

Abstract
The first barrier that an antimicrobial agent must overcome when interacting with its target is the microbial cell wall. In the case of Gram-negative bacteria, additional to the cytoplasmic membrane and the peptidoglycan layer, an outer membrane (OM) is the outermost barrier. The OM has an asymmetric distribution of the lipids with phospholipids and lipopolysaccharide (LPS) located in the inner and outer leaflets, respectively. In contrast, Gram-positive bacteria lack OM and possess a much thicker peptidoglycan layer compared to their Gram-negative counterparts. An additional class of amphiphiles exist in Gram-positives, the lipoteichoic acids (LTA), which may represent important structural components. These long molecules cross-bridge the entire cell envelope with their lipid component inserting into the outer leaflet of the cytoplasmic membrane and the teichoic acid portion penetrating into the peptidoglycan layer. Furthermore, both classes of bacteria have other important amphiphiles, such as lipoproteins, whose importance has become evident only recently.
It is not known yet whether any of these amphiphilic components are able to stimulate the immune system under physiological conditions as constituents of intact bacteria. However, all of them have a very high pro-inflammatory activity when released from the cell. Such a release may take place through the interaction with the immune system, or with antibiotics (particularly with those targeting cell wall components), or simply by the bacterial division. Therefore, a given antimicrobial agent must ideally have a double character, namely, it must overcome the bacterial cell wall barrier, without inducing the liberation of the pro-inflammatory amphiphiles. Here, new data are presented which describe the development and use of membrane-active antimicrobial agents, in particular antimicrobial peptides (AMPs) and lipopolyamines. In this way, essential progress was achieved, in particular with respect to the inhibition of deleterious consequences of bacterial infections such as severe sepsis and septic shock.
Keywords: Antimicrobial peptides, endotoxin shock, lipopolyamines, lipopolysaccharides, tumor-necrosis-factorα
Introduction
Bacterial infections are still one of the major threats to human health worldwide. Bacterial pathogens frequently cause severe diseases not only as primary agents, but also subsequent to pathologies caused by other agents (superinfection such as ‘Spanish flu’, [1]). This fact is, due, at least in part, to the increasing occurrence of bacterial resistance to antibiotics. In turn, the spread of antibiotic resistant clones is greatly enhanced by the improper use of antibiotics in daily medicinal practise and by the massive application of antibiotics in animal husbandry [2]. Furthermore, in the last decades only two antibiotic classes with novel mechanism of action have been marketed, and none of them are effective against Gram-negative bacteria.
On the other hand, the antimicrobial activity of a given antibiotic does not correlate with its ability to inactivate bacterial pathogenicity factors (PF) and interaction of some antibiotics with their bacterial targets in vivo even enhance the release of PF into the extracellular milieu. This frequently results in bacterial sepsis [8], a pathology that is the leading cause of death in the critical care units of hospitals worldwide.
The cell wall is the first barrier that an antimicrobial agent must counteract [12]. In contrast to Gram positive bacteria, Gram-negative organisms have an additional membrane, the so called outer membrane (OM), that lies over and covers both the cytoplasmic membrane and the peptidoglycan layer. The OM has an asymmetric distribution of the lipids with phospholipids and lipopolysaccharide (LPS) located in the inner and outer leaflets, respectively [13].
Gram-positive organisms lack OM and possess a much thicker peptidoglycan layer compared to their Gram-negative counterparts [12]. The additional class of amphiphiles in Gram-positives, the lipoteichoic acids (LTA), representing important structural components [14], cross-bridge the entire cell envelope with their lipid component inserting into the outer leaflet of the cytoplasmic membrane and the teichoic acid portion penetrating into the peptidoglycan layer. The importance of another class of amphiphiles, the lipoproteins, has become evident only recently [15]. Summarized, the main PF of Gram-negative bacteria is the endotoxin (LPS, lipopolysaccharide), whereas lipoproteins (LP) rank among the most important PF in Gram-positive. Therefore, an ideal antibiotic should combine a high antimicrobial activity with a potent ability to bind and neutralize PF.
A promising approach to develop new effective drugs since the 1990’s is the use of AMPs derived from binding domains of host defence proteins such as lactoferrin, NK-lysin, and Limulus anti-LPS factor (LALF) [9], [10], [11]. Successive cycles of improvement rendered compounds endowed with considerable antimicrobial and anti-endotoxic activity. In addition, pathogens are frequently unable to develop resistance to these rapidly bactericidal agents [3]. For instance, some pathogens such as Pseudomonas aeruginosa remain sensitive only to polymyxins [4], although the systemic use of these AMPs is not widely accepted by clinicians due to their alleged nephro- and neurotoxicity [5]. An important feature of the majority of AMPs is their ability to bind to LPS and permeabilize the outer membrane (OM) of Gram-negative bacteria thus sensitizing bacterial cells to co-administered compounds [6]. In addition, the ability of AMPs to kill cells in stationary phase enables them to be bactericidal not only for planktonic cells but also for bacteria growing in biofilms [7]. Up to now, however, available peptides lack a sufficiently high therapeutical index, which is necessary to avoid detrimental side effects.
Whereas it is clear that amphiphilic components such as LPS and LTA provide important permeability barrier functions, it has not been demonstrated that these molecules stimulate the innate immune system as constituents of intact bacterial cell wall structures. However, all of them are highly active when released from the cell and this occurs either by the action of the immune effector functions (such as complement-mediated lysis, opsonophagocytosis, etc.), or by antibiotics, or simply by cell division of the bacteria. The clinical significance of antibiotic-mediated release of these immunostimulatory and proinflammatory amphiphiles is of high relevance [16], [17]. It is therefore desirable that antimicrobial regimens not only exert bactericidal activity, but also either avoid release of proinflammatory cell wall-derived amphiphiles, or sequester and neutralize them even as they are being released as a consequence of microbial lysis and death.
