

Abstract
Prion diseases are fatal neurodegenerative disorders characterized by misfolding of the cellular prion protein (PrPc) into the disease-associated isoform (PrPSc) that has increased β-sheet content and partial resistance to proteolytic digestion. Prion diseases from different mammalian species have varying propensities for transmission upon exposure of an uninfected host to the infectious agent. Chronic Wasting Disease (CWD) is a highly transmissible prion disease that affects free ranging and farmed populations of cervids including deer, elk and moose, as well as other mammals in experimental settings. The molecular mechanisms allowing CWD to maintain comparatively high transmission rates have not been determined. Previous work has identified a unique structural feature in cervid PrP, a rigid loop between β-sheet 2 and α-helix 2 on the surface of the protein. This study was designed to test the hypothesis that the rigid loop has a direct influence on the misfolding process. The rigid loop was introduced into murine PrP as the result of two amino acid substitutions: S170N and N174T. Wild-type and rigid loop murine PrP were expressed in E. coli and purified. Misfolding propensity was compared for the two proteins using biochemical techniques and cell free misfolding and conversion systems. Murine PrP with a rigid loop misfolded in cell free systems with greater propensity than wild type murine PrP. In a lipid-based conversion assay, rigid loop PrP converted to a PK resistant, aggregated isoform at lower concentrations than wild-type PrP. Using both proteins as substrates in real time quaking-induced conversion, rigid loop PrP adopted a misfolded isoform more readily than wild type PrP. Taken together, these findings may help explain the high transmission rates observed for CWD within cervids.
Introduction
Prion diseases or transmissible spongiform encephalopathies (TSE's) are a class of fatal infectious neurodegenerative disorders [1]–[5]. Prion diseases are characterized by conformational conversion of the normal, cellular isoform of the prion protein (PrPc) into the disease-associated isoform (PrPSc) which has an increased β-sheet content and a partial resistance to proteolytic digestion [6]–[8]. Different mammalian hosts are affected by various prion diseases, including scrapie in sheep and goat, bovine spongiform encephalopathy (BSE) in cattle, chronic wasting disease (CWD) in cervids, and Creutzfeldt-Jakob disease (CJD), Gerstmann-Straussler Scheinker disease (GSS), fatal familial insomnia (FFI), and kuru in humans. Transmission propensity upon exposure of a naïve host to prions varies for TSE's from different species and is modulated by species barrier and prion strain issues [9]–[11]. Compared to prion diseases in other species, CWD is highly transmissible [12]–[19]. CWD infectivity is found in a variety of tissues, body fluids and excretions, and is shed into the environment [12]–[19]. CWD is also the only prion disease that affects both free ranging and farmed animals, including deer, elk and moose, as well as other mammals in experimental settings [18], [19]. CWD poses currently insurmountable challenges to wildlife management strategies aimed at controlling the disease [18], [19]. While there have been recent gains in understanding about routes of transmission and the epidemiology of CWD, there has been little investigation into the molecular and cellular factors that may help CWD maintain such high transmission rates. [9], [18], [19].
Although both primary sequences and three-dimensional structures of PrP as determined by NMR are highly conserved among mammals [20], previous NMR work has identified a unique secondary structural feature in cervid PrP and PrP from a few other mammalian species, a rigid loop between β-sheet 2 and α-helix 2 on the surface of the molecule [21]. Although other amino acid substitutions provide loop rigidity in species such as the wallaby, horse, pig and rabbit [22]–[27], two amino acid substitutions in cervid PrPc as compared to other species provides rigidity in the β-sheet 2 - α-helix 2 loop: S170N and N174T [21]. Introducing these two amino acid substitutions into murine PrPc stabilizes its loop structure. Transgenic mice over-expressing mouse PrPc with the two rigid loop associated substitutions developed spontaneous prion disease by an undetermined mechanism [28]. While the rigid loop region has been determined as an important determinant of the species barrier [14], [27], [29]–[31] and has been proposed to control susceptibility to prion disease [32], [33], its role in protein misfolding which is central to prion disease development has not been explored in full detail.
Our aim in this study was to test in vitro the hypothesis that the rigid loop has a direct influence on the prion protein misfolding process. Differences in misfolding with the introduction of the rigid loop could help to explain the high levels of transmission observed for CWD within cervid populations. Misfolding propensities for recombinant mouse PrP and a chimeric mouse/elk PrP with an introduced rigid loop were compared by biochemical techniques and cell free misfolding and conversion systems. We found that introducing the rigid loop increased the propensity of PrP to misfold in cell free systems.
Methods
Cloning and recombinant PrP expression
For expression of wild type mouse PrP (moPrPWT), PCR was used to amplify the coding sequence of mouse PrP (23–231). The murine prnp gene was inserted into pET30b. A plasmid for expression of mouse PrP with an introduced rigid loop (moPrPRL) was produced using the Quikchange site directed mutagenesis kit from Stratagene as per the manufacturer's recommendations. The S170N and N174T coding changes were introduced sequentially using the following primer pairs: GCCAGTGGATCAGTACAACAACCAGAACACCTTCG and CGAAGGTGTTCTGGTTGTTGTACTGATCCACTGGC (S170N), GTACAACAACCAGAACACCTTCGTGCACGACTGCG and CGCACGCGTGCACGAAGGTGTTCTGGTTGTTGTAC (N174T), and the moPrPWT pET30b plasmid as template. Rosetta (DE3) E. coli were used for expression of recombinant PrP (rPrP). Proteins were produced as previously described [34]. Briefly, cells were induced for 22–24 hours, using the Overnight Express Autoinduction System in LB media. Cells were harvested and subjected to two freeze/thaw cycles in liquid nitrogen before storage at −20°C. Before purification, inclusion bodies were washed twice with 0.1×BugBuster Master Mix (EMD), pelleted by centrifugation, and stored at −20°C. Alternatively, proteins were expressed in BL21 (DE3) E. coli using induction with 1 mM IPTG. Cells were lysed in lysis buffer (20 mM Tris HCl pH 7.4, 150 mM NaCl, 2 mM EDTA, 100 µg/mL lysozyme, 0.1% Triton X-100 and 10 mM β-mercapto-ethanol) and sonication (Misonix 3000 sonicator), followed by subsequent washes and pelleting by centrifugation in (20 mM Tris HCl pH 7.4, 2 M NaCl, 0.5% Triton X-100) containing 2, 4 and 6 M urea solutions. Purified inclusion bodies were then stored at -20°C. Proteins produced by this protocol were used for circular dichroism (CD) and for replications of the work presented here with the same conclusions. Both types of preparations were subjected to purification using the same protocol.
Protein purification
Proteins were purified as previous described [34]. Proteins denatured in 8 M guanidine-HCl were incubated with 20 g Qiagen Ni-NTA superflow resin pre-equilibrated in denaturing buffer (100 mM sodium phosphate, 10 mM Tris, 6 M guanidine-HCl, pH 8.0). The slurry was loaded into an XK-16 column (GE Healthcare) for purification using an AKTA Explorer chromatography system at room temperature. Recombinant PrP was refolded on the column using a linear gradient of denaturing buffer to refolding buffer (100 mM sodium phosphate, 10 mM Tris, pH 8.0). Protein was eluted from the column using a linear gradient from refolding buffer to elution buffer (100 mM sodium phosphate, 10 mM Tris, 500 mM imidazole, pH 5.8). Eluted protein was collected in 2 ml fractions and diluted into 2 ml dialysis buffer (10 mM sodium phosphate, pH 5.8). rPrP containing fractions were pooled, 0.22 µm filtered, and then dialyzed into dialysis buffer using 7,000 MWCO SnakeSkin dialysis tubing (Pierce-Thermo) overnight at 4°C. Samples were filtered again following dialysis. Protein concentration was determined using a BCA Protein Assay Kit (Pierce-Thermo).
