

Abstract
Obtaining a proper fold of affinity tagged chimera proteins can be difficult. Frequently, the protein of interest aggregates after the chimeric affinity tag is cleaved off, even when the entire chimeric construct is initially soluble. If the attached protein is incorrectly folded, chaperone proteins such as GroEL bind to the misfolded construct and complicate both folding and affinity purification. Since chaperonin/osmolyte mixtures facilitate correct folding from the chaperonin, we explored the possibility that we could use this intrinsic binding reaction to advantage to refold two difficult-to-fold chimeric constructs. In one instance, we were able to recover activity from a properly folded construct after the construct was released from the chaperonin in the presence of osmolytes. As an added advantage, we have also found that this method involving chaperonins can enable researchers to decide 1) if further stabilization of the folded product is required and 2) if the protein construct in question will ever be competent to fold with osmolytes.
Introduction
Protein production efforts have been greatly aided by protein engineering efforts where specific affinity tags are constructed with the protein of interest to help with protein folding and protein purification during and following overexpression. While the specific affinity tag correctly folds and binds to its specific ligand on the affinity column, a correctly folded tag does not insure that the tagged protein of interest will also fold correctly. Thus it is not surprising that the chaperonin GroEL, known to bind with high affinity to misfolded proteins and commonly found in crude protein extracts (1), is a frequent contaminant in the isolation and purification of chimeric proteins in E. coli. In many affinity tag purification schemes, efforts are made to remove the GroEL and refold column-bound protein by adding GroES and ATP (1). However, in addition to being expensive, such approaches often fail to either remove the GroEL or induce proper folding. Removing GroEL by the addition of ATP is generally ineffective because release of tightly bound polypeptides, particularly those that require GroES for folding, is slow with ATP alone (2,3). These folding failures are primarily due to the inability of GroES to bind to GroEL to sequester the protein of interest inside the nanostructured GroEL-GroES folding cavity (cis folding) when the substrate protein is bound to the affinity column. It has been repeatedly shown that sequestered folding (cis folding) is more efficient than folding from the opposite ring (referred to as trans folding) (4–7). In the latter instance, GroES will bind to the GroEL double ring but opposite the ring that is occupied by the folding substrate. Although in vivo trans folding can and does occur (8,9) particularly for large substrates, in vitro trans folding for smaller sized proteins (≥ 60 kDa) has been repeatedly shown to be less optimal (10).
It is possible however, to use GroEL binding to one’s advantage. Work in our laboratory has shown that chaperonin-facilitated folding and stabilization of the protein product can be accomplished and is even enhanced when folding osmolytes are added to chaperonin-protein substrate complexes in the presence of ATP alone (11–14). Most importantly when folding very large affinity tagged proteins, it is conceivable that the chaperonin/osmolyte folding protocol would also work without requiring GroES addition.
In this work we summarize our recent experiences with using the GroEL/osmolytes protocol to refold two chimeric protein constructs, glutathione-S-transferase-linked human mitochondrial phosphoenolpyruvatecarboxykinase (hmPEPCK-GST) and a truncated construct derived from the cytoplasmic C terminus of the murine Polycystic kidney disease 1 protein engineered with his-tagged SUMO (mPKD11–193 –SUMO). We observed that when these overexpressed chimeric proteins were simultaneously overexpressed with GroEL and GroES chaperonins, there was an increase in both soluble product and GroEL contamination and copurification. The isolation and refolding of overexpressed GST-PEPCK with the GroEL/osmolyte refolding protocol resulted in correct folding and activity regain. The short term stability of these proteins and their subsequent rebinding to GroEL gave us the unique opportunity to determine optimal conditions to not only fold proteins but to identify conditions that may ultimately be used to maintain long-term folding competence and stability.
Materials and Methods
Extremely pure GroEL (>99%) was provided as a gift from Edge BioSystems. Mitochondrial porcine malate dehydrogenase was purchased from Sigma. The chaperonin-protein folding intermediates were prepared as previously described (15). The E. coli BL21strain (DE3) that was used in these experiments contained the plasmids pGro7 (Takara bio inc) and PEPCK-GST encoding the entire GroE chaperonin system and phosphoenolpyruvate carboxykinase-glutathione-S-transferase (PEPCK-GST) respectively. For the overproduction and purification of the mPolycystin-11–193 –SUMO construct, the E coli strain DE3 was used. All other assay reagents listed in these studies were purchased from Sigma
PEPCK-GST Purification
E. Coli BL21(DE3) containing the plasmids pGro7 and PEPCK-GST construct was cultured overnight in 10 ml Luria broth containing 100 ug/ml ampicillin and 20ug/ml chloramphenicol at 300 rpm and 37°C. The entire contents were added to 500ml Luria broth with 100ug/ml ampicillin and 20 ug/ml chloramphenicol and incubated at 37 °C and 300 rpm on Excella E25 shaking incubator(New Brunswick Scientific). Arabinose was added to final concentration of 1mg/ml at 3 hours, and IPTG was added to final concentration of 1 mM at 5 hours. At 8 hours, cells were pelleted by 10 minutes spin at 10,000 rpm with an SLA-3000 rotor in a Sorvall RB-5 centrifuge. The cell pellet was resuspended in 10 mL buffer 50 mM TRIS pH 7.5, 5mM EDTA, 1 mM DTT, 1 ug/ml DNase, and 1 ug/ml lysozyme. Cells were lysed in SLM Aminco french pressure with a 20K cell, and the lysate was centrifuged 10 minutes at 20,000 rpm with an SS-34 rotor in a Sorvall RB-5 centrifuge. The soluble fraction was added to 5 ml glutathione uniflow resin (Clontech) and incubated at 25 C for 10 minutes with stirring. The resin suspension was transferred to a small glass column (Kontes) and washed twice with 50 ml 50 mM TRIS pH 7.5 and once with 50 ml refolding buffer (50 mM TRIS, pH 7.5, 100 mM KCl, 10 mM MgCl2, 5 mM DTT, with or without 4M Glycerol). The refolding experiments were begun after the column was equilibrated with refolding buffer.