We present data describing the development and use of dual-purpose membrane-active antimicrobial agents and LPS/LP sequestrants, focusing on AMPs and lipopolyamines. Our long-term goal is to evaluate these compounds either as single-agent antimicrobials or as adjunctive chemotherapeutic agents that may have value in combination with bactericidal antibiotics. These compounds have been shown to effectively neutralize LPS in vitro and to protect mice from a lethal endotoxic shock in an animal model of acute sepsis. Different aspects of the mechanisms of action in in vitro as well as in vivo systems are described in this paper.
Peptide-induced inhibition of inflammation reaction
Lactoferricin-derived peptides
It has been previously shown that acylation of a lactoferricin-derived AMP leads to an improvement both of the antimicrobial and LPS detoxification activities of the resultant compound [18].
To confirm and expand these observations, we used LF11, an 11-amino acid (FQWQRNIRKVR) peptide derived from human lactoferricin, as a template to design a library of peptides and lipopeptides as described elsewhere [19, 20]. To study the influence of acylation on the antimicrobial activity of the compounds we measured their minimum inhibitory concentration (MIC) on Gram-negative and Gram-positive bacterial pathogens grown under conventional (planktonic) culture conditions. In addition, we tested the ability of the peptides and their acylated derivatives to reduce the viability of biofilms formed by Pseudomonas aeruginosa PAO1 and/or to remove them. Results obtained with peptides P2-15, P2-27, P2-29 and P2-33 (see Table 1) exemplified to a large extent the behaviour observed with the entire library. In general, acylation neither increased the potency nor the spectrum of antimicrobial activity of the compounds since lipopeptides had equal or worse MIC than their parent molecules. These results show that sometimes acylation of a lactoferricin-derived peptide can be detrimental for its antimicrobial performance as opposed to previous observations with LF11 and its N-acyl derivative lauryl-LF11 [18]. In addition, our data confirm conclusions of Japelj and collaborators reporting that in synthetic lipopeptides the acyl chain represents an integral part of the molecular structure thus influencing the shape of the lipopeptide for optimal interaction with bacterial membrane [21]. Notably, the only organism that was more sensitive to the acylated than to the parent compounds was Brucella abortus, a pathogen endowed with a lipopolysaccharide and a cell envelope unconventional for a Gram-negative bacterium [22].
Table 1.
Antibacterial and antiendotoxic activities of the peptides and their N-acyl derived Lipopeptides
| Compound | Sequence1 | MIC (μg/mL) of compound on the indicated bacterial strain
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ec | Ec-2 | Pa | Pa-2 | Ab | Sm | Bb | Ba | Sa | ||
| P2-15 | F W R I R I R R -NH2 | 8 | 8 | 32 | 16 | 128 | 64 | 2 | 64 | 32 |
| LP3-55 | octanoil-P2-15 | 32 | 16 | 64 | 64 | 64 | 256 | 8 | 8 | 16 |
| LP3-56 | 2-etilhexanoil-P2-15 | 16 | 8 | 32 | 64 | 64 | 128 | 8 | 8 | 16 |
| LP3-57 | 2,2-dimetilbutanoil-P2-15 | 8 | 8 | 32 | 8 | 64 | 128 | 4 | 8 | 8 |
| LP3-58 | 6-metiloctanoil-P2-15 | 32 | 16 | 64 | 256 | 64 | 256 | 16 | 8 | 64 |
| LP3-79 | diciclohexilacetil-P2-15 | 32 | 64 | 256 | 64 | 256 | >256 | 8 | 8 | 32 |
|
| ||||||||||
| P2-27 | F W R R F W R R -NH2 | 16 | 64 | 64 | 32 | 128 | 256 | 4 | 16 | 16 |
| LP3-60 | octanoil-P2-27 | 64 | 128 | 128 | 128 | 256 | 256 | 8 | 8 | 128 |
| LP3-63 | 6-metiloctanoil-P2-27 | 64 | 64 | 256 | 128 | 256 | 128 | 8 | 16 | 16 |
|
| ||||||||||
| P2-29 | F W R I W R W R -NH2 | 16 | 16 | 32 | 8 | 32 | 128 | 4 | 16 | 8 |
|
| ||||||||||
| P2-33 | P F W R I R I R R -NH2 | 16 | 64 | 64 | 32 | 128 | 256 | 4 | 16 | 16 |
| LP3-53 | octanoil-P2-33 | 64 | 128 | 128 | 128 | 256 | 256 | 8 | 8 | 128 |
Parent peptides and their N-acyl derivatives were synthesized with an amidated C-terminus
Minimum inhibitory concentration of the compound determined by a microbroth-based assay in Mueller-Hinton medium. Abbreviations: Ec, Escherichia coli ATCC 25922; Ec-2; Escherichia coli DC2; Pa, Pseudomonas aeruginosa 4158-02 CUN; Pa-2, Pseudomonas aeruginosa ATCC 27853; Ab, Acinetobacter baumannii ATCC 19606; Sm, Stenotrophomonas maltophilia 3998-00 CUN; Bb, Bordetella bronchiseptica 11844-99 CUN; Ba, Brucella abortus 9,49 perA; Sa, Staphylococcus aureus ATCC 25923; MRSA, Staphylococcus aureus ATCC 43300 (MRSA)
novobiocin enhancing activity of peptides on Pseudomonas aeruginosa Ps4 expressed as Fractional Inhibitory Concentration (FIC) index. FIC index was calculated according to the following formula: [(A) /MICA] + [(P) /MICP]= FICA + FICP = FIC index, where MICA and MICP are the MICs of the antibiotic and peptide determined separately, and (A) and (P) are the MICs of the antibiotic and peptide when determined in combination
number on the left: percentage of killing by the compound (MTT test; 4 h of treatment) when the peptide was added at 10 times its MIC on a biofilm of Pseudomonas aeruginosa PAO1; number on the right: percentage of biofilm removal determined by crystal violet staining under the same conditions of the killing assay. Values of standard deviation were always below 11%. n.d. not determined
survival of CD-1 mice after 7 days of being inoculated intraperitoneally (i.p) with 0.3 μg of lipopolysaccharide from Pseudomonas aeruginosa PAO1 followed 1 min later by the compound (150 μg, i.p.); galactosamine (18 mg) was co-inoculated with the lipopolysaccharide to sensitize the animals to endotoxin. −, +, ++, +++ denotes the level of protection (null, low, intermediate, high, respectively) afforded by the compound with respect to a control group of animals receiving no treatment
Testing the antimicrobial activity of compounds on biofilms instead of on planktonic cells heightened the differences between peptides and lipopeptides. Thus, whereas the former agents killed at least 95 % of cells and showed a significant biofilm-removing activity, most lipopeptides displayed a very modest performance under the same conditions. The low anti-biofilm activity of lipopeptides may be due to their higher affinity for alginate, a component of the extracellular matrix of P. aeruginosa biofilm that has been reported to bind hydrophobic compounds [23]. Additionally, it is possible that the higher molecular weight of the acylated compounds may hinder their penetration into the extracellular matrix of the biofilm.
