

Abstract
Retroviral vectors based on foamy viruses (FV) are efficient gene delivery vehicles for therapeutic and research applications. While previous studies have shown that FV vectors transduce quiescent cell cultures more efficiently than oncoviral vectors, their specific cell cycle requirements have not been determined. Here we compare the transduction frequencies of FV vectors with those of onco- and lentiviral vectors in nondividing and dividing normal human fibroblasts by several methods. FV vectors transduced serum-deprived fibroblast cultures more efficiently than oncoretroviral vectors and at rates comparable to those of lentiviral vectors. However, in these cultures FV vectors only transduced a subpopulation of proliferating cells, as determined by bromodeoxyuridine staining for DNA synthesis. In contrast to lentiviral vectors, FV vectors were unable to transduce human fibroblasts arrested by aphidicolin (G1/S phase) or γ-irradiation (G2 phase), and a partial cell cycle that included mitosis but not DNA synthesis was required. We could not determine if mitosis facilitated nuclear entry of FV vectors, since cell-free vector preparations contained long terminal repeat circles, precluding their use as nuclear markers. In contrast to oncoviral vectors, both FV and lentiviral vectors efficiently transduced G0 fibroblasts that were later stimulated to divide. In the case of FV vectors, this was due to the persistence of a stable transduction intermediate in quiescent cells. Our findings support the use of FV vectors as a safe and effective alternative to lentiviral vectors for ex vivo transduction of stem cells that are quiescent during culture but divide following transplantation.
Retroviral vectors are efficient gene transfer vehicles that deliver transgenes by precise integration into the genome of host cells. This ability has been exploited for many gene delivery applications, including gene therapy. The foamy viruses (FVs) (Spumavirinae) comprise a distinct genus of retroviruses endemic in a number of mammalian species, including nonhuman primates, cats, and cows (24). Despite the name of the prototype spumaretrovirus, human FV, this isolate is now thought to be a chimpanzee virus (4, 21, 51, 61), and an extensive survey has demonstrated that FVs are not endemic in human populations (62). FV-based vectors have been developed (2, 19, 58, 60, 68, 69) that have several desirable properties. Regarding safety, FVs have not been associated with any pathology (36, 41), humans infected with FVs by exposure to nonhuman primates have not developed any FV-associated disease (20, 41), and human-to-human transmission has never been documented. Replication-defective FV vectors can be produced in the absence of detectable helper virus and at titers sufficient for ex vivo gene therapy applications (67, 69). In addition to safety considerations, FV vectors have a broad host and tissue tropism (22, 24, 43, 58, 78), and the virions are stable enough for concentration by centrifugation (71), increasing their utility. Finally, FVs have the largest genomes of all retroviruses, and FV vectors can efficiently package up to 9.2 kb of foreign genetic material (67), making them useful for large therapeutic transgenes. FV vectors efficiently transduce murine (71) and human (26, 33, 82) hematopoietic stem cells (HSCs), suggesting that they will be useful for the treatment of blood diseases by ex vivo gene therapy.
One limitation of retroviral vectors is their reduced transduction efficiency in quiescent cells. Early studies with oncoviruses defined blocks to infection prior to the completion of reverse transcription in quiescent cells (7, 13, 18, 70). In the case of murine leukemia virus (MLV), mitosis is required for infection, allowing preintegration complexes to enter the nucleus without traversing the nuclear membrane (35, 57). Consequently MLV-based vectors require cell proliferation for efficient transduction (45). Lentiviruses such as human immunodeficiency virus (HIV) can enter the nucleus via redundant mechanisms in the absence of mitosis (reviewed in reference 48) and infect nondividing macrophages and growth-arrested cells in vitro (34, 73). Thus, HIV vectors transduce quiescent cells more efficiently than MLV vectors (50, 55), albeit at lower frequencies than those obtained in dividing cells (52; also see below). In addition, low levels of deoxynucleoside triphosphates in quiescent cells can block infection by both MLV (16) and HIV (14, 30, 44, 52), and although the addition of exogenous nucleosides to quiescent T cells enhances reverse transcription of HIV, it does not rescue productive infection (31), demonstrating that there are other potential blocks to infection besides nuclear entry. These studies illustrate that both the activation state of the cell and progression through the cell cycle regulate the ability of retroviral vectors to efficiently deliver transgenes.
The replication strategy of FVs differs significantly from those of onco- and lentiviruses (32, 37, 76, 79) and may be relevant to their efficacy as vectors. Reverse transcription of FV genomes occurs in the cells producing virions rather than in infected target cells, so the functional genome is double-stranded DNA (47, 76, 79). Thus, FV vectors may be more stable than onco- or lentiviral vectors, which must complete reverse transcription in the host cell and depend on host cell nucleotide pools for this to occur. FV vectors transduce contact-inhibited, confluent cell cultures at higher rates than MLV vectors (58), but a small percentage of proliferating cells are present in these cultures, so the cell cycle requirements for transduction are not clear. Bieniasz et al. (3) demonstrated with wild-type FV that expression of viral proteins requires mitosis. However, this may not be true of vector-encoded transgenes, since simian FV vectors that expressed β-galactosidase (β-Gal) from an internal promoter reportedly transduced cells treated with aphidicolin and arrested at G1/S phase (42). Similarly, Saïb et al. (59) found that both one-long-terminal-repeat (LTR) and two-LTR circles were present in aphidicolin-treated cells, suggesting that FV preintegration complexes can enter the nucleus in the absence of mitosis. Given the importance of cell cycle effects on the use of FV vectors for gene therapy and other applications, we compared the transduction frequencies of onco-, lenti-, and spumavirus vectors containing the same transgene and promoter in cell cycle-arrested human fibroblasts and characterized the specific cell cycle requirements for FV vector transduction.
MATERIALS AND METHODS
Cells.
Human embryonic kidney 293T cells (11) and Phoenix-GP cells (29) were used to generate vector stocks. Rat 208F cells (54) and MHF2 normal human fibroblasts (obtained from the Coriell Institute for Medical Research [repository number GM05387]) were used for transduction assays. BHK-21 cells (40) and FAB cells (77) were used for wild-type FV production and titration, respectively. Human CD34+-mobilized peripheral blood cells were isolated from granulocyte colony-stimulating factor-primed healthy volunteers by leukapheresis under an Institutional Review Board-approved protocol and were purified to >98% purity by magnetic cell sorting (Miltenyi CliniMacs; Miltenyi Biotec).
Cells were cultured at 37°C in Dulbecco's modified Eagle's medium containing penicillin G sodium (100 IU/ml), streptomycin sulfate (100 IU/ml), amphotericin B (1.25 μg/ml), and 10% heat-inactivated fetal bovine serum unless otherwise stated. Human CD34+ cells were cultured in medium with 20% fetal bovine serum and stem cell factor (100 ng/ml), flt-3 ligand (100 ng/ml), and thrombopoietin (Peprotech).
Vector and virus constructs and preparations.