Lipid vesicle preparation
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) was purchased from Avanti Polar Lipids and dried at room temperature under a stream of nitrogen. Lipids were processed and stored under a nitrogen atmosphere. Lipids were rehydrated to a concentration of 2.5 mg/mL in 20 mM Tris-HCl, pH 7.4 and allowed to rehydrate at room temperature for a minimum of one hour before sonication in a cup horn using a Misonix 3000 sonicator until clear.
Lipid based conversion assay, PK digestion and Western blot
For lipid based conversion assays, moPrPWT or moPrPRL were incubated with lipids or lipid free buffer (20 mM Tris-HCl, pH 7.4) at a ratio of 3 parts PrP to one part lipid or lipid free buffer in a volume to volume ratio for varying lengths of time. Proteinase K (PK) digestion was performed at a concentration of 5 µg/mL PK for 30 minutes at 37°C. The reaction was stopped using pefablock inhibitor and samples were centrifuged at 100,000×g for one hour (Beckman Optima TL ultra-centrifuge, TLA 45 rotor, 4°C). The resulting pellet was dissolved in 8 M urea and loading buffer. Samples not treated with PK were centrifuged as above. Both pellet fraction containing insoluble aggregates and the supernatant containing the soluble fraction of PrP were analyzed via SDS-PAGE and Western blot. The undigested pellet was dissolved in urea and loading buffer as above. All samples were boiled for five minutes previous to SDS-PAGE. Samples were run on 4–20% gradient gels from Pierce-Thermo and transferred to nitrocellulose membranes. Samples were probed with 4H11 anti-PrP monoclonal antibody at a concentration of 1∶5,000 for one hour. A goat, anti-mouse, alkaline phosphatase (AP) conjugated antibody from Promega was used as a secondary antibody. For visualization, BCIP/NBT from Pierce-Thermo was used as an AP substrate according to manufacturer's recommendations.
Circular dichroism
Using a Jasco 720 circular dichroism (CD) spectrometer, far-UV spectra were obtained for moPrPWT or moPrPRL. Lipid based conversion reactions and lipid free reactions were prepared as above. Measurements were obtained at a response time of 1 s, a resolution of 0.5 nm and standard sensitivity (100 mdeg). Ten scans were averaged for each protein and treatment. Readings were baseline subtracted, meaning that the background spectra from the buffer (buffer with or without lipid) was subtracted from the spectrum. Meaningful data could not be obtained below 200 nm due to high noise to signal ratio.
Real time quaking-induced conversion (RT-QuIC)
RT-QuIC was performed as previously described [34]. Briefly, each reaction containing 0.1 mg/ml rPrP substrate, 20 mM sodium phosphate (pH 6.9), 300 mM NaCl, 1 mM EDTA, and 10 µM Thioflavin T were prepared in quadruplicate in a 96 well optical bottom plate (Nalge Nunc International). 10% w/v brain homogenates were prepared from the brains of mock infected and terminally ill 22L mouse adapted scrapie infected C57Bl/6 mice, mock infected and CWD infected transgenic mice expressing deer PrPc (1536 Tg[CerPrP]) [35] and brain stem slices from uninfected white-tailed deer and white-tailed deer experimentally infected with CWD as described previously [36]. Ninety eight µL of substrate cocktail was seeded with 2 µl of brain homogenate serially diluted to various concentrations using 20 mM sodium phosphate (pH 6.9), 130 mM NaCl, 0.1% (w/v) SDS, 1X N2 Supplement (Invitrogen). The plate was sealed with Nunc Amplification Tape (Nalge Nunc International). Using a BMG Labtech FLUOstar Omega fluorescence plate reader pre-heated to 42°C, the reactions were subjected to a cycle of 1 min rest followed by 1 min shaking (700 rpm, double orbital) with fluorescence readings (450 nm excitation, 480 nm emission) every 15 minutes. This cycling was continued for 50 hours (200 readings). Data was plotted as the average of quadruplicate reactions using every third time point (45 min) with GraphPad Prism software. Time at beginning of conversion was determined as the time point at which fluorescence rose from baseline (17–20,000 relative fluorescence units) to above 24,000 fluorescence units. Time to start of conversion was analyzed statistically using GraphPad Prism software.
Ethics statement
Original animal work that produced brain samples from terminal ill CWD infected and uninfected transgenic mice was performed at Lab Animal Resources, Colorado State University. Brains were kindly provided by Dr. M. Zabel and Dr. G. Telling. Mice were bred and maintained in animal facilities accredited by the Association for Assessment and Accreditation of Lab Animal Care International, in accordance with protocols approved by the Institutional Animal Care and Use Committee at Colorado State University. Intracerebral inoculations were performed under Isoflurane anesthesia, and mice euthanized using CO2 inhalation followed by decapitation. All efforts were made to minimize suffering.
Results
moPrPRL converts to a lipid-induced, aggregated, proteinase K resistant isoform at lower concentrations than moPrPWT
It has been previously documented that anionic lipids can be used to stimulate spontaneous conversion and aggregation of rPrP expressed from a variety of mammalian genes in the absence of contact with infectious prions [37]–[40]. To investigate the role of the rigid loop on protein misfolding, we used a cell free system in which interactions with POPG lipid vesicles cause rPrP to undergo conformational changes to a PK resistant isoform. This method operates under physiological conditions in a simplified system and has been shown to produce misfolded rPrP with physical properties similar to naturally occurring prions [38]. Utilizing this method, we compared the propensity for misfolding in vitro in an unseeded manner for moPrPWT and moPrPRL. Instead of comparing cervid rPrP with rPrP from another species such as murine rPrP, we decided to introduce the rigid loop (RL) structure into mouse PrP in order to be able to reduce observed effects to the RL structure alone. It was found that moPrPRL converts at lower concentrations when incubated with POPG vesicles than moPrPWT.
By Western blot analysis, aggregated, PK resistant isoforms were detectible for both moPrPWT and moPrPRL after treatment with POPG lipid vesicles, even after an incubation of only five minutes in duration ( Fig. 1 E). PK resistant material is observed by 14.5, 15 and 24 kDa signals, consistent with previous findings [38]. While the 14.5 and 15 kDa bands represent PK resistant core particles, the presence of the 24 kDa signal may result from one of two possibilities. The first possibility relies on the observation that lipids induce aggregation of rPrP [40]. As aggregation may shield full length rPrP from digestion by PK, it is plausible that full length protein remains undigested under conditions of lipid induced aggregation. Another explanation is that small molecular weight, PK resistant fragments form dimers as has been observed for newly converted misfolded isomers produced using QuIC [41]. Importantly, in the absence of lipids, no PK resistant, insoluble PrP was detectible ( Fig. 1 A–D), nor was PK resistant PrP detectible in the soluble fraction.
Figure 1. moPrPRL converts to a PK resistant isoform at lower concentrations than moPrPWT.

moPrPWT and moPrPRL at three different concentrations were incubated in the presence (E–H) or absence (A–D) of POPG lipid vesicles. Samples were removed for proteolytic digestion by proteinase K (PK) followed by ultra-centrifugation and Western blot analysis at four different time points. Generation of protease resistant, insoluble PrP was compared over time between moPrPWT and moPrPRL. In POPG vesicle containing samples, generation of material that is both insoluble and resistant to PK digestion was detectible after only five minutes (E) as a 15 kDa PrP band (denoted by large arrow). A smaller molecular weight band of approximately 14.5 kDa (denoted by small arrow) was detectible by 24 hours (F). Conversion to a PK resistant, insoluble form at a concentration of 100 µg/mL is observed for only moPrPRL, consistently at all time periods analyzed (E–H, lanes 3 vs 6). Over time, PK treated, ultracentrifuged samples accumulated a 24 kDa band (denoted by double arrow) (E–H). This band may represent undigested rPrP or a dimer of smaller molecular weight, PK cleaved fragments. Generation of aggregated, PK resistant material in the absence of lipids was not detected for either moPrPWT or moPrPRL (A–D). PK resistant material was also undetectable in the soluble fraction (A–H). Data shown is representative of results from four independent experiments.