To study if acylation improved the OM-permeabilizing activity of peptides, we measured the ability of all the compounds to act in synergy with novobiocin against P. aeruginosa. This hydrophobic antibiotic cannot reach its intracellular target, gyrase, due to its inability to cross an intact outer membrane. To quantify the synergistic effect, we determined the Fractional Inhibitory Concentration (FIC) index of the peptide-novobiocin combinations. The majority of authors consider a combination as synergistic when its FIC index is ≤ 0.5. As shown in Table 1, acylation systematically resulted in compounds devoid of the potent novobiocin-enhancing activity displayed by the parent peptides. In agreement with this observation, Vaara and Vaara demonstrated that OM-permeabilizing activity does not depend on acylation since PMBN, a deacylated derivative of PMB, is one of the most potent permeabilizers known to date [24]. The exception to this behaviour was peptide P2-29, the compound rendering the lowest MIC on the test strain (31.25 μg/mL). Lack of synergistic activity of P2-29 could be due to the fact that its lethal (bactericidal) activity is so great that it is not possible to observe non-lethal (permeabilization) activity, as it has been reported previously [20].
In light of our results, one could conclude that acylation of peptides may be of minor importance since it appears to lead to compounds lacking any meaningful biological property. To the contrary, our data demonstrate that the ability to bind LPS in vivo and to protect animals from a lethal endotoxic shock is strongly dependent on acylation (see Table 1). In all the cases, protection was associated with a very significant drop in TNFα serum levels 60 min after inoculation of the lipopeptide (data not shown). These observations are in good correlation with those mentioned above by Andrä and collaborators [18] and by Tsubery and collaborators [25] reporting that lipopeptides display a much higher LPS neutralizing activity compared to their non-acylated counterparts. The degree of protection conferred by the lipopeptides was very high and homogenous with very few exceptions (Table 1 and our unpublished results). Therefore, it was not possible to establish relationships between antiendotoxic activity and the presence of a particular amino acid sequence or acyl groups in the compounds. Our results demonstrate that acylation is sufficient in most of the cases to turn a peptide with poor affinity towards LPS into a compound endowed with potent anti-endotoxic activity in vivo.
Peptides based on Limulus anti-LPS factor and derivatives
For the development of suitable compounds to neutralize bacterial endotoxins a peptide library was constructed which were originally based on the binding region of the Limulus anti-LPS factor (LALF) [26]. The LALF was crystallized and the LPS-binding domain was identified [27]. Ried et al [28] were the first authors reporting synthesis of peptides based on this domain. They found that the complete binding sequence cLALF22 (amino acid sequence 31–52 of LALF: GCHYRIKPTFRRLKWKYKGKFWCG) had the highest ability to bind to the lipid A part of LPS, its ‘endotoxic principle’, whereas the shortened analogs down to cLALF10 (LALF sequence 38–45, GCTFRRLKKCG) were much less active. Andrä et al performed biophysical studies of the interaction of LALF (in recombinant form called ENP = endotoxin-neutralizing protein) and part structures with endotoxins [9], [29], [30], and found considerable inhibition of the biological activity of LPS at [ENP]:[LPS] 20 to 200:1 molar: Parallel to this, data on the aggregate supramolecular structure of LPS, performed with synchrotron radiation small-angle X-ray scattering (SAXS), were indicative of a change of the lipid A cubic structure into a more lamellar one. Importantly, determination of the surface potential of LPS, the Zeta potential, which is a measure of the accessible negative charges, showed an increase from −70 mV to positive values for lipid A and LPS Re. LALF-derived cyclic peptides were analysed in subsequent studies [31], [32], starting with the compound with the complete LPS-binding domain (cLALF22) and various shortened analogues. In all these studies the data consistently showed a change of the endotoxin aggregate structure from preferentially cubic into a multilamellar one, as well as a change of the morphologies of the LPS Re aggregates, as evidenced by freeze-fracture electron microscopy, from ‘open egg-shells’ (i.e., spherical particles in the range 100 to 200 nm), into large stacks of some 1000 nm. The latter structures corresponded to the multilamellae in the SAXS experiment with periodicities of 6.3 to 7.5 nm. With isothermal titration calorimetry (ITC) the binding of the peptides to LPS in all cases was characterized as an exothermic process, driven by the Coulomb interaction of the positive charges of peptides with the negative charges of the endotoxins.
The terminal part of the shrimp anti-LPS-factor, a synthetic peptide with 24 AA, cyclic and linear with a sequence Ac- ECKFTVKPYLKRFQVYYKGRMWCP, was analysed by Pan et al [31]. Pre-treatment of mice with the cyclic compound led to a considerable enhancement of survival of mice which had been infected with 1.28 · 106 cfu from Pseudomonas aeruginosa, and the peptides were able to decrease the bacteria-induced production of TNFα in the animals. Unfortunately, the peptides exhibited at a concentration of already 2 μg/ml significant cytotoxicity in HeLa, MCF-7, and HT1080 cell lines, as measured by the MTT test.
Due to the inability of the above described peptides to get a complete anti-se3ptic action, we have designed and constructed a new series of 19’mer peptides. The biophysical analysis of peptide 19-2 (sequence see Table 2) exhibited strong endotoxin binding, as deduced from ITC experiments, resulting in a nearly complete blocking of the LPS-induced TNFα production of human MNC at [Pep]:[LPS] 100:1 weight% [32]. A slightly shortened variant exhibited much less anti-endotoxic activity, but considerably higher antibacterial activity. This illustrates that the two parameters, antibacterial and anti-endotoxin activity, not necessarily correlate.