The FV vector plasmid pCGPMAPΔBel (69), infectious FV proviral plasmid pHSRV13 (39), and MLV vector plasmid pLAPSN (8) have been previously described. The HIV vector and helper plasmids pHR'CMVlacZ, pCMVΔR8.2, and pCMVΔR8.9 (49, 50, 83) were a gift of Didier Trono (University of Geneva, Geneva, Switzerland). The vesicular stomatitis virus glycoprotein (VSV-G)-expressing plasmid pCI-VSVG was kindly provided by Garry Nolan (Stanford University, Stanford, Calif.). The HIV vector plasmid pHR'cPPTMAP was constructed by replacing the cytomegalovirus promoter and lacZ gene of plasmid pHR'CMVlacZ with the MLV LTR promoter and alkaline phosphatase (AP) gene from pCGPMAPΔBel using standard cloning techniques. In addition, the central polypurine tract (cPPT) described by Follenzi et al. (12) was amplified by PCR from pCMVΔR8.2 and inserted immediately upstream of the internal promoter.
FV-based CGPMAPΔBel vector preparations were generated by transient cotransfection of 293T cells with pCGPMAPΔBel as previously described (68). The MLV-based LAPSN vectors were generated by transient cotransfection of Phoenix-GP packaging cells with pLAPSN and pCI-VSVG. HIV vector preparations were generated by three-plasmid cotransfection of 293T cells with pHR'cPPTMAP, pCI-VSVG, and either helper plasmid pCMVRΔ8.2 to generate HIV vectors with accessory genes (HIV+Acc), or helper plasmid pCMVRΔ8.9 to generate HIV vectors without the accessory genes vif, vpr, vpu, and nef (HIV−Acc). The MLV and HIV cotransfections were performed as described elsewhere for the FV vector preparations (68) by using 125 μg of helper and/or vector plasmid(s) and an additional 31.25 μg of pCI-VSVG per three 15-cm-diameter-plate transfections, and vectors were harvested at 48 h posttransfection. All vector preparations were concentrated 60-fold by ultracentrifugation as described previously (68), resuspended in medium with 0.5% serum, and stored at −80°C in the presence of 5% dimethyl sulfoxide. The wild-type FV preparation used for LTR circle analysis was prepared by transfection of BHK-21 cells with the infectious FV proviral plasmid pHSRV13 followed by three serial passages of filtered culture supernatants (filter pore size, 0.45 μm) on BHK-21 cells and concentration by ultracentrifugation. Wild-type FV was used fresh (not frozen).
All vector stocks were titrated by infecting dividing MHF2 cultures and staining for AP expression as described previously (68), except that for human CD34+ cells the solution changes were performed by centrifugation of the cells at 2,000 × g for 5 min and removal of the supernatant by aspiration (23). Polybrene (hexadimethrine bromide; Sigma) was added to a final concentration of 4 μg/ml immediately before MLV and HIV vector infection of human fibroblasts. Polybrene was not necessary for FV vector transduction (58) and did not affect the relative transduction frequencies of stationary and dividing cells (data not shown). Transduction of CD34+ cells was carried out on fibronectin fragment CH296 (Retronectin)-treated plates prepared as described previously (68). Wild-type FV was titrated on FAB indicator cells as described previously (68).
Determination of DNA content and BrdU incorporation.
For DNA content analysis, cells were stained with propidium iodide as described previously (80) and analyzed by flow cytometry using a Becton Dickinson Facstar. For bromodeoxyuridine (BrdU) analysis, cells were analyzed using the Cell Proliferation kit (catalog no. RPN20) from Amersham Biosciences (Piscataway, N.J.) according to the manufacturer's directions, except cells were fixed in acid-ethanol fixative (90% ethanol, 5% glacial acetic acid, and 5% H2O [vol/vol]) for 10 min, the primary antibody-nuclease treatment was reduced to 30 min, and 3,3′-diaminobenzidine was purchased from Sigma (catalog no. D5905) and used without substrate intensifier to yield a brown-orange nuclear stain that contrasted with the purple histochemical staining of AP. For BrdU-AP double staining experiments, staining for AP expression was performed after the BrdU staining as previously described (68). To calculate the number of unlabeled nuclei following BrdU staining, unstained cell nuclei were detected by incubating the cells in 4′,6-diamidino-2-phenylindole (DAPI) (2 μg/ml) in Dulbecco's phosphate-buffered saline at 4°C and viewing under UV illumination. The percentage of BrdU-stained cells in the culture was determined in >500 cells in multiple fields per sample.
Transduction of serum-deprived and dividing cultures.
To generate quiescent serum-deprived normal human fibroblast cultures, 2.5 × 105 MHF2 cells were plated per well in six-well tissue culture plates and cultured overnight. The following morning the medium was replaced and the cultures were grown an additional 4 days until they were confluent, at which time the concentration in serum was reduced to 0.5% for 4 days before transduction. For dividing MHF2 cultures, cells were arrested as explained above and then plated in six-well dishes at 2.5 × 105/ml per well 16 h before transduction. The MLV, FV, and HIV vectors were added to stationary MHF2 cells at multiplicities of infection (MOIs) of 0.01 and 0.001 and to dividing cells at a MOI of 0.001.
To determine whether transduced cells had undergone DNA synthesis using the BrdU-AP double stain, quiescent MHF2 cells were prepared as described above except that 24-well plates were used with 5 × 104 cells initially plated per well, or quiescent rat 208F cells were prepared by plating 5 × 104 cells per well in 24-well plates, replacing the media and culturing for 3 days, then reducing the concentration in serum to 5%, adding 10−6 M dexamethasone and culturing for an additional 7 days before vector addition (58). Arrested cells were transduced at MOIs of 0.1 (MHF2 cells) and 0.5 (208F cells) and then were double stained for AP expression and BrdU incorporation 2 days later.
Transduction of aphidicolin-arrested and irradiated cultures.
To compare the transduction of drug-arrested or γ-irradiated cultures, 1 × 105 MHF2 or CD34+ cells were plated per well in 12-well plates, then exposed to 40 Gy of γ-irradiation from a dual cesium-137 gamma source (GammaCell 40; AEC, Kanata, Ontario, Canada), treated with aphidicolin at 5 μg/ml the next day, or left untreated (dividing). Twenty-four hours later, vectors were added at MOIs of 0.01 for the fibroblasts and 0.05 for the CD34+ cells and stained for AP expression at 40 h postinfection.
Transduction during partial cell cycles.
To investigate the transduction of cells that did not traverse mitosis, 1.5 × 105 MHF2 cells were plated per well in 12-well plates and allowed to attach for 8 h, aphidicolin was then added at 5 μg/ml, and vector was added 20 h later at an MOI of 0.01. For the S-cycle treatment, cells were washed 4 h after vector administration and then cultured without aphidicolin. For the S-G2 γ treatment cells were washed 4 h after vector administration and then were cultured without aphidicolin and treated with 40 Gy of γ-irradiation 3 h later. For the S-G2 γ treatment, cells were treated with 40 Gy of γ-irradiation 1 h before vector administration and then were washed 4 h after vector administration and cultured without aphidicolin. For the arrested treatment, cells were washed in medium with aphidicolin (5 μg/ml) 4 h after vector administration and then cultured in medium with aphidicolin (5 μg/ml).