GST-PEPCK-GroEL Protein Refolding
Unless specified, the refolding experiments were generally performed as follows. The glutathione resin with bound GST-PEPCK-GroEL was resuspended in 15 ml refolding buffer and divided into 500 ul aliquots. 25 ul 100 mM ATP stock (pH 7.5 at room temperature) was added at time zero to each aliquot and the suspension was mixed on a Biorad rotator at 10 rpm for the duration of the experiment. 25 uL reduced glutathione solution (200 mM GSH, 50 mM TRIS pH 7.5) was added at each time point (Figure 4) and incubated for 5 minutes on a rotator. The resin suspension was centrifuged for 10 sec at 10,000 rpm using a table top Labnet Spectrafuge 24. The supernatant was used for activity assays.
Figure 4.

After a GroEL PEPCK-GST complex was loaded onto a glutathione column (isolated directly from cells that overexpressed GroEL and PEPCK-GST, the column material was split and one portion was incubated with ATP alone while the other portion was incubated with the osmolyte glycerol (4 M) and ATP. As our initial test, we assayed the PEPCK activity using approximately equal volumes of whole beads. While no bead bound activity was observed when ATP alone was added to the preformed complex, a substantially larger activity regain was observed when the bead bound complexes were incubated in the presence of the osmolyte/ATP mixture. However, activity declines between 2 and 6 hours of refolding. The resulting activity for two split samples at 6 hours (± ATP) were compared immediately after ATP addition (~ 1 min) and 2 hours later. The activity remain higher in the sample that contained ATP in agreement with the notion that ATP added to GroEL complexes with rebound PEPCK show a moderate amount of PEPCK rerelease, refolding and regain of activity.
PEPCK Activity assay
The PEPCK activity assay follows the carboxylation of PEP to form OAA as described in Hebda and Nowak (16). Assay mix contains final concentration of 50 mM Hepes, 200 mM KCL, 4mM MnCl2, 2 mM PEP, 2 mM IDP, 1% mercaptoethanol, 120 ug/ml NADH, 200 mM KHCO3, 40 ug/ml Malate dehydrogenase (MDH). KHCO3 and MDH were added just before mixing with sample. 50 uL sample was added to the 1 mL final volume of the assay mix. Absorbance at 340 nm was monitored. The assay reactions were performed at 25°C.
Electron Microscopy
PEPCK-GST/GroEL complex was purified following the same procedure used for refolding except that the sample was eluted after washing without splitting the resin for refolding. Negative stain grids were prepared with 10 diluted elution with 2% uranyl acetate stain. Images were recorded using a minimal-dose protocol at a magnification of 60,000 with a JEOL 1200EX electron microscope at a defocus of 0.78 μm and digitized them with a Microtek ScanMaker i900 scanner at a pixel size of 3.53 Å on the specimen. For our initial data set, 160 GroEL particles were selected and sorted into 92 free and 68 bound particles by correspondence analysis and hierarchical clustering. Image analysis was performed using SPIDER (17).
mPolycystin-11–193 –SUMO construct
A The truncated Polycystic kidney Disease protein encoding the C-terminal 193 amino acids of murine polycystin-1 and tagged at the N-terminus with his-tagged (PKD1) SUMO tagged affinity protein was constructed as follows: mPKD1 sequence was PCR amplified from an expression plasmid encoding the C-terminal 193 amino acids of murine polycystin (provided by Stephen C. Parnell, unpublished) using the following primers: 5′-GGAATTCGGTCTC GAGGTCACGCCTTGCGTGGCGAGCTC- 3′ and --> <--5′-GATGCGGCCGCCTAAGTGCTGCTGGGGTGGAC- 3′
Following gel electrophoresis, the resulting_0.6 kb PCR product was extracted using the QIAquick® Gel Extraction Kit (QIAGEN), digested with BsaI and NotI (bold; New England BioLabs) and ligated into pE-SUMO (Life Sensors) that had been previously digested with BsaI and NotI. Resulting plasmids were transformed into DH5αand plated on LB agar containing 30 ug/ml kanamycin. After 24 hours, individual colonies were picked and cultured overnight in LB + 30 ug/ml kanamycin. Plasmid DNA was purified from the overnight cultures, and positive constructs were verified by restriction analysis and DNA sequencing.