Table 2.
Sequences of the Pep19 series
| • Pep9 | FRRLKWKFW |
| • Pep12-2 | FRRLKWKKFWFW |
| • Pep17-1 | KKFRRLKWKYKGKFWFW |
| Pep17-2 | KKYRRFRWKFKGKFWFW |
| Pep17-3 | RRYKKFKWRYRGRFWFW |
| • Pep19-2: | GCKKYRRFRWKFKGKFWFWCG |
| • Pep19-2.2 | GCKKYRRFRWKFKGKFWFW |
| • Pep19-2.5 | GCKKYRRFRWKFKGKFWFWG |
| • Pep19-5: | GKKYRRFRWKFRKGRFWFWG |
| • Pep19-6: | GCKKFRRFKLKCKQKLWLWCG |
| Pep19-7: | GKKYRRFWKFKGKWFWFWG |
| • Pep19-12: | GCRRFKKFKKWRYRGRFWFWCFG |
| • Pep19-12.1 | GCKKFRRFRRWKYKGKFWFWCFG |
| • Pep19-12.1cyclic | GCKKFRRFRRWKYKGKFWFWCFG |
The systematic study of various peptides differing in their amino acid chain lengths from 9 over 12 and 17 to 19, showed that the basic sequence, as given by Pep9, is already able to confer a significant inhibition of the LPS-induced cytokine production (Fig. 1). However, relatively high concentrations of the peptides were still necessary to inhibit cytokine production and we found that we could improve peptide’s activity in this respect by prolonging the peptide length with suitable amino acids. As can be seen in the Figures 1–2, the increase in the AA acid chain lengths must be adapted to the physico-chemistry of the lipid A part. To achieve this, the N-terminal side of the peptide must be composed essentially of polar and basic AA, the C-terminal must be more hydrophobic. This alone, however, is not enough, the exact number and type and sequence of AA is important, as deduced from the comparison of the peptides with different chain lengths (Fig. 1) as well as the comparison of Pep19-5, -6, and -7 (Fig. 2).
Figure 1.

Inhibition of the LPS-induced production of tumor-necrosis-factorα in human mononuclear cells by peptides with different AA chain lengths at different [LPS]:[Pep] weight ratios.
Figure 2.

Inhibition of the LPS-induced production of tumor-necrosis-factorα in human mononuclear cells by peptides Pep19-5, Pep19-6, and Pep19-7 at different [LPS]:[Pep] weight ratios.
The importance of an exact length and composition of the AA to neutralize LPS is illustrated by the results shown in Table 3, in which the action of two peptides with 17 AA is compared with those of Pep19-2 in a mouse model of sepsis, yielding only protection for the longer peptide.
Table 3.
Survival of mice
| Peptide | Dead mice/Number of mice after 48 h |
|---|---|
| LPS alone | 10/10 |
| LPS + Polymyxin B | 0/10 |
| LPS + Pep17-1 | 7/10 |
| LPS + Pep17-2 | 7/10 |
| LPS + Pep19-2 | 0/10 |
Further improvements were obtained by modifications of the sequences (AA sequences see Table 2), which led to a nearly complete inhibition of the LPS-induced cytokine secretion at a [Pep]:[LPS] 3:1 molar ratio [33]. This is particularly evident in the case of the peptide Pep19-2.5, our lead structure, which could protect animals in the mouse model of sepsis already at a [Pep]:[LPS] ratio of 50:1 weight % (25 ng LPS and 1250 ng peptide). Also, peptides Pep19-2.2 and Pep19-12.1 were very effective in protecting animals in the animal model of sepsis at peptide concentrations far below their threshold of cytotoxicity (Fig. 3). These investigations were performed with a rough mutant LPS Ra from S. minnesota strain R60. To assure that such strong inhibition activity takes also place for naturally occurring heterogeneous smooth mutant LPS, the inhibition of the cytokine secretion induced by LPS from E. coli O8:K27 was monitored by using the lead structure Pep19-2.5 (Fig. 4). As can be seen, also for this endotoxin a considerable reduction of the LPS-induced concentration of TNFα takes place already at [Pep]:[LPS] 3:1 weight %.
Figure 3.

Survival of mice in the galactosamine model of endotoxin shock for Pep12-2 and Pep12.1 in comparison to the gold standard| polymyxin B (PMB). LPS from Pseudomnas aeruginosa was administered i.p. at a concentration of, and the peptides were added at a concentration of
Figure 4.

Inhibition of the tumor-necrosis-factorα in human mononuclear cells, induced by LPS from E.coli O8:K27, by the lead peptide Pep19-2.5 at three different [LPS]:[Pep] weight ratios.
Since it was reported in literature, that peptides in a cyclic form, made by connecting two cysteins via disulfide bridges, had higher cytokine inhibitory activity than the corresponding linear peptides [34], we have constructed peptide Pep19-12.1 besides in its linear also in a cyclic form by bridging the second Cys at the N-terminal with the third Cys as seen from the C-terminal side (see Tab. 2). The comparison of the two peptides to inhibit the LPS-induced cytokine reaction showed unequivocally (Fig. 5) that the cyclic form, although still active, had significantly less ability to suppress the inflammation reaction.
Figure 5.

Inhibition of the LPS-induced production of tumor-necrosis-factorα in human mononuclear cells by peptides Pep12.1 in linear and cycled form at different [LPS]:[Pep] weight ratios.
Finally, for therapeutical use it would be of essential importance that some decrease of the inflammation reaction takes place also, when the peptide is administered in a time-delayed mode. Therefore, the peptide Pep19-2.2 was added at times 0 up to 3 h after the LPS administration (whole incubation time 4 h). As can be deduced from Fig. 6, there is still a considerable inhibition of the TNFα production even when the peptide is added 3 h after LPS addition, despite the fact that the action of the peptide can take place only in the remaining stimulation time of 1 h.