To investigate the transduction of cells that did not traverse S phase, MHF2 cells were synchronized by using the method of Tobey et al. (66). A total of 1.5 × 105 MHF2 cells were plated per well in 12-well plates and cultured in minimal essential medium α with 0.1% serum for 48 h, followed by culture for 24 h in medium with 10% serum and aphidicolin (5 μg/ml) to synchronize the cells at the G1/S border at time zero. For the S-cycle treatment vector was added at time zero and cells were cultured without aphidicolin until AP staining at 45 h postinfection. For the M-cycle treatment vector was added at 9 h and cells were cultured without aphidicolin until AP staining at 36 h postinfection. For the S-G2-M-G1 treatment vector was added at time zero and at 9 h the medium was changed to aphidicolin (5 μg/ml) until AP staining at 45 h postinfection. For the G2-M-G1 treatment, vector was added at 9 h and cells were cultured in aphidicolin (5 μg/ml) until AP staining at 36 h postinfection. For all treatments FV vector was added at MOIs of 0.001 and 0.01 μg/ml.
Transduction after resuming cell proliferation.
To assess the persistence of transduction intermediates in quiescent cultures, serum-deprived and dividing MHF2 cultures were prepared in six-well plates as described above. Two days after vector addition, cultures were washed with Dulbecco's phosphate-buffered saline and either replated (see below) or maintained in medium with 0.5% serum. At 2, 4, 6, or 10 days after vector addition, cells were dissociated with trypsin and replated into 10-cm-diameter dishes in medium with 10% serum, then stained for AP expression 2 days later.
Analysis of LTR circles.
Concentrated vector and virus preparations were extracted with phenol-chloroform and precipitated with ethanol. DNA representing either 106 transducing units of vector or 5 × 105 wild-type FV units was digested with restriction enzymes and analyzed by Southern blotting with a 686-nucleotide (nt) probe corresponding to nt 315 to 1000 of the FV Gag open reading frame.
RESULTS
Transduction of serum-deprived normal human fibroblasts.
To compare transduction properties of vectors derived from the onco-, lenti-, and spumaviruses, we used three vectors that express a placental AP reporter gene (Fig. 1). The MLV vector LAPSN and the HIV vector HR'cPPTMAP were pseudotyped with the VSV-G envelope, and the FV vector CGPMAPΔBel used the FV envelope. Both the VSV-G and FV envelopes allow for concentration of vector preparations by ultracentrifugation (6, 71). The HIV vector was prepared with (HIV+Acc) and without (HIV−Acc) the accessory genes vif, vpr, vpu, and nef. The cPPT and central termination sequence (CTS) were included in the HIV vector to enhance transduction of both dividing and quiescent cells (12, 81). The vectors used were all replication-incompetent, and did not express viral gene products. The AP gene was expressed from the MLV LTR promoter in all three vector types, which was located internally in the FV and HIV vectors. Transcription from the FV LTR does not occur in the vector provirus, since this requires the transcriptional transactivator Tas (56, 72), which is absent from the vector (69). Expression from the HIV LTR does not occur in the vector provirus in the absence of the transacting factors Tat and Rev (50).
FIG. 1.

Retroviral vectors used in this study. All vectors are shown in their integrated proviral form with the locations of LTRs, packaging signals (ψ), internal MLV LTR promoters (M), AP transgenes, cPPTs, and the Rev response element (RRE) indicated. Arrows indicate the start sites of AP transcription. The MLV vector LAPSN contains a downstream simian virus 40 promoter (S) and neomycin resistance gene (Neo) not relevant to the experiments described here.
Each vector type was used to transduce serum-deprived and dividing normal human fibroblast cultures. In these experiments, BrdU was added to duplicate cultures 16 h prior to vector addition and was present throughout the 48 h transduction period to determine the percentage of cells that underwent DNA synthesis just prior to, and during the transduction period. Within this 64 h period, 3.7% of cells in serum-deprived cultures had undergone DNA synthesis, compared to 89% in dividing cultures (data not shown). Propidium iodide staining confirmed that the serum-deprived cultures were quiescent, with 1.2% of cells containing an intermediate DNA content indicative of DNA synthesis (Fig. 2A). For each vector the transduction frequencies in stationary and dividing cultures were expressed as ratios (S/D ratio; Fig. 2B). The S/D ratios were compared using Student's t test and the ability of the MLV vector to transduce serum-deprived cultures was significantly lower than that of the FV vector (P = 0.025) and the HIV vector with accessory proteins (P = 0.024). The HIV accessory proteins did not significantly affect the S/D transduction ratio. These data demonstrate that all three vector types preferentially transduce dividing cultures, but the preference is most pronounced for MLV vectors. In addition, the S/D ratios were similar to what one might expect if only dividing cells present in the serum-deprived cultures had been transduced.
FIG. 2.

Transduction of serum-deprived cultures. Normal human fibroblasts were grown to confluence and serum starved for 4 days (Stationary) or were serum-starved for 3 days and then plated in medium with 10% serum 16 h prior to the addition of vector (Dividing). (A) Flow cytometry analysis of the DNA content by propidium iodide staining. (B) The number of AP+ foci in stationary cultures divided by the number of AP+ foci in dividing cultures (S/D ratio) is reported for three independent experiments with standard errors (error bars). HIV vector preparations were prepared with (HIV+Acc) and without (HIV−Acc) the accessory genes vif, vpr, vpu, and nef. An asterisk indicates significant differences (P < 0.05) compared to the MLV S/D ratio using Student's t test.
Single-cell analysis of serum-deprived fibroblast cultures transduced by FV vectors.
To determine which cells in the serum-deprived cultures had been transduced by FV vectors, BrdU was added 16 h before vector addition and maintained throughout the transduction period to detect cells that had undergone DNA synthesis (Fig. 3A to C). When these cultures were stained both for AP expression and BrdU incorporation, every AP+ fibroblast identified with a distinct nucleus in three separate experiments had also incorporated BrdU (654 of 654 cells). This experiment was also performed on quiescent rat 208F fibroblasts, which have a morphology that allows facile determination of BrdU and AP staining (Fig. 3D to F). In these cultures 10.2% of cells underwent S phase during the 64 h transduction period based on BrdU uptake (data not shown). Once again, every AP + 208F cell identified in three independent experiments had also incorporated BrdU (277 of 277 cells). Thus, the cells transduced by FV vectors in stationary cultures are actually a subset that underwent DNA synthesis and possibly other cell cycle phases.
FIG. 3.
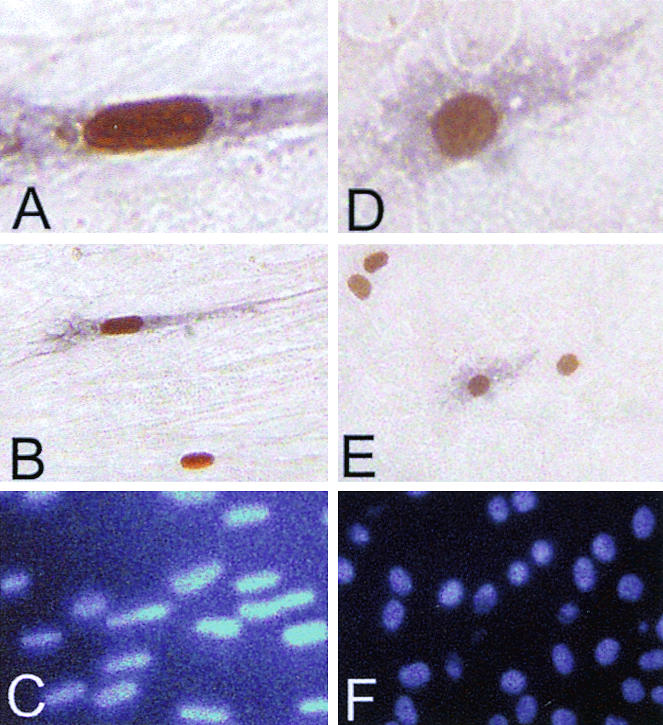
Transduction by FV vectors occurs only in cells that have undergone DNA synthesis. (A and D) High-power views (magnification, ×400) of a transduced human fibroblast and a rat 208F cell, respectively, after staining for BrdU incorporation as a marker of S phase (orange-brown nucleus) and for AP expression as a marker of transduction (purple cytoplasm). (B and E) Low-power views (magnification, ×100) of panels A and B, respectively. (C and F) Same fields as those in panels B and E, respectively, under UV illumination to detect all DAPI-stained nuclei.