Previous studies have shown that POPG induced conversion is protein concentration dependent [38]. Therefore, we analyzed lipid based conversion at different rPrP concentrations. Differences were detected in amounts of aggregated, PK resistant material produced between the two proteins when reactions were assembled with decreasing protein concentrations ( Fig. 1 E–H). Using 125 and 100 µg/mL protein concentrations, moPrPWT only poorly produced PK resistant fragments as compared to moPrPRL.
While both proteins were shown to convert to a PK-resistant, aggregated conformation, the observation that moPrPRL converts at lower concentrations than moPrPWT provides evidence that the rigid loop enhances PrP misfolding.
Levels of lipid induced aggregated protein do not differ between moPrPWT and moPrPRL
Having seen that moPrPRL converts to a PK resistant, aggregated form in the presence of POPG vesicles at lower concentrations than moPrPWT, we decided to compare levels of aggregated proteins between the two molecules. Basing our hypothesis on the previously published observation that aggregation leads to misfolding [40], we assessed the possibility that moPrPRL undergoes lipid induced misfolding at lower concentrations because it has a greater tendency to aggregate than moPrPWT. To investigate this possibility, moPrPWT and moPrPRL samples incubated in the presence or absence of lipids were subjected to ultracentrifugation to separate aggregated from soluble, monomeric prion protein ( Fig. 2 ).
Figure 2. Altered lipid induced misfolding does not result from differences in aggregation between moPrPWT and moPrPRL.

Lipid- (left panels, A–C) and lipid+ reactions (right panels, D–F) containing 100 µg/mL of moPrPWT or moPrPRL were assembled such that all incubations were completed simultaneously. Untreated samples (A and D) were removed and the remaining sample was subjected to centrifugation to separate aggregated (C and F) from soluble PrP (B and E). Samples were then analyzed by Western blot analysis using mAb 4H11. Lipid- samples showed lower intensity than lipid+ samples despite equal starting concentration of protein reflecting that POPG lipid vesicles stabilize PrP at 37°C. moPrPWT and moPrPRL both showed equivalent amounts of soluble (B and E) and insoluble PrP (C and F), indicating that differences in lipid induced generation of PK resistant particles are obviously not the result of differing tendencies to aggregate between the two molecules.
After separation by ultra-centrifugation, differences were revealed between lipid containing reactions and reactions without lipids. When analyzed via Western blot, samples without lipids had consistently lower signal intensity than samples incubated in the presence of lipids, although both showed bands of the same molecular size (compare Fig. 2 A–C and Fig. 2 D–F). As conversion reactions with and without lipids had identical starting concentrations and were prepared from the same solutions, this finding suggests that rPrP is stabilized by the presence of POPG vesicles at 37°C. While differences were observed for lipid containing reactions and reactions without lipids, no differences were observed in amounts of soluble or aggregated protein between moPrPWT and moPrPRL conversion reactions, whether in the presence of lipids ( Fig. 2 E–F) or absence of lipids ( Fig. 2 B–C).
As levels of aggregation were comparable between moPrPWT and moPrPRL for a given treatment, we conclude that differences in aggregation are not the likely cause of differences observed in lipid based conversion between the two proteins.
Lipid induced generation of PK-resistant, aggregated rPrP is accompanied by a shift in secondary structure toward increased β-sheet content
After analysis of lipid induced aggregation, we next verified that generation of PK resistant, aggregated rPrP is accompanied by a structural change indicative of protein misfolding. Previous studies have shown that association of rPrP with POPG vesicles induces a structural change from an α-helical conformation to a structure with increased β-sheet content [38]–[40]. To ensure that our experimental protocol produces PK resistant, aggregated rPrP with an altered conformation, lipid induced changes in secondary structure were investigated by far-UV circular dichroism (CD).
In analyzing secondary structure via CD, we found that both proteins shift from an α-helix enriched conformation in the absence of lipids to a conformation enriched in β-sheet content in the presence of lipids ( Fig. 3 ). The spectra indicative of a conformation enriched in α-helices observed for both moPrPWT and moPrPRL without lipids are consistent with previous spectroscopic characterizations of rPrP [38], [42]. Also the shift in spectra observed for both molecules when in contact with POPG vesicles indicative of an increase in β-sheet content was as expected [38]–[40]. However, moPrPWT and moPrPRL did not exhibit identical spectra after lipid induced structural conversion.
Figure 3. Generation of lipid induced, PK-resistant isoform is accompanied by a structural change increasing β-sheet content.
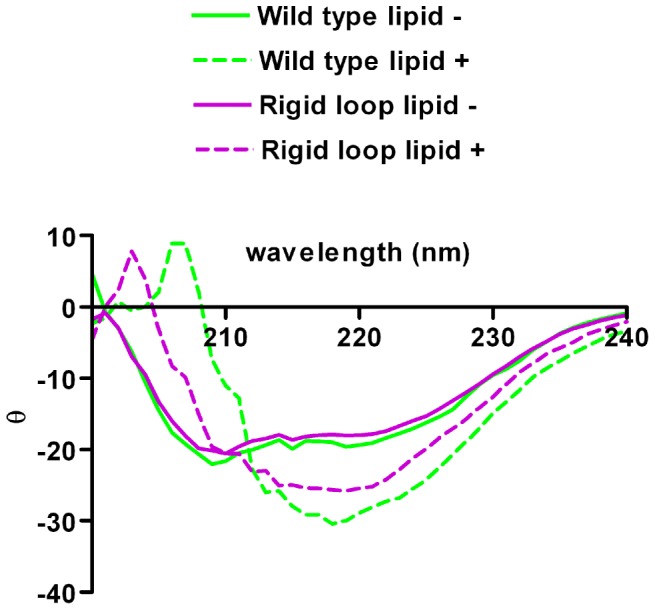
Differences in secondary structure between moPrPWT and moPrPRL are not detectible by circular dichroism (CD) as shown by their nearly identical CD spectra (solid lines). Upon addition of POPG lipid vesicles, the secondary structure of both moPrPWT and moPrPRL changed to reflect an increase in β-sheet content (dashed lines). In the presence of lipids, moPrPWT and moPrPRL do not exhibit identical CD spectra (dashed lines), possibly indicating differences in lipid induced secondary structural changes between these molecules.
Although the details of the different conformational state of moPrPWT and moPrPRL in the presence of lipids cannot be fully explored in the absence of higher resolution structural data, it implies that the rigid loop structural feature influences the outcome of lipid induced changes in secondary structure.
Introduction of the rigid loop to moPrPWT increases the speed at which it converts to a Thioflavin-T positive isoform in RT-QuIC
As it was found in unseeded conversion assays that introduction of the rigid loop increases the propensity of moPrPWT to misfold, we next compared seeded conversion of moPrPWT and moPrPRL via RT-QuIC. Consistent with an increased propensity for misfolding seen in lipid induced, unseeded misfolding, moPrPRL converted earlier than moPrPWT to an isoform that binds Thioflavin-T, a dye that emits fluorescence after binding to amyloid-like structures.