Figure 6.

Inhibition of the LPS-induced production of tumor-necrosis-factorα in human mononuclear cells by peptide Pep19-2.2, at t=0 and added time-delayed up to 3 h later (entire stimulation time 4 hours). [LPS]:[Pep19-2.2] 1:100 weight%.
As mechanism of action for the ability of the peptides to neutralize endotoxins, the change of lipid A / LPS cubic aggregate into a multilamellar structure was proposed, connected with an extremely low saturation value of binding at [peptide]:[LPS] = 0.3 M/M, corresponding to the binding saturation of 3 peptides with 10 LPS molecules [34]. Thus, human binding proteins such as LBP and CD14 are no more able to bind to the LPS epitopes and thus cannot initiate the inflammation reaction. Furthermore, the ability of the peptides to act even after LPS administration is indicative of a membrane process of the interaction. This is presently under investigation.
Neutralization of endotoxin by lipopolyamines
Recognition that membrane-active antimicrobial compounds may also possess LPS-sequestering and -neutralizing activities was first identified with polymyxin B [36]. As described above, a variety of cationic, amphipathic peptides share these properties, as was evident with melittin, a basic, amphipathic peptide constituent of bee venom [37]. as well as structurally unrelated peptides such as gramicidin S, tyrocidin, efrapeptin as well as de novo designed peptides [38],[39],[40]. Hypothesizing that polycationicity and amphipathicity are necessary and sufficient requirements for bacterial membrane perturbation and for LPS sequestration, we turned our attention to small molecules embodying the above-mentioned physical properties. Upon finding that bis-arg Gemini compounds indeed displayed microbicidal and LPS-sequestering [41] activities, and that DOSPER, a model lipopolyamine afforded protection in murine models of both purified LPS- and antibiotic-induced LPS release models [42] led us eventually to a careful examination of the lipopolyamine class of compounds typified by N-acyl or N-alkyl spermine or homospermine [43], [44], [45],[46], [47], [48], [49],[50].
In examining a homologous series of mono- and bis-acyl polyamines, it was noted that the carbon number (hydrophobicity) was a critical structural determinant of LPS-neutralizing potency [50]; for the mono-acyl 4 series of compounds, there was a progressive increase in LPS-neutralizing potency (Fig. 7), while for the bis-acyl 8 series, the activity progressively decreased with acyl chains longer than dodecyl (C12). It was of interest to probe possible correlations with MICs against E. coli versus S. aureus. The results shown in Fig. 8 indicate that against both organisms, a very similar structure-activity correlation is observed. Thus, for the 4 compounds, acyl chain lengths from C12 to C16 result in maximal antimicrobial efficacy against S. aureus, while maximal antibacterial effects are observed between C12 to C14 against E. coli, with the activity falling off at C16, suggesting that the structural requisites for optimal interaction with the Gram-negative outer membrane are rather specific. For the 8 series, however, the converse is true with the short chain (C8–11) analogues exhibiting maximal antibacterial effect (Fig. 9); the decline in activity in the higher homologues in the 8 series is ascribable to progressive loss of aqueous solubility. These structure-activity relationships of these compounds closely mirror that of LPS-induced TNF-α and nitric oxide (NO) production in murine macrophages.[46] The similarities of the 4 and 8 series between antimicrobial activity against Gram-negative bacteria on the one hand, and sequestration of LPS on the other, suggest that the antimicrobial activity is likely mediated via the interaction of these compounds with the outer membrane [50].
Figure 7.
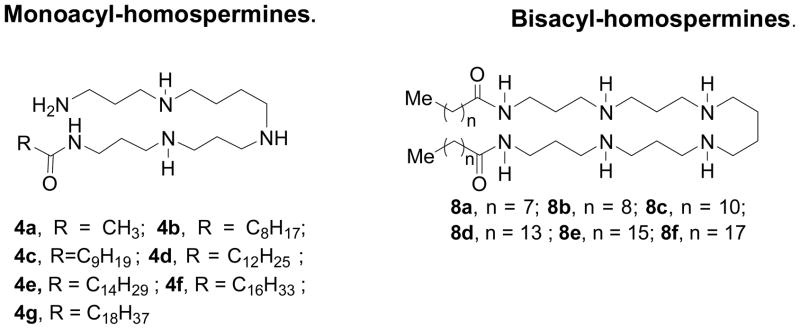
Structures of the mono-acyl (4 series) and bis-acyl (8 series) lipopolyamines (top). Correlation of MIC against S. aureus and E. coli with length of the acyl group.
Figure 8.

Antimicrobial activity of various lipopolyamines against E. coli and S. aureus.
Figure 9.

Correlation of outer- (OM) and inner-membrane (IM) permeabilizing activity. OM permeabilizing activity was determined using E. coli ML-35p (parent ATCC 43827 transformed with pBR322 vector encoding periplasmic β-lactamase); the leakage of periplasmic β-lactamase activity was quantified using nitrocefin as a chromogenic substrate. IM permeabilizing activity was determined using E. coli ML-35 using o-nitrophenyl-α-D-galactopyranoside as the substrate to determine the α-galactosidase activity. Polymyxin B, Polymyxin B nonapeptide (PMBN) and melittin were used as reference compounds.
Our primary interest in examining these compounds, however, lay not so much in evaluating their intrinsic antimicrobial properties, but rather in understanding the mechanisms and structure-activity relationships underlying their putative membrane permeabilizing action, and in exploring the possibility of employing such compounds as adjuncts to conventional antimicrobial chemotherapy for purposes of sequestering endotoxin released as a consequence of Gram-negative bacterial lysis. We first investigated if the acylpolyamines would act on both the inner membrane (IM) and the outer membrane (OM) [50], [51], presumably as a consequence of nonspecific membranophilic effects as has been reported for a variety of cationic amphipathic peptides such as melittin [37], defensins [51], bactenecins [52] or, selectively perturb the OM in the manner of polymyxin B [53]. This is shown in Fig. 8, a direct linear relationship was observed between OM and IM permeabilizing activities [50]. Furthermore, these two events seem tightly coupled with near-identical kinetics even under conditions of high osmotic strength. It is to be noted that IM damage would necessarily require antecedent OM lysis.