FV vectors do not transduce G1/S or G2 arrested cells.
We also compared the transduction of cells arrested by means other than serum deprivation. Aphidicolin is a reversible inhibitor of DNA polymerases α, δ, and ɛ, arresting cells in S phase or at the G1/S boundary (25). γ-Irradiation damages DNA and causes cell cycle arrest in G2 (74, 75). Normal human fibroblasts were either treated with aphidicolin (5 μg/ml) or irradiated with 40 Gy of γ-irradiation 24 h before infection with vectors. Under these conditions transduction by FV or MLV vectors was almost completely eliminated, while the HIV vectors still transduced a significant proportion of cells (Table 1). We also investigated the transduction of human CD34+ cells, a population enriched for HSCs and an important target for gene therapy. As with fibroblasts, transduction of aphidicolin-arrested CD34+ cells by FV and MLV vectors was significantly reduced in comparison to that by HIV vectors (Table 1).
TABLE 1.
Transduction of G1/S and G2 arrested human fibroblasts and CD34+ cellsa
| Vector | % Transduction of dividing cells
|
||
|---|---|---|---|
| Aphidicolin (G1/S)
|
γ-Irradiation (G2) fibroblasts | ||
| Fibroblasts | CD34+ | ||
| FV | 0 | 0b | 0.2 |
| MLV | 0.05 | 0.07 | 0 |
| HIV−Acc | 45 | 130b | 60 |
| HIV+Acc | 43 | 89 | 61 |
Human fibroblasts or CD34+ cells were treated with either aphidicolin (5 μg/ml) or 40 Gy of γ-irradiation 24 h before exposure to the indicated vectors and were analyzed for AP expression 64 h later. Transduction frequencies are expressed as a percentage of those obtained with the same amount of vector in dividing cells. Aphidicolin treatment of CD34+ cells was toxic and decreased the number of cells ultimately available for analysis.
Fewer than 100 cells were enumerated after AP staining, with 0 of 75 (FV) and 5 of 88 (HIV−Acc) cells being AP positive.
Since HIV vectors have been shown to express transgenes as episomes (17, 50), we established that transduction was due to integrated vector proviruses by using an integrase-deficient HIV vector preparation, which transduced arrested fibroblast cultures at much lower rates (data not shown). These results confirm previous studies demonstrating that MLV vectors require mitosis for transduction (45, 57), while HIV vectors do not (50, 55), and they suggest that FV vectors may resemble MLV vectors in this regard.
A partial cell cycle that includes mitosis but not DNA synthesis is sufficient for transduction with FV vectors.
To determine if mitosis was required for transduction by FV vectors, human fibroblasts were allowed to progress through partial cell cycles that included or lacked mitosis during the transduction period (Fig. 4). Cells were first synchronized at the G1/S border with aphidicolin, exposed to vector, and then released from aphidicolin block and either allowed to continue through all phases of the cell cycle (S-cycle) or arrested at G2 prior to mitosis by irradiation before or after vector addition (S-G2 γ or γ S-G2). In addition, control cultures were maintained in aphidicolin throughout the transduction period (Arrested). In this assay both MLV and FV vectors were unable to transduce cells in cultures that did not pass through mitosis, despite the fact that DNA synthesis occurred in approximately one-third of the cells in both the S-G2 γ and γ S-G2 cultures as determined by BrdU incorporation. HIV vectors transduced cells at similar rates in all culture conditions.
FIG. 4.

Transduction by FV vectors requires passage through mitosis. (A) Aphidicolin-arrested human fibroblasts were exposed to retroviral vectors and either allowed to progress through the cell cycle following removal of aphidicolin (S-cycle), released from aphidicolin arrest and irradiated 3 h later (S-G2 γ), irradiated 1 h prior to infection and then released from aphidicolin arrest (γ S-G2), or left in the presence of aphidicolin (Arrested). Solid lines delineate phases of the cell cycle traversed by the cells during each treatment, and dotted lines delineate blockage of these phases during the transduction period. Aphidicolin treatment is depicted by a white bar. The durations of cell cycle phases during the experiment are indicated at the top. (B) The percent transduction relative to that obtained in the S-cycle treatment is shown for each treatment in panel A. DNA synthesis was monitored by BrdU staining, and the percentage of cells that underwent DNA synthesis for each treatment is given in brackets. Progression (+M) or lack of progression (−M) through mitosis is indicated.
We next tested whether FV vector transduction could occur in cells that traversed a partial cell cycle that included mitosis but not DNA synthesis (Fig. 5). For this experiment human fibroblasts were synchronized at the G1/S border with aphidicolin, then released from the cell cycle block and exposed to vector before or after the next S phase. Under these conditions 51% of the human fibroblasts progress through S phase within 9 h of removing aphidicolin as determined by BrdU incorporation (data not shown). For the S-G2-M-G1 and S-cycle treatments, vector was added at the time of release from aphidicolin block. For the G2-M-G1 and M-cycle treatments vector was added 9 h later when 13% of cells remained in S phase based on propidium iodide analysis (data not shown). Aphidicolin was added to the medium in the G2-M-G1 or S-G2-M-G1 treatments from 9 h after aphidicolin release to the end of the transduction period (45 h), so that no additional DNA synthesis would occur. BrdU staining showed that there was no DNA synthesis (0%) in these cultures following aphidicolin treatment. The S-cycle and M-cycle treatments were allowed to continue through subsequent phases of the cell cycle. Transduction by MLV and FV vectors occurred at about 10% of the rate for cycling cells (S-cycle) when cells traversed only the G2-M-G1 portion of the cell cycle. This demonstrates that a partial cell cycle that includes mitosis but not DNA synthesis is sufficient for transduction by FV vectors. Taken together the experiments in Fig. 4 and 5 show that mitosis is required and likely sufficient for transduction with FV vectors as G1 and G2 are not expected to play an essential role.
FIG. 5.