RT-QuIC was developed as a highly sensitive assay for detection of infectious prions [34]. In this assay, prion sources of unknown titers are used to seed conversion of rPrP into a Thioflavin-T positive isoform through cycles of quaking and incubation [34]. Rather than using RT-QuIC to detect seeding differences between two sources of potential infectivity, RT-QuIC was utilized in this study to detect differences in misfolding between two different rPrP substrates when seeded with the same infectious prions. Generation of misfolded PrP is considered to be most efficient when there is a high level of primary sequence homology between infecting prions and the native PrP being recruited into a misfolded conformation. 22 L prions used here and moPrPWT have identical primary amino acid sequences while moPrPRL differs from the 22 L seed by two residues, S170N and N174T. Although moPrPWT has greater sequence homology with 22 L prions, moPrPRL converted to a Thioflavin-T positive isoform quicker than moPrPWT when RT-QuIC reactions were seeded with brain homogenate containing 22 L mouse adapted scrapie prions ( Fig. 4 ). Notably, faster misfolding was also observed for moPrPRL when QuIC reactions were seeded with lower seed dilutions of 22 L and when reactions were seeded with other prion strains including CWD from cervidized transgenic mice ( Fig.5 ) and CWD from white-tailed deer ( Fig. 6 ).
Figure 4. Generation of a Thioflavin T positive isoform via RT-QuIC occurs faster for moPrPRL than moPrPWT.
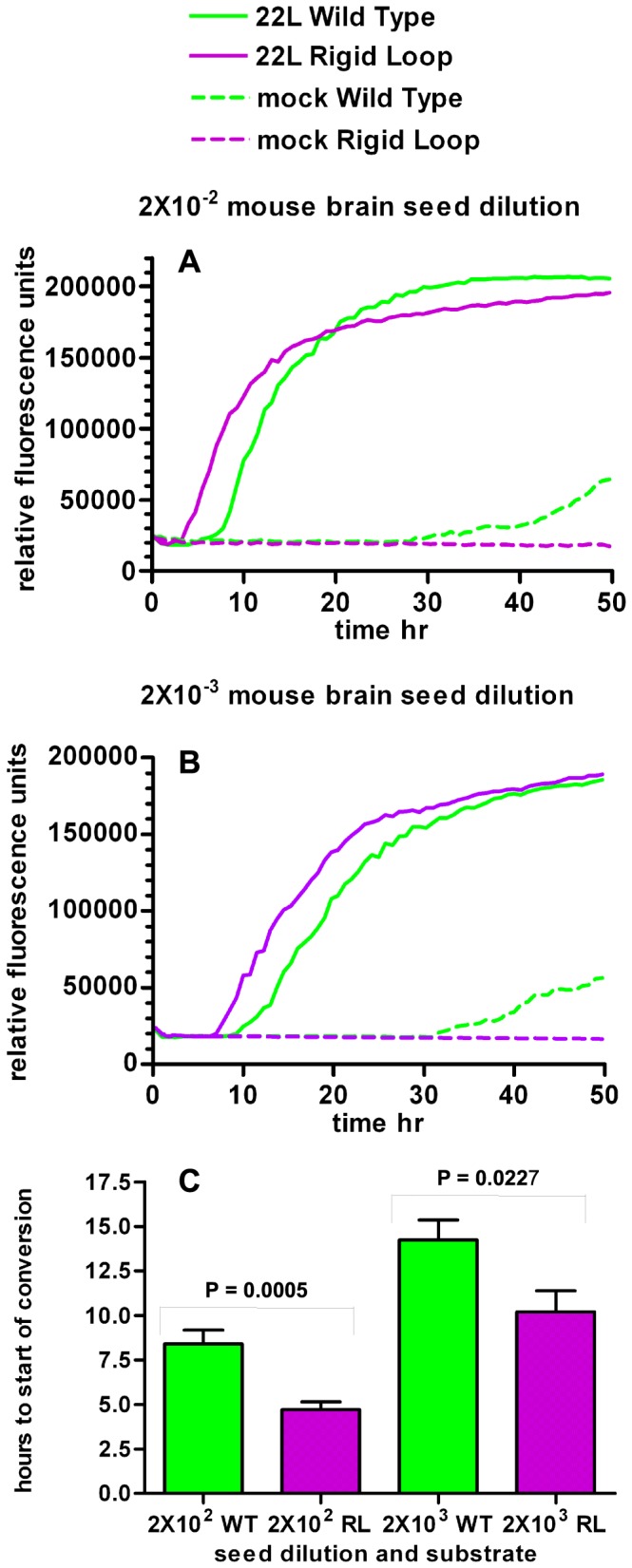
RT-QuIC reactions containing moPrPWT (green lines) and moPrPRL (purples lines) were seeded with brain homogenate from terminally ill 22 L prion infected mice (solid lines) or brain homogenate from mock infected controls (dashed lines) at a dilution of 2×10−2 (A) or 2×10−3 (B). Relative fluorescence over time was averaged for quadruplicate reactions. Time at beginning of conversion determined as the time point at which fluorescence rose from baseline to above 24,000 fluorescence units was compared for both dilutions of 22 L prion seed using GraphPad Prism software (shown in hours; data from 3 independent experiments) (C). Reactions with moPrPRL as a substrate exhibited earlier conversion to a Thioflavin T positive amyloid structure than reactions with moPrPWT substrate.
Figure 5. moPrPRL converts faster than moPrPWT in RT-QuIC reactions seeded with CWD prions from transgenic mice.
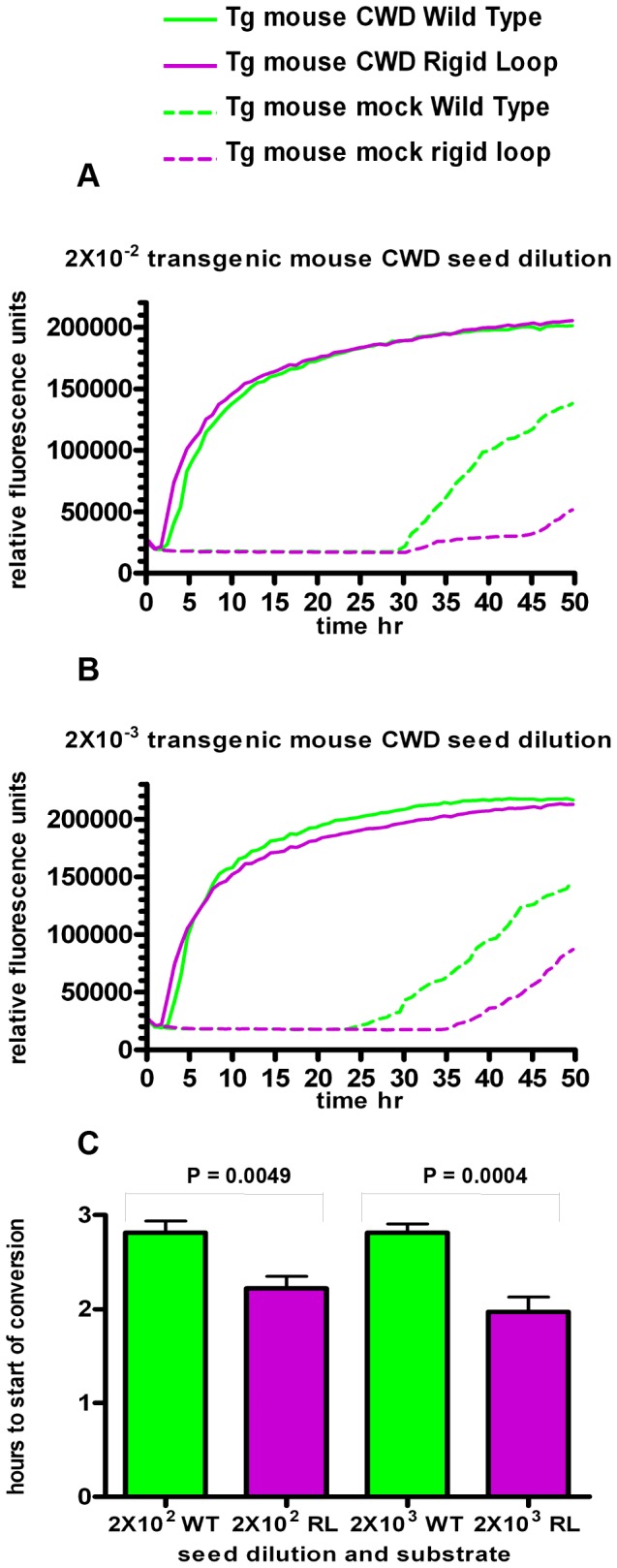
RT-QuIC reactions containing moPrPWT (green lines) and moPrPRL (purples lines) were seeded with brain homogenate from terminally ill CWD prion infected transgenic mice expressing deer PrPc (solid lines) or brain homogenate from mock infected control mice (dashed lines) at a dilution of 2×10−2 (A) or 2×10−3 (B). Data shown is the average of relative fluorescence over time for quadruplicate samples from two independent experiments. Reactions with moPrPRL as a substrate exhibited earlier conversion when seeded with mouse CWD prions than reactions with moPrPWT substrate (C).