Hypothesizing that compounds that sequester LPS could also bind to and inhibit LTA-induced cellular activation, we screened congeneric series of polyamine sulfonamides which we had previously shown effectively neutralized LPS both in vitro and in animal models of endotoxemia. We observed that these compounds indeed bind and neutralize LTA, as reflected by the inhibition of TLR2-mediated NF-κB induction in reporter gene assays. Structure-activity studies showed a clear dependence of the acyl chain length on activity against LTA in compounds with spermine and homospermine scaffolds [54]. We then sought to examine possible correlations between the neutralizing potency toward LTA and antimicrobial activity in S. aureus. A linear relationship between LTA sequestration activity and antimicrobial activity for compounds with a spermine backbone was observed, while all compounds with a homospermine backbone were equally active against S. aureus, regardless of their neutralizing potency toward LTA [54]. These results suggest that the number of protonatable charges is a key determinant of the activity toward the membranes of Gram-positive bacteria.
The development of resistance to membrane-active antibiotics has been relatively slower than that to conventional antibiotics, and it is possible that compounds such as the acylpolyamines may be useful clinically, provided that they have an acceptable safety profile and margin of safety. A more detailed understanding of the mechanisms of interactions of these compounds with LPS and LTA, as well as the Gram-negative and -positive bacterial cell surfaces, will be instructive and should allow the rational design of analogues which combine antisepsis and antibacterial properties.
Acknowledgments
The authors are indebted the German ministry (Ministerium für Bildung und Forschung) BMBF for financial help in the frame of a preclinical study ‘Therapy of infectious diseases with special regards to bacterial sepsis‘ (project: 01GU0824). G.M.T. was funded by a grant from Ministerio de Sanidad y Consumo (FIS-PI050768), from Proyectos de Investigación Universidad de Navarra (PIUNA-2008-11) Spain, and received financial support from the Commission of the European Communities, specific RTD program ‘Quality of Life and Management of Living Resources’ (QLCK2-CT-2002-01001, Antimicrobial Endotoxin Neutralizing Peptides To Combat Infectious Diseases). S. A. David gratefully acknowledges NIH support via grants 5U01AI077947, 5U01AI054785, and 5U01AI056476.
References
- 1.Springer J, Safley M, Huber S, Troxclair D, Craver R, Newman WP, et al. Histopathological findings in fatal novel H1N1: an autopsy case series from September-November 2009 in New Orleans. Louisiana J La State Med Soc. 2010;162:88–91. [PubMed] [Google Scholar]
- 2.Guillemot D. Antibiotic use in humans and bacterial resistance. Curr Opin Microbiol. 1999;2:494–498. doi: 10.1016/s1369-5274(99)00006-5. [DOI] [PubMed] [Google Scholar]
- 3.Jenssen H, Hamill P, Hancock RE. Peptide antimicrobial agents. Clin Microbiol Rev. 2006;19:491–511. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mesaros N, Nordmann P, Plesiat P, Roussel-Delvallez M, Van Eldere J, Glupczynski Y, et al. Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin Microbiol Infect. 2007;13:560–578. doi: 10.1111/j.1469-0691.2007.01681.x. [DOI] [PubMed] [Google Scholar]
- 5.Garidel P, Brandenburg K. Current understanding of polymyxin B applications in bacteraemia/sepsis therapy prevention: clinical, pharmaceutical, structural and mechanistic aspects. Antiinfect Agents Medic Chem. 2009;8:367–385. [Google Scholar]
- 6.Savage PB. Multidrug-resistant bacteria: overcoming antibiotic permeability barriers of gram-negative bacteria. Ann Med. 2001;33:167–171. doi: 10.3109/07853890109002073. [DOI] [PubMed] [Google Scholar]
- 7.Wei GX, Campagna AN, Bobek LA. Effect of MUC7 peptides on the growth of bacteria and on Streptococcus mutans biofilm. J Antimicrob Chemother. 2006;57 :1100–1109. doi: 10.1093/jac/dkl120. [DOI] [PubMed] [Google Scholar]
- 8.Cross AS, Opal SM. Endotoxin’s role in Gram-negative bacterial infection. Curr Opin Infect Dis. 1995;8:156–163. [Google Scholar]
- 9.Andrä J, Garidel P, Majerle A, Jerala R, Ridge R, Paus E, et al. Biophysical characterization of the interaction of Limulus polyphemus endotoxin neutralizing protein with lipopolysaccharide. Eur J Biochem. 2004;271:2037–2046. doi: 10.1111/j.1432-1033.2004.04134.x. [DOI] [PubMed] [Google Scholar]
- 10.Brandenburg K, Jürgens G, Müller M, Fukuoka S, Koch MHJ. Biophysical characterization of lipopolysaccharide and lipid A inactivation by lactoferrin. Biol Chem. 2001;382:1215–1225. doi: 10.1515/BC.2001.152. [DOI] [PubMed] [Google Scholar]
- 11.Andrä J, Koch MHJ, Bartels R, Brandenburg K. Biophysical characterization of endotoxin inactivation by NK-2, an antimicrobial peptide derived from mammalian NK-lysin. Antimicrob Agents Chemother. 2004;48:1593–1599. doi: 10.1128/AAC.48.5.1593-1599.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baddiley J. Bacterial cell walls and membranes. Discovery of the teichoic acids BioEssays. 1989;10:207–210. doi: 10.1002/bies.950100607. [DOI] [PubMed] [Google Scholar]
- 15.Hayashi S, Wu HC. Lipoproteins in bacteria. J Bioenerg Biomembr. 1990;22:451–471. doi: 10.1007/BF00763177. [DOI] [PubMed] [Google Scholar]