A partial cell cycle that includes mitosis but lacks S phase is sufficient for transduction by FV vectors. (A) Human fibroblasts were synchronized at the G1/S border and exposed to retroviral vectors at the beginning of S phase (S-cycle and S-G2-M-G1) or 9 h later (M-cycle and G2-M-G1). Aphidicolin was added 9 h after the initial withdrawal to prevent additional DNA synthesis in the S-G2-M-G1 and G2-M-G1 treatments. Solid lines delineate phases of the cell cycle traversed by the cells, and dotted lines delineate blockage of these phases during the transduction period. Aphidicolin treatment is depicted by a white bar. The durations of cell cycle phases during the experiment are indicated at the top. (B) The percent transduction relative to that obtained in the S-cycle treatment is shown for each treatment in panel A.
Although transduction was increased for all vectors in the S-G2-M-G1 and S-cycle treatment groups compared to that for those in G2-M-G1 and M-cycle treatment groups, respectively, this does not necessarily indicate that S phase increased transduction frequencies. It may simply reflect the earlier addition of vector in S-G2-M-G1 and S-cycle groups.
Both FV vector and wild-type FV preparations contain LTR circles.
We next sought to determine whether FV vectors were able to enter the nuclei of cell cycle-arrested cells or if this required mitosis, as in the case of MLV (57). Circular DNA molecules containing one or two LTRs are by-products of retroviral infection, and their presence correlates with nuclear entry, although the basis for this correlation has not been determined (9). LTR circles have been widely used as a marker for nuclear entry of many retroviruses including FVs (59). We detected LTR circles in aphidicolin-arrested cells infected with FV vectors, but they were also present in control samples spiked with vector stock during DNA isolation (data not shown), suggesting that these forms exist in vector particles before they infect cells. To confirm that LTR circles were present in vector preparations, DNA was purified directly from a vector stock by phenol-chloroform extraction and ethanol precipitation, digested with restriction enzymes, and analyzed by Southern blotting (Fig. 6). The FV vector preparation contained DNA fragments of the predicted size for one-LTR circles, in addition to the expected linear cDNA genomes. Similarly, DNA purified from a wild-type FV stock contained fragments consistent with both one- and two-LTR circles. Thus, LTR circles are present in FV vector and wild-type FV particles, and are not reliable markers of nuclear entry.
FIG. 6.

Southern blot analysis of LTR circles. Purified DNA from FV vector and wild-type FV stocks was digested with StuI, SwaI, EagI, PacI, and/or NheI as indicated and analyzed by Southern blotting. The vector preparation DNA was also digested with DpnI to remove transfected plasmid DNA. Diagrams of the predicted LTR-containing fragments (not to scale) are shown below the corresponding Southern blots with the predicted sizes of linear, one-LTR circle (1-LTRc), and two-LTR circle (2-LTRc) DNA fragments. The position of the FV probe used is shown with a black bar.
Persistence of transduction intermediates in quiescent cells.
We hypothesized that greater stability of FV vector genomes in quiescent cells could explain why FV vectors transduce serum-deprived cultures more efficiently than MLV vectors, despite the requirement for mitosis. The transduction of serum-deprived fibroblast cultures that were later stimulated to divide was examined to assess the stability of FV, MLV, and HIV transduction intermediates. We first confirmed by BrdU analysis that these cultures remain quiescent for up to 16 days, the longest time point tested, with 1.5% of the cells undergoing DNA synthesis during a 64-h period after 16 days of serum deprivation. Cultures were maintained under serum deprivation for 2, 4, 6, and 10 days after vector exposure and then were stimulated to divide by replating in larger wells and increasing the serum concentration from 0.5 to 10%. Forty-eight hours later the cells were stained for AP expression and compared with transduced dividing cultures (Fig. 7). In contrast to the MLV vector, the FV and HIV vectors efficiently transduced cells that were released from cell cycle arrest up to 10 days after vector exposure, the longest time point tested. Given that both FV and MLV vectors require mitosis for transduction, this suggests that a transduction intermediate of the FV vector is stable in quiescent cells for at least 10 days, while the analogous MLV vector intermediate survives less than 2 days.
FIG. 7.

Transduction after release from serum deprivation. Retroviral vectors were added to confluent, serum-deprived human fibroblasts, and 2, 4, 6, or 10 days later (days before release) the cells were stimulated to divide by replating in larger wells and increasing the concentration in serum from 0.5 to 10%. Forty-eight hours after replating, AP staining was performed; the number of AP+ foci in released cultures divided by the number of AP foci in dividing cultures (R/D ratio) is reported for three independent experiments with standard errors (error bars).
DISCUSSION
In this report we compared the cell cycle requirements for transduction by FV, MLV, and HIV vectors expressing the same transgene from the same promoter in normal human cells. Although prior studies have focused on the effects of cell proliferation on retroviral life cycles, ours is the first direct comparison of vectors from all three classes of retroviruses: oncoviruses, lentiviruses, and spumaviruses. We find that MLV vectors require mitosis for transduction and HIV vectors do not, consistent with earlier results (45, 50, 55, 57). FV vectors have distinct requirements for transduction in that a partial cell cycle including mitosis is ultimately required for transgene expression, but a transduction intermediate is stable and persists in nondividing cells.
Our finding that mitosis is a critical phase in the cell cycle for FV transduction confirms those of Bieniasz et al. (3), who showed that G1/S or G2 arrest blocked FV protein expression in infected cells. In addition, we can rule out indirect cell cycle effects that might act through the FV transactivator required for viral gene expression, since we measured transactivator-independent transgene expression from an internal promoter. We also confirm earlier observations that FV vectors infect quiescent cultures more efficiently than MLV vectors do (58) but show that this is due to transduction over time of the subpopulation of cycling cells present in the culture. Our results conflict with a prior study that concluded that simian FV vectors can efficiently transduce aphidicolin-treated cells (42); however, an alternative interpretation of the data may explain these differences. In the simian FV vector study, the β-Gal transgene was only detected in aphidicolin-treated cells when high vector MOIs were used, and stable expression was observed if arrested cells were allowed to divide after infection. Transfer of β-Gal protein present in the vector preparation could have produced β-Gal+ cells at high MOIs in a process of pseudotransduction (1, 38), and stable expression could have been due to transduction of cycling cells after the cultures were released from cell cycle arrest, as we observed in our experiments. Thus, while we cannot rule out strain-specific differences of the vector systems used, it is likely that simian FV vectors also require mitosis for transduction.
MLV vectors require mitosis for nuclear entry and thus transduction (57), while lentiviral vectors can enter using nuclear localization signals in nonmitotic cells (5, 53). Although our data demonstrate that mitosis is important for FV vector transduction, we could not attribute this to nuclear entry, since the LTR circles often used as nuclear markers were present in FV vector preparations. This is not unexpected as FV virions contain DNA genomes formed by reverse transcription before cellular entry (47, 79). One-LTR circles are thought to form by homologous recombination of retroviral cDNAs (15, 63) or from an intermediate in reverse transcription (10, 27), and two-LTR circles are thought to arise by direct ligation of cDNA ends (28, 64). Retroviruses are known to package cellular proteins nonspecifically, and DNA ligase activity has been demonstrated in virions (65), so these steps may occur in extracellular FV virions that contain cDNAs or before virions are released from cells. Given that a prior study detected FV LTR circles by PCR in aphidicolin-arrested but not serum-deprived cultures (59), this issue needs to be addressed further with additional experiments and by other methods. It is possible that FV vectors use distinct mechanisms to enter the nucleus, as both the viral genome and Gag proteins accumulate near the centrosome (59).