Figure 6. moPrPRL converts faster than moPrPWT in RT-QuIC reactions seeded with CWD prions from deer.
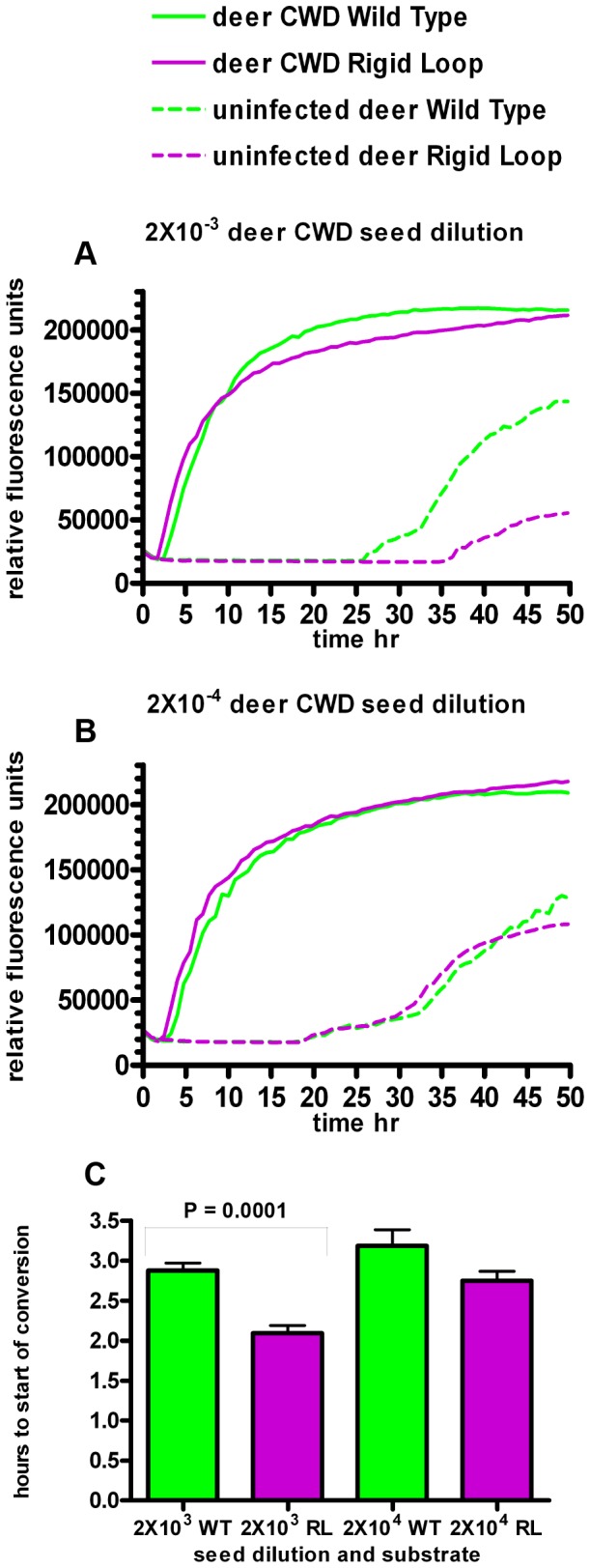
RT-QuIC reactions containing moPrPWT (green lines) and moPrPRL (purples lines) were seeded with brain homogenate from terminally ill CWD prion infected white-tailed deer (solid lines) or brain homogenate from uninfected controls (dashed lines) at a dilution of 2×10−3 (A) or 2×10−4 (B). Reactions were prepared in quadruplicate and average fluorescence over time was averaged for two independent experiments (C). Reactions with moPrPRL as a substrate exhibited earlier conversion than reactions with moPrPWT substrate when seeded with CWD prions from the brain of an infected white-tailed deer.
In summary, introduction of the rigid loop results in earlier conversion of recombinant mouse PrP in real time quaking-induced conversion.
Discussion
This study was designed to assess the influence of the rigid, β-sheet 2 - α-helix 2 loop from cervid PrP on prion protein misfolding. Using cell free conversion techniques, a key difference between moPrPWT and moPrPWT with an introduced rigid loop (moPrPRL) was identified. moPrPRL was shown to convert to a misfolded isoform in cell free systems more readily than moPrPWT. Both unseeded conversion via association with POPG lipid vesicles and seeded conversion using RT-QuIC were utilized in this study. The use of RT-QuIC to explore differences in conversion dynamics between two PrP variants is a novel application for QuIC which was developed originally as a diagnostic technique. Chronic wasting disease is highly transmissible among cervids [12]–[19] and the finding that introduction of the rigid loop increases the propensity by which moPrPWT misfolds could be relevant to the high transmission and shedding rates observed for CWD.
In investigating the role of the rigid loop on prion protein misfolding, we opted to study murine PrP with an introduced rigid loop rather than wild type cervid PrP. Comparative analysis of moPrPWT and moPrPRL allowed us to directly draw conclusions on the influence of the rigid loop on PrP misfolding without the need to detangle it from the influence of other amino acid variations observed for cervid PrP and species barrier issues. Importantly, the use of moPrPRL allowed the use of moPrPWT as a control. This study would have lacked this critical control if the experiments were performed with cervid PrP. As such, we were able to determine that the introduction of the rigid loop increases the propensity for moPrPWT to misfold in both seeded and unseeded conversion reactions.
Although both the S170N and N174T substitutions are required for full stabilization of the β-sheet 2 - α-helix 2 loop region, there is evidence from NMR and modeling studies that S170 is the primary determinant of mobility of the loop region between β-sheet 2 and α-helix 2 [21], [43]. In simulations, mobility of the loop was noted to disrupt hydrophobic stacking interactions as well as hydrogen bonds that help to connect separated regions of PrP, potentially facilitating conformational change to a isoform with increased β-sheet content that was observed during simulations using the moPrPWT sequence [43]. Lack of mobility in the loop region improved the electrostatic interaction energy of the molecule in simulations [43]. Additionally, NMR calculations indicated slightly higher stability for moPrPRL than moPrPWT [21] and moPrPRL has been reported to be less thermally labile [33].