mPolycystin-11–193 –SUMO Purification
Small-scale purification trials showed that mPolycystin-11–193 –SUMO was expressed in inclusion bodies. In order to get soluble mPolycystin-11–193 –SUMO induction temperatures in the range 16 – 37 °C were tested, as well as induction at different optical densities at 600 nm (OD600). Various concentrations of IPTG were also used. Numerous lysis buffers were tried, and also two lysis techniques, lysis by French press and by the freeze and thaw method. SDS-PAGE was used to check protein expression. As none of these procedures gave soluble protein, denaturation procedures were used to extract the protein from inclusion bodies. In this study, the extracted 6-His tagged mPolycystin-11–193 –SUMO protein was purified using Nickel affinity chromatography under denaturing conditions.
The following procedure gave goods yields of denatured mPolycystin-11–193 –SUMO: 10 ml overnight culture was added to 500ml LB broth with 30 μg ml−1 kanamycin and incubated at 37°C with 250 rpm shaking. Protein expression was induced, at the same temperature, by addition of IPTG to a final concentration of 1 mM when OD600 reached 0.7. After 6 hours the cells were harvested from the culture medium by centrifugation (4 °C, 10 minutes, 3500 rpm). Cells were frozen and kept at −80°C for later purification procedures. The cell pellet was resuspended in 50 mL resuspension solution (25 mM HEPES, 20 mM imidazole, 300 mM NaCl, pH 7.5) and the cells were lysed by the freeze and thaw method. The lysate was centrifuged at 4 °C, 7,000 rpm for 15 minutes. The supernatant (soluble proteins) yielded very small quantities of mPolycystin-11–193 –SUMO, but removal of soluble proteins proved to be a good purification step in this procedure. The pellet containing inclusion bodies was resuspended in 15 mL of denaturing buffer (8M urea, 0.1 M NaH2PO4, 0.01 M Tris-Cl, pH 8.0) at room temperature and treated with protamine sulphate to final 0.1%. After centrifugation for 15 min at 10,000 rpm the supernatant contained denatured mPolycystin-11–193 –SUMO. It was then added to 6 ml of pre-equilibrated Ni-NTA resin (PerfectPro Ni-NTA Agarose, 5 Prime) and incubated for 60 minutes with stirring. The resin was then poured in a column and washed with denaturing buffer, then with wash buffer (same as denaturing, only pH 6.3), and eluted with elution buffer (same as denaturing, only pH 4.5). This way denatured 6-His tagged mPolycystin-11–193 –SUMO of high purity was obtained. When refolding was attempted the final eluting buffer contained glycerol (0.05 M Tris-Cl, pH 7.5, 0.4 M NaCl, 0.05 M KCl, 0.1 M MgCl2, 4 M glycerol, 0.2 M imidazole). Under these latter conditions, the 6-His tagged mPolycystin-11–193 –SUMO remained in solution. This was the starting material for the GroEL/sumolyase experiments (Figure 9)
Figure 9.

Sumo tag is from the mPKD11–193 –Sumo-his can be removed even in the presence of GroEL. The partially folded polycystin-1 remains bound to GroEL (no evidence of aggregation).
Lane 1 – Retentate on Microcon YM-10 before addition of ATP (final concentration 5 mM): GroEL-PKD1- -complex, treated with SUMO-protease (~31kDa)(sumolyase): GroEL (~60 kDa); mPKD11–193 –SUMO (~37 kDa); mPKD11–193 –(~24 kDa); SUMO-tag (~12 kDa);
Lane 2 – Filtrate through Microcon YM-10 after addition of ATP: SUMO-tag (~12 kDa) was the only protein observed in the filtrate;
Lane 3 – In a separate experiment mPKD11–193 SUMO partially cleaved with SUMO-protease: mPKD11–193 –SUMO(~37 kDa)- (PKD1-SUMO-his); mPKD11–193 –(~24 kDa) (PKD1); SUMO-tag (~12 kDa) (SUMO-His). This latter experiment provides reference molecular mass markers for the experiments with GroEL, mPKD11–193 – Sumo-his and all of the cleavage products.
Results and Discussion
A. Folding the Glutathione-S-Transferase (GST) chimera construct of human mitochondrial phosphoenolpyruvate carboxykinase (hmPEPCK)
Protein samples from the E. coli extract containing hmPEPCK-GST that eluted from the glutathione column was initially subjected to SDS gel electrophoresis. Two major bands were observed, one with an estimated mass at the expected 82 kDa molecular weight of GST-hmPEPCK and another with an estimated mass at the expected 57kDa molecular weight of GroEL. Two separate samples of upper band (~ 87 kDa) and the GroEL band (~ 57 kDa) were submitted for trypsin digestion, reduction and alkylation using a ProGest robot (Genomic solutions). The resulting peptides were desalted and analyzed by MALDI TOF on a 4700 poteomics analyzer. The peptide mass maps were compared with the NCBI protein databank and both samples confirmed that the proteins isolated from the upper SDS-PAGE was hmPEPCK and the lower band was identified as GroEL.