Although both FV and HIV vectors efficiently transduced serum-deprived fibroblasts that were later stimulated to divide, this presumably occurred by different mechanisms. In the case of FV vectors, transgene expression requires mitosis, so a stable intermediate must persist for at least 10 days until the majority of cells in the culture resume cell division. This is likely due to the fact that the FV vector completes reverse transcription before cellular entry, so unlike MLV vectors, the FV vector has no dependency on cellular nucleotide pools for DNA synthesis, and no potentially unstable RNA genome is exposed to the intracellular environment. In the case of HIV vectors, nuclear entry of the RNA genome in the absence of mitosis may provide improved stability relative to MLV vectors. However, despite nuclear entry, efficient transduction by HIV vectors did not occur in serum-deprived cultures, perhaps due to the inadequacy of nucleotide pools for reverse transcription. Similar results were obtained by Naldini et al. (50), who demonstrated that as fibroblasts were maintained under low-serum conditions for longer times and division rates presumably decreased, transduction frequencies were reduced.
Our findings have implications for the use of FV vectors in gene therapy applications. The persistence of FV vectors in G0 cells suggests that they are well-suited for the transduction of quiescent stem cells that divide later on and may explain why FV vectors transduce human HSCs efficiently when assayed in immunodeficient mice (26, 33, 46). However, FV vectors may not work well in nondividing cell types that remain quiescent, such as postmitotic myotubes or neurons. Our comparison of all three classes of retroviral vectors suggests that FV vectors may be the best choice for ex vivo stem cell gene therapy, as they were equal to HIV vectors and superior to MLV vectors in their ability to transduce transiently quiescent cells and they lack safety concerns associated with wild-type MLV and HIV. Further studies will be needed to directly compare all three classes of vectors in preclinical gene therapy experiments and to define the nature of the stable FV vector transduction intermediate in quiescent cells.
Acknowledgments
We thank Andrew Leavitt, Garry Nolan, and Didier Trono for providing plasmid DNAs and/or cell lines and Richard Newton and Roli Hirata for technical assistance. We thank Shelly Heimfeld for providing human CD34+ cells through the Cell Processing Core of the NHLBI Programs of Excellence in Gene Therapy.
This work was supported by grants from the National Institutes of Health.
REFERENCES
- 1.Alexander, I. E., D. W. Russell, and A. D. Miller. 1997. Transfer of contaminants in adeno-associated virus vector stocks can mimic transduction and lead to artifactual results. Hum. Gene Ther. 8:1911-1920. [DOI] [PubMed] [Google Scholar]
- 2.Bieniasz, P. D., O. Erlwein, A. Aguzzi, A. Rethwilm, and M. O. McClure. 1997. Gene transfer using replication-defective human foamy virus vectors. Virology 235:65-72. [DOI] [PubMed] [Google Scholar]
- 3.Bieniasz, P. D., R. A. Weiss, and M. O. McClure. 1995. Cell cycle dependence of foamy retrovirus infection. J. Virol. 69:7295-7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown, P., G. Nemo, and D. C. Gajdusek. 1978. Human foamy virus: further characterization, seroepidemiology, and relationship to chimpanzee foamy viruses. J. Infect. Dis. 137:421-427. [DOI] [PubMed] [Google Scholar]
- 5.Bukrinsky, M., S. Haggerty, M. Dempsey, N. Sharova, A. Adzhubel, L. Spitz, P. Lewis, D. Goldfarb, M. Emerman, and M. Stevenson. 1993. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature 365:666-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burns, J. C., T. Friedmann, W. Driever, M. Burrascano, and J. K. Yee. 1993. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA 90:8033-8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, I. S., and H. M. Temin. 1982. Establishment of infection by spleen necrosis virus: inhibition in stationary cells and the role of secondary infection. J. Virol. 41:183-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clowes, M. M., C. M. Lynch, A. D. Miller, D. G. Miller, W. R. Osborne, and A. W. Clowes. 1994. Long-term biological response of injured rat carotid artery seeded with smooth muscle cells expressing retrovirally introduced human genes. J. Clin. Investig. 93:644-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coffin, J. M., S. H. Hughes, and H. E. Varmus. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [PubMed]
- 10.Dina, D., and E. W. Benz, Jr. 1980. Structure of murine sarcoma virus DNA replicative intermediates synthesized in vitro. J. Virol. 33:377-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DuBridge, R. B., P. Tang, H. C. Hsia, P. M. Leong, J. H. Miller, and M. P. Calos. 1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 7:379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Follenzi, A., L. E. Ailles, S. Bakovic, M. Geuna, and L. Naldini. 2000. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat. Genet. 25:217-222. [DOI] [PubMed] [Google Scholar]
- 13.Fritsch, E. F., and H. M. Temin. 1977. Inhibition of viral DNA synthesis in stationary chicken embryo fibroblasts infected with avian retroviruses. J. Virol. 24:461-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao, W. Y., A. Cara, R. C. Gallo, and F. Lori. 1993. Low levels of deoxynucleotides in peripheral blood lymphocytes: a strategy to inhibit human immunodeficiency virus type 1 replication. Proc. Natl. Acad. Sci. USA 90:8925-8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilboa, E., S. Goff, A. Shields, F. Yoshimura, S. Mitra, and D. Baltimore. 1979. In vitro synthesis of a 9 kbp terminally redundant DNA carrying the infectivity of Moloney murine leukemia virus. Cell 16:863-874. [DOI] [PubMed] [Google Scholar]
- 16.Goulaouic, H., F. Subra, J. F. Mouscadet, S. Carteau, and C. Auclair. 1994. Exogenous nucleosides promote the completion of MoMLV DNA synthesis in G0-arrested Balb c/3T3 fibroblasts. Virology 200:87-97. [DOI] [PubMed] [Google Scholar]
- 17.Haas, D. L., S. S. Case, G. M. Crooks, and D. B. Kohn. 2000. Critical factors influencing stable transduction of human CD34(+) cells with HIV-1-derived lentiviral vectors. Mol. Ther. 2:71-80. [DOI] [PubMed] [Google Scholar]
- 18.Harel, J., E. Rassart, and P. Jolicoeur. 1981. Cell cycle dependence of synthesis of unintegrated viral DNA in mouse cells newly infected with murine leukemia virus. Virology 110:202-207. [DOI] [PubMed] [Google Scholar]
- 19.Heinkelein, M., M. Dressler, G. Jarmy, M. Rammling, H. Imrich, J. Thurow, D. Lindemann, and A. Rethwilm. 2002. Improved primate foamy virus vectors and packaging constructs. J. Virol. 76:3774-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heneine, W., W. M. Switzer, P. Sandstrom, J. Brown, S. Vedapuri, C. A. Schable, A. S. Khan, N. W. Lerche, M. Schweizer, D. Neumann-Haefelin, L. E. Chapman, and T. M. Folks. 1998. Identification of a human population infected with simian foamy viruses. Nat. Med. 4:403-407. [DOI] [PubMed] [Google Scholar]