It has long been proposed that reduced structural stability of prion proteins is associated with increased propensity for misfolding. This is supported by the observation that some mutations in human PRNP which are associated with familiar forms of prion diseases disrupt salt bridges, alter hydrophobic reactions or otherwise reduce the global stability of PrPc [44]–[47], and that partial denaturation of PrP stimulates misfolding in vitro [33], [48]–[54]. As such, it would be expected that moPrPWT would exhibit a greater misfolding propensity than moPrPRL as the rigid loop per se increases the structural stability of the molecule. In our study, introduction of the rigid loop clearly enhanced the propensity of the resulting recombinant protein to misfold in vitro. Although structural instability is sometimes correlated with misfolding, there is evidence that familial prion diseases cannot entirely be defined by thermodynamic destabilization of human PrPc by amino acid mutations [47], [55], [56]. Some of the disease-associated mutations in humans and polymorphisms in animals that modulate susceptibility to disease do not alter the thermodynamic stability of PrPc or are even associated with increased stability [32], [33], [47], [55]–[61]. Furthermore, increased stability has been observed to correlate to increased misfolding in vitro [32], [33]. Further investigation of the influence of structural stability on misfolding of cervid PrP molecules as compared to those from other species may aid in understanding the mechanism by which the rigid loop structure enhances misfolding in our in vitro system.
As differences in structural stability may not be the only explanation for enhanced misfolding as observed for moPrPRL, we next investigated whether introduction of the rigid loop altered lipid induced aggregation of moPrPWT. Enhanced aggregation upon the introduction of the rigid loop has previously been suggested as a possible explanation for development of de novo prion disease in mice over-expressing mouse/elk chimeric PrP with an introduced rigid loop [28]. In our in vitro studies, we did not detect differences in lipid induced aggregation between moPrPWT and moPrPRL. Therefore, we do not consider differences in aggregation as the likely cause of increased misfolding propensity observed here in vitro for moPrPRL.
Although a mechanistic explanation of enhanced misfolding upon the introduction of the rigid loop has yet to be determined, we conjecture that perhaps the rigid loop confers differences in ability to form lipid associations in a manner that enhances misfolding. There is increasing evidence that lipids play a direct role in prion protein misfolding [6], [39], [40], [51], [62]–[66], perhaps functioning as a lipo-chaperone modulating the conformation of PrP [48]. Our conversion assays heavily utilized lipids (POPG in lipid based conversion assays and brain derived lipids in RT-QuIC). A conformation with increased β-sheet content is expected to be a thermodynamically favorable state for rPrP [48], [50], [67] and it has long been suggested that unknown cofactors could lower the activation energy for misfolding, thus enhancing misfolding [33], [67]–[69]. PrPc variants could differ in conversion efficiency as a result of alterations in the ability of cofactors to modulate stability and folding [33]. Alterations in surface charge distribution have been proposed to alter interactions with entities that can modulate misfolding such as membrane lipids and other potential cofactors [44], [56]. Therefore, it is plausible that moPrPRL exhibits a greater propensity for misfolding than moPrPWT due to alterations in interactions with critical lipid cofactors. There is experimental evidence that the rigid loop region along with the C-terminal portion of helix 3 are the putative site for cofactor binding [68], [70], which adds support to this hypothesis.
Transmission studies on mice expressing wild-type and mouse/elk chimeric PrP have provided evidence that the β-sheet 2 – α-helix 2 loop region is also an important determinant for the species barrier [14], [27], [29]–[31]. Further support for this theory has been provided by studies using protein misfolding cyclic amplification (PMCA) [27], [31]. With few exceptions, it was observed that prions with rigid loop residues 170N and 174T recruit PrPc into a misfolded form if the PrPc also has the rigid loop, while prions without the rigid loop variations do not [14], [27], [29]–[31]. The reverse has also been found: prions without the rigid loop substitutions convert PrPc that also lacks the substitutions while prions with the rigid loop do not [14], [27], [29]–[31]. Slightly in contrast with these previous findings, moPrPRL misfolded earlier than moPrPWT in our study when RT-QuIC reactions were seeded with 22 L mouse adapted scrapie prions. Earlier conversion for moPrPRL occurred even though the 22 L seed prions lack the rigid loop associated variations. However, classical species barriers based on PrP homology are not consistently preserved in in vitro conversion systems such as RT-QuIC. Notably, the failure of our experimental system to recapitulate previously observed species barriers strengthens our argument that the enhanced misfolding propensity of moPrPRL is based on an intrinsic structural feature. Nevertheless, we wanted to verify our findings using other prion strains. Earlier conversion for moPrPRL than for moPrPWT was consistent when RT-QuIC reactions were seeded with 22 L mouse prions, CWD prions from brains of transgenic mice as well as CWD prions from infected deer brain. Stronger conversion of moPrPRL by CWD prions was anticipated based on previous studies. However, stronger conversion of moPrPRL when seeded with 22 L prions was not anticipated. The finding that moPrPRL converted more readily when seeded with 22 L prions although moPrPWT has identical primary sequence homology and a homologous loop structure with 22 L prions is a very strong indication that introduction of the rigid loop increases the propensity of moPrPWT to misfold.
In summary, introduction of the rigid loop, a structure inherent to cervid PrP, increases the propensity for recombinant mouse PrP to misfold. This was assessed in vitro using both an unseeded, lipid based conversion assay and seeded conversion using RT-QuIC to detect differences in conversion rates between two PrP variants. This finding may have relevance to the high levels of shedding of CWD prions and transmission of CWD among cervids as compared to prion diseases in other species.
Acknowledgments
We would like to than Dr. Byron Caughey (NINDS, NIH, Rocky Mountain Laboratories, Hamilton, MT) for allowing TRJ's instruction on RT-QuIC methods. We would also like to thank Dr. Sheena D'Arcy (Colorado State University) and Dr. Milan Balaz (University of Wyoming) for allowing use of their CD instruments, and Dr. Glenn Telling and Dr. Mark Zabel (Colorado State University) for providing brains of CWD infected transgenic mice. Thanks to Dr. Jean Jewell for her generous gift of CWD infected white-tailed deer brains. Additional thanks to Dr. Sabine Gilch, Missy Stuart, Dr. Yuzuru Taguchi and Dr. Suzette Priola for helpful discussions and to Justin Jones for assistance with CD experiments.