The eluted samples that clustered around the sole protein peak (qualitative identification by Bradford assay) from the GST column were then separated on native PHAST gels (Figure 1). Prominent bands that migrate in the position of the GroEL tetradecamer and two upper bands were resolved. Typically such band shifts above GroEL indicate that GroEL is binding a polypeptide substrate (18,19). To confirm this, we incubated the GSH column eluate with anti-GST antibodies to determine if the larger migrating bands would super shift in a native gel upon antibody binding. The results indicate that these upper bands do indeed shift to higher molecular masses suggesting that the antibodies are binding to the folded GST of the PEPCK-GST construct bound to GroEL. Thus, our results suggest that the two upper protein bands correspond to GroEL-PEPCK-GST complexes. Since GST can form a dimer, it is conceivable that the upper most bands are GroEL-(PCK-GST)2 complex. In data not shown, refolding PEPCK-GST from inclusion bodies in the presence of GroEL results in a substantial increase in the amount of the highest migrating GroEL-PEPCK-GST complex (illustrated in Lane 1).
Figure 1.

Lane 1-Native gel depicts fractions containing GroEL and GroEL-PEPCK-GST complexes eluted from glutathione column. GroEL appears in three bands. Two upper Bands contain GST-PEPCK. The lowest band corresponds to free GroEL. Lane 2 examines consequences of a antibody induced shift in molecular mass that results when Monoclonal antibodies to GST are added to the GroEL-PEPCK-GST samples before they are run on the native gel. (upper band GroEL-PEPCK-GST shifts to higher molecular masses (lone arrow upper right and resulting upper smear).
A further highly definitive confirmation that the other major contaminant is tetradecameric GroEL was achieved when the purified glutathione eluted samples were applied onto copper grids for uranyl acetate negative-staining and examined by electron microscopy. The predominant species that appears in the electron micrograph fields is has all the molecular characteristics of a low resolution structure of GroEL (Figure 2). Using the single particle analysis program SPIDER (17), a classification analysis of collected images (initial combined set of ~ 300 images) was performed and a substantial set of images were classified into GroEL-substrate protein populations that are reminiscent of previously confirmed GroEL-glutamine synthetase monomer averages (20,21). The total density from the protein substrates that contributes to the entire GroEL-protein substrate complex in both cases is small (~ 7–8%) and is manifested as a slight increase in overall density on one end of the GroEL complex. The protein bound forms of GroEL population do however show a characteristic doming of density at one end of the GroEL molecule (20,21).
Figure 2.

Negative stain EM Field: GroEL complexes A) Electron Micrograph of the PEPCK-GST sample eluted with glutathione clearly shows the presence of GroEL and additional density at the protein substrate binding site.. Inset: GroEL after treatment with ATP removes the complexed protein. B) Initial correspondence analysis of negatively stained GroEL-glutamine synthetase samples (20,21) revealing uncomplexed GroEL and GroEL complexed with protein is compared with a similar analysis from the current study showing uncomplexed GroEL and GroEL complexed with protein, presumed to be GroEL-PEPCK-GST. Arrow points to area of increased electron density in current study. For GroEL-PEPCK-GST, the distribution of images in a typical population was ~ 60% free, 40% bound) see methods for more details).
We exploited the intrinsic GroEL- [PEPCK-GST] interaction and designed a column based refolding protocol (Figure 3) that relies on the intrinsic folding of the chaperonin/osmolyte refolding system. In this procedure, when one adds an osmolyte such as glycerol along with ATP, the GroEL-[PEPCK-GST] complex dissociates and PEPCK folds to an active conformer. There are two variations of this folding scheme that can be used. In one instance, the GroEL can be eluted off with the osmolyte and ATP and PEPCK-GST activity could be assessed either on column or after the PEPCK-GST is eluted off with glutathione and its activity assessed (top path Figure 3). In the other variation, one could elute the entire sample (as was done for EM analysis) and follow this procedure with a glycerol and ATP addition to analyze PEPCK refolding. In both instances, since the chimeric protein attaches to the affinity column by binding glutathione, the GST portion of the chimera acquires its proper fold regardless of the folding state of the engineered construct.
Figure 3.

Scheme for folding a chimeric protein attached to a column using the GroEL/osmolyte folding system with a chimeric protein construct (PEPCK-GST as our example).