- 21.Herchenroder, O., R. Renne, D. Loncar, E. K. Cobb, K. K. Murthy, J. Schneider, A. Mergia, and P. A. Luciw. 1994. Isolation, cloning, and sequencing of simian foamy viruses from chimpanzees (SFVcpz): high homology to human foamy virus (HFV). Virology 201:187-199. [DOI] [PubMed] [Google Scholar]
- 22.Hill, C. L., P. D. Bieniasz, and M. O. McClure. 1999. Properties of human foamy virus relevant to its development as a vector for gene therapy. J. Gen. Virol. 80:2003-2009. [DOI] [PubMed] [Google Scholar]
- 23.Hirata, R. K., A. D. Miller, R. G. Andrews, and D. W. Russell. 1996. Transduction of hematopoietic cells by foamy virus vectors. Blood 88:3654-3661. [PubMed] [Google Scholar]
- 24.Hooks, J. J., and C. J. Gibbs, Jr. 1975. The foamy viruses. Bacteriol. Rev. 39:169-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huberman, J. A. 1981. New views of the biochemistry of eucaryotic DNA replication revealed by aphidicolin, an unusual inhibitor of DNA polymerase alpha. Cell 23:647-648. [DOI] [PubMed] [Google Scholar]
- 26.Josephson, N. C., G. Vassilopoulos, G. Trobridge, G. V. Priestly, B. L. Wood, T. Papayannopoulou, and D. W. Russell. 2002. Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors. Proc. Natl. Acad. Sci. USA 99:8295-8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Junghans, R. P., L. R. Boone, and A. M. Skalka. 1982. Products of reverse transcription in avian retrovirus analyzed by electron microscopy. J. Virol. 43:544-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katz, R. A., C. A. Omer, J. H. Weis, S. A. Mitsialis, A. J. Faras, and R. V. Guntaka. 1982. Restriction endonuclease and nucleotide sequence analyses of molecularly cloned unintegrated avian tumor virus DNA: structure of large terminal repeats in circle junctions. J. Virol. 42:346-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinsella, T. M., and G. P. Nolan. 1996. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7:1405-1413. [DOI] [PubMed] [Google Scholar]
- 30.Kootstra, N. A., B. M. Zwart, and H. Schuitemaker. 2000. Diminished human immunodeficiency virus type 1 reverse transcription and nuclear transport in primary macrophages arrested in early G1 phase of the cell cycle. J. Virol. 74:1712-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korin, Y. D., and J. A. Zack. 1999. Nonproductive human immunodeficiency virus type 1 infection in nucleoside-treated G0 lymphocytes. J. Virol. 73:6526-6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lecellier, C. H., and A. Saib. 2000. Foamy viruses: between retroviruses and pararetroviruses. Virology 271:1-8. [DOI] [PubMed] [Google Scholar]
- 33.Leurs, C., M. Jansen, K. E. Pollok, M. Heinkelein, M. Schmidt, M. Wissler, D. Lindemann, C. Von Kalle, A. Rethwilm, D. A. Williams, and H. Hanenberg. 2003. Comparison of three retroviral vector systems for transduction of nonobese diabetic/severe combined immunodeficiency mice repopulating human CD34(+) cord blood cells. Hum. Gene Ther. 14:509-519. [DOI] [PubMed] [Google Scholar]
- 34.Lewis, P., M. Hensel, and M. Emerman. 1992. Human immunodeficiency virus infection of cells arrested in the cell cycle. EMBO J. 11:3053-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewis, P. F., and M. Emerman. 1994. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 68:510-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linial, M. 2000. Why aren't foamy viruses pathogenic? Trends Microbiol. 8:284-289. [DOI] [PubMed] [Google Scholar]
- 37.Linial, M. L. 1999. Foamy viruses are unconventional retroviruses. J. Virol. 73:1747-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu, M. L., B. L. Winther, and M. A. Kay. 1996. Pseudotransduction of hepatocytes by using concentrated pseudotyped vesicular stomatitis virus G glycoprotein (VSV-G)-Moloney murine leukemia virus-derived retrovirus vectors: comparison of VSV-G and amphotropic vectors for hepatic gene transfer. J. Virol. 70:2497-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lochelt, M., H. Zentgraf, and R. M. Flugel. 1991. Construction of an infectious DNA clone of the full-length human spumaretrovirus genome and mutagenesis of the bel 1 gene. Virology 184:43-54. [DOI] [PubMed] [Google Scholar]
- 40.Macpherson, I., and M. Stoker. 1962. Polyoma transformation of hamster cell clones: an investigation of genetic factors affecting cell competence. Virology 16:147-151. [DOI] [PubMed] [Google Scholar]
- 41.Meiering, C. D., and M. L. Linial. 2001. Historical perspective of foamy virus epidemiology and infection. Clin. Microbiol. Rev. 14:165-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mergia, A., S. Chari, D. L. Kolson, M. M. Goodenow, and T. Ciccarone. 2001. The efficiency of simian foamy virus vector type-1 (SFV-1) in nondividing cells and in human PBLs. Virology 280:243-252. [DOI] [PubMed] [Google Scholar]
- 43.Mergia, A., N. J. Leung, and J. Blackwell. 1996. Cell tropism of the simian foamy virus type 1 (SFV-1). J. Med. Primatol. 25:2-7. [DOI] [PubMed] [Google Scholar]
- 44.Meyerhans, A., J. P. Vartanian, C. Hultgren, U. Plikat, A. Karlsson, L. Wang, S. Eriksson, and S. Wain-Hobson. 1994. Restriction and enhancement of human immunodeficiency virus type 1 replication by modulation of intracellular deoxynucleoside triphosphate pools. J. Virol. 68:535-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller, D. G., M. A. Adam, and A. D. Miller. 1990. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell. Biol. 10:4239-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyoshi, H., K. A. Smith, D. E. Mosier, I. M. Verma, and B. E. Torbett. 1999. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors. Science 283:682-686. [DOI] [PubMed] [Google Scholar]
- 47.Moebes, A., J. Enssle, P. D. Bieniasz, M. Heinkelein, D. Lindemann, M. Bock, M. O. McClure, and A. Rethwilm. 1997. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J. Virol. 71:7305-7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naldini, L. 1998. Lentiviruses as gene transfer agents for delivery to non-dividing cells. Curr. Opin. Biotechnol. 9:457-463. [DOI] [PubMed] [Google Scholar]
- 49.Naldini, L., U. Blomer, F. Gage, D. Trono, and I. Verma. 1996. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 93:11382-11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naldini, L., U. Blomer, P. Gallay, D. Ory, R. Mulligan, F. H. Gage, I. M. Verma, and D. Trono. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263-267. [DOI] [PubMed] [Google Scholar]