Funding Statement
This work was supported by grants from the Department of Defense (DOD 030542, to RVL), the National Institute of Health (R01 NS076853-01A1, to HMS), and was performed within the framework of the Wyoming Endowed Excellence Chair. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Prusiner SB (1998) Prions. Proceedings of the National Academy of Sciences 95: 13363–13383 doi:10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weissmann C, Li J, Mahal SP, Browning S (2011) Prions on the move. EMBO Rep 12: 1109–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aguzzi A, Polymenidou M (2004) Mammalian Prion Biology: One Century of Evolving Concepts. Cell 116: 313–327. [DOI] [PubMed] [Google Scholar]
- 4. Collinge J (2001) Prion diseases of humans and animals: their causes and molecular basis. Annual review of neuroscience 24: 519–550 doi:10.1146/annurev.neuro.24.1.519 [DOI] [PubMed] [Google Scholar]
- 5. Tatzelt J, Schätzl HM (2007) Molecular basis of cerebral neurodegeneration in prion diseases. FEBS Journal 274: 606–611 doi:10.1111/j.1742-4658.2007.05633.x [DOI] [PubMed] [Google Scholar]
- 6. Ma J (2012) The role of cofactors in prion propagation and infectivity. PLoS pathogens 8: e1002589 doi:10.1371/journal.ppat.1002589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science (New York, NY) 216: 136–144. [DOI] [PubMed] [Google Scholar]
- 8. Cohen F, Pan K, Huang Z, Baldwin M, Fletterick R, et al. (1994) Structural clues to prion replication. Science 264: 530–531 doi:10.1126/science.7909169 [DOI] [PubMed] [Google Scholar]
- 9.Telling G (2011) Transgenic Mouse Models and Prion Strains. In: Tatzelt J, editor. Prion Proteins SE - 166. Springer Berlin Heidelberg, Vol. 305 79–99 doi:10.1007/128_2011_166 [DOI] [PubMed] [Google Scholar]
- 10. Lasmezas CI, Deslys J-P, Demaimay R, Adjou KT, Lamoury F, et al. (1996) BSE transmission to macaques. Nature 381: 743–744. [DOI] [PubMed] [Google Scholar]
- 11. Priola SA, Vorberg I (2006) Molecular aspects of disease pathogenesis in the transmissible spongiform encephalopathies. Molecular biotechnology 33: 71–88 doi:∶10.1385/MB:33∶1:71 [DOI] [PubMed] [Google Scholar]
- 12. Angers RC, Browning SR, Seward TS, Sigurdson CJ, Miller MW, et al. (2006) Prions in skeletal muscles of deer with chronic wasting disease. Science (New York, NY) 311: 1117 doi:10.1126/science.1122864 [DOI] [PubMed] [Google Scholar]
- 13. Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, et al. (2006) Infectious prions in the saliva and blood of deer with chronic wasting disease. Science (New York, NY) 314: 133–136 doi:10.1126/science.1132661 [DOI] [PubMed] [Google Scholar]
- 14. Tamgüney G, Giles K, Bouzamondo-Bernstein E, Bosque PJ, Miller MW, et al. (2006) Transmission of elk and deer prions to transgenic mice. Journal of virology 80: 9104–9114 doi:10.1128/JVI.00098-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haley NJ, Mathiason CK, Carver S, Zabel M, Telling GC, et al. (2011) Detection of Chronic Wasting Disease Prions in Salivary, Urinary, and Intestinal Tissues of Deer: Potential Mechanisms of Prion Shedding and Transmission. Journal of Virology 85: 6309–6318 doi:10.1128/JVI.00425-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tamguney G, Miller MW, Wolfe LL, Sirochman TM, Glidden DV, et al. (2009) Asymptomatic deer excrete infectious prions in faeces. Nature 461: 529–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nichols TA, Pulford B, Wyckoff AC, Meyerett C, Michel B, et al. (2009) Detection of protease-resistant cervid prion protein in water from a CWD-endemic area. prion 3: 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilch S, Chitoor N, Taguchi Y, Stuart M, Jewell J, et al. (2011) Chronic Wasting Disease. In: Tatzelt J, editor. Prion Proteins SE - 159. Springer Berlin Heidelberg, Vol. 305 . pp. 51–77 LA – English.doi:10.1007/128_2011_159 [DOI] [PubMed] [Google Scholar]
- 19. Saunders SE, Bartelt-Hunt SL, Bartz JC (2012) Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerging infectious diseases 18: 369–376 doi:10.3201/eid1803.110685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wopfner F, Weidenhöfer G, Schneider R, Von Brunn A, Gilch S, et al. (1999) Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. Journal of Molecular Biology 289: 1163–1178 doi http://dx.doi.org/10.1006/jmbi.1999.2831. [DOI] [PubMed] [Google Scholar]
- 21. Gossert AD, Bonjour S, Lysek D a, Fiorito F, Wüthrich K (2005) Prion protein NMR structures of elk and of mouse/elk hybrids. Proceedings of the National Academy of Sciences of the United States of America 102: 646–650 doi:10.1073/pnas.0409008102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Christen B, Hornemann S, Damberger FF, Wüthrich K (2009) Prion Protein NMR Structure from Tammar Wallaby (Macropus eugenii) Shows that the β2–α2 Loop Is Modulated by Long-Range Sequence Effects. Journal of Molecular Biology 389: 833–845 doi http://dx.doi.org/10.1016/j.jmb.2009.04.040. [DOI] [PubMed] [Google Scholar]
- 23. Vorberg I, Groschup MH, Pfaff E, Priola SA (2003) Multiple Amino Acid Residues within the Rabbit Prion Protein Inhibit Formation of Its Abnormal Isoform. Journal of Virology 77: 2003–2009 doi:–10.1128/JVI.77.3.2003–2009.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wen Y, Li J, Yao W, Xiong M, Hong J, et al. (2010) Unique structural characteristics of the rabbit prion protein. The Journal of biological chemistry 285: 31682–31693 doi:10.1074/jbc.M110.118844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khan MQ, Sweeting B, Mulligan VK, Arslan PE, Cashman NR, et al. (2010) Prion disease susceptibility is affected by β-structure folding propensity and local side-chain interactions in PrP. Proceedings of the National Academy of Sciences 107: 19808–19813 doi:10.1073/pnas.1005267107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pérez DR, Damberger FF, Wüthrich K (2010) Horse prion protein NMR structure and comparisons with related variants of the mouse prion protein. Journal of molecular biology 400: 121–128 doi:10.1016/j.jmb.2010.04.066 [DOI] [PubMed] [Google Scholar]
- 27. Bett C, Fernández-Borges N, Kurt TD, Lucero M, Nilsson KPR, et al. (2012) Structure of the β2-α2 loop and interspecies prion transmission. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 26: 2868–2876 doi:10.1096/fj.11-200923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sigurdson CJ, Nilsson KPR, Hornemann S, Heikenwalder M, Manco G, et al. (2009) De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proceedings of the National Academy of Sciences of the United States of America 106: 304–309 doi:10.1073/pnas.0810680105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sigurdson CJ, Nilsson KPR, Hornemann S, Manco G, Fernández-borges N, et al. (2010) A molecular switch controls interspecies prion disease transmission in mice. Journal of Clinical Investigation 120: 2590–2599 doi:10.1172/JCI42051.2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tamgüney G, Giles K, Oehler A, Johnson NL, DeArmond SJ, et al. (2013) Chimeric elk/mouse prion proteins in transgenic mice. Journal of General Virology 94: 443–452 doi:10.1099/vir.0.045989-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kurt TD, Telling GC, Zabel MD, Hoover E a (2009) Trans-species amplification of PrP(CWD) and correlation with rigid loop 170N. Virology 387: 235–243 doi:10.1016/j.virol.2009.02.025 [DOI] [PubMed] [Google Scholar]
- 32. Kirby L, Agarwal S, Graham JF, Goldmann W, Gill AC (2010) Inverse correlation of thermal lability and conversion efficiency for five prion protein polymorphic variants. Biochemistry 49: 1448–1459 doi:10.1021/bi901855z [DOI] [PubMed] [Google Scholar]
- 33. Gill AC, Agarwal S, Pinheiro TJT, Graham JF (2010) Structural requirements for efficient prion protein conversion: cofactors may promote a conversion-competent structure for PrP(C). Prion 4: 235–242 doi:10.4161/pri.4.4.13394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilham JM, Orrú CD, Bessen R, Atarashi R, Sano K, et al. (2010) Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS pathogens 6: e1001217 doi:10.1371/journal.ppat.1001217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seelig DM, Mason GL, Telling GC, Hoover EA (2010) Pathogenesis of chronic wasting disease in cervidized transgenic mice. The American journal of pathology 176: 2785–2797 doi:10.2353/ajpath.2010.090710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saá P, Castilla J, Soto C (2006) Ultra-efficient Replication of Infectious Prions by Automated Protein Misfolding Cyclic Amplification. Journal of Biological Chemistry 281: 35245–35252 doi:10.1074/jbc.M603964200 [DOI] [PubMed] [Google Scholar]