Our very first attempts to obtain an enzymatically active PEPCK moiety from the initial GroEL-PEPCK-GST complex proved successful. The folding of PEPCK-GST was initially assessed after Glycerol/ATP was added to the column and incubated for a period of time. In our first two series of experiments, GroEL was not removed from the column. In the first instance, compared with the ATP control in the absence of osmolytes, PEPCK activity could be detected on the beads (Figure 4). In this case, the specific activities of this particular sample were not estimated because the PEPCK-GST protein was not purified away from the column. To insure that folding can occur, GroEL was not removed during the refolding reaction because some proteins have to undergo numerous rebinding and refolding cycles before they attain a folded state (3). Curiously, however, we also noted that when GroEL was allowed remain in contact with PEPCK-GST during ATP depletion (i.e. ATP is hydrolyzed), the PEPCK activity appear over time but then declined after ~ 2 hours at room temperature. If another bolus of ATP was added to the glutathione beads a later time point (~6 hour), some of the PEPCK activity increased within one minute and remained (Figure 4). With this observation, the most likely reason for the decline in activity is due to the probable rebinding of GroEL onto the PEPCK portion of the GST-PEPCK construct. It is common for the high affinity ADP form of GroEL to bind to previously folded proteins if that particular protein of interest is in equilibrium with misfolded or partially folded states (22,23).
In the course of the initial experiments it was also noted that there was a steady loss of GroEL from the GST column during the initial wash steps after the [GST-PEPCK]-GroEL complex binds to the column. If refolding and activity regain depends on the initial concentration of GroEL present, it was reasoned that one should observe an increase in activity if GroEL was deliberately added back onto the glutathione column containing bound GST-PEPCK. For these experiments, the column slurry was equally divided into two smaller separate column fractions. On one column fraction, the refolding was examined in the presence of the intrinsically bound GroEL (black trace). In the other fraction, an extra bolus of GroEL (1 mg/ml ~ 1.2 μM oligomer) was added directly to the column (Figure 5) prior to elution (red trace). For these experiments, the entire GroEL-PEPCK-GST complex was eluted from the column by the adding glutathione, then glycerol and ATP are added to the eluted extract, and finally PEPCK activity was monitored for the GroEL PEPCK-GST mixture. The activity of PEPCK is clearly higher when additional GroEL was added back onto the column prior to elution. However, again as observed previously, the GST-PEPCK activity also declines as a function of time when GroEL is present. Again, the reason for the decline is most likely due to a partitioning reaction of PEPCK-SST back onto GroEL (24). To prevent this kinetic partition onto the high affinity nucleotide free (or in this particular example, the ADP bound form) of GroEL, the next logical approach is to determine conditions that can stabilize the protein of interest (addition of a stabilizing ligand or removal of oxidizing conditions respectively) (22,23).
Figure 5.

The column based refolding and regain of GST- PEPCK activity was compared with the intrinsically bound GroEL and the GST-PEPCK bound sample that contained an extra bolus of GroEL added to the column slurry (~ 1 mg/ml or ~ 1.2 uM GroEL oligomer). After ATP and glycerol addition, the refolded proteins and GroEL were eluted from the column by adding glutathione. As is readily apparent, the sample with the extra GroEL added exhibited higher PEPCK activity. In both cases, with either intrinsic GroEL or extra GroEL present, the PEPCK activities begin to show a decline after 2 hours.
Interestingly, these studies indicate that one could use GroEL to advantage to monitor protein stability conditions by observing the extent of protein rebinding onto GroEL. Apparently in vitro, the mitochondrial PEPCK enzyme appears to be most stable at alkaline pH values (>8.0) (25). Refolding and repartitioning of folded PEPCK onto GroEL at pH 7.5 (previous refolding conditions) and at pH 8.5 were compared. GroEL/osmolyte dependent folding can occur over a broad range of pH conditions (~7– 8.8). These preliminary experiments show that the activity is attained at pH 8.5 and most importantly, the activity of the refolded protein does not decline in the presence of the copurified GroEL (Figure 6).
Figure 6.

The refolding of PEPCK was carried out in either a pH 7.5 solution or the pH was elevated to pH 8.5. As shown above, our preliminary results indicate that the PEPCK activity is stabilized at pH 8.5, even in the presence of GroEL.
Thus, the utility of the chaperonin/osmolyte refolding scheme can be expanded to identify solution conditions or ligands that will stabilize a protein once it acquires a stable highly populated fold. This approach is particularly helpful in situations where proteins exhibit a transient burst of refolded activity followed by an unexpected decline. In addition to identifying appropriate stabilizing folding conditions, many long term storage issues may be resolved using the chaperonin as a detection system to identify slow misfolding, oxidation or aggregation reactions.
B. Using the chaperonin as an identification tool to rapidly screen for protein stabilizers
Based on the experimental results presented above, we explored the possibility of designing an easy-to-use chaperonin based system to identify and screen for stabilizing conditions or ligands for folding proteins. This procedure will lead to broader applications for proteins in general because maintaining adequate protein stability is a common problem in all aspects of protein chemistry. The ability to rapidly identify stability conditions will be extremely useful, particularly during protein folding reactions or even after the protein has acquired its active fold. Identifying the best stabilization conditions are usually very difficult and time consuming. Since all folded proteins are in a dynamic equilibrium with partially folded intermediates, the inherent promiscuous binding behavior of the chaperonin enabled us to devise a rapid kinetic process and broad method monitor general protein stability, particularly if there is a substantial steady-state transient population. Building on the GroEL assisting PEPCK-GST folding and stabilization observations, the tight binding affinity of the nucleotide free chaperonin can serve a binding platform for a very general method to identify either stabilizing ligands or solution conditions that stabilize any natively folded protein. To demonstrate the feasibility of this process, we tested the utility of using the chaperonin as a general binder of protein undergoing a shift in equilibrium misfoldiing populations due to a slight increase in temperature. It was predicted that we could prevent this partitioning reaction from occurring if we performed this partitioning reaction in the presence of known protein stabilizers. Fortunately, the GroEL oligomer is very heat stable and resists heat denaturation at moderately elevated temperatures. Sot et al., (26) found that the protein substrate free chaperonin is stable up to 60°C. With this in mind, we reasoned that proteins which rapidly partition onto the chaperonin at moderately elevated temperatures (~50°C) will show a significantly slower partitioning kinetics if well known stabilizers such as sucrose and/or trehalose were present during the heat denaturation step.