- 51.Nemo, G. J., P. W. Brown, C. J. Gibbs, Jr., and D. C. Gajdusek. 1978. Antigenic relationship of human foamy virus to the simian foamy viruses. Infect. Immun. 20:69-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Brien, W. A., A. Namazi, H. Kalhor, S. H. Mao, J. A. Zack, and I. S. Chen. 1994. Kinetics of human immunodeficiency virus type 1 reverse transcription in blood mononuclear phagocytes are slowed by limitations of nucleotide precursors. J. Virol. 68:1258-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Popov, S., M. Rexach, G. Zybarth, N. Reiling, M. A. Lee, L. Ratner, C. M. Lane, M. S. Moore, G. Blobel, and M. Bukrinsky. 1998. Viral protein R regulates nuclear import of the HIV-1 pre-integration complex. EMBO J. 17:909-917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quade, K. 1979. Transformation of mammalian cells by avian myelocytomatosis virus and avian erythroblastosis virus. Virology 98:461-465. [DOI] [PubMed] [Google Scholar]
- 55.Reiser, J., G. Harmison, S. Kluepfel-Stahl, R. O. Brady, S. Karlsson, and M. Schubert. 1996. Transduction of nondividing cells using pseudotyped defective high-titer HIV type 1 particles. Proc. Natl. Acad. Sci. USA 93:15266-15271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rethwilm, A., O. Erlwein, G. Baunach, B. Maurer, and V. ter Meulen. 1991. The transcriptional transactivator of human foamy virus maps to the bel 1 genomic region. Proc. Natl. Acad. Sci. USA 88:941-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roe, T., T. C. Reynolds, G. Yu, and P. O. Brown. 1993. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 12:2099-2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Russell, D. W., and A. D. Miller. 1996. Foamy virus vectors. J. Virol. 70:217-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saïb, A., F. Puvion-Dutilleul, M. Schmid, J. Périès, and H. de Thé. 1997. Nuclear targeting of incoming human foamy virus Gag proteins involves a centriolar step. J. Virol. 71:1155-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmidt, M., and A. Rethwilm. 1995. Replicating foamy virus-based vectors directing high level expression of foreign genes. Virology 210:167-178. [DOI] [PubMed] [Google Scholar]
- 61.Schweizer, M., and D. Neumann-Haefelin. 1995. Phylogenetic analysis of primate foamy viruses by comparison of pol sequences. Virology 207:577-582. [DOI] [PubMed] [Google Scholar]
- 62.Schweizer, M., R. Turek, H. Hahn, A. Schliephake, K. O. Netzer, G. Eder, M. Reinhardt, A. Rethwilm, and D. Neumann-Haefelin. 1995. Markers of foamy virus infections in monkeys, apes, and accidentally infected humans: appropriate testing fails to confirm suspected foamy virus prevalence in humans. AIDS Res. Hum. Retrovir. 11:161-170. [DOI] [PubMed] [Google Scholar]
- 63.Shank, P. R., S. H. Hughes, H. J. Kung, J. E. Majors, N. Quintrell, R. V. Guntaka, J. M. Bishop, and H. E. Varmus. 1978. Mapping unintegrated avian sarcoma virus DNA: termini of linear DNA bear 300 nucleotides present once or twice in two species of circular DNA. Cell 15:1383-1395. [DOI] [PubMed] [Google Scholar]
- 64.Swanstrom, R., W. J. DeLorbe, J. M. Bishop, and H. E. Varmus. 1981. Nucleotide sequence of cloned unintegrated avian sarcoma virus DNA: viral DNA contains direct and inverted repeats similar to those in transposable elements. Proc. Natl. Acad. Sci. USA 78:124-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Temin, H. M., and D. Baltimore. 1972. RNA-directed DNA synthesis and RNA tumor viruses. Adv. Virus Res. 17:129-186. [DOI] [PubMed] [Google Scholar]
- 66.Tobey, R. A., J. G. Valdez, and H. A. Crissman. 1988. Synchronization of human diploid fibroblasts at multiple stages of the cell cycle. Exp. Cell Res. 179:400-416. [DOI] [PubMed] [Google Scholar]
- 67.Trobridge, G., N. Josephson, G. Vassilopoulos, J. Mac, and D. W. Russell. 2002. Improved foamy virus vectors with minimal viral sequences. Mol. Ther. 6:321-328. [DOI] [PubMed] [Google Scholar]
- 68.Trobridge, G., G. Vassilopoulous, N. Josephson, and D. W. Russell. 2002. Gene transfer with foamy virus vectors. Methods Enzymol. 346:628-648. [DOI] [PubMed] [Google Scholar]
- 69.Trobridge, G. D., and D. W. Russell. 1998. Helper-free foamy virus vectors. Hum. Gene Ther. 9:2517-2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Varmus, H. E., T. Padgett, S. Heasley, G. Simon, and J. M. Bishop. 1977. Cellular functions are required for the synthesis and integration of avian sarcoma virus-specific DNA. Cell 11:307-319. [DOI] [PubMed] [Google Scholar]
- 71.Vassilopoulos, G., G. Trobridge, N. C. Josephson, and D. W. Russell. 2001. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood 98:604-609. [DOI] [PubMed] [Google Scholar]
- 72.Venkatesh, L. K., P. A. Theodorakis, and G. Chinnadurai. 1991. Distinct cis-acting regions in U3 regulate trans-activation of the human spumaretrovirus long terminal repeat by the viral bel1 gene product. Nucleic Acids Res. 19:3661-3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weinberg, J. B., T. J. Matthews, B. R. Cullen, and M. H. Malim. 1991. Productive human immunodeficiency virus type 1 (HIV-1) infection of nonproliferating human monocytes. J. Exp. Med. 174:1477-1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whitmore, G., C. Stanners, J. Till, and S. Gulyas. 1961. Nucleic acid synthesis and the division cycle in X-irradiated L-strain mouse cells. Biochim. Biophys. Acta 47:66-77. [DOI] [PubMed] [Google Scholar]
- 75.Yamada, M., and T. Puck. 1961. Action of radiation on mammalian cells. IV. Reversible mitotic lag in the S3 Hela cell produced by low doses of X-rays. Proc. Natl. Acad. Sci. USA 47:1181-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu, S. F., D. N. Baldwin, S. R. Gwynn, S. Yendapalli, and M. L. Linial. 1996. Human foamy virus replication: a pathway distinct from that of retroviruses and hepadnaviruses. Science 271:1579-1582. [DOI] [PubMed] [Google Scholar]
- 77.Yu, S. F., and M. L. Linial. 1993. Analysis of the role of the bel and bet open reading frames of human foamy virus by using a new quantitative assay. J. Virol. 67:6618-6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu, S. F., J. Stone, and M. L. Linial. 1996. Productive persistent infection of hematopoietic cells by human foamy virus. J. Virol. 70:1250-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu, S. F., M. D. Sullivan, and M. L. Linial. 1999. Evidence that the human foamy virus genome is DNA. J. Virol. 73:1565-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zbigniew, Z., and G. Juan. 1997. DNA content measurement for DNA ploidy and cell cycle analysis, p. 7.5.1-7.5.24. In J. P. Robinson (ed.), Current protocols in cytometry. John Wiley & Sons, Inc., Hoboken, N.J. [DOI] [PubMed]
- 81.Zennou, V., C. Petit, D. Guetard, U. Nerhbass, L. Montagnier, and P. Charneau. 2000. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell 101:173-185. [DOI] [PubMed] [Google Scholar]
- 82.Zucali, J. R., T. Ciccarone, V. Kelley, J. Park, C. M. Johnson, and A. Mergia. 2002. Transduction of umbilical cord blood CD34+ NOD/SCID-repopulating cells by simian foamy virus type 1 (SFV-1) vector. Virology 302:229-235. [DOI] [PubMed] [Google Scholar]
- 83.Zufferey, R., D. Nagy, R. J. Mandel, L. Naldini, and D. Trono. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15:871-875. [DOI] [PubMed] [Google Scholar]