- 16.Holzheimer RG. The significance of endotoxin release in experimental and clinical sepsis in surgical patients--evidence for antibiotic-induced endotoxin release? Infection. 1998;26:77–84. doi: 10.1007/BF02767765. [DOI] [PubMed] [Google Scholar]
- 17.Kirikae T, Nakano M, Morrison DC. Antibiotic-induced endotoxin release from bacteria and its clinical significance. Microbiol Immunol. 1997;41:285–294. doi: 10.1111/j.1348-0421.1997.tb01203.x. [DOI] [PubMed] [Google Scholar]
- 18.Andrä J, Lohner K, Blondelle SE, Jerala R, Moriyon I, Koch MHJ, et al. Enhancement of endotoxin neutralization by coupling of a C12-alkyl chain to a lactoferricin-derived peptide. Biochem J. 2005;385:135–143. doi: 10.1042/BJ20041270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez-Gomez S, Lamata M, Leiva J, Blondelle SE, Jerala R, Andrä J, et al. Comparative analysis of selected methods for the assessment of antimicrobial and membrane-permeabilizing activity: a case study for lactoferricin derived peptides. BMC Microbiol. 2008;8:196. doi: 10.1186/1471-2180-8-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanchez-Gomez S, Japelj B, Jerala R, Moriyon I, Fernandez AM, Leiva J, et al. Structural features governing the activity of lactoferricin-derived peptides that act in synergy with antibiotics against Pseudomonas aeruginosa in vitro and in vivo. Antimicrob Agents Chemother. 2011;55:218–228. doi: 10.1128/AAC.00904-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Japelj B, Pristovsek P, Majerle A, Jerala R. Structural origin of endotoxin neutralization and antimicrobial activity of a lactoferrin-based peptide. J Biol Chem. 2005;280:16955–16961. doi: 10.1074/jbc.M500266200. [DOI] [PubMed] [Google Scholar]
- 22.Martinez deTejada G, Moriyon I. The outer membranes of Brucella spp. are not barriers to hydrophobic permeants. J Bacteriol. 1993;175:5273–5275. doi: 10.1128/jb.175.16.5273-5275.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan C, Burrows LL, Deber CM. Alginate as an auxiliary bacterial membrane: binding of membrane-active peptides by polysaccharides. J Pept Res. 2005;65:343–351. doi: 10.1111/j.1399-3011.2005.00217.x. [DOI] [PubMed] [Google Scholar]
- 24.Vaara M, Vaara T. Polycations sensitize enteric bacteria to antibiotics. Antimicrob Agents Chemother. 1983;24:107–113. doi: 10.1128/aac.24.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsubery H, Ofek I, Cohen S, Fridkin M. Structure-function studies of polymyxin B nonapeptide: implications to sensitization of gram-negative bacteria. J Med Chem. 2000;43:3085–3092. doi: 10.1021/jm0000057. [DOI] [PubMed] [Google Scholar]
- 26.Vallespie MG, Glaria LA, Reyes O, Garay HE, Ferrero J, Arana MJ. Limulus antilipopolysaccharide factor-derived peptide exhibits a new immunological activity with potential applicability in infectious diseases. Clin DiagnLab Immunol. 2000;4:669–675. doi: 10.1128/cdli.7.4.669-675.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoess A, Watson S, Siber GR, Liddington R. Crystal structure of an endotoxin-neutralizing protein from the horseshoe crab, Limulus anti-LPS factor, at 1. 5 A resolution. EMBO J. 1993;12:3351–3356. doi: 10.1002/j.1460-2075.1993.tb06008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ried C, Wahl C, Miethke T, Wellnhofer G, Landgraf C, Schneider-Mergener J, et al. High affinity endotoxin-binding and neutralizing peptides based on the crystal structure of recombinant Limulus anti-lipopolysaccharide factor. J Biol Chem. 1996;271:28120–28127. doi: 10.1074/jbc.271.45.28120. [DOI] [PubMed] [Google Scholar]
- 29.Andrä J, Lamata M, Martinez deTejada G, Bartels R, Koch MHJ, Brandenburg K. Cyclic antimicrobial peptides based on Limulus anti-lipopolysaccharide factor for neutralization of lipopolysaccharide. Biochem Pharmacol. 2004;68:1297–1307. doi: 10.1016/j.bcp.2004.05.054. [DOI] [PubMed] [Google Scholar]
- 30.Andrä J, Howe J, Garidel P, Rössle M, Richter W, Leiva-Leon J, et al. Mechanism of interaction of optimized Limulus-derived cyclic peptides with endotoxins: thermodynamic, biophysical and microbiological analysis. Biochem J. 2007;406:297–307. doi: 10.1042/BJ20070279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan CY, Chao TT, Chen JC, Chen JY, Liu WC, Lin CH, et al. Shrimp (Penaeus monodon) anti-lipopolysaccharide factor reduces the lethality of Pseudomonas aeruginosa sepsis in mice. Int Immunopharmacol. 2007;7:687–700. doi: 10.1016/j.intimp.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Kowalski I, Kaconis Y, Andra J, Razquin-Olazaran I, Gutsmann T, Martinez de Tejada G, et al. Physicochemical and biological characterization of anti-endotoxin peptides and their influence on lipid properties. Protein Pept Lett. 2010;17 doi: 10.2174/0929866511009011328. (in press) [DOI] [PubMed] [Google Scholar]
- 33.Gutsmann T, Razquin-Olazaran I, Kowalski I, Kaconis Y, Howe J, Bartels R, et al. New antiseptic peptides to protect against endotoxin-mediated shock. Antimicrob Agents Chemother. 2010;54:3817–3824. doi: 10.1128/AAC.00534-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dankesreiter S, Hoess A, Schneider-Mergener J, Wagner H, Mietke T. Synthetic endotoxin-binding peptides block endotoxin-triggered TNF-α production by macrophages in vitro and in vivo and prevent endotoxin-mediated toxic shock. J Immunol. 2000;164:4804–4811. doi: 10.4049/jimmunol.164.9.4804. [DOI] [PubMed] [Google Scholar]