Using a well known chaperonin substrate, malate dehydrogenase (MDH), it is important to first demonstrate that this protein will rapidly partition onto GroEL at these temperatures (Figure 7 Panel B). This partitioning is not due to irreversible denaturation because the heat-denatured MDH that is bound to GroEL can be refolding once the temperature is lowered and folding osmolytes such as 4 M Glycerol and ATP are added to the immobilized GroEL support system.
Figure 7.
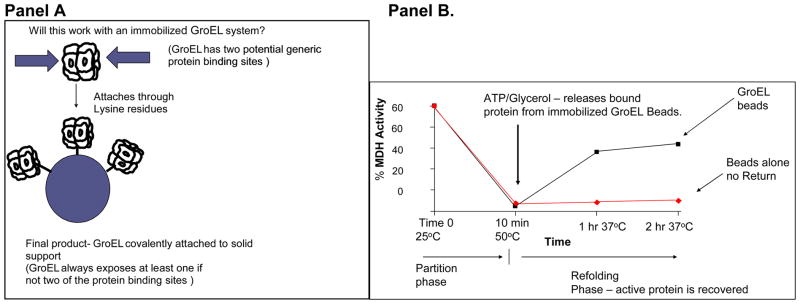
Demonstration of the partitioning of a Native folded Protein onto GroEL beads – Malate dehydrogenase. Panel A. Immobilization scheme for GroEL shows that orientation will not matter. A single or double binding site will always be presented. Panel B. After a temperature pulse of 50°C for 10 min, activity of MDH decreases to zero within ten minutes (partitioning phase). After the temperature is returned to 37°C, glycerol and ATP facilitate refolding of partitioned malate dehydrogenase.
Stabilization of MDH against thermal denaturation
Once conditions were established that demonstrated efficient partitioning occurs, it is apparent that reversing this partitioning or at least slowing the kinetics of partitioning will allow one to rapidly identify solution conditions (e.g. the protein stabilizer trehalose) would lead to enhanced stability, thus decreasing the intrinsic unfolding rate constant, leading to a slower or negligible partitioning reaction onto the immobilized GroEL. The measurable parameter is the observation that protein or activity remains in solution. One could also monitor protein depletion from solution by either employing standard protein assays or ELISA based assays (data available upon request). The latter method of detection will avoid possible spectroscopic inferences that arise because of absorption characteristics that are encountered when one uses typical small molecule based chemical combinatorial screens. These results demonstrate that one can rapidly identify stabilizing conditions (in this case within 10–20 minutes).
In summary, a numerous laboratories (including ours) have documented a variety of instances where natively folded protein substrates will kinetically partition onto the GroEL chaperonin due to the presence of a preexisting unfolding ↔ refolding equilibrium (22, 23). Although we used a temperature perturbation in increase the extend of misfolded protein, one could just as easily perturb this equilibrium with an non-denaturing additive such as 1 M urea. It turns out that GroEL is also stable under these conditions (23). Thus, simply monitoring a decrease or complete prevention of a kinetic partitioning reaction onto the GroEL chaperonin can be used as a general method for identifying ligands or conditions that stabilize natively folded populations. Therefore, in addition to folding proteins, the chaperonin utility can be broadened to aid in the identification of specific ligand/solution stabilization conditions for specific proteins. The beauty of this approach is that identifying stabilizing conditions will simply depend on evaluating the amount of protein that remains in solution in the presence of the high-affinity immobilized chaperonin.