- 37. Lührs T, Zahn R, Wüthrich K (2006) Amyloid formation by recombinant full-length prion proteins in phospholipid bicelle solutions. Journal of molecular biology 357: 833–841 doi:10.1016/j.jmb.2006.01.016 [DOI] [PubMed] [Google Scholar]
- 38. Wang F, Yang F, Hu Y, Wang X, Wang X, et al. (2007) Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry 46: 7045–7053 doi:10.1021/bi700299h [DOI] [PubMed] [Google Scholar]
- 39. Sanghera N, Pinheiro TJT (2002) Binding of prion protein to lipid membranes and implications for prion conversion. Journal of molecular biology 315: 1241–1256 doi:10.1006/jmbi.2001.5322 [DOI] [PubMed] [Google Scholar]
- 40. Kazlauskaite J, Sanghera N (2003) Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry 42: 3295–3304. [DOI] [PubMed] [Google Scholar]
- 41. Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, et al. (2007) Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nature methods 4: 645–650 doi:10.1038/nmeth1066 [DOI] [PubMed] [Google Scholar]
- 42. Hornemann S, Korth C, Oesch B, Riek R, Wider G, et al. (1997) Recombinant full-length murine prion protein, mPrP(23–231): purification and spectroscopic characterization. FEBS Letters 413: 277–281 doi:10.1016/S0014-5793(97)00921-6 [DOI] [PubMed] [Google Scholar]
- 43. Gorfe AA, Caflisch A (2007) Ser170 controls the conformational multiplicity of the loop 166–175 in prion proteins: implication for conversion and species barrier. The FASEB Journal 21: 3279–3287 doi:10.1096/fj.07-8292com [DOI] [PubMed] [Google Scholar]
- 44. Van der Kamp MW, Daggett V (2009) The consequences of pathogenic mutations to the human prion protein. Protein engineering, design & selection: PEDS 22: 461–468 doi:10.1093/protein/gzp039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vanik DL, Surewicz WK (2002) Disease-associated F198S Mutation Increases the Propensity of the Recombinant Prion Protein for Conformational Conversion to Scrapie-like Form. Journal of Biological Chemistry 277: 49065–49070 doi:10.1074/jbc.M207511200 [DOI] [PubMed] [Google Scholar]
- 46. Hasegawa K, Mohri S, Yokoyama T (2010) Fragment molecular orbital calculations reveal that the E200K mutation markedly alters local structural stability in the human prion protein. prion 4: 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liemann S, Glockshuber R (1999) Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry 38: 3258–3267 doi:10.1021/bi982714g [DOI] [PubMed] [Google Scholar]
- 48. Fasano C, Campana V, Zurzolo C (2006) Protein Only or Something More? Overview of Potential Prion Cofactors. Journal of molecular neuroscience 29: 195–214 doi:10.1385/JMN/29 [DOI] [PubMed] [Google Scholar]
- 49. Hornemann S, Glockshuber R (1998) A scrapie-like unfolding intermediate of the prion protein domain PrP(121–231) induced by acidic pH. Proceedings of the National Academy of Sciences 95: 6010–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baskakov I, Legname G (2004) The peculiar nature of unfolding of the human prion protein. Protein Science 13: 586–595 doi:10.1110/ps.03457204.Spontaneous [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morillas M, Swietnicki W, Gambetti P, Surewicz WK (1999) Membrane Environment Alters the Conformational Structure of the Recombinant Human Prion Protein. Journal of Biological Chemistry 274: 36859–36865 doi:10.1074/jbc.274.52.36859 [DOI] [PubMed] [Google Scholar]
- 52. Surewicz WK, Jones EM, Apetri AC (2006) The emerging principles of mammalian prion propagation and transmissibility barriers: Insight from studies in vitro. Accounts of chemical research 39: 654–662 doi:10.1021/ar050226c [DOI] [PubMed] [Google Scholar]
- 53. Apetri A, Maki K, Roder H, Surewicz WK (2006) Early intermediate in human prion protein folding as evidenced by ultrarapid mixing experiments. Journal of the American 128: 11673–11678 doi:10.1021/ja063880b.Early [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Apetri AC, Surewicz WK (2002) Kinetic Intermediate in the Folding of Human Prion Protein. Journal of Biological Chemistry 277: 44589–44592 doi:10.1074/jbc.C200507200 [DOI] [PubMed] [Google Scholar]
- 55. Swietnicki W, Petersen RB, Gambetti P, Surewicz WK (1998) Familial Mutations and the Thermodynamic Stability of the Recombinant Human Prion Protein. Journal of Biological Chemistry 273: 31048–31052 doi:10.1074/jbc.273.47.31048 [DOI] [PubMed] [Google Scholar]
- 56. Zhang Y, Swietnicki W, Zagorski MG, Surewicz WK, Sönnichsen FD (2000) Solution Structure of the E200K Variant of Human Prion Protein: IMPLICATIONS FOR THE MECHANISM OF PATHOGENESIS IN FAMILIAL PRION DISEASES. Journal of Biological Chemistry 275: 33650–33654 doi:10.1074/jbc.C000483200 [DOI] [PubMed] [Google Scholar]
- 57. Apetri AC, Surewicz K, Surewicz WK (2004) The Effect of Disease-associated Mutations on the Folding Pathway of Human Prion Protein. Journal of Biological Chemistry 279: 18008–18014 doi:10.1074/jbc.M313581200 [DOI] [PubMed] [Google Scholar]
- 58. Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, et al. (1998) Prion protein NMR structure and familial human spongiform encephalopathies. Proceedings of the National Academy of Sciences 95: 11667–11672 doi:10.1073/pnas.95.20.11667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Paludi D, Thellung S, Chiovitti K, Corsaro A, Villa V, et al. (2007) Different structural stability and toxicity of PrPARR and PrPARQ sheep prion protein variants. Journal of Neurochemistry 103: 2291–2300 doi:10.1111/j.1471-4159.2007.04934.x [DOI] [PubMed] [Google Scholar]
- 60. Bujdoso R, Burke DF, Thackray AM (2005) Structural differences between allelic variants of the ovine prion protein revealed by molecular dynamics simulations. Proteins 61: 840–849 doi:10.1002/prot.20755 [DOI] [PubMed] [Google Scholar]
- 61. Fitzmaurice TJ, Burke DF, Hopkins L, Yang S, Yu S, et al. (2008) The stability and aggregation of ovine prion protein associated with classical and atypical scrapie correlates with the ease of unwinding of helix-2. The Biochemical journal 409: 367–375 doi:10.1042/BJ20071122 [DOI] [PubMed] [Google Scholar]
- 62. Deleault NR, Kascsak R, Geoghegan JC, Supattapone S (2010) Species-Dependent Differences in Cofactor Utilization for Formation of the Protease-Resistant Prion Protein in Vitro. Biochemistry 49: 3928–3934 doi:10.1021/bi100370b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Deleault NNR, Walsh DDJ, Piro JR, Wang F, Wang X, et al. (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proceedings of the National Academy of Sciences 109: E1938–E1946 doi:10.1073/pnas.1206999109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Supattapone S (2012) Phosphatidylethanolamine as a prion cofactor. Prion 6: 417–419 doi:10.4161/pri.21826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Deleault NR, Harris BT, Rees JR, Supattapone S (2007) Formation of native prions from minimal components in vitro. Proceedings of the National Academy of Sciences 104: 9741–9746 doi:10.1073/pnas.0702662104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang F, Wang X, Yuan C–G, Ma J (2010) Generating a prion with bacterially expressed recombinant prion protein. Science . 327: 1132–1135 doi:10.1126/science.1183748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang H, Stockel J, Mehlhorn I, Groth D, Baldwin M, et al. (1997) Physical studies of conformational plasticity in a recombinant prion protein. Biochemistry 36: 3543–3553 doi:10.1021/bi961965r [DOI] [PubMed] [Google Scholar]
- 68. Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, et al. (1997) Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proceedings of the National Academy of Sciences 94: 10069–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Caughey B, Baron G (2009) Getting a grip on prions: oligomers, amyloids and pathological membrane interactions. Annual review of … 78: 177–204 doi:10.1146/annurev.biochem.78.082907.145410.Getting [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zahn R, Liu A, Lührs T, Riek R, Von Schroetter C, et al. (2000) NMR solution structure of the human prion protein. Proceedings of the National Academy of Sciences 97: 145–150 doi:10.1073/pnas.97.1.145 [DOI] [PMC free article] [PubMed] [Google Scholar]