- 35.Bhattacharjya S, David SA, Mathan VI, Balaram P. Polymyxin B nonapetide: Conformations in water and in the lipopolysaccharide-bound state determined by two-dimensional NMR and molecular dynamics. Biopolymers. 1997;41:251–265. [Google Scholar]
- 36.Morrison DC, Jacobs DM. Binding of polymyxin B to the Lipid A portion of bacterial lipopolysaccharides. Immunochemistry. 1976;13:813–818. doi: 10.1016/0019-2791(76)90181-6. [DOI] [PubMed] [Google Scholar]
- 37.David SA, Mathan VI, Balaram P. Interaction of melittin with endotoxic lipid A. Biochim Biophys Acta. 1992;1123:269–274. doi: 10.1016/0005-2760(92)90006-h. [DOI] [PubMed] [Google Scholar]
- 38.David SA, Balaram P, Mathan VI. Interaction of basic amphiphilic polypeptide antimicrobials, Gramicidin S, Tyrocidin, and Efrapeptin, with endotoxic lipid A. Med Microbiol Lett. 1993;2:42–47. [Google Scholar]
- 39.David SA, Awasthi SK, Wiese A, Ulmer AJ, Lindner B, Brandenburg K, et al. Characterization of the interactions of a polycationic, amphiphilic, terminally branched oligopeptide with lipid A and lipopolysaccharide from the deep rough mutant of Salmonella minnesota. J Endotoxin Res. 1996;3:369–379. [Google Scholar]
- 40.Bhunia A, Chua GL, Domadia PN, Warshakoon H, Cromer JR, David SA, et al. Interactions of a designed peptide with lipopolysaccharide: Bound conformation and anti-endotoxic activity. Biochem Biophys Res Commun. 2008;369:853–857. doi: 10.1016/j.bbrc.2008.02.105. [DOI] [PubMed] [Google Scholar]
- 41.David SA, Perez L, Infante MR. Sequestration of bacterial lipopolysaccharide by bis(Args) gemini compounds. Bioorg Med Chem Lett. 2002;12:357–360. doi: 10.1016/s0960-894x(01)00749-1. [DOI] [PubMed] [Google Scholar]
- 42.David SA, Silverstein R, Amura CR, Kielian T, Morrison DC. Lipopolyamines: novel antiendotoxin compounds that reduce mortality in experimental sepsis caused by gram-negative bacteria. Antimicrob Agents Chemother. 1999;43:912–919. doi: 10.1128/aac.43.4.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blagbrough IS, Geall AJ, David SA. Lipopolyamines incorporating the tetraamine spermine, bound to an alkyl chain, sequester bacterial lipopolysaccharide. Bioorg Med Chem Lett. 2000;10:1959–1962. doi: 10.1016/s0960-894x(00)00380-2. [DOI] [PubMed] [Google Scholar]
- 44.Burns MR, Wood SJ, Miller KA, Nguyen T, Cromer JR, David SA. Lysine-spermine conjugates: hydrophobic polyamine amides as potent lipopolysaccharide sequestrants. Bioorg Med Chem. 2005;13:2523–2536. doi: 10.1016/j.bmc.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 45.Burns MR, Jenkins SA, Wood SJ, Miller K, David SA. Structure-activity relationships in lipopolysaccharide neutralizers: design, synthesis, and biological evaluation of a 540-membered amphipathic bisamide library. J Comb Chem. 2006;8:32–43. doi: 10.1021/cc0500755. [DOI] [PubMed] [Google Scholar]
- 46.Miller KA, Suresh Kumar EV, Wood SJ, Cromer JR, Datta A, David SA. Lipopolysaccharide sequestrants: structural correlates of activity and toxicity in novel acylhomospermines. J Med Chem. 2005;48:2589–2599. doi: 10.1021/jm049449j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khownium K, Wood SJ, Miller KA, Balakrishna R, Nguyen TB, Kimbrell MR, et al. Novel endotoxin-sequestering compounds with terephthalaldehyde-bis-guanylhydrazone scaffolds. Bioorg Med Chem Lett. 2006;16:1305–1308. doi: 10.1016/j.bmcl.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 48.Shrestha A, Sil D, Malladi SS, Warshakoon HJ, David SA. Structure-activity relationships of lipopolysaccharide sequestration in N-alkylpolyamines. Bioorg Med Chem Lett. 2009;19:2478–2481. doi: 10.1016/j.bmcl.2009.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sil D, Shrestha A, Kimbrell MR, Nguyen TB, Adisechan AK, Balakrishna R, et al. Bound to shock: protection from lethal endotoxemic shock by a novel, nontoxic, alkylpolyamine lipopolysaccharide sequestrant. Antimicrob Agents Chemother. 2007;51:2811–2819. doi: 10.1128/AAC.00200-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balakrishna R, Wood SJ, Nguyen TB, Miller KA, Suresh Kumar EV, Datta A, et al. Structural correlates of antibacterial and membrane-permeabilizing activities in acylpolyamines. Antimicrob Agents Chemother. 2006;50:852–861. doi: 10.1128/AAC.50.3.852-861.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jackson JJ, Kropp H, Hurley JC. Influence of antibiotic class and concentration on the percentage of release of lipopolysaccharide from Escherichia coli. J Infect Dis. 1994;169:471–472. doi: 10.1093/infdis/169.2.471. [DOI] [PubMed] [Google Scholar]
- 52.Skerlavaj B, Romeo D, Gennaro R. Rapid membrane permeabilization and inhibition of vital functions of gram-negative bacteria by bactenecins. Infect Immun. 1990;58:3724–3730. doi: 10.1128/iai.58.11.3724-3730.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lehrer RI, Barton A, Ganz T. Concurrent assessment of inner and outer membrane permeabilization and bacteriolysis in E. coli by multiple-wavelength spectrophotometry. J Immunol Methods. 1988;108:153–158. doi: 10.1016/0022-1759(88)90414-0. [DOI] [PubMed] [Google Scholar]
- 54.Warshakoon HJ, Burns MR, David SA. Structure-activity relationships of antimicrobial and lipoteichoic acid-sequestering properties in polyamine sulfonamides. Antimicrob Agents Chemother. 2009;53:57–62. doi: 10.1128/AAC.00812-08. [DOI] [PMC free article] [PubMed] [Google Scholar]