C. Assessing folding competence of a truncated protein construct, mPKD11–193 –SUMO
Another protein construct was examined where folding was particularly refractory following removal of a chimeric affinity tag. The protein construct in question is the putative cytoplasmic C terminal domain of the polycystic kidney disease-1 (PKD1) protein (polycystin-1) engineered with a combination His tagged - Sumo affinity tag yielding the mPKD11–193 –SUMO construct. This is a particularly interesting region of the protein because some single site mutations within the C terminal region lead to some forms of polycystic kidney disease. Previous overexpression of this truncated protein region alone either resulted in inclusion body formation or fragmentation. Addition of a GST tag increased protein yields but most all of the protein was found to ultimately reside in the insoluble pellet following cell lysis. To improve the chances for folding this C terminal region, it is possible that one could construct a soluble form of this truncated product when a His-SUMO tagged polycystin-1 fragment where it is possible to introduce the chaperonin GroEL to facilitate folding. It was found that overexpression of this truncated C terminal protein construct results in a substantial increase in soluble protein if inclusion body refolding is performed in the an osmolyte such as 4 M glycerol (Methods section). However, even in the presence of this osmolyte, the removal of the His-SUMO tag with sumoylase resulted in a rapid precipitation of the polycystin-1 portion from solution. In order to determine if GroEL would be helpful at both preventing aggregation of the free PKD1 C- terminus and in identifying conditions to fold this protein, pure GroEL was added to the complete construct mPKD11–193 –SUMO prior to removing the SUMO tag with sumolyase. Importantly, in the presence of GroEL, the sumolyase cleavage of the SUMO tag protein was successful and no visible precipitation of the remaining protein construct was observed. At this point, the cleaved PKD1 product binds to GroEL and the cleaved and folded SUMO tag portion can be easily separated from the GroEL PKD1 complex via ultrafiltration (see lane 2 Figure 9). Starting from the GroEL-PKD1 complex, ATP and a number of other test osmolytes (glycerol, TMAO and proline) unfortunately all failed to achieve a soluble folded PKD1 product because a folded protein of this molecular mass and identity did not appear in the filtrate following ultrafiltration separation. Results indicate that 1) it is possible to remove the folded chimera affinity tag even in the presence of GroEL and 2) the appearance of a stable folded form that does not partition or repartition onto GroEL will be useful in analyzing folding competence. This is particularly important for evaluating generic folding efforts associated with the structural genomics initiative where unknown proteins are routinely being tested for a homogenous folding population. Upon further reflection, it is perhaps not surprising that the PKD1 cytoplasmic domain alone is unable to acquire a stable fold without its interaction partner, PKD2 (polycystin-2). It turns out that the C-terminal portions of the PKD1 and PKD2 proteins presumably interact in vivo to form a hetero-oligomeric complex (27). At present, the stoichiometry for this association is not known. The association domains between PKD1 and PKD2 are proposed to interact through coiled-coil domains (28).
Summary
Folding chimera proteins with chaperonin/osmolyte solutions either on column or in solution illustrates a new and previously unanticipated use for this folding system. Thus far, the best performing osmolytes for chaperonin/osmolyte folding appear to be the following mixtures: 2–4 M glycerol, 1 M sucrose, 1 M proline or 1 M trehalose in our respective refolding buffers (11–13). In this method, the purified chaperonin can be considered to be an additional folding aid and reagent that one utilize to fold column bound chimeric proteins in the presence of a folding compatible osmolyte. In addition, the observation that a transiently folded protein will repartition onto the chaperonin allows one use this method to screen for solutions or conditions that lead to the improved stability of the final released folded protein. Because the binding is reversible, one can potentially use this system repeatedly. The ability to easily assess protein stability by simply monitoring the amount of natively folded protein that remains in solution has broad applications in the protein folding and protein stability fields. We have demonstrated that the utility of the chaperonin/osmolyte folding system can be expanded to aid in evaluating on-column folding propensities of various chimeric constructs and determine if the complete refolding reaction requires, for example, additional cofactors or protein interaction partners to achieve their final stable folded states.
Figure 8. Partitioning reaction of MDH in the absence and presence of the stabilizers trehalose/sucrose.

In the absence of stabilizing osmolytes (closed triangles), the thermally denatured MDH will partition onto the immobilized GroEL support. If thermal denaturation occurs in the presence of stabilizers such as a trehalose/sucrose mixture, a substantial amount of MDH activity remains in solution and is readily detectable (closed circles). After 20 min at 46°C, 70 percent of the original activity of MDH (double arrow) remains in solution while the unprotected sample is no longer active or detected in the assay solution.
Acknowledgments
This research was supported in part by NSF MCB-0445936 (MTF), NIH R41 GM080074 (MTF), NIH P50 DK057301 (JPC), and a grant from the Kansas Technology Enterprise Corporation KTEC (MTF). MB and AK are undergraduates supported by the NSF grant. AK was specifically supported by an NSF REU supplement NSF MCB-0735909 (MTF). A portion of this research was presented at the “Protein Structure Inititative “Bottlenecks” Workshop. The authors gratefully acknowledge Stephen C. Parnell for generating the pSUMO-HT193 plasmid and for helpful discussions with D. M.-C. D. M.-C. was supported by a Fulbright Scholarship.
