RS, McDougal NT, Schaus SE, Porco JA., Jr Org. Lett. 2010;12(14):3222–3225. doi: 10.1021/ol101144k. Dandapani S, Lowe JT, Comer E, Marcaurelle LA. J. Org. Chem. 2011;76(19):8042–8048. doi: 10.1021/jo2011957. Painter TO, Wang L, Majumder S, Xie X-Q, Brummond KM. ACS Comb. Sci. 2011;13(2):166–174. doi: 10.1021/co100052s. Samarakoon TB, Loh JK, Rolfe A, Le LS, Yoon SY, Lushington GH, Hanson PR. Org. Lett. 2011;13(19):5148–5151. doi: 10.1021/ol201962n. Attanasi OA, Bartoccini S, Favi G, Giorgi G, Perrulli FR, Santeusanio S. J. Org. Chem. 2012;77(2):1161–1167. doi: 10.1021/jo2021949. Loh JK, Yoon SY, Samarakoon TB, Rolfe A, Porubsky P, Neuenswander B, Lushington GH, Hanson PR. Beilstein J. Org. Chem. 2012;8:1293–1302. doi: 10.3762/bjoc.8.147.
- 2.For selected recent examples, see: Kumagai N, Muncipinto G, Schreiber SL. Angew. Chem. Int. Ed. 2006;45(22):3635–3638. doi: 10.1002/anie.200600497. Wyatt EE, Fergus S, Galloway WRJD, Bender A, Fox DJ, Plowright AT, Jessiman AS, Welch M, Spring DR. Chem. Commun. 2006;(31):3296–3298. doi: 10.1039/b607710b. Sunderhaus JD, Dockendorff C, Martin SF. Org. Lett. 2007;9(21):4223–4226. doi: 10.1021/ol7018357. Muncipinto G, Kaya T, Wilson JA, Kumagai N, Clemons PA, Schreiber SL. Org. Lett. 2010;12(22):5230–5233. doi: 10.1021/ol102266j. Pizzirani D, Kaya T, Clemons PA, Schreiber SL. Org. Lett. 2010;12(12):2822–2825. doi: 10.1021/ol100914b. Murrison S, Maurya SK, Einzinger C, McKeever-Abbas B, Warriner S, Nelson A. Eur. J. Org. Chem. 2011;(12):2354–2359. O'Connor CJ, Beckmann HSG, Spring DR. Chem. Soc. Rev. 2012;41(12):4444–4456. doi: 10.1039/c2cs35023h.
- 3.For recent reviews on spirocycle synthesis, see: Kotha S, Deb AC, Lahiri K, Manivannan E. Synthesis. 2009;(2):165–193. Rosenberg S, Leino R. Synthesis. 2009;(16):2651–2673. Kang F-A, Sui Z. Tetrahedron Lett. 2011;52(32):4204–4206. Rios R. Chem. Soc. Rev. 2012;41(3):1060–1074. doi: 10.1039/c1cs15156h.
- 4.For selected recent examples of spirocycle syntheses, see: Badillo JJ, Arevalo GE, Fettinger JC, Franz AK. Org. Lett. 2011;13(3):418–421. doi: 10.1021/ol1027305. Dohi T, Nakae T, Ishikado Y, Kato D, Kita Y. Org. Biomol. Chem. 2011;9(20):6899–6902. doi: 10.1039/c1ob06199b. Li D-B, Rogers-Evans M, Carreira EM. Org. Lett. 2011;13(22):6134–6136. doi: 10.1021/ol2025313. Burkhard JA, Guerot C, Knust H, Carreira EM. Org. Lett. 2012;14(1):66–69. doi: 10.1021/ol2028459. Liu T-L, He Z-L, Tao H-Y, Wang C-J. Chem. Eur. J. 2012;18(26):8042–8046. doi: 10.1002/chem.201103876.
- 5.(a) Yao T, Yue D, Larock RC. J. Comb. Chem. 2005;7(6):809–812. doi: 10.1021/cc050062r. [DOI] [PubMed] [Google Scholar]; (b) Yao T, Yue D, Larock RC. J. Org. Chem. 2005;70(24):9985–9989. doi: 10.1021/jo0517038. [DOI] [PubMed] [Google Scholar]; (c) Yue D, Yao T, Larock RC. J. Org. Chem. 2005;70(25):10292–10296. doi: 10.1021/jo051299c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cho C-H, Neuenswander B, Lushington GH, Larock RC. J. Comb. Chem. 2008;10(6):941–947. doi: 10.1021/cc800120y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Okitsu T, Nakazawa D, Taniguchi R, Wada A. Org. Lett. 2008;10(21):4967–4970. doi: 10.1021/ol8020463. [DOI] [PubMed] [Google Scholar]; (f) Godoi B, Schumacher RF, Zeni G. Chem. Rev. 2011;111(4):2937–2980. doi: 10.1021/cr100214d. [DOI] [PubMed] [Google Scholar]
- 6.Wenkert E, Bookser BC, Arrhenius TS. J. Am. Chem. Soc. 1992;114(2):644–654. [Google Scholar]
- 7.Imamoto T, Kusumoto T, Tawarayama Y, Sugiura Y, Mita T, Hatanaka Y, Yokoyama M. J. Org. Chem. 1984;49(21):3904–3912. [Google Scholar]
- 8.Ma X, Wang J-X, Li S, Wang K-H, Huang D. Tetrahedron. 2009;65(42):8683–8689. [Google Scholar]
- 9.(a) Baldwin JE. J. Chem. Soc. Chem. Commun. 1976;(18):734–736. [Google Scholar]; (b) Baldwin JE, Cutting J, Dupont W, Kruse L, Silberman L, Thomas RC. J. Chem. Soc. Chem. Commun. 1976;(18):736–738. [Google Scholar]
- 10.Zhang X, Zhou Y, Wang H, Guo D, Ye D, Xu Y, Jiang H, Liu H. Adv. Synth. Catal. 2011;353(9):1429–1437. [Google Scholar]
- 11.Horvath L, Petz A, Kollar L. Lett. Org. Chem. 2010;7(1):54–60. [Google Scholar]
- 12.Ye L, Cui L, Zhang G, Zhang L. J. Am. Chem. Soc. 2010;132(10):3258–3259. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The syntheses of compounds 11 and 18b were previously reported via a three step route: Moskalenko AI, Belopukhov SL, Ivlev AA, Boev VI. Russ. J. Org. Chem. 2011;47(7):1073–1077. An online search for compound 18a returns a commercial supplier but not a published description of its preparation.
- 14.Tang F, Moeller KD. Tetrahedron. 2009;65(52):10863–10875. [Google Scholar]
- 15.Burchat AF, Chong JM, Nielsen N. J. Organomet. Chem. 1997;542:281–283. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.