Abbreviations
- IPTG
isopropyl-β-D-thiogalactopyranoside
- Ni-NTA
nickel-nitrilotriacetic acid
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SUMO
small ubiquitin-like modifier
- DTT
dithiothreitol
- PKD1
polycystin-1
- PKD2
polycystin-2
- PEPCK
Phosphoenolpyruvate carboxykinase
- GST
Glutathione –S-transferase
References
- 1.Thain A, Gaston K, Jenkins O, Clarke AR. Trends Genet. 1996;12:209–210. doi: 10.1016/s0168-9525(96)90022-0. [DOI] [PubMed] [Google Scholar]
- 2.Horwich AL, Fenton WA. Prot Sci. 1997;6:743–60. doi: 10.1002/pro.5560060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher MT, Yuan X. J Biol Chem. 1994;269:29598–29601. [PubMed] [Google Scholar]
- 4.Weissman JS, Kashi Y, Fenton WA, Horwich AL. Cell. 1994;78:693–702. doi: 10.1016/0092-8674(94)90533-9. [DOI] [PubMed] [Google Scholar]
- 5.Weissman JS, Rye HS, Fenton WA, Beechem JM, Horwich AL. Cell. 1996;84:481–490. doi: 10.1016/s0092-8674(00)81293-3. [DOI] [PubMed] [Google Scholar]
- 6.Rye HS, Roseman AM, Chen S, Furtak K, Fenton WA, Saibil HR, Horwich AL. Cell. 1999;97:325–328. doi: 10.1016/s0092-8674(00)80742-4. [DOI] [PubMed] [Google Scholar]
- 7.Mayhew M, da Silva ACR, Martin J, Erdjument-Bromage H, Tempst P, Hartl FU. Nature. 1996;379:420–426. doi: 10.1038/379420a0. [DOI] [PubMed] [Google Scholar]
- 8.Dubaquie Y, Looser R, Funfschilling U, Jeno P, Rospert S. EMBO J. 1998;17:5868–5876. doi: 10.1093/emboj/17.20.5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaudhuri TK, Farr GW, Fenton WA, Rospert S, Horwich AL. Cell. 2001;107:235–46. doi: 10.1016/s0092-8674(01)00523-2. [DOI] [PubMed] [Google Scholar]
- 10.Sigler PB, Xu Z, Rye HS, Burston SG, Fenton WA, Horwich AL. Annu Rev Biochem. 1998;67:581–608. doi: 10.1146/annurev.biochem.67.1.581. [DOI] [PubMed] [Google Scholar]
- 11.Voziyan PA, Jadhav L, Fisher MT. J of Pharm Sci. 2000;89:1036–1045. doi: 10.1002/1520-6017(200008)89:8<1036::aid-jps8>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 12.Tieman BC, Johnston M, Fisher MT. J Biol Chem. 2001;276:44541–44550. doi: 10.1074/jbc.M106693200. [DOI] [PubMed] [Google Scholar]
- 13.Voziyan PA, Johnston M, Chao A, Bomhoff G, Fisher MT. J of Struc Func Genomics. 2005;6:183–188. doi: 10.1007/s10969-005-2646-6. [DOI] [PubMed] [Google Scholar]
- 14.Katayama H, Janowiak BE, Brzozowski M, Jurcyk J, Falke S, Gogol EP, Collier RJ, Fisher MT. Nat Struc and Mol Biol. 2008;15:754–60. doi: 10.1038/nsmb.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voziyan PA, Fisher MT. Prot Sci. 2000;9:2405–2415. doi: 10.1110/ps.9.12.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hebda CA, Nowak T. J Biol Chem. 1982;257:5503–5514. [PubMed] [Google Scholar]
- 17.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. J Struct Biol. 116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 18.Fisher MT. Biochemistry. 1992;31:3955–3963. doi: 10.1021/bi00131a010. [DOI] [PubMed] [Google Scholar]
- 19.Fisher MT. J Biol Chem. 1994;269:13629–13636. [PubMed] [Google Scholar]
- 20.Falke SF, Fisher MT, Gogol EG. J Mol Biol. 2001a;308:569–577. doi: 10.1006/jmbi.2001.4613. [DOI] [PubMed] [Google Scholar]
- 21.Falke SF, Fisher MT, Gogol EG. J Struct Biol. 2001b;133:203–213. doi: 10.1006/jsbi.2001.4354. [DOI] [PubMed] [Google Scholar]
- 22.Viitanen PV, Donaldson GK, Lorimer GH, Lubben TH, Gatenby AA. Biochemistry. 1991;30:9716–23. doi: 10.1021/bi00104a021. [DOI] [PubMed] [Google Scholar]
- 23.Smith KE, Voziyan PA, Fisher MT. J Biol Chem. 1998;273:28677–28681. doi: 10.1074/jbc.273.44.28677. [DOI] [PubMed] [Google Scholar]
- 24.Walter S, Lorimer GH, Schmid FX. Proc Natl Acad Sci U S A. 1996;93:9425–30. doi: 10.1073/pnas.93.18.9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holyoak T, Sullivan SM, Nowak T. Biochemistry. 2006;45:8254–63. doi: 10.1021/bi060269g. [DOI] [PubMed] [Google Scholar]
- 26.Sot B, Bañuelos S, Valpuesta JM, Muga A. J Biol Chem. 2003;278:32083–90. doi: 10.1074/jbc.M303958200. [DOI] [PubMed] [Google Scholar]
- 27.Gallagher AR, Cedzich A, Gretz N, Somlo S, Witzgall R. Proc Natl Acad Sci U S A. 2000;97:4017–22. doi: 10.1073/pnas.97.8.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian F, Germino J, Cai Y, Zhang X, Somlo S, Germino GG. 1997;16:179–